

ABSTRACT
Pulmonary infections caused by Mycobacterium abscessus are emerging as a global threat, especially in cystic fibrosis patients. Further intensifying the concern of M. abscessus infection is the recent evidence of human-to-human transmission of the infection. M. abscessus is a naturally multidrug-resistant fast-growing pathogen for which pharmacological options are limited. Repurposing antitubercular drugs represents an attractive option for the development of chemotherapeutic alternatives against M. abscessus infections. Bedaquiline (BDQ), an ATP synthase inhibitor, has recently been approved for the treatment of multidrug-resistant tuberculosis. Herein, we show that BDQ has a very low MIC against a vast panel of clinical isolates. Despite being bacteriostatic in vitro, BDQ was highly efficacious in a zebrafish model of M. abscessus infection. Remarkably, a very short period of treatment was sufficient to protect the infected larvae from M. abscessus-induced killing. This was corroborated with reduced numbers of abscesses and cords, considered to be major pathophysiological signs in infected zebrafish. Mode-of-action studies revealed that BDQ triggered a rapid depletion of ATP in M. abscessus in vitro, consistent with the drug targeting the FoF1 ATP synthase. Importantly, despite a failure to select in vitro for spontaneous mutants that are highly resistant to BDQ, the transfer of single nucleotide polymorphisms leading to D29V or A64P substitutions in atpE conferred high resistance, thus resolving the target of BDQ in M. abscessus. Overall, this study indicates that BDQ is active against M. abscessus in vitro and in vivo and should be considered for clinical use against the difficult-to-manage M. abscessus pulmonary infections.
KEYWORDS: Mycobacterium abscessus, bedaquiline, zebrafish, therapeutic activity, resistance, ATP synthase, AtpE, drug resistance mechanisms
INTRODUCTION
Mycobacterium abscessus pulmonary disease is a significant cause of morbidity and mortality among patients with preexisting lung conditions, such as bronchiectasis, chronic obstructive pulmonary disease (COPD), and cystic fibrosis (CF). Whole-genome sequencing of M. abscessus in CF patients indicated human-to-human transmission of this infection (1, 2). Together with members of the Mycobacterium avium complex (MAC), M. abscessus represents the most frequent nontuberculous mycobacterium (NTM) respiratory pathogen. Unfortunately, there are no predictable or reliably effective treatment strategies for pulmonary infections caused by M. abscessus (3). MAC and M. abscessus lung diseases, similarly to multidrug-resistant tuberculosis (MDR-TB), are very difficult to treat due to limited therapeutic options, particularly when current therapy fails. The American Thoracic Society recommends a treatment regimen consisting of a combination of a macrolide (clarithromycin or azithromycin), an aminoglycoside (amikacin), and a β-lactam (cefoxitin or imipenem) for a period of 1 year (4). M. abscessus subsp. abscessus and M. abscessus subsp. bolletii possess an erm(41) RNA methylase gene that confers inducible resistance to macrolides (5). Intravenous antibiotics are an essential element for M. abscessus lung disease treatment. Unfortunately, oral medication options are limited against M. abscessus diseases. Therefore, new chemotherapeutic options, particularly those given orally, are urgently needed to improve M. abscessus treatment outcomes, particularly for clarithromycin-resistant M. abscessus lung diseases.
To alleviate the initiation of new de novo chemical screens against M. abscessus, “cross-screen” approaches based on existing data from previous TB screens have recently been applied to identify new pharmacological entities that are active against M. abscessus. This confirmed that TB drug discovery platforms could easily be exploited to screen for M. abscessus-active compounds. This led, for instance, to the identification of a piperidinol-based compound (6) or thiacetazone (TAC) derivatives (7), which exhibit potent activity against M. abscessus. However, the quest for discovering potent novel active drugs against M. abscessus could also rely on repurposing TB drugs.
Bedaquiline (BDQ; code names TMC207 and R207910), which was approved by the Food and Drug Administration and the European Medicines Agency for the treatment of MDR-TB (8), is a diarylquinoline antibiotic that acts through inhibition of the essential FoF1 ATP synthase. There is now clear evidence that BDQ targets the c subunit of the ATP synthase (9–11). Biochemical and X-ray crystallographic studies indicated that BDQ is likely to prevent the rotor ring from acting as an ion shuttle (12). Recent biochemical and genetic studies also proposed that BDQ inhibits mycobacterial F-ATP synthase via another mechanism involving the ε subunit of the enzyme in addition to binding to its c subunit (13, 14). Interestingly, BDQ has also been shown to be more effective than currently existing antimycobacterial agents in treating Mycobacterium ulcerans in a mouse model of infection and has shown promise as a salvage therapy for M. avium and M. abscessus (15, 16). Very few experimental models have been developed for rapidly growing mycobacterial infections, which hampers in vivo drug susceptibility testing. Hence, a limited number of animal models for evaluating antibiotic activity against M. abscessus infection have been reported recently, mostly based on the use of immunocompromised mice (17–21). Two independent studies that evaluated the efficacy of BDQ to reduce the M. abscessus loads in those models led to conflictual conclusions. Studies conducted in gamma interferon knockout (GKO) mice have shown a potent benefit of BDQ treatment in reducing bacterial loads (19), whereas work in nude mice failed to show a decrease in bacillary loads in the lungs and prevention of death, in contrast to cefoxitin (20). We previously reported the usefulness of zebrafish as a preclinical model to evaluate in real time the efficacy of antibiotics, particularly clarithromycin and imipenem, against M. abscessus in living infected vertebrates (22–24). This biological system can be complementary to murine models, as it permits in vivo imaging, at the spatiotemporal level, of the effects of drug treatment on the infection process.
Thus, to determine whether BDQ has a place in the management of M. abscessus infection, we investigated the potential antimicrobial efficacy of BDQ against the M. abscessus complex in vitro against M. abscessus clinical isolates, particularly from CF patients, as well as in zebrafish embryos. This study was also undertaken to determine the mode of action and the mechanism(s) of resistance of BDQ in M. abscessus. We provide evidence that, as observed in M. tuberculosis, BDQ triggers rapid ATP depletion in M. abscessus, and high levels of resistance are associated with mutations in atpE.
RESULTS
BDQ inhibits M. abscessus growth in vitro.
The exposure of exponentially growing M. abscessus to increasing concentrations of BDQ, corresponding to 2×, 8×, and 64× the MIC (0.125 μg/ml), showed important growth inhibition (Fig. 1). However, since the CFU numbers over the 4-day period of treatment were only slightly below those of the inoculum and remained constant over time, these results suggest that BDQ exerts a bacteriostatic effect in vitro against M. abscessus. Exposure to imipenem, an active β-lactam drug against M. abscessus (25), was associated with a more pronounced killing effect (Fig. 1).
FIG 1.

In vitro activity of bedaquiline. M. abscessus CIP104536T (S) was exposed either to 8 μg of imipenem (IMP) or to increasing concentrations of BDQ (from 2 to 64× the MIC) in CaMH broth at 30°C. At various time points, bacteria were plated on LB agar and further incubated at 30°C for 4 days prior to CFU determination. Results are expressed as the mean ± standard error of the mean (SEM) of triplicates and are representative of two independent experiments. UN, untreated cultures.
Activity of BDQ against M. abscessus isolates.
The activity of BDQ was next tested using a large set of clinical isolates. The M. abscessus group is classified into three groups: M. abscessus subsp. abscessus, M. abscessus subsp. bolletii, and M. abscessus subsp. massiliense, and this distinction is of clinical relevance, as these subspecies respond differently to antibiotics (26, 27). BDQ exhibited potent activity against the different strains isolated from either CF patients or non-CF patients, with MICs ranging from 0.031 to 0.125 μg/ml (Table 1). The same strains also exhibited various susceptibility profiles to imipenem, one of the most widely used drugs in clinical settings, ranging from 8 to 32 μg/ml. The smooth and rough morphotypes of M. abscessus, here referred to as S and R, respectively, were also equally sensitive to BDQ. Overall, these results demonstrate that BDQ exerts very strong activity against the M. abscessus complex, including isolates from CF patients.
TABLE 1.
Comparison of activities of BDQ and imipenem against clinical isolates from CF and non-CF patients
| Strain | Morphotype | Source | MIC (μg/ml)a |
|
|---|---|---|---|---|
| BDQ | IMP | |||
| M. abscessus subsp. abscessus | ||||
| CIP104536T | S | Non-CF | 0.062 | 16 |
| 3321 | S | Non-CF | 0.062 | 32 |
| 1298 | S | CF | 0.031 | 16 |
| 2587 | S | CF | 0.062 | 16 |
| 2069 | S | Non-CF | 0.062 | 8 |
| CF | S | CF | 0.062 | 16 |
| 2524 | R | CF | 0.125 | 32 |
| 2648 | R | CF | 0.062 | 32 |
| 3022 | R | Non-CF | 0.062 | 16 |
| 5175 | R | CF | 0.062 | 32 |
| CIP104536T | R | Non-CF | 0.062 | 16 |
| M. abscessus subsp. massiliense | ||||
| CIP108297T | R | Addison disease | 0.062 | 32 |
| 210 | R | CF | 0.125 | 32 |
| 179 | R | CF | 0.062 | 8 |
| CIP108297T | S | Addison disease | 0.062 | 32 |
| 140 | S | CF | 0.062 | 32 |
| 185 | S | CF | 0.125 | 16 |
| 107 | S | CF | 0.125 | 32 |
| 122 | S | CF | 0.125 | 16 |
| 120 | S | CF | 0.062 | 16 |
| 212 | S | CF | 0.125 | 32 |
| 100 | S | CF | 0.062 | 16 |
| 111 | S | CF | 0.062 | 16 |
| M. abscessus subsp. bolletii | ||||
| CIP108541T | S | Nonreported | 0.062 | 8 |
| 114 | S | CF | 0.125 | 16 |
| 17 | S | CF | 0.031 | 32 |
| 116 | S | CF | 0.125 | 16 |
| 97 | S | CF | 0.125 | 16 |
| 112 | R | CF | 0.062 | 16 |
| 19 | R | Non-CF | 0.125 | 32 |
| 10 | R | Nonreported | 0.062 | 32 |
| 108 | R | CF | 0.125 | 32 |
The MIC was determined in cation-adjusted Mueller-Hinton broth for different subspecies belonging to the M. abscessus complex. BDQ, bedaquiline; IMP, imipenem.
In vivo susceptibility of M. abscessus to bedaquiline.
Animal models are currently limited to study host immunity and pathogenesis unless very large doses of bacilli are given intravenously. If small doses are given, there is little evidence that a productive infection is even fully established. Consequently, better models are required to elucidate pathogenesis and to enable new drugs for M. abscessus infections to be tested. This led to the recent development of the zebrafish model to assess the suitability and sensitivity of clinically relevant drugs in M. abscessus-infected embryos (6, 21–24). Small bacterial doses can be used in this model to allow visualizing, in a dose- and time-dependent manner, the dynamics of infection and physiopathological markers, such as cords and abscesses, in the presence of an active compound (22). The injection of a small inoculum allows administration of homogenous bacterial suspensions without obstructing the needle during the microinjection procedure. Therefore, we adapted a previously designed protocol (22) to assess the activity of BDQ against M. abscessus in zebrafish larvae. Red fluorescent tdTomato-expressing M. abscessus (R variant) was injected in the caudal vein of embryos at 30 h postfertilization (hpf) and transferred to 24-well plates. BDQ was then directly added at 1 day postinfection (dpi) to the water containing the infected zebrafish, and the BDQ-supplemented water was then changed on a daily basis for 3 days. In preliminary experiments, noninfected embryos were exposed to increasing concentrations of BDQ and observed under a microscope. No signs of toxicity-induced killing or developmental abnormalities were recorded in the presence of 3 μg/ml BDQ (data not shown). When infected embryos were exposed for 3 days to the lowest (1 μg/ml) concentration of BDQ tested, a significant increased survival rate was observed compared to the untreated group of embryos and was as efficient as treatment with 360 μg/ml imipenem (Fig. 2A). Exposure to a higher dose of BDQ (3 μg/ml) further extended the life span of infected zebrafish and protected around 80% of the infected embryos at 13 dpi (Fig. 2A). This indicates that BDQ is very efficient in this zebrafish test system against M. abscessus infection. We next investigated whether reducing the duration of treatment would affect the protective efficacy of BDQ. Infected embryos were treated with 3 μg/ml BDQ for 1, 2, or 3 days, and the efficacy of the treatment was determined by monitoring the killing curves of the various treatments. Figure 2B clearly indicates that 24 or 48 h of treatment resulted in high survival rates against M. abscessus, although the percentage of survival was lower than for embryos treated for 3 days. Overall, these results suggest that short treatment with BDQ is sufficient to confer high protection levels against M. abscessus infection.
FIG 2.

Activity of BDQ against M. abscessus in infected zebrafish embryos. Survival curve of embryos infected with 50 to 270 CFU of rough M. abscessus CIP104536T expressing tdTomato without treatment (UN) or treated with 1 or 3 μg/ml BDQ or with 360 μg/ml imipenem (IMP) for 3 days (A) and with 3 μg/ml BDQ or for 1, 2, or 3 days (B). Embryos injected with PBS were used as controls for injections. Treatment started at 1 dpi. The duration of the treatment is indicated with a colored line along the x axis. Curves are representative of two independent experiments. Data were subjected to the log rank statistical test (n = 20 to 30); ***, P < 0.001. (C) Frequency of abscesses in whole untreated or drug-treated embryos was recorded at 5 dpi (50 to 270 CFU, n = 30). Data are representative of three experiments, and statistical analysis was done using Fisher's exact test; **, P < 0.01; ***, P < 0.001. (D) Frequency of cords in whole untreated or drug-treated embryos was recorded at 5 dpi (50 to 270 CFU, n = 30). Data are representative of three experiments, and statistical analysis was done using Fisher's exact test; *, P < 0.05; ***, P < 0.001. (E) Spatiotemporal visualization of the infection by M. abscessus expressing tdTomato in untreated or BDQ-treated embryos at 2, 4, and 6 dpi. The representative fluorescence and transmission overlays of whole embryos are shown (scale bar, 200 μm), with the inset displaying a cord at higher resolution (scale bar, 10 μm). White arrowheads at 2 dpi indicate individual bacilli or small bacterial clumps.
Previous studies have shown that virulence of the rough strain of M. abscessus in zebrafish is correlated with the presence of abscesses, particularly in the central nervous system, and extracellular cords, which due to their size prevent the bacilli from being phagocytosed by macrophages (28, 29). To investigate whether the high survival rates are associated with decreased pathophysiological symptoms upon drug treatment, the percentages of abscesses and cords were determined. In agreement with previous studies, exposure to imipenem was associated with a significant reduction in the numbers of abscesses (Fig. 2C) and cords (Fig. 2D) (22). Importantly, and similarly to imipenem, exposure of infected embryos to 3 μg/ml BDQ for either 24, 48, or 72 h was accompanied by a significant decrease in the frequency of abscesses (Fig. 2C) and cords (Fig. 2D). This decrease in the physiopathological signs of BDQ-treated larvae was corroborated by whole-embryo imaging (Fig. 2E).
Together, these results suggest that BDQ exerts a therapeutic effect by preventing the development of abscesses/cords and protecting the embryos from bacterial killing.
Bedaquiline inhibits ATP production in M. abscessus.
BDQ has been identified as a potent and specific inhibitor of mycobacterial ATP synthase, thereby validating oxidative phosphorylation as a target pathway for antimycobacterial drug development (9–11). Thus, we investigated whether BDQ inhibits ATP homeostasis in M. abscessus and found that BDQ induced a rapid dose-dependent depletion of the intracellular ATP pool as soon as 180 min posttreatment (Fig. 3). Under similar experimental conditions, amikacin did not have any effect on ATP synthesis, indicating that rapid ATP depletion is specific to BDQ. Comparable results were obtained in both the rough and smooth variants of M. abscessus. Thus, it can be inferred that the FoF1 ATP synthase is the primary target of BDQ in M. abscessus.
FIG 3.
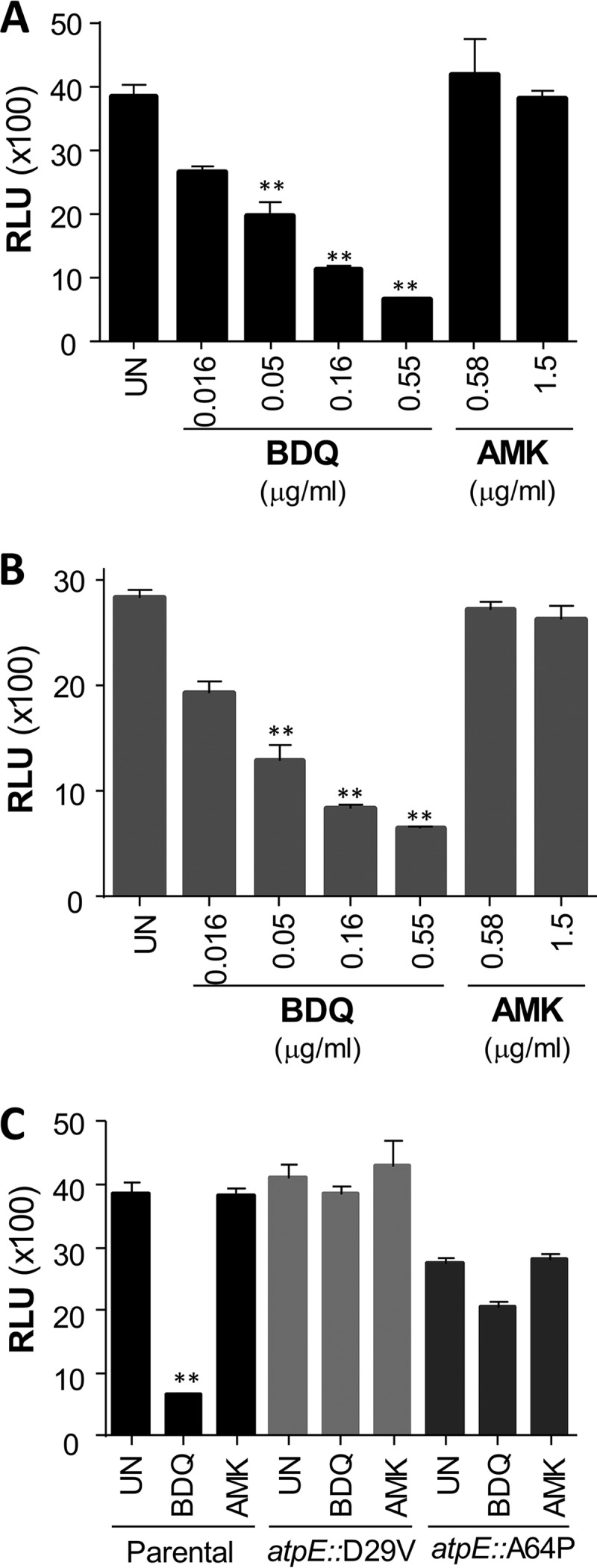
Inhibition of ATP synthesis by BDQ in M. abscessus. ATP levels were measured in the M. abscessus rough (A) and smooth (B) variants incubated with a dose range of either BDQ or amikacin (AMK) for 180 min. (C) ATP levels were determined in the parental CIP104536T smooth strain and the CIP104536T_atpE::D29V and CIP104536T_atpE::A64P derivatives treated with 0.55 μg/ml BDQ or 1.5 μg/ml AMK. Statistical analysis was performed using one-way analysis of variance (ANOVA), with Dunnett's multiple-comparison test. **, P < 0.01.
Genetic validation of AtpE as the target of BDQ in M. abscessus.
The c subunit of the ATP synthase of M. abscessus (MAB_1448) shares a very high sequence identity with its M. tuberculosis (92%) and M. smegmatis (85%) homologues, and particularly, a full conservation of the sequence in the transmembrane region where BDQ has been proposed to bind (9, 12). In addition, Ala63 and Asp32, the major residues previously shown to participate in BDQ resistance in M. tuberculosis and M. smegmatis (9, 10), respectively, are conserved in M. abscessus (Fig. 4A). To validate atpE as a specific target of BDQ, several atpE alleles were cloned under the control of the constitutive hsp60 promoter in the replicative pMV261 to allow overexpression of the wild-type (pAtpE) or mutated versions (pAtpED29V and pAtpEA64P carrying the D29V and A64P replacements, respectively) of the protein in M. abscessus. However, neither overproduction of the wild type nor of the D29V or A64P variants resulted in increased MIC values compared to the parental CIP104536T strain or the control strain carrying the empty pMV261 (Table 2). This may be explained by the fact that in these transformed strains, overexpression of the different AtpE subunits is not sufficient to titrate the drug to the extent that higher drug concentrations are needed to inhibit the ATP synthase activity. Preiss et al. hypothesized that BDQ may block rotation of the c ring at the interface between the a subunit–c-ring interface of the ATP synthase Fo motor unit, implying that binding of BDQ to only one wild-type c subunit per complex can fully inhibit ion exchange and ATP synthesis activity (12). This hypothesis is supported by our observations that overexpression of mutated alleles in a wild-type background does not result in resistance. However, the definitive determination of a clinically relevant drug target requires the ability to transfer a single point mutation to a chromosomal gene that encodes the putative drug target and to demonstrate that this transfer is sufficient to confer drug resistance. Unexpectedly, when plating the various transformed strains densely (± 108 CFU) on agar plates supplemented with BDQ at 2 μg/ml, we observed an unusually high number of colonies with apparently acquired resistance in the case of the strains transformed with pAtpED29V or with pAtpEA64P, but not with pAtpE (Fig. 4B). This was surprising given the lack of MIC upshift in liquid medium. We hypothesized that a recombinogenic event between the episomal copies of pAtpED29V (or pAtpEA64P) and the chromosomal AtpE-encoding gene may have resulted in the introduction of a single nucleotide polymorphism (SNP) leading to BDQ resistance, by way of a mechanism illustrated in Fig. 4C. To verify this assumption, the chromosomal atpE gene from 10 randomly selected colonies of each plate was PCR amplified and sequenced. This analysis revealed that, indeed, all resistant colonies had acquired an SNP conferring either a D29V or an A64P mutation. Remarkably, this genetic event occurred at a very high frequency in the presence of BDQ, since the acquisition of the SNP leading to the D29V and A64P mutations happened at a frequency of 5.6 × 10−5 to 9.4 × 10−5 and 1.1 × 10−5 to 3.5 × 10−5, respectively (Table 3). In sharp contrast, no spontaneous resistant mutants in the strain harboring the episomal copy of pAtpE were selected at 2 μg/ml, even when almost 109 CFU were plated. To further explore whether the transfer of the SNP leading to the D29V and A64P mutations in the chromosomal atpE locus is sufficient to confer resistance to BDQ, the strains were cured of their pAtpED29V and pAtpEA64P plasmids by subculturing the colonies in the absence of kanamycin and BDQ. PCR and sequencing analyses demonstrated that the plasmids were lost, and the resulting strains differed from the parental CIP104536T strain only by one SNP in atpE (Fig. 4D). Curing was further confirmed by the absence of growth on kanamycin of the two resulting strains, designated CIP104536T_atpE::D29V and CIP104536T_atpE::A64P (Fig. 4E). Importantly, these isogenic strains, but not the CIP104536T parental strain, grew in the presence of 2 μg/ml BDQ (Fig. 4E). This high resistance profile was further confirmed by their MIC of 16 μg/ml, corresponding to more than 100 times the MIC of the parental strain (Table 2). In contrast, the MIC values of clofazimine or imipenem against CIP104536T_atpE::D29V and CIP104536T_atpE::A64P were unaffected, indicating that the transfer of these mutations in atpE conferred specific resistance to BDQ. Complementation of the CIP104536T_atpE::D29V and CIP104536T_atpE::A64P strains with a wild-type copy of atpE on the pAtpE plasmid partially restored sensitivity to BDQ, while transformation of these strains with pAtpED29V and pAtpEA64P, respectively, did not (Fig. 4E), confirming BDQ sensitivity as a dominant phenotype in these merodiploid strains. Furthermore, BDQ did not induce ATP depletion in the CIP104536T_atpE::D29V and CIP104536T_atpE::A64P strains (Fig. 3C), thus confirming the BDQ resistance profile of the atpE mutant strains.
FIG 4.

Generation of M. abscessus strains that are highly resistant to BDQ through homologous recombination in the atpE locus. (A) Multiple alignment showing high amino acid identity between homologues of the ATP synthase c subunit in M. abscessus (M. abs), M. tuberculosis (M. tub), and M. smegmatis (M. smeg). The red rectangle comprises the peptide sequence of the c subunit, previously shown to interact with BDQ via numerous Van der Waals interactions. The proton-binding glutamic acid is indicated in red. Nonsynonymous mutations of the conserved D29 and A64 residues were previously identified in M. tuberculosis and M. smegmatis and shown to confer resistance in these strains probably by creating steric clashes with BDQ, preventing it from binding. (B) Selection of acquired resistance to BDQ in M. abscessus carrying the pMV261 derivatives with wild-type (pAtpE) or mutated atpE alleles (pAtpED29V and pAtpEA64P). Ten out of 10 such colonies arising from either plasmid were confirmed to have undergone homologous recombination between plasmid-borne and chromosomal atpE loci. (C) Introduction of SNPs into the chromosomal atpE locus conferring BDQ resistance is accomplished through the following steps: in step 1, M. abscessus is first transformed with a multicopy vector harboring a mutated atpE allele (red arrow). In step 2, propagation on selective medium (kanamycin) maintains several copies of this plasmid in the cytosol. In step 3, after a few passages in liquid broth, homologous exchange between the wild-type chromosomal atpE gene and the mutated plasmid-borne alleles transfers the mutation onto the chromosomal allele. In step 4, in such a cell which has undergone homologous exchange, the plasmid now harboring the wild-type atpE allele is outnumbered by multiple copies of plasmid harboring the mutated allele, and reversion of the chromosomal atpE allele to wild-type is thus expected to occur at a low frequency. Plating the bacterial suspension on agar plates containing BDQ only allows the recombinant cells to form colonies. Sequencing analysis confirms that all BDQ-resistant colonies carry the mutations originally brought by the plasmid-borne atpE in the chromosomal atpE. In step 5, serial passage of bacteria in the absence of the selective antibiotic (kanamycin) cures the bacteria of the plasmid, resulting in the presence of only a mutated atpE allele in the chromosome. (D) PCR analysis of the chromosomal and plasmid-borne copy of atpE and sequencing verification of the D29V mutation in the different strains. Top (line drawing), open and closed arrows indicate where primers used in the PCR and sequencing analysis of chromosomal atpE and plasmid-borne atpE, respectively, bind. Middle (agarose gels), an open arrow indicates the band obtained for the chromosomal atpE locus (820 bp), and a solid arrow indicates the band obtained for the plasmid-borne atpE locus (386 bp). The same results were obtained regarding the A64P mutation (not shown). Bottom, representative chromatographs obtained during sequencing analysis of wild-type atpE alleles (left) and mutated atpE alleles (right). During sequencing analysis for the presence of the mutation, careful attention was paid to verify that the junctions between the atpE coding sequence and the sequences of chromosomal or plasmid origin surrounding it were indeed present in the sequence obtained from the chromosomal atpE and plasmid-borne atpE PCR products, respectively. (E) Susceptibility/resistance profiles of the various strains studied to antibiotics. Bacterial cultures were spotted on LB medium alone or supplemented with either 100 μg/ml kanamycin or 2 μg/ml BDQ. Plates were incubated for 3 days at 37°C.
TABLE 2.
MICs of BDQ against M. abscessus S strains carrying single point mutations in atpE either on a pMV261-derived multicopy plasmid or in the chromosome
| Strain, plasmid, or mutation | MIC (μg/ml)a |
||
|---|---|---|---|
| BDQ | CFZ | IMP | |
| CIP104536T (S) | 0.06 | 0.5 | 8 |
| Multicopy plasmid | |||
| pMV261 | 0.125 | 0.5 | 8 |
| pAtpE | 0.125 | 0.5 | 8 |
| pAtpED29V | 0.125 | 1 | 8 |
| pAtpEA64P | 0.125 | 1 | 8 |
| Chromosomal mutation | |||
| atpE_D29V | 16 | 0.5 | 8 |
| atpE_A64P | 16 | 0.5 | 8 |
The MIC was determined in cation-adjusted Mueller-Hinton broth. Data are representative of two independent experiments. BDQ, bedaquiline; CFZ, clofazimine; IMP, imipenem.
TABLE 3.
Frequency of BDQ resistance in strains carrying or not a plasmid with wild-type or mutated atpE alleles
| Strain or plasmid | BDQ (2 μg/ml) |
|
|---|---|---|
| Expt 1 | Expt 2 | |
| CIP104536T (S) | <1.1 × 10−8 | <7.8 × 10−9 |
| pAtpE | <4.7 × 10−8 | <9,8 × 10−8 |
| pAtpED29V | 5.6 × 10−5 | 9.4 × 10−5 |
| pAtpEA64P | 3.5 × 10−5 | 1.1 × 10−5 |
Overall, these results indicate that the single transfer of the D29V and A64P mutations into the chromosomal atpE locus is sufficient to confer high BDQ resistance, thus validating AtpE as the primary target of BDQ in M. abscessus.
DISCUSSION
Recognized as a cause of chronic pulmonary infections, especially in individuals with altered host defenses or disrupted airway clearance mechanisms, M. abscessus appears as a major infectious threat to the airway in CF patients, and reports suggest increased prevalence in recent years (27, 30). This situation is worsened by the fact that antibiotherapy against M. abscessus is often unsuccessful and/or poorly tolerated by patients. M. abscessus is notorious for being intrinsically resistant to most antibiotics (31), thus rendering these infections particularly complicated, difficult to treat, and associated with a high rate of therapeutic failure (32). Because of the absence of new active molecules, recent studies have focused on exploring the synergy of already-available drug combinations against M. abscessus (33) or on repurposing approved drugs (34, 35). In this study, we evaluated the in vitro and in vivo activity of BDQ against M. abscessus. In agreement with a recent study addressing the activity of BDQ against several NTMs in China (36), we found that BDQ exhibited low MIC values against a collection of clinical isolates from France. The efficacy was not dependent on the bacterial morphotype (rough versus smooth) and was equal in the different M. abscessus subspecies, which is of interest since M. abscessus, M. massiliense, and M. bolletii can respond differently to some antibiotics (37).
While two previous studies have evaluated the activity of BDQ against M. abscessus in immunocompromised mice, one conducted in GKO mice showing the efficacy of BDQ treatment in reducing bacterial loads (19) and another one in nude mice with no decrease in pulmonary loads (20), we report here a robust and sustained effect of BDQ in infected zebrafish. Our results highlight the in vivo efficacy of BDQ in this animal model, allowing visualizing in a dose- and time-dependent manner the dynamics of cord and abscess formation/resorption. BDQ exerted a very strong and positive impact on embryo survival (around 80% at 13 dpi). However, treatment with BDQ did not fully abrogate the development of infection cords, which in turn initiate the formation of infection foci (28). This may ultimately lead to the killing of the remaining 20% infected embryos observed at 13 dpi, which were not protected by the treatment. The notion of cords being associated with killing has recently been emphasized by a deletion mutant of MAB_4780, encoding a dehydratase, which exhibited a pronounced defect in cording, correlating with an extremely attenuated phenotype not only in wild-type but also in immunocompromised zebrafish (38). Overall, the present study reports the usefulness of zebrafish as a preclinical model to evaluate in real time the efficacy of BDQ against M. abscessus infection in the sole context of innate immunity. Since zebrafish embryos have been successfully used in the past to test the efficacy of the two clinically relevant drugs, clarithromycin and imipenem (22), and to demonstrate the potential of a combination consisting of a β-lactam (amoxicillin or imipenem) and a β-lactamase inhibitor (avibactam) (23, 24), future studies should address the in vivo efficacies of these drugs given in combination with BDQ using the zebrafish model of M. abscessus infection.
In vitro assessments of BDQ suggest that the drug exerts a bacteriostatic effect by targeting the FoF1 ATP synthase. This is in contrast to the reported bactericidal potency of BDQ in M. tuberculosis (9). However, similar to M. abscessus, it was reported earlier that inhibition of the ATP synthase of M. avium is also not bactericidal (39). It remains to be determined why BDQ is bacteriostatic against some NTM species despite being an excellent growth inhibitor at low doses in vitro. Nevertheless, this work indicates that oxidative phosphorylation is an attractive target space for future drug development in M. abscessus, as recently proposed in the case of M. tuberculosis (52, 53). A recent high-throughput screen in mycobacterial inverted membrane vesicles and subsequent biochemical deconvolution identified a squaramide series as a new class of ATP synthase inhibitors (40). The notion that these compounds may also inhibit the growth of M. abscessus is an attractive hypothesis, which requires further investigations. In addition, inhibitors of energy metabolism may prove highly suitable in drug combination regimens due to interference with drug efflux (53). Efflux pumps have emerged recently as important determinants in drug resistance mechanisms in M. tuberculosis (41, 42), as well as in M. abscessus (7). Because a sufficiently high proton motive force and ATP levels have to be maintained for drug extrusion mechanisms, draining the energy supply of efflux pumps may represent an alternative strategy to impede drug efflux. Since BDQ blocks oxidative phosphorylation, it may indirectly interfere with efflux pump function. Because TAC analogues are actively transported by an MmpL5-like efflux pump mechanism (7), the combination of these compounds with BDQ may lead to increased intracellular levels of the TAC analogues, thereby enhancing their potency against M. abscessus.
Another major finding of this study is the demonstration that the ATP synthase represents the primary target of BDQ in M. abscessus, thanks to the construction of genetically isogenic mutant strains. We report here a powerful method to introduce single point mutations into genes encoding potential drug targets. This recombineering strategy involves transformation of a BDQ-susceptible strain with a multicopy plasmid carrying atpE and harboring the desired mutation(s) to introduce these into the chromosomal atpE locus. Maintaining the BDQ pressure allowed the selection of double homologous recombination events between the episomal and chromosomal atpE locus. Subsequent curing of the plasmid leads to an isogenic strain differing from the parental strain by only one SNP in atpE. The generation of the CIP10453T_atpE::D29V and CIP104536T _atpE::A64P isogenic mutant strains revealed that these amino acid substitutions led to high resistance to BDQ, probably because of the structural interference of these mutations with BDQ binding, as previously established by biochemical and X-ray crystallographic studies in M. tuberculosis (12). The genetic approach described here by introducing independently two single point mutations in AtpE (D29V and A64P) provides a proof of concept of a simple approach that could be applied to characterize other antimycobacterial drug targets and single point mutations and assessment of these consequences for antibiotic resistance in M. abscessus.
To gain further insights into the mechanisms of resistance to BDQ in M. abscessus, nine spontaneous resistant mutants exhibiting low levels of resistance at 4- to 8-fold the MIC to BDQ were selected and sequenced for the presence of eventual mutations in genes previously correlated with BDQ resistance in M. tuberculosis (data not shown). The genes sequenced included atpE (9, 10), atpC (13), and pepQ (43), as well as three genes encoding TetR repressors (MAB_4384, MAB_4312, and MAB_4709c) of MmpL5-like proteins (7). However, no SNPs were identified in any of these genes, suggesting the existence of additional mechanisms of resistance to BDQ in M. abscessus. Moreover, M. abscessus mutants that were highly resistant to TAC analogues (7) due to increased expression of the MmpL5-like MAB_4382c efflux system were not resistant to BDQ (data not shown). Although these results exclude MAB_4382c as an efflux pump in BDQ resistance, it remains possible that other MmpL members contribute to low BDQ resistance levels. This view is emphasized by the unusual abundance of the MmpL class of efflux pumps in the M. abscessus clade, compared to most other mycobacterial species, presumably contributing to the intrinsic resistance of M. abscessus to many antibiotics (44). Further investigations are needed to decipher whether drug efflux-mediated mechanisms participate in BDQ resistance, as proposed recently in BDQ-resistant M. tuberculosis strains through the upregulation of MmpL5 (45–47).
In summary, this work suggests that BDQ may have a place in new drug regimens for M. abscessus infections, especially in CF patients where current therapeutic outcomes are very poor. Since BDQ can be given orally, the use of this drug may therefore improve the M. abscessus treatment outcomes, particularly for clarithromycin-resistant M. abscessus lung diseases. Future studies are now required to identify new combinations of oxidative phosphorylation inhibitors for the design of completely new drug regimens against M. abscessus.
MATERIALS AND METHODS
Bacterial strains.
M. abscessus subsp. abscessus CIP104536T, M. abscessus subsp. bolletii CIP108541T, and M. abscessus subsp. massiliense CIP108297T reference strains and clinical isolates from CF and non-CF patients were reported previously (7, 33). Strains were routinely grown and maintained at 30°C in Middlebrook 7H9 broth (BD Difco) supplemented with 0.05% Tween 80 (Sigma-Aldrich) and 10% oleic acid-albumin-dextrose-catalase (OADC enrichment; BD Difco) (7H9T/OADC) or on Middlebrook 7H10 agar (BD Difco) containing 10% OADC enrichment (7H10OADC) and in the presence of antibiotics, when required. For drug susceptibility testing, bacteria were grown in cation-adjusted Mueller-Hinton broth (CaMHB; Sigma-Aldrich).
Drug susceptibility testing.
The CLSI guidelines (48) were followed to determine the MICs based on the broth microdilution method in CaMHB using an inoculum containing 5 × 106 CFU/ml in the exponential-growth phase. Bacteria (100 μl) were seeded in 96-well plates, and 2 μl of drug at its highest concentration was added to the first wells containing double the volume of bacterial suspension (200 μl). Twofold serial dilutions were then carried out, and incubation with drugs was performed at 30°C for 3 to 5 days. MICs were recorded by visual inspection and by absorbance at 560 nm to confirm visual recording. Experiments were done in triplicate on three independent occasions.
Time-kill assay.
Microtiter plates were set up as for MIC determination. Serial dilutions of the bacterial suspension were plated after 0, 24, 48, 72, and 96 h of exposure to different drug concentrations. CFU were enumerated after 4 days of incubation at 30°C.
Zebrafish care and ethics statements.
All zebrafish experiments were approved by the Direction Sanitaire et Vétérinaire de l'Hérault et Comité d'Ethique pour l'Expérimentation Animale de la Région Languedoc Roussillon under reference no. CEEA-LR-1145. Experiments were done using the golden mutant (49) crossed with wild-type AB zebrafish, maintained as described earlier (28). The ages of the embryos are expressed in hours postfertilization (hpf).
Assessment of BDQ efficacy in infected zebrafish.
Rough M. abscessus CIP104536T (ATCC 19977T) carrying pTEC27 (plasmid 30182; Addgene) and expressing the red fluorescent protein tdTomato was prepared and microinjected in zebrafish embryos, according to procedures described earlier (28, 29). Briefly, mid-log-phase cultures of M. abscessus expressing tdTomato were centrifuged, washed, and resuspended in phosphate-buffered saline (PBS) supplemented with 0.05% Tween 80 (PBS-T). Bacterial suspensions were then homogenized through a 26-gauge needle and sonicated, and the remaining clumps were allowed to settle down for 5 to 10 min. Bacteria were concentrated to an optical density at 600 nm (OD600) of 1 in PBS-T and injected intravenously (≈2 to 5 nl containing 50 to 300 CFU) into the caudal vein in 30-h-postfertilization (hpf) embryos previously dechorionated and anesthetized. To follow infection kinetics and embryo survival, infected larvae were transferred into 24-well plates (2 embryos/well) and incubated at 28.5°C. The CFU numbers in the inoculum were determined by injection of 2 nl of the bacterial suspension in sterile PBS-T and plating on 7H10 with 500 μg/ml hygromycin.
Drugs were added at 1 day postinfection (dpi) directly into the water containing the embryos. Three doses were tested, corresponding to 1, 2, and 3 μg/ml BDQ. In vivo drug efficacy was determined by following the survival of embryos as well as by monitoring the evolution of the abscesses and cords within whole embryos under the microscope (22). Survival curves were determined by recording dead embryos (no heartbeat) every day for up to 13 days.
Microscopy and image analysis.
In order to visualize the infection foci, abscesses, and cords, infected larvae were anesthetized with tricaine, positioned on 35-mm dishes, and then immobilized in 1% low-melting-point agarose and covered with water containing tricaine. Bright-field and fluorescence pictures of live infected embryos were taken with a Zeiss microscope equipped with a Zeiss Plan Neofluar Z 1×/0.25 FWD objective and an Axiocam503 monochrome (Zeiss) camera, with acquisition and processing using ZEN 2 (blue edition). The final image analysis was carried out using GIMP 2.6 to merge fluorescent and bright-field images and to adjust levels and brightness and to remove out-of-focus background fluorescence.
Selection of spontaneous resistant mutants.
Exponentially growing M. abscessus cultures were plated on 7H10OADC containing 0.5 and 1 μg/ml BDQ, corresponding to 4× and 8× the MIC, respectively. After 1 week of incubation at 30°C, single colonies were selected and grown in liquid medium and individually subjected to MIC determination and scored for resistance to BDQ.
DNA constructs and homologous recombination.
All oligonucleotides used in this study are listed in Table 4. For cloning, PCR amplifications were performed using purified M. abscessus genomic DNA and Phusion polymerase (Finnzymes, Finland). The atpE coding sequence was PCR amplified using the primers atpE_cloning_Fw and atpE_cloning_HindIII_Rev, and the product was digested with HindIII and ligated to MscI-HindIII-linearized pMV261, yielding pAtpE. To introduce D29V and A64P mutations in atpE, the overlap extension PCR method was followed (50). Briefly, the primers atpE_cloning_Fw and atpE_D29V_Rev and pAtpE as the template were first used to generate a PCR product. In parallel, the primers atpE_D29V_Fw and atpE_cloning_Rev and pAtpE were used to generate a second PCR product. Subsequently, the two PCR products were mixed in a single fresh Phusion PCR lacking any primers and cycled once with the following conditions: 40 s at 98°C, 10 min at 60°C, and 2 min at 72°C. The primers atpE_cloning_Fw and atpE_cloning_HindIII_Rev were then added, and a standard Phusion PCR cycling protocol was executed. The product was digested with HindIII and ligated to MscI-HindIII-linearized pMV261 to produce pAtpED29V. The same procedure was followed to generate pAtpEA64P using the appropriate mutagenic primers. For sequencing reactions of spontaneous BDQ-resistant strains, including the strains CIP104536T::atpED29V and CIP104536T::atpEA64P, PCRs were performed using the GoldStar ready-to-use PCR mix (Eurogentec, France), using the primers listed in Table 4 and 1 μl of a small aliquot of boiled bacterial culture as the template.
TABLE 4.
Oligonucleotides used in this study
| Primera | Sequenceb |
|---|---|
| atpE_Fw | GTAAGACCGCCGAACTCTTG |
| atpE_Rev | TGGGGATGAGGAAGTTGTTC |
| pepQ_Fw | AAGCCATTGGGTTGTGAGAG |
| pepQ_Rev | GCGGTACAGGTAGGTGGTGT |
| MAB_4312_Fw | GTTATGGGCCGACAGTAGGA |
| MAB_4312_Rev | GATGTGCTTCGGGTTGAAAT |
| MAB_4384_Fw | TTCTGAGTTGGATGTCACGGGCCGGATGA |
| MAB_4384_Rev | CTGCCACGAGATCGACGCCGCTGA |
| MAB_4709c_Fw | CTGGGTCCGAGTAGAAGCTG |
| MAB_4709c_Rev | AGATGCGAAGCGTTCTTGAT |
| atpE_cloning_Fw | TGGCGGACCCCACAATTGTTG |
| atpE_cloning_HindIII_Rev | GAGTGAAAGCTTTTAGCTGGCGCCGGGAGT |
| atpE_D29V_Fw | CCGGTATCGGTGTCGGTATCGCCGGTAA |
| atpE_D29V_Rev | TTACCGGCGATACCGACACCGATACCGG |
| atpE_A64P_Fw | GTCTGGTTGAGGCTCCGTACTTCATCAACCT |
| atpE_A64P_Rev | AGGTTGATGAAGTACGGAGCCTCAACCAGAC |
| pMV5' | CGCCCGGCCAGCGTAAGTAGC |
| pMV3' | GCCTGGCAGTCGATCGTACG |
Fw, forward; Rev, reverse.
Restriction sites are underlined, and mutated residues are in bold type.
Intracellular ATP quantification.
Intracellular ATP levels were determined using a 96-well flat-bottom plate, as described previously for M. tuberculosis (51). M. abscessus was exposed to BDQ or amikacin (negative control) and incubated for 180 min at 32°C. Twenty-five microliters of M. abscessus culture was mixed with an equal volume of the BacTiter-Glo reagent in 96-well flat-bottom white plates and incubated for 5 min in the darkness. Luminescence was detected using a BioTek Cytation 3 multimode reader, and the values obtained were plotted using GraphPad Prism 6 software.
ACKNOWLEDGMENTS
We thank B. Heym, A.-L. Roux, and J.-L. Gaillard for clinical isolates of the M. abscessus complex.
We declare no conflicts of interest.
L.K. acknowledges the support by the Fondation pour la Recherche Médicale (FRM) (DEQ20150331719). K.P. acknowledges support from the Singapore Ministry of Health's National Medical Research Council under its Cooperative Basic Research Grant (project award NMRC/CBRG/0083/2015) and the Lee Kong Chian School of Medicine, Nanyang Technological University start-up grant. The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Bryant JM, Grogono DM, Greaves D, Foweraker J, Roddick I, Inns T, Reacher M, Haworth CS, Curran MD, Harris SR, Peacock SJ, Parkhill J, Floto RA. 2013. Whole-genome sequencing to identify transmission of Mycobacterium abscessus between patients with cystic fibrosis: a retrospective cohort study. Lancet 381:1551–1560. doi: 10.1016/S0140-6736(13)60632-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bryant JM, Grogono DM, Rodriguez-Rincon D, Everall I, Brown KP, Moreno P, Verma D, Hill E, Drijkoningen J, Gilligan P, Esther CR, Noone PG, Giddings O, Bell SC, Thomson R, Wainwright CE, Coulter C, Pandey S, Wood ME, Stockwell RE, Ramsay KA, Sherrard LJ, Kidd TJ, Jabbour N, Johnson GR, Knibbs LD, Morawska L, Sly PD, Jones A, Bilton D, Laurenson I, Ruddy M, Bourke S, Bowler ICJW, Chapman SJ, Clayton A, Cullen M, Dempsey O, Denton M, Desai M, Drew RJ, Edenborough F, Evans J, Folb J, Daniels T, Humphrey H, Isalska B, Jensen-Fangel S, Jönsson B, Jones AM, et al. 2016. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science 354:751–757. doi: 10.1126/science.aaf8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Griffith DE, Aksamit T, Brown-Elliott BA, Catanzaro A, Daley C, Gordin F, Holland SM, Horsburgh R, Huitt G, Iademarco MF, Iseman M, Olivier K, Ruoss S, von Reyn CF, Wallace RJ Jr, Winthrop K, ATS Mycobacterial Diseases Subcommittee, American Thoracic Society, Infectious Disease Society of America. 2007. An official ATS/IDSA statement: diagnosis, treatment, and prevention of nontuberculous mycobacterial diseases. Am J Respir Crit Care Med 175:367–416. doi: 10.1164/rccm.200604-571ST. [DOI] [PubMed] [Google Scholar]
- 4.Floto RA, Olivier KN, Saiman L, Daley CL, Herrmann J-L, Nick JA, Noone PG, Bilton D, Corris P, Gibson RL, Hempstead SE, Koetz K, Sabadosa KA, Sermet-Gaudelus I, Smyth AR, van Ingen J, Wallace RJ, Winthrop KL, Marshall BC, Haworth CS. 2016. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of nontuberculous mycobacteria in individuals with cystic fibrosis: executive summary. Thorax 71:88–90. doi: 10.1136/thoraxjnl-2015-207983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nash KA, Brown-Elliott AB, Wallace RJ Jr. 2009. A novel gene, erm(41), confers inducible macrolide resistance to clinical isolates of Mycobacterium abscessus but is absent from Mycobacterium chelonae. Antimicrob Agents Chemother 53:1367–1376. doi: 10.1128/AAC.01275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dupont C, Viljoen A, Dubar F, Blaise M, Bernut A, Pawlik A, Bouchier C, Brosch R, Guérardel Y, Lelièvre J, Ballell L, Herrmann J-L, Biot C, Kremer L. 2016. A new piperidinol derivative targeting mycolic acid transport in Mycobacterium abscessus. Mol Microbiol 101:515–529. doi: 10.1111/mmi.13406. [DOI] [PubMed] [Google Scholar]
- 7.Halloum I, Viljoen A, Khanna V, Craig D, Bouchier C, Brosch R, Coxon G, Kremer L. 2017. Resistance to thiacetazone derivatives active against Mycobacterium abscessus involves mutations in the MmpL5 transcriptional repressor MAB_4384. Antimicrob Agents Chemother 61:e02509-16. doi: 10.1128/AAC.02509-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matteelli A, Carvalho AC, Dooley KE, Kritski A. 2010. TMC207: the first compound of a new class of potent anti-tuberculosis drugs. Future Microbiol 5:849–858. doi: 10.2217/fmb.10.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Andries K, Verhasselt P, Guillemont J, Göhlmann HWH, Neefs J-M, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, Williams P, de Chaffoy D, Huitric E, Hoffner S, Cambau E, Truffot-Pernot C, Lounis N, Jarlier V. 2005. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 10.Koul A, Dendouga N, Vergauwen K, Molenberghs B, Vranckx L, Willebrords R, Ristic Z, Lill H, Dorange I, Guillemont J, Bald D, Andries K. 2007. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat Chem Biol 3:323–324. doi: 10.1038/nchembio884. [DOI] [PubMed] [Google Scholar]
- 11.Haagsma AC, Podasca I, Koul A, Andries K, Guillemont J, Lill H, Bald D. 2011. Probing the interaction of the diarylquinoline TMC207 with its target mycobacterial ATP synthase. PLoS One 6:e23575. doi: 10.1371/journal.pone.0023575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Preiss L, Langer JD, Yildiz Ö Eckhardt-Strelau L, Guillemont JEG, Koul A, Meier T. 2015. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci Adv 1:e1500106. doi: 10.1126/sciadv.1500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biuković G, Basak S, Manimekalai MSS, Rishikesan S, Roessle M, Dick T, Rao SPS, Hunke C, Grüber G. 2013. Variations of subunit ε of the Mycobacterium tuberculosis F1Fo ATP synthase and a novel model for mechanism of action of the tuberculosis drug TMC207. Antimicrob Agents Chemother 57:168–176. doi: 10.1128/AAC.01039-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kundu S, Biukovic G, Grüber G, Dick T. 2016. Bedaquiline Targets the ε subunit of mycobacterial F-ATP synthase. Antimicrob Agents Chemother 60:6977–6979. doi: 10.1128/AAC.01291-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji B, Lefrançois S, Robert J, Chauffour A, Truffot C, Jarlier V. 2006. In vitro and in vivo activities of rifampin, streptomycin, amikacin, moxifloxacin, R207910, linezolid, and PA-824 against Mycobacterium ulcerans. Antimicrob Agents Chemother 50:1921–1926. doi: 10.1128/AAC.00052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Philley JV, Wallace RJ Jr, Benwill JL, Taskar V, Brown-Elliott BA, Thakkar F, Aksamit TR, Griffith DE. 2015. Preliminary results of bedaquiline as salvage therapy for patients with nontuberculous mycobacterial lung disease. Chest 148:499–506. doi: 10.1378/chest.14-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Groote MA, Johnson L, Podell B, Brooks E, Basaraba R, Gonzalez-Juarrero M. 2014. GM-CSF knockout mice for preclinical testing of agents with antimicrobial activity against Mycobacterium abscessus. J Antimicrob Chemother 69:1057–1064. doi: 10.1093/jac/dkt451. [DOI] [PubMed] [Google Scholar]
- 18.Ordway D, Henao-Tamayo M, Smith E, Shanley C, Harton M, Troudt J, Bai X, Basaraba RJ, Orme IM, Chan ED. 2008. Animal model of Mycobacterium abscessus lung infection. J Leukoc Biol 83:1502–1511. doi: 10.1189/jlb.1007696. [DOI] [PubMed] [Google Scholar]
- 19.Obregón-Henao A, Arnett KA, Henao-Tamayo M, Massoudi L, Creissen E, Andries K, Lenaerts AJ, Ordway DJ. 2015. Susceptibility of Mycobacterium abscessus to antimycobacterial drugs in preclinical models. Antimicrob Agents Chemother 59:6904–6912. doi: 10.1128/AAC.00459-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lerat I, Cambau E, Roth Dit Bettoni R, Gaillard J-L, Jarlier V, Truffot C, Veziris N. 2014. In vivo evaluation of antibiotic activity against Mycobacterium abscessus. J Infect Dis 209:905–912. doi: 10.1093/infdis/jit614. [DOI] [PubMed] [Google Scholar]
- 21.Bernut A, Herrmann J-L, Ordway D, Kremer L. 2017. The diverse cellular and animal models to decipher the physiopathological traits of Mycobacterium abscessus infection. Front Cell Infect Microbiol 7:100. doi: 10.3389/fcimb.2017.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernut A, Le Moigne V, Lesne T, Lutfalla G, Herrmann J-L, Kremer L. 2014. In vivo assessment of drug efficacy against Mycobacterium abscessus using the embryonic zebrafish test system. Antimicrob Agents Chemother 58:4054–4063. doi: 10.1128/AAC.00142-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dubée V, Bernut A, Cortes M, Lesne T, Dorchene D, Lefebvre A-L, Hugonnet J-E, Gutmann L, Mainardi J-L, Herrmann J-L, Gaillard J-L, Kremer L, Arthur M. 2015. β-Lactamase inhibition by avibactam in Mycobacterium abscessus. J Antimicrob Chemother 70:1051–1058. [DOI] [PubMed] [Google Scholar]
- 24.Lefebvre A-L, Le Moigne V, Bernut A, Veckerlé C, Compain F, Herrmann J-L, Kremer L, Arthur M, Mainardi J-L. 2017. Inhibition of the β-lactamase BlaMab by avibactam improves the in vitro and in vivo efficacy of imipenem against Mycobacterium abscessus. Antimicrob Agents Chemother 61:e02440-16. doi: 10.1128/AAC.02440-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lefebvre A-L, Dubée V, Cortes M, Dorchêne D, Arthur M, Mainardi J-L. 2016. Bactericidal and intracellular activity of β-lactams against Mycobacterium abscessus. J Antimicrob Chemother 71:1556–1563. doi: 10.1093/jac/dkw022. [DOI] [PubMed] [Google Scholar]
- 26.Bastian S, Veziris N, Roux A-L, Brossier F, Gaillard J-L, Jarlier V, Cambau E. 2011. Assessment of clarithromycin susceptibility in strains belonging to the Mycobacterium abscessus group by erm(41) and rrl sequencing. Antimicrob Agents Chemother 55:775–781. doi: 10.1128/AAC.00861-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Prevots DR, Shaw PA, Strickland D, Jackson LA, Raebel MA, Blosky MA, Montes de Oca R, Shea YR, Seitz AE, Holland SM, Olivier KN. 2010. Nontuberculous mycobacterial lung disease prevalence at four integrated health care delivery systems. Am J Respir Crit Care Med 182:970–976. doi: 10.1164/rccm.201002-0310OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bernut A, Herrmann J-L, Kissa K, Dubremetz J-F, Gaillard J-L, Lutfalla G, Kremer L. 2014. Mycobacterium abscessus cording prevents phagocytosis and promotes abscess formation. Proc Natl Acad Sci U S A 111:E943–E952. doi: 10.1073/pnas.1321390111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernut A, Dupont C, Sahuquet A, Herrmann J-L, Lutfalla G, Kremer L. 2015. Deciphering and imaging pathogenesis and cording of Mycobacterium abscessus in zebrafish embryos. J Vis Exp 103:e53130. doi: 10.3791/53130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martiniano SL, Nick JA, Daley CL. 2016. Nontuberculous mycobacterial infections in cystic fibrosis. Clin Chest Med 37:83–96. doi: 10.1016/j.ccm.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. 2012. Mycobacterium abscessus: a new antibiotic nightmare. J Antimicrob Chemother 67:810–818. doi: 10.1093/jac/dkr578. [DOI] [PubMed] [Google Scholar]
- 32.Ferro BE, Srivastava S, Deshpande D, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2016. Failure of the amikacin, cefoxitin, and clarithromycin combination regimen for treating pulmonary Mycobacterium abscessus infection. Antimicrob Agents Chemother 60:6374–6376. doi: 10.1128/AAC.00990-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh S, Bouzinbi N, Chaturvedi V, Godreuil S, Kremer L. 2014. In vitro evaluation of a new drug combination against clinical isolates belonging to the Mycobacterium abscessus complex. Clin Microbiol Infect 20:O1124–O1127. doi: 10.1111/1469-0691.12780. [DOI] [PubMed] [Google Scholar]
- 34.Maurer FP, Bruderer VL, Ritter C, Castelberg C, Bloemberg GV, Böttger EC. 2014. Lack of antimicrobial bactericidal activity in Mycobacterium abscessus. Antimicrob Agents Chemother 58:3828–3836. doi: 10.1128/AAC.02448-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aziz DB, Low JL, Wu M-L, Gengenbacher M, Teo JWP, Dartois V, Dick T. 2017. Rifabutin is active against Mycobacterium abscessus complex. Antimicrob Agents Chemother 61:e00155-17. doi: 10.1128/AAC.00155-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pang Y, Zheng H, Tan Y, Song Y, Zhao Y. 2017. In vitro activity of bedaquiline against nontuberculous mycobacteria in China. Antimicrob Agents Chemother 61:e02627-16. doi: 10.1128/AAC.02627-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harada T, Akiyama Y, Kurashima A, Nagai H, Tsuyuguchi K, Fujii T, Yano S, Shigeto E, Kuraoka T, Kajiki A, Kobashi Y, Kokubu F, Sato A, Yoshida S, Iwamoto T, Saito H. 2012. Clinical and microbiological differences between Mycobacterium abscessus and Mycobacterium massiliense lung diseases. J Clin Microbiol 50:3556–3561. doi: 10.1128/JCM.01175-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halloum I, Carrère-Kremer S, Blaise M, Viljoen A, Bernut A, Le Moigne V, Vilchèze C, Guérardel Y, Lutfalla G, Herrmann J-L, Jacobs WR, Kremer L. 2016. Deletion of a dehydratase important for intracellular growth and cording renders rough Mycobacterium abscessus avirulent. Proc Natl Acad Sci U S A 113:E4228–E4237. doi: 10.1073/pnas.1605477113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lounis N, Gevers T, Van den Berg J, Vranckx L, Andries K. 2009. ATP synthase inhibition of Mycobacterium avium is not bactericidal. Antimicrob Agents Chemother 53:4927–4929. doi: 10.1128/AAC.00689-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tantry SJ, Markad SD, Shinde V, Bhat J, Balakrishnan G, Gupta AK, Ambady A, Raichurkar A, Kedari C, Sharma S, Mudugal NV, Narayan A, Naveen Kumar CN, Nanduri R, Bharath S, Reddy J, Panduga V, Prabhakar KR, Kandaswamy K, Saralaya R, Kaur P, Dinesh N, Guptha S, Rich K, Murray D, Plant H, Preston M, Ashton H, Plant D, Walsh J, Alcock P, Naylor K, Collier M, Whiteaker J, McLaughlin RE, Mallya M, Panda M, Rudrapatna S, Ramachandran V, Shandil R, Sambandamurthy VK, Mdluli K, Cooper CB, Rubin H, Yano T, Iyer P, Narayanan S, Kavanagh S, Mukherjee K, Balasubramanian V, et al. 2017. Discovery of imidazo[1,2-a]pyridine ethers and squaramides as selective and potent inhibitors of mycobacterial adenosine triphosphate (ATP) synthesis. J Med Chem 60:1379–1399. doi: 10.1021/acs.jmedchem.6b01358. [DOI] [PubMed] [Google Scholar]
- 41.Adams KN, Takaki K, Connolly LE, Wiedenhoft H, Winglee K, Humbert O, Edelstein PH, Cosma CL, Ramakrishnan L. 2011. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 145:39–53. doi: 10.1016/j.cell.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gupta S, Tyagi S, Bishai WR. 2015. Verapamil increases the bactericidal activity of bedaquiline against Mycobacterium tuberculosis in a mouse model. Antimicrob Agents Chemother 59:673–676. doi: 10.1128/AAC.04019-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Almeida D, Ioerger T, Tyagi S, Li S-Y, Mdluli K, Andries K, Grosset J, Sacchettini J, Nuermberger E. 2016. Mutations in pepQ confer low-level resistance to bedaquiline and clofazimine in Mycobacterium tuberculosis. Antimicrob Agents Chemother 60:4590–4599. doi: 10.1128/AAC.00753-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Viljoen A, Dubois V, Girard-Misguich F, Blaise M, Herrmann J-L, Kremer L. 2017. The diverse family of MmpL transporters in mycobacteria: from regulation to antimicrobial developments. Mol Microbiol 104:889–904. doi: 10.1111/mmi.13675. [DOI] [PubMed] [Google Scholar]
- 45.Hartkoorn RC, Uplekar S, Cole ST. 2014. Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob Agents Chemother 58:2979–2981. doi: 10.1128/AAC.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Andries K, Villellas C, Coeck N, Thys K, Gevers T, Vranckx L, Lounis N, de Jong BC, Koul A. 2014. Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS One 9:e102135. doi: 10.1371/journal.pone.0102135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Villellas C, Coeck N, Meehan CJ, Lounis N, de Jong B, Rigouts L, Andries K. 2017. Unexpected high prevalence of resistance-associated Rv0678 variants in MDR-TB patients without documented prior use of clofazimine or bedaquiline. J Antimicrob Chemother 72:684–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Woods GL, Brown-Elliott BA, Conville PS, Desmond EP, Hall GS, Lin G, Pfyffer GE, Ridderhof JC, Siddiqi SH, Wallace RJ. 2011. Susceptibility testing of mycobacteria, nocardiae and other aerobic actinomycetes: approved standard, 2nd ed CLSI document M24-A2. Clinical and Laboratory Standards Institute, Wayne, PA. [PubMed] [Google Scholar]
- 49.Lamason RL, Mohideen M-APK, Mest JR, Wong AC, Norton HL, Aros MC, Jurynec MJ, Mao X, Humphreville VR, Humbert JE, Sinha S, Moore JL, Jagadeeswaran P, Zhao W, Ning G, Makalowska I, McKeigue PM, O'donnell D, Kittles R, Parra EJ, Mangini NJ, Grunwald DJ, Shriver MD, Canfield VA, Cheng KC. 2005. SLC24A5, a putative cation exchanger, affects pigmentation in zebrafish and humans. Science 310:1782–1786. doi: 10.1126/science.1116238. [DOI] [PubMed] [Google Scholar]
- 50.Aiyar A, Xiang Y, Leis J. 1996. Site-directed mutagenesis using overlap extension PCR, p 177–191. In Trower MK. (ed), In vitro mutagenesis protocols. Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- 51.Rao SPS, Alonso S, Rand L, Dick T, Pethe K. 2008. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 105:11945–11950. doi: 10.1073/pnas.0711697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cook GM, Hards K, Dunn E, Heikal A, Nakatani Y, Greening C, Crick DC, Fontes FL, Pethe K, Hasenoehrl E, Berney M. 2017. Oxidative phosphorylation as a target space for tuberculosis: success, caution, and future directions. Microbiol Spectr 10.1128/microbiolspec.TBTB2-0014-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bald D, Villellas C, Lu P, Koul A. 2017. Targeting energy metabolism in Mycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. mBio 8:e00272-17. doi: 10.1128/mBio.00272-17. [DOI] [PMC free article] [PubMed] [Google Scholar]