

ABSTRACT
Changes in the pharmacokinetics of piperacillin in febrile neutropenic patients have been reported to result in suboptimal exposures. This study aimed to develop a population pharmacokinetic model for piperacillin and perform dosing simulation to describe optimal dosing regimens for hematological malignancy patients with febrile neutropenia. Concentration-time data were obtained from previous prospective observational pharmacokinetic and interventional therapeutic drug monitoring studies. Nonparametric population pharmacokinetic analysis and Monte Carlo dosing simulations were performed with the Pmetrics package for R. A two-compartment model, with between-subject variability for clearance (CL), adequately described the data from 37 patients (21 males, age of 59 ± 12 years [means ± standard deviations] and weight of 77 ± 16 kg). Parameter estimates were CL of 18.0 ± 4.8 liters/h, volume of distribution of the central compartment of 14.3 ± 7.3 liters, rate constant for piperacillin distribution from the central to peripheral compartment of 1.40 ± 1.35 h−1, and rate constant for piperacillin distribution from the peripheral to central compartment of 4.99 ± 7.81 h−1. High creatinine clearance (CLCR) was associated with reduced probability of target attainment (PTA). Extended and continuous infusion regimens achieved a high PTA of >90% for an unbound concentration of piperacillin remaining above the MIC (fT>MIC) of 50%. Only continuous regimens achieved >90% PTA for 100% fT>MIC when CLCR was high. The cumulative fraction of response (FTA, for fractional target attainment) was suboptimal (<85%) for conventional regimens for both empirical and directed therapy considering 50% and 100% fT>MIC. FTA was maximized with prolonged infusions. Overall, changes in piperacillin pharmacokinetics and the consequences on therapeutic dosing requirements appear similar to those observed in intensive care patients. Guidelines should address the altered dosing needs of febrile neutropenic patients exhibiting high CLCR or with known/presumed infections from high-MIC bacteria.
KEYWORDS: febrile neutropenia, piperacillin, population pharmacokinetics
INTRODUCTION
Altered antibiotic dosing requirements in febrile neutropenic patients have been documented previously (1–3). Changes in the pharmacokinetics (PK) of hydrophilic antibiotics appear common and can result in low plasma and tissue antibiotic exposure when conventional dosing regimens are used. The drivers for altered PK of antibiotics are thought to be the pathophysiologic phenomena associated with systemic inflammation, including increased cardiac output and organ blood flow and movement of fluid into the interstitial space (3). There are also additional causes of altered PK associated with iatrogenic factors, such as high intravenous fluid loading, a common intervention in patients with hematological malignancies, which can affect drug volume of distribution (4).
In a single-dose pilot study (5), we previously described the altered PK of piperacillin in febrile neutropenic patients with hematological malignancy. We observed markedly altered PK with elevations in volume of distribution as well as clearance leading to suboptimal exposure. This was reflected in a low percentage of the dosing interval for which the unbound concentration of piperacillin remained above the MIC (fT>MIC). We observed that 10 (83%) and eight (67%) participants had less than 50% fT>MIC against Pseudomonas aeruginosa and Enterobacteriaceae, respectively.
Subsequently, we hypothesized that, given the unpredictability of PK alterations, therapeutic drug monitoring (TDM)-guided dose optimization is required to ensure adequate exposure in all patients. Accordingly, we conducted a randomized controlled study to test the utility of piperacillin TDM in febrile neutropenic patients (6). We found that TDM was able to increase the success rate of target attainment from a baseline of 19% to 73%, in contrast to a decrease from 25% to 7% in the control group. Although TDM was able to significantly improve drug exposure, there was a large delay for most patients in achieving therapeutic concentrations, and some patients never achieved the targets. The results of this study highlight the difficulty with empirical prediction of dosing requirements based simply on a measured TDM concentration which may not achieve ideal concentrations until better and quicker real-time TDM is available. To this end, a population pharmacokinetic model that is able to individualize initial dosing based on patient covariate data, e.g., renal function and/or body weight, or that can be used for Bayesian forecasting in combination with TDM would enable more accurate dosing (7). Unfortunately, there is a paucity of such models for piperacillin in adult patients with febrile neutropenia and hematological malignancies.
Therefore, the aim of this work was to develop a population PK model for piperacillin and perform dosing simulations to describe optimized dosing regimens for piperacillin-tazobactam for the treatment of febrile neutropenia.
RESULTS
Demographic and clinical data.
Data from 37 patients were used for population PK analysis. Patient demographics and clinical characteristics are presented in Table 1. PK samples during the first dosing interval were available from 12 patients, and steady-state samples during intermittent dosing were available from 25 patients. A total of 184 concentration-time data points were included in the analysis.
TABLE 1.
Characteristics of study participants
| Characteristica | n (%) or median (IQRb) |
|---|---|
| Age (yr) | 61 (53–66) |
| Sex | |
| Male | 21 (57) |
| Female | 16 (43) |
| Body mass index (kg/m2) | 25.7 (23.0–28.1) |
| Weight (kg) | 78 (63–87) |
| Creatinine clearance (ml/min/1.73 m2) | 94 (71–132) |
| Albumin (g/liter) | 27 (23–28) |
| Malignancy | |
| Acute myeloid leukemia | 14 (38) |
| Multiple myeloma | 10 (27) |
| Hodgkin's lymphoma | 4 (11) |
| Non-Hodgkin's lymphoma | 4 (11) |
| Diffuse large B-cell lymphoma | 2 (5) |
| B-lymphoblastic lymphoma | 1 (3) |
| Follicular lymphoma | 1 (3) |
| Primary CNS lymphoma | 1 (3) |
| Positive blood culture | 14 (38) |
| Organisms isolated | |
| Bacillus cereus | 2 (5) |
| Escherichia coli | 1 (3) |
| Enterobacter cloacae | 1 (3) |
| Enterobacter aerogenes | 1 (3) |
| Klebsiella pneumonia | 2 (5) |
| Pseudomonas aeruginosa | 1 (3) |
| Staphylococcus sp. | 3 (8) |
| Viridans streptococci | 3 (8) |
| Additional antibiotics | |
| Gentamicin | 36 (97) |
| Vancomycin | 4 (11) |
| Ciprofloxacin | 1 (3) |
| Metronidazole | 1 (3) |
| Trimethoprim-sulfamethoxazole | 1 (3) |
CNS, central nervous system.
IQR, interquartile range.
Pharmacokinetic model building.
The concentration-time data were adequately described by a two-compartment model with linear elimination from the central compartment and linear intercompartmental distribution. Only creatinine clearance (CLCR) resulted in significant reduction in the log likelihood ratio and showed improved model fit as assessed by goodness-of-fit plots. The lowest value of objective function and better goodness-of-fit plots (Fig. 1; see also Fig. S1 and S2 in the supplemental material) were observed when CLCR was normalized to 100 ml/min/1.73 m2. The final covariate model thus was given by the equation piperacillin clearance = TVCL × (CLCR/100), where TVCL is the typical value of clearance. Parameter estimates for the final covariate model are given in Table 2.
FIG 1.

Diagnostic plots for the final covariate model, observed versus individual-predicted (left) and population-predicted (right) concentrations.
TABLE 2.
Estimates of piperacillin pharmacokinetic parameters for the final covariate model
| Parametera | Mean (SD) | %CVb |
|---|---|---|
| CL (liters/h) | 18.02 (4.80) | 26.63 |
| V (h) | 14.30 (7.31) | 51.09 |
| Kcp (h−1) | 1.40 (1.35) | 96.28 |
| Kpc (h−1) | 4.99 (7.81) | 156.49 |
CL, clearance; V, volume of distribution of central compartment; Kcp, rate constant for piperacillin distribution from central to peripheral compartment; Kpc, rate constant for piperacillin distribution from peripheral to central compartment.
CV, coefficient of variation.
Dosing simulations.
The final covariate model was used for Monte Carlo dosing simulations. The PTA during the first 24 h for conventional intermittent dosing regimens of piperacillin for PK/pharmacodynamic (PD) targets of 50% fT>MIC and 100% fT>MIC at various levels of CLCR are presented in Fig. 2. Similar PTA was observed at steady state (data not shown).The results show that for patients with normal CLCR, the probability of attaining 50% fT>MIC is low even for MIC values as low as 1 to 2 mg/liter. The PTA is very low when CLCR is higher (140 and 160 ml/min/1.73 m2). PTA for 100% fT>MIC is low even for very low MIC values (0.125 mg/liter) with normal to high CLCR values (>100 ml/min/1.73 m2). The PTA from prolonged infusion dosing regimens is given in Tables 3 and 4. Generally, extended infusion (EI) and continuous infusion (CI) regimens achieve a high PTA (>90%) for 50% fT>MIC, except for MIC values of ≥16 mg/liter in patients with a high CLCR. However, only CI regimens achieve a high PTA for 100% fT>MIC for a wide range of renal function and MIC values.
FIG 2.

Probability of target attainment for conventional intermittent dosing regimens of piperacillin for PK/PD targets of 50% fT>MIC and 100% fT>MIC. CLCR, creatinine clearance in ml/min/1.73 m2; PTA, probability of target attainment; q8h, every 8 h intermittent infusion; q6h, every 6 h intermittent infusion.
TABLE 3.
PTA for alternative prolonged infusion dosing regimens during the first 24 ha
| Dosing regimen and CLCR (ml/min/1.73 m2) | PTA for 50% fT>MIC by MIC (mg/liter) |
PTA for 100% fT>MIC by MIC (mg/liter) |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 | 0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 | |
| 4.0-g EI over 4 h q8h | ||||||||||||||||
| 40 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | − | − |
| 60 | + | + | + | + | + | + | + | + | + | + | + | + | − | − | − | − |
| 80 | + | + | + | + | + | + | + | + | + | + | − | − | − | − | − | − |
| 100 | + | + | + | + | + | + | + | − | − | − | − | − | − | − | − | − |
| 120 | + | + | + | + | + | + | + | − | − | − | − | − | − | − | − | − |
| 140 | + | + | + | + | + | + | + | − | − | − | − | − | − | − | − | − |
| 160 | + | + | + | + | + | + | + | − | − | − | − | − | − | − | − | − |
| 4.0-g EI over 3 h q6h | ||||||||||||||||
| 40 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | − |
| 60 | + | + | + | + | + | + | + | + | + | + | + | + | + | − | − | − |
| 80 | + | + | + | + | + | + | + | + | + | + | + | + | − | − | − | − |
| 100 | + | + | + | + | + | + | + | + | + | + | + | − | − | − | − | − |
| 120 | + | + | + | + | + | + | + | + | + | − | − | − | − | − | − | − |
| 140 | + | + | + | + | + | + | + | + | − | − | − | − | − | − | − | − |
| 160 | + | + | + | + | + | + | + | − | − | − | − | − | − | − | − | − |
| 4.0-g LD + 8.0-g CI | ||||||||||||||||
| 40 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 60 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 80 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 100 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 120 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | − | − |
| 140 | + | + | + | + | + | + | − | − | + | + | + | + | + | + | − | − |
| 160 | + | + | + | + | + | + | − | − | + | + | + | + | + | + | − | − |
| 4.0-g LD + 12.0-g CI | ||||||||||||||||
| 40 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 60 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 80 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 100 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 120 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 140 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 160 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 4.0-g LD + 16.0-g CI | ||||||||||||||||
| 40 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 60 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 80 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 100 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 120 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 140 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 160 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
+, PTA of ≥0.9; −, PTA of <0.9.
TABLE 4.
PTA for alternative infusional dosing regimens at pharmacokinetic steady state measured at 48 to 72 ha
| Dosing regimen and CLCR (ml/min/1.73 m2) | PTA for 50% fT>MIC by MIC (mg/liter) |
PTA for 100% fT>MIC by MIC (mg/liter) |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 | 0.125 | 0.25 | 0.5 | 1 | 2 | 4 | 8 | 16 | |
| 4.0-g EI over 4 h q8h | ||||||||||||||||
| 40 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | − | − |
| 60 | + | + | + | + | + | + | + | + | + | + | + | + | − | − | − | − |
| 80 | + | + | + | + | + | + | + | + | + | + | − | − | − | − | − | − |
| 100 | + | + | + | + | + | + | + | − | − | − | − | − | − | − | − | − |
| 120 | + | + | + | + | + | + | + | − | − | − | − | − | − | − | − | − |
| 140 | + | + | + | + | + | + | + | − | − | − | − | − | − | − | − | − |
| 160 | + | + | + | + | + | + | − | − | − | − | − | − | − | − | − | − |
| 4.0-g EI over 3 h q6h | ||||||||||||||||
| 40 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | − |
| 60 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | − | − |
| 80 | + | + | + | + | + | + | + | + | + | + | + | + | − | − | − | − |
| 100 | + | + | + | + | + | + | + | + | + | + | − | − | − | − | − | − |
| 120 | + | + | + | + | + | + | + | − | + | − | − | − | − | − | − | − |
| 140 | + | + | + | + | + | + | + | − | − | − | − | − | − | − | − | − |
| 160 | + | + | + | + | + | + | + | − | − | − | − | − | − | − | − | − |
| 4.0-g LD + 8.0-g CI | ||||||||||||||||
| 40 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 60 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | − |
| 80 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 100 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 120 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | − | − |
| 140 | + | + | + | + | + | + | − | − | + | + | + | + | + | + | − | − |
| 160 | + | + | + | + | + | + | − | − | + | + | + | + | + | + | − | − |
| 4.0-g LD + 12.0-g CI | ||||||||||||||||
| 40 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 60 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 80 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 100 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 120 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 140 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 160 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
| 4.0-g LD + 16.0-g CI | ||||||||||||||||
| 40 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 60 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 80 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 100 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + |
| 120 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | − |
| 140 | + | + | + | + | + | + | + | + | + | + | + | + | + | + | + | − |
| 160 | + | + | + | + | + | + | + | − | + | + | + | + | + | + | + | − |
+, PTA of ≥0.9; −, PTA of <0.9.
The fraction of response (FTA, for fractional target attainment) for various dosing regimens of piperacillin against the EUCAST MIC distributions of Escherichia coli, Klebsiella pneumoniae, and Pseudomonas aeruginosa for the different simulated renal functions (CLCR) is given in Tables 5 and 6. For E. coli, the FTA for intermittent dosing regimens was suboptimal (<85%) for both empirical and directed therapy considering 50% and 100% fT>MIC, particularly at high CLCR. Both EI and CI regimens achieved high coverage at all CLCR levels for empirical and directed therapy targeting 50% fT>MIC. However, only CI achieved optimal FTA for 100% fT>MIC at all CLCR levels. For K. pneumoniae, high-dose CI (4.0-g loading dose over 1 h [LD] plus 16.0-g CI) or more frequent EI (4.0 g every 6 h [q6h] with 3-h EI) was required for empirical coverage at 50% and 100% fT>MIC. For directed therapy, all of the EI or CI regimens achieved optimal FTA for 50% fT>MIC, but only CI regimens achieved optimal FTA for 100% fT>MIC. For P. aeruginosa, none of the tested dosing regimens achieved optimal FTA for empirical therapy, but for directed therapy, EI regimens and CI regimens (except the 8-g CI) provided good coverage at 50% fT>MIC. At 100% fT>MIC, only two of the CI regimens (12.0 g and 16.0 g with 4.0-g LD) achieved optimal FTA.
TABLE 5.
FTA for various dosing regimens of piperacillin against the EUCAST MIC distributions of E. coli, K. pneumoniae, and P. aeruginosa during the first 24 h
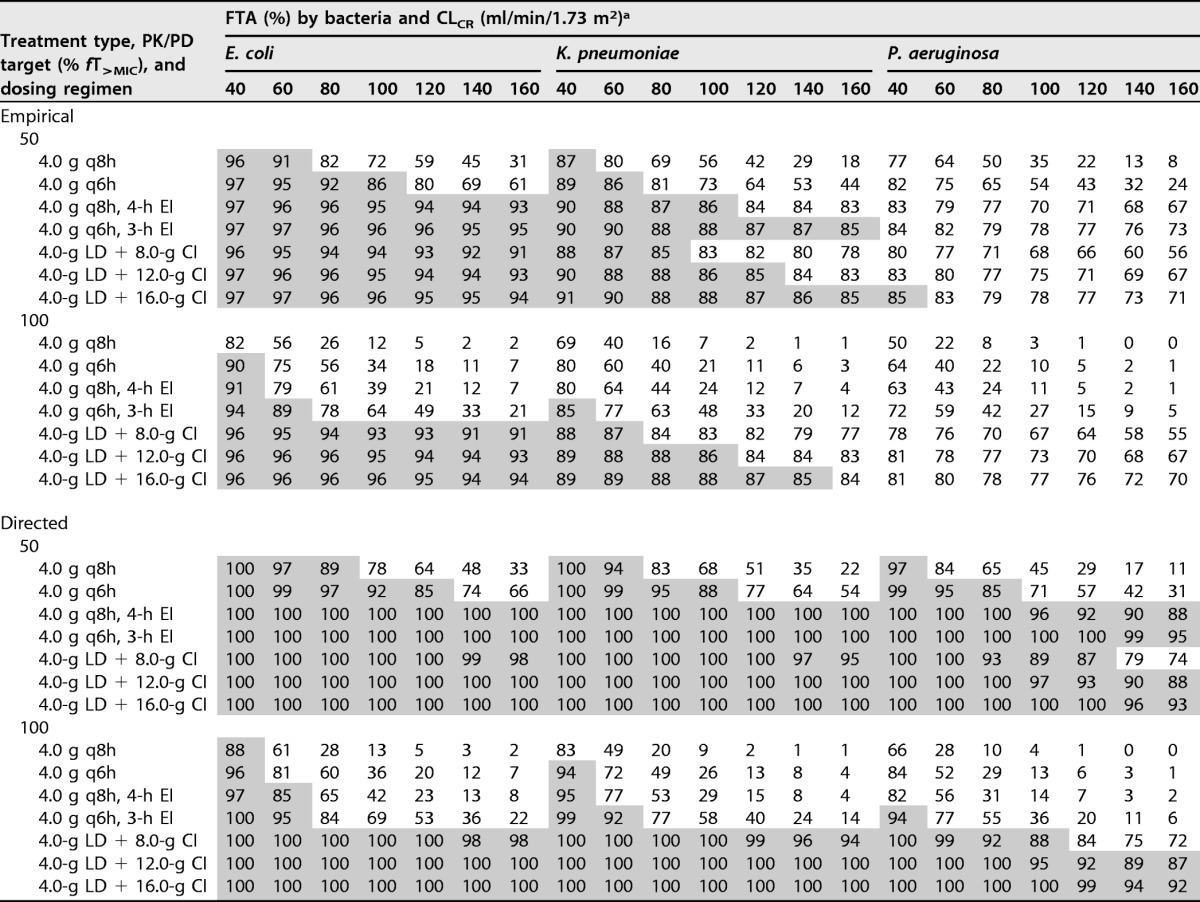
aShaded area indicates optimal FTA of greater than or equal to 85%.
TABLE 6.
Steady-state FTA for various dosing regimens of piperacillin against the EUCAST MIC distributions of E. coli, K. pneumoniae, and P. aeruginosa
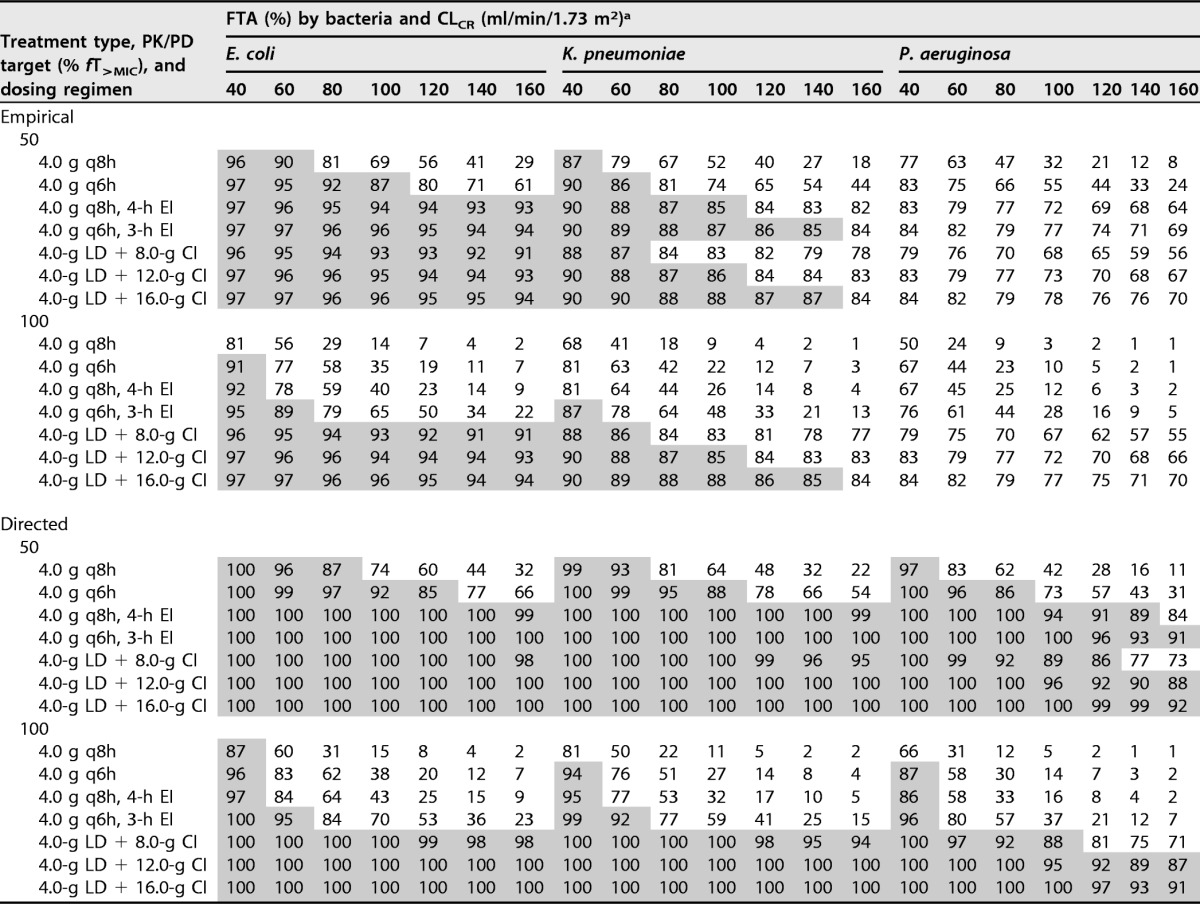
aShaded area indicates optimal FTA of greater than or equal to 85%.
Table 7 presents a comparison of the model-predicted steady-state concentrations (for continuous infusions) and steady-state trough concentrations at 72 h (for extended infusion and intermittent infusion) for a low and high simulated CLCR.
TABLE 7.
Comparison of model-predicted steady-state concentrations for low and high creatinine clearance
| CLCR (ml/min/1.73 m2) and simulated dosing regimen | Model-predicted Cssa |
||
|---|---|---|---|
| Median (mg/liters) | IQR (mg/liters) | Maximum Css/trough (mg/liters) | |
| 40 | |||
| 4.0 g q8h | 14.2 | 6.1–22.8 | 68.9 |
| 4.0 g q6h | 28 | 16.5–42.7 | 87.2 |
| 4.0 g q8h, 4-h EI | 25.8 | 5.0–39.8 | 74.8 |
| 4.0 g q6h, 3-h EI | 43.1 | 13.1–62.3 | 107.5 |
| 4.0-g LD + 8.0-g CI | 56.3 | 34.1–56.3 | 88.2 |
| 4.0-g LD + 12.0-g CI | 67.8 | 51.2–84.3 | 132.0 |
| 4.0-g LD + 16.0-g CI | 88.3 | 67.7–112.1 | 172.2 |
| 140 | |||
| 4.0 g q8h | 0.1 | BLOQ of −0.4 | 10.6 |
| 4.0 g q6h | 0.4 | 0.1–1 | 15.8 |
| 4.0 g q8h, 4-h EI | 0.591556 | 0.2–1.3 | 11.3 |
| 4.0 g q6h, 3-h EI | 1.803293 | 0.7–3.3 | 16.9 |
| 4.0-g LD + 8.0-g CI | 12.91229 | 10.8–16.1 | 25.6 |
| 4.0-g LD + 12.0-g CI | 19.36153 | 16.1–24.2 | 38.1 |
| 4.0-g LD + 16.0-g CI | 25.25549 | 21.3–32.0 | 49.5 |
Data for continuous infusion regimens are for steady-state concentration (Css) between 48 and 72 h and trough concentrations at 72 h for intermittent and extended infusion regimens. BLOQ, below the limit of quantification of the assay method for the model data (<0.1 mg/liter).
DISCUSSION
Compared to data from healthy volunteers, changes in the pharmacokinetic parameters of piperacillin and other hydrophilic antibiotics have been described in some special patient populations, such as febrile neutropenic patients receiving cancer chemotherapy (1, 3), surgical and medical intensive care unit patients (8), patients with cystic fibrosis (9), and obese patients (10). The change in primary pharmacokinetic parameters (volume of distribution and clearance) have been attributed to disease-specific physiological perturbances and iatrogenic interventions (1, 11). The estimated mean population central volume of distribution in the current study (14.3 ± 7.3 liters) is comparable with a previously reported population estimate (14.5 ± 6.6 liters) for critically ill patients (12). These values are only slightly higher than reported values for healthy individuals, e.g., 12.7 liters (13) and 10.4 liters (14). However, other population pharmacokinetic analyses have reported significant expansion of the volume of distribution in critically ill and hospitalized patients, e.g., 19.4 liters (15) and 21.7 liters (16), which appears highest in obese critically ill patients (49.0 ± 19.0 liters) (17).
On the other hand, the estimated mean population clearance of piperacillin in this analysis (18.2 ± 4.8 liters/h) is higher than that reported for healthy volunteers (10.1 liters/h [13] and 11.3 liters/h [13]. The increased clearance in this study is consistent with the study cohort which included a substantial proportion of patients with high CLCR (32% of patients had CLCR greater than 120 ml/min/1.73 m2). Indeed, clearance of piperacillin is highly dependent on the renal function of the study cohort, and this in many ways explains the different reported mean piperacillin clearances in different patient populations. In the critically ill, for instance, clearance could be elevated (e.g., 17.1 liters [18]) or similar/slightly higher (e.g., 13.8 liters/h [15, 16] and 14.0 ± 7.1 liters [17]) or may even be reduced (e.g., 3.6 liters/h [19] and 5.6 ± 3.2 liters/h [12]) compared to healthy volunteers (13). It could be particularly high in those patients with burns and trauma (20). For example, a pharmacokinetic model for burn patients predicts elevated clearance of 16 liters/h for patients when CLCR is 130 ml/min and 20 ml/min when CLCR is 160 ml/min (21). This is comparable to the elevated mean clearance observed in the current study (18.2 ± 4.8 liters/h) and, consistent with other studies, contributes to suboptimal therapeutic exposure from traditional dosing regimens.
Indeed, results of this pharmacometric analysis suggest that commonly employed intermittent piperacillin doses (4.0 g every 6 to 8 h) are highly likely to result in subtherapeutic exposures, particularly when higher-MIC Gram-negative bacteria are targeted in patients with high CLCR (Fig. 2). Although the prevalence of Gram-negative infection in febrile neutropenia is low in the developed world, e.g., 10.7% in a French hematology center (22), in other parts of the world the majority of bacteremia in these patients are attributed to Gram-negative pathogens, e.g., 60.3% in Malaysia (23) and 78.8% in Lebanon (24). Given the relatively high MIC of Gram-negative pathogens, subtherapeutic exposure is highly likely in patients with high CLCR, as observed in this study (Tables 5 to 7). Increased renal elimination of antibiotics, termed augmented renal clearance (ARC), has been described previously for piperacillin and other antibiotics predominantly eliminated by renal excretion (6, 25). In a previous trial of febrile neutropenic patients, we observed that 31% of the study cohort exhibited ARC. This is comparable to other reports for intensive care unit (ICU) patients: 33% by Ruiz et al. (26), 38.7% by Kawano et al. (27), and 28% by Campassi et al. (28). A higher incidence in ICU patients was also reported by other authors, e.g., 55.8% (29) and 65.1% (30). In patients with ARC (commonly defined as CLCR of ≥130 ml/min/1.73 m2), intermittent dosing regimens of piperacillin are highly likely to result in poor exposure (31). While this is in agreement with the findings of the current study, our results also suggest that underexposure occurs even in patients without ARC. The FTA of piperacillin-directed therapy at the traditional dosing regimen of 4.0 g every 8 h (q8h) against the common pathogens E. coli, K. pneumoniae, and P. aeruginosa was inadequate (<85%) for CLCR values as low as 100, 80, and 60 ml/min/1.73 m2, respectively, even for the conservative target 50% fT>MIC (Tables 5 and 6). Similarly, dosing with 4.0 g every 6 h also fails to provide adequate FTA for normal to high CLCR values. Importantly, both intermittent regimens show poor FTA for a wide range of low to high CLCR values if 100% fT>MIC is the desired treatment target (Tables 5 and 6). These findings are consistent with previous observations of altered antibiotic PK in febrile neutropenic patients that result in frequent failure of conventional intermittent dosing regimens to meet PK/PD dosing targets (3). Thus, the use of traditional intermittent dosing regimens of piperacillin-tazobactam in the treatment of febrile neutropenia should be critically reevaluated in clinical studies, particularly if it is to be used as a monotherapy either for directed therapy against pathogens with higher MIC breakpoints (≥8 mg/liter) or for initial empirical coverage.
On the other hand, in accordance with previous observations (2), the dosing simulations presented in this paper suggest that for optimal piperacillin exposure covering common pathogens isolated from patients with febrile neutropenia (E. coli, K. pneumoniae, and P. aeruginosa), particularly those with higher MICs, prolonged infusion dosing regimens (EI or CI) are necessary. High PTA values for 50% fT>MIC were observed for EI regimens (Tables 3 and 4) with high FTA against susceptible strains of E. coli, K. pneumoniae, and P. aeruginosa. Similar results were observed with CI regimens, except that low-dose (8-g) CI may result in underexposure against P. aeruginosa when patients have high CLCR (≥140 ml/min/1.73 m2) (Tables 5 and 6). These findings are in accordance with previous studies which reported that EI or CI regimens achieve better PK/PD exposure for the time-dependent beta-lactam antibiotics, including piperacillin (32–34).
However, the current PK analysis shows that despite the high PTA for EI or CI regimens within the susceptible MIC range, the FTA for empirical therapy, especially against the MIC distribution of P. aeruginosa, was very poor across the wide range of simulated CLCR (Tables 5 and 6). Therefore, monotherapy with piperacillin-tazobactam even with prolonged infusion is unlikely to provide consistent empirical coverage against P. aeruginosa. Similarly for K. pneumoniae, CI may not provide empirical coverage in those patients with high CLCR, even at the maximum recommended total daily doses. Although some guidelines suggest routine combination of piperacillin-tazobactam with an aminoglycoside (e.g., gentamicin), other guidelines still recommend monotherapy with conventional intermittent dosing regimens of piperacillin-tazobactam as a first-line empirical therapy for febrile neutropenia (35, 36). When culture results are available and the susceptibility of the pathogen is known, directed therapy with EI or CI regimens of piperacillin-tazobactam monotherapy could provide adequate exposure against high-MIC pathogens in the susceptible range if the conservative target of 50% fT>MIC is considered the optimal target. However, there is as yet no clear data on which PK/PD target is optimal for beta-lactams in general, although for the immunocompromised febrile neutropenic patients, studies suggested a more aggressive 100% fT>MIC is prudent. For the higher exposure of 100% fT>MIC, results of this study suggest CI is necessary to cover for high-MIC pathogens. FTA was optimal (≥85%) only for the CI and was suboptimal both for intermittent and EI regimens when 100% fT>MIC was targeted (Tables 5 and 6). Given that most beta-lactams share similar PK properties, these findings also highlight that for all beta-lactams, CI may be necessary in neutropenic patients with ARC when exposure at 100% fT>MIC is considered to maximize outcomes.
On the other hand, in patients with low creatinine clearance, CI regimens may result in high sustained steady-state concentrations (Table 7; see also Fig. S3 in the supplemental material). Although piperacillin and the beta-lactams in general have a wide safety margin, very high concentrations remain a concern due to potential neurotoxicity (37). Nevertheless, there is a lack of clearly defined cutoffs for steady-state concentrations that mark the risk of toxicity. Centers that perform beta-lactam TDM use an arbitrary cutoff of 6 to 10 times the MIC to denote concentrations above which greater effectiveness is unlikely, rather than where toxicity is more likely (i.e., toxicity is likely to be related to a concentration threshold rather than a concentration/MIC ratio) (38). Considering the EUCAST breakpoints for Enterobacteriaceae (8 mg/liter) and P. aeruginosa (16 mg/liter), this would mean concentrations as high as 80 mg/liter and 160 mg/liter, respectively. Steady-state concentrations predicted for the CI regimens when CLCR is 40 ml/min/1.73 m2 were generally within this range (Table 7). However, low-dose intermittent infusions (e.g., 4.5 g piperacillin-tazobactam every 8 hours) may be safe and provide adequate exposure in patients with low CRCL (Tables 3 to 7).
This study has several limitations. First, only total concentrations of piperacillin were available for modeling, and plasma protein binding was assumed to be similar in all patients (30%). Of note, however, patients in the study cohort did not exhibit high variability in albumin concentrations except for a slight hypoalbuminemia (5, 6). Further, a protein binding study by Wong et al. (39) showed that 30% binding is a reasonable assumption for piperacillin. Second, CLCR was calculated using the Cockcroft–Gault formula or the Jelliffe equation and was not directly measured. Mathematical equations generally provide poor estimates at extremes of CLCR and may not be optimal for accurate dosing, although they are commonly used clinically (40). Third, the current analysis provides only the effect of different dosing regimens on PK/PD exposure and not clinical outcome.
Conclusions.
The traditional intermittent dosing regimens of piperacillin-tazobactam are unlikely to provide adequate exposure for empirical management of febrile neutropenia. Subtherapeutic exposure is highly likely if Gram-negative pathogens with susceptibility close to clinical breakpoints are causative of the underlying infection. Patients with ARC are particularly vulnerable to underexposure even with the use of EI or CI regimens. Directed therapy with EI or CI regimens are highly likely to achieve adequate exposure in the majority of patients. At least for patients at high risk of subtherapeutic exposure (presence of high-MIC pathogens and/or ARC), guidelines should consider the altered dosing requirements of piperacillin-tazobactam. We suggest the use of EI or CI regimens unless TDM is performed to confirm appropriateness of exposures from intermittent regimens, particularly when used alone as a monotherapy.
MATERIALS AND METHODS
Patients and study setting.
Patient data were retrieved from a previously described prospective observational PK study (5) and a prospective interventional TDM study (6). The study population and settings were similar in both studies and included febrile neutropenic patients aged ≥18 years and undergoing treatment for hematological malignancies at The Queen Elizabeth Hospital (TQEH) in Adelaide, Australia. Febrile neutropenia was defined as the presence of a single oral temperature of ≥38.4°C (101°F) or a temperature of ≥38.0°C (100.4°F) for 1 h, with a neutrophil count of <500 cells/mm3 or a count of <1,000 cells/mm3 with a predicted decrease to <500 cells/mm3 (41). Ethics approval was granted from local institutional human research ethics committees (HREC/13/TQEHLMH/301; HREC/12/TQEHLMH/157).
Population pharmacokinetic modeling.
Piperacillin concentration-time data after intermittent dosing of piperacillin-tazobactam in the aforementioned studies (5, 6) were included in the population PK analysis. For patients who participated in both studies, only data from the prospective PK study with more frequent sampling was used. All included patients received 4.5 g piperacillin-tazobactam every 8 h. For 12 patients receiving intensive serial blood sampling, the concentration-time data were collected after the first dose: first sample at the end of line flushing (45 min) and then at 1 h, 2 h, 3 h, 4 h, 5 h, 6 h, and 7 h after the start of infusion, with a final sample just before the second dose (7.8 h). For the remaining 25 patients, blood samples were collected at steady state. Available data were included from mid-dose intervals and/or trough samples collected after the third dose (20 h and 23.8 h), sixth dose (44 h and 47.8 h), and ninth dose (71.8 h).
An R package for nonparametric adaptive grid algorithms, Pmetrics version 1.5.0, was used for pharmacometric analysis. A stepwise approach was followed in the model-building process: (i) determination of the structural base model, (ii) selection of the best-fit statistical error model, (iii) development of covariate model, and (iv) model evaluation.
Determination of the structural base model.
Different structural models, based on one, two, or three compartments, were fitted to the concentration-time data. The elimination of piperacillin from the central compartment and intercompartmental distribution were modeled as linear processes.
Selection of statistical error models.
Both the additive and multiplicative error models available in the Pmetrics package were tested. The multiplicative error model takes the form of Error = SD × γ, where SD is standard deviations of observations and γ is the process noise associated with observations. The additive error model is given by Error = (SD2 + λ2)0.5. The SD was further modeled by a second-degree polynomial function, beginning with coefficients deduced from assay data and further iterative optimization based on model diagnostics.
Development of covariate model.
Available clinical covariates were assessed for biological plausibility and subsequently evaluated in the covariate analysis. Selected covariates that were tested on the structural model parameters (volume[s] of distribution and clearance) include age, sex, weight, and creatinine clearance (calculated by the Cockcroft-Gault formula if renal function was stable or the Jelliffe equation if not; expressed in ml/min/1.73 m2). A standard covariate evaluation algorithm was followed through forward addition and backward elimination in a stepwise fashion.
Model evaluation.
Diagnostic plots and statistical examination through objective function values were used for comparison and selection of models. Diagnostic plots included scatter plots of observed concentrations versus predicted concentrations, scatter plots of residuals versus predicted concentrations, scatter plots of residuals versus time, a histogram of residuals with a test of normality (D'Agostino test), and visual predictive checks. The log-likelihood ratio test for the nested model, Akaike information criterion (AIC), and Bayesian information criterion (BIC) were each examined. In addition, model bias and imprecision were examined to discriminate between models. Model bias in Pmetrics is defined as the mean weighted error of predicted minus observed concentrations, ∑(predicted-observed/standard deviation)/N, and imprecision is defined as the bias-adjusted, mean weighted squared error of predicted minus observed concentration, i.e., ∣∑[(predicted-observed)2/(standard deviation)2]/N∣ − ∣∑(predicted-observed)/standard deviations/N∣, where N is the number of observations/predictions.
Dosing simulations.
Monte Carlo simulations (n = 1,000) were performed to determine the probability of target attainment (PTA) for different dosing regimens and renal function (CLCR) for PK/PD targets of 50% and 100% fT>MIC and 30% plasma protein binding (39). The dosing regimens were simulated up to steady state from 0 to 72 h for CLCR of 40, 60, 80, 100, 120, 140, and 160 ml/min/1.73 m2 and included 4.0-g intermittent infusion (II) over 30 min q8h, 4.0-g II over 30 min q6h, 4.0-g extended infusion (EI) over 4 h q8h, 4.0-g EI over 3 h q6h, 4.0-g loading dose over 1 h (LD) plus 8.0-g continuous infusion (CI), 4.0-g LD plus 12-g CI, and 4.0-g LD plus 16.0-g CI. PTA was determined at two stages of therapy, during the first 24 h and at steady state from 48 h to 72 h.
The fractional target attainment (FTA), during the first 24 h and at steady state from 48 h to 72 h, was calculated for Escherichia coli, Klebsiella pneumoniae, and P. aeruginosa based on the MIC distribution of the European Committee for Antimicrobial Susceptibility and Testing (EUCAST) database (available at www.eucast.org). FTA describes the proportion of the bacterial population for which the selected PK/pharmacodynamics (PD) target is attained given the Monte Carlo simulation and the MIC distribution. FTA was calculated both for empirical therapy, i.e., considering all of the categories of the entire MIC distribution, and for directed therapy, i.e., considering those categories of MIC distribution within the susceptibility range defined by clinical breakpoints (8 mg/liter for Enterobacteriaceae and 16 mg/liter for P. aeruginosa). Doses were considered optimal if the FTA was greater than 85%.
Supplementary Material
ACKNOWLEDGMENTS
F.B.S. acknowledges funding from a University of Queensland Postdoctoral Fellowship. J.A.R. recognizes funding from the Australian National Health and Medical Research Council for a Centre of Research Excellence (APP1099452) and a Practitioner Fellowship (APP1117065).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00311-17.
REFERENCES
- 1.Theuretzbacher U. 2012. Pharmacokinetic and pharmacodynamic issues for antimicrobial therapy in patients with cancer. Clin Infect Dis 54:1785–1792. doi: 10.1093/cid/cis210. [DOI] [PubMed] [Google Scholar]
- 2.Abbott IJ, Roberts JA. 2012. Infusional beta-lactam antibiotics in febrile neutropenia: has the time come? Curr Opin Infect Dis 25:619–625. doi: 10.1097/QCO.0b013e32835915c2. [DOI] [PubMed] [Google Scholar]
- 3.Lortholary O, Lefort A, Tod M, Chomat AM, Darras-Joly C, Cordonnier C, Club de Reflexion sur les Infections en Onco-Hematologie. 2008. Pharmacodynamics and pharmacokinetics of antibacterial drugs in the management of febrile neutropenia. Lancet Infect Dis 8:612–620. doi: 10.1016/S1473-3099(08)70228-7. [DOI] [PubMed] [Google Scholar]
- 4.McBride A, Westervelt P. 2012. Recognizing and managing the expanded risk of tumor lysis syndrome in hematologic and solid malignancies. J Hematol Oncol 5:75. doi: 10.1186/1756-8722-5-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sime FB, Roberts MS, Warner MS, Hahn U, Robertson TA, Yeend S, Phay A, Lehman S, Lipman J, Peake SL, Roberts JA. 2014. Altered pharmacokinetics of piperacillin in febrile neutropenic patients with hematological malignancy. Antimicrob Agents Chemother 58:3533–3537. doi: 10.1128/AAC.02340-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sime FB, Roberts MS, Tiong IS, Gardner JH, Lehman S, Peake SL, Hahn U, Warner MS, Roberts JA. 2015. Can therapeutic drug monitoring optimize exposure to piperacillin in febrile neutropenic patients with haematological malignancies? A randomized controlled trial. J Antimicrob Chemother 70:2369–2375. doi: 10.1093/jac/dkv123. [DOI] [PubMed] [Google Scholar]
- 7.Felton TW, Roberts JA, Lodise TP, Van Guilder M, Boselli E, Neely MN, Hope WW. 2014. Individualization of piperacillin dosing for critically ill patients: dosing software to optimize antimicrobial therapy. Antimicrob Agents Chemother 58:4094–4102. doi: 10.1128/AAC.02664-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts JA, Abdul-Aziz MH, Lipman J, Mouton JW, Vinks AA, Felton TW, Hope WW, Farkas A, Neely MN, Schentag JJ, Drusano G, Frey OR, Theuretzbacher U, Kuti JL, International Society of Anti-Infective Pharmacology and the Pharmacokinetics and Pharmacodynamics Study Group of the European Society of Clinical Microbiology and Infectious Diseases. 2014. Individualised antibiotic dosing for patients who are critically ill: challenges and potential solutions. Lancet Infect Dis 14:498–509. doi: 10.1016/S1473-3099(14)70036-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Touw DJ. 1998. Clinical pharmacokinetics of antimicrobial drugs in cystic fibrosis. Pharm World Sci 20:149–160. doi: 10.1023/A:1008634911114. [DOI] [PubMed] [Google Scholar]
- 10.Alobaid AS, Hites M, Lipman J, Taccone FS, Roberts JA. 2016. Effect of obesity on the pharmacokinetics of antimicrobials in critically ill patients: a structured review. Int J Antimicrob Agents 47:259–268. doi: 10.1016/j.ijantimicag.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Blot SI, Pea F, Lipman J. 2014. The effect of pathophysiology on pharmacokinetics in the critically ill patient–concepts appraised by the example of antimicrobial agents. Adv Drug Deliv Rev 77:3–11. doi: 10.1016/j.addr.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 12.Tsai D, Stewart P, Goud R, Gourley S, Hewagama S, Krishnaswamy S, Wallis SC, Lipman J, Roberts JA. 2016. Pharmacokinetics of piperacillin in critically ill australian indigenous patients with severe sepsis. Antimicrob Agents Chemother 60:7402–7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bulitta JB, Kinzig M, Jakob V, Holzgrabe U, Sorgel F, Holford NH. 2010. Nonlinear pharmacokinetics of piperacillin in healthy volunteers–implications for optimal dosage regimens. Br J Clin Pharmacol 70:682–693. doi: 10.1111/j.1365-2125.2010.03750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bulitta JB, Duffull SB, Kinzig-Schippers M, Holzgrabe U, Stephan U, Drusano GL, Sorgel F. 2007. Systematic comparison of the population pharmacokinetics and pharmacodynamics of piperacillin in cystic fibrosis patients and healthy volunteers. Antimicrob Agents Chemother 51:2497–2507. doi: 10.1128/AAC.01477-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li C, Kuti JL, Nightingale CH, Mansfield DL, Dana A, Nicolau DP. 2005. Population pharmacokinetics and pharmacodynamics of piperacillin/tazobactam in patients with complicated intra-abdominal infection. J Antimicrob Chemother 56:388–395. doi: 10.1093/jac/dki243. [DOI] [PubMed] [Google Scholar]
- 16.Chen R, Qian Q, Sun MR, Qian CY, Zou SL, Wang ML, Wang LY. 2016. Population pharmacokinetics and pharmacodynamics of piperacillin/tazobactam in patients with nosocomial infections. Eur J Drug Metab Pharmacokinet 41:363–372. doi: 10.1007/s13318-015-0276-3. [DOI] [PubMed] [Google Scholar]
- 17.Alobaid AS, Wallis SC, Jarrett P, Starr T, Stuart J, Lassig-Smith M, Mejia JL, Roberts MS, Roger C, Udy AA, Lipman J, Roberts JA. 2017. Population pharmacokinetics of piperacillin in nonobese, obese, and morbidly obese critically ill patients. Antimicrob Agents Chemother 61:e01276-16. doi: 10.1128/AAC.01276-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts JA, Kirkpatrick CM, Roberts MS, Dalley AJ, Lipman J. 2010. First-dose and steady-state population pharmacokinetics and pharmacodynamics of piperacillin by continuous or intermittent dosing in critically ill patients with sepsis. Int J Antimicrob Agents 35:156–163. doi: 10.1016/j.ijantimicag.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 19.Obrink-Hansen K, Juul RV, Storgaard M, Thomsen MK, Hardlei TF, Brock B, Kreilgaard M, Gjedsted J. 2015. Population pharmacokinetics of piperacillin in the early phase of septic shock: does standard dosing result in therapeutic plasma concentrations? Antimicrob Agents Chemother 59:7018–7026. doi: 10.1128/AAC.01347-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Udy AA, Roberts JA, Shorr AF, Boots RJ, Lipman J. 2013. Augmented renal clearance in septic and traumatized patients with normal plasma creatinine concentrations: identifying at-risk patients. Crit Care 17:R35. doi: 10.1186/cc12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jeon S, Han S, Lee J, Hong T, Paek J, Woo H, Yim DS. 2014. Population pharmacokinetic analysis of piperacillin in burn patients. Antimicrob Agents Chemother 58:3744–3751. doi: 10.1128/AAC.02089-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cordonnier C, Herbrecht R, Buzyn A, Leverger G, Leclercq R, Nitenberg G, Bastuji-Garin S, Club de Réflexion sur les Infections en Onco-Hématologie Group . 2005. Risk factors for Gram-negative bacterial infections in febrile neutropenia. Haematologica 90:1102–1109. [PubMed] [Google Scholar]
- 23.Baskaran ND, Gan GG, Adeeba K, Sam IC. 2007. Bacteremia in patients with febrile neutropenia after chemotherapy at a university medical center in Malaysia. Int J Infect Dis 11:513–517. doi: 10.1016/j.ijid.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 24.Kanafani ZA, Dakdouki GK, El-Chammas KI, Eid S, Araj GF, Kanj SS. 2007. Bloodstream infections in febrile neutropenic patients at a tertiary care center in Lebanon: a view of the past decade. Int J Infect Dis 11:450–453. doi: 10.1016/j.ijid.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Udy AA, Roberts JA, De Waele JJ, Paterson DL, Lipman J. 2012. What's behind the failure of emerging antibiotics in the critically ill? Understanding the impact of altered pharmacokinetics and augmented renal clearance. Int J Antimicrob Agents 39:455–457. [DOI] [PubMed] [Google Scholar]
- 26.Ruiz S, Minville V, Asehnoune K, Virtos M, Georges B, Fourcade O, Conil JM. 2015. Screening of patients with augmented renal clearance in ICU: taking into account the CKD-EPI equation, the age, and the cause of admission. Ann Intensive Care 5:49. doi: 10.1186/s13613-015-0090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kawano Y, Morimoto S, Izutani Y, Muranishi K, Kaneyama H, Hoshino K, Nishida T, Ishikura H. 2016. Augmented renal clearance in Japanese intensive care unit patients: a prospective study. J Intensive Care 4:62. doi: 10.1186/s40560-016-0187-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Campassi ML, Gonzalez MC, Masevicius FD, Vazquez AR, Moseinco M, Navarro NC, Previgliano L, Rubatto NP, Benites MH, Estenssoro E, Dubin A. 2014. Augmented renal clearance in critically ill patients: incidence, associated factors and effects on vancomycin treatment. Rev Bras Ter Intensiva 26:13–20. doi: 10.5935/0103-507X.20140003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.De Waele JJ, Dumoulin A, Janssen A, Hoste EA. 2015. Epidemiology of augmented renal clearance in mixed ICU patients. Minerva Anestesiol 81:1079–1085. [PubMed] [Google Scholar]
- 30.Udy AA, Baptista JP, Lim NL, Joynt GM, Jarrett P, Wockner L, Boots RJ, Lipman J. 2014. Augmented renal clearance in the ICU: results of a multicenter observational study of renal function in critically ill patients with normal plasma creatinine concentrations. Crit Care Med 42:520–527. doi: 10.1097/CCM.0000000000000029. [DOI] [PubMed] [Google Scholar]
- 31.Udy AA, Varghese JM, Altukroni M, Briscoe S, McWhinney BC, Ungerer JP, Lipman J, Roberts JA. 2012. Subtherapeutic initial beta-lactam concentrations in select critically ill patients: association between augmented renal clearance and low trough drug concentrations. Chest 142:30–39. doi: 10.1378/chest.11-1671. [DOI] [PubMed] [Google Scholar]
- 32.Roberts JA, Abdul-Aziz MH, Davis JS, Dulhunty JM, Cotta MO, Myburgh J, Bellomo R, Lipman J. 2016. Continuous versus intermittent beta-lactam infusion in severe sepsis: a meta-analysis of individual patient data from randomized trials. Am J Respir Crit Care Med 194:681–691. doi: 10.1164/rccm.201601-0024OC. [DOI] [PubMed] [Google Scholar]
- 33.Abdul-Aziz MH, Sulaiman H, Mat-Nor MB, Rai V, Wong KK, Hasan MS, Abd Rahman AN, Jamal JA, Wallis SC, Lipman J, Staatz CE, Roberts JA. 2016. Beta-lactam infusion in severe sepsis (BLISS): a prospective, two-centre, open-labelled randomised controlled trial of continuous versus intermittent beta-lactam infusion in critically ill patients with severe sepsis. Intensive Care Med 42:1535–1545. doi: 10.1007/s00134-015-4188-0. [DOI] [PubMed] [Google Scholar]
- 34.De Waele J, Carlier M, Hoste E, Depuydt P, Decruyenaere J, Wallis SC, Lipman J, Roberts JA. 2014. Extended versus bolus infusion of meropenem and piperacillin: a pharmacokinetic analysis. Minerva Anestesiol 80:1302–1309. [PubMed] [Google Scholar]
- 35.Freifeld AG, Bow EJ, Sepkowitz KA, Boeckh MJ, Ito JI, Mullen CA, Raad II, Rolston KV, Young JA, Wingard JR, Infectious Diseases Society of America. 2011. Clinical practice guideline for the use of antimicrobial agents in neutropenic patients with cancer: 2010 update by the Infectious Diseases Society of America. Clin Infect Dis 52:e56–93. doi: 10.1093/cid/cir073. [DOI] [PubMed] [Google Scholar]
- 36.Averbuch D, Orasch C, Cordonnier C, Livermore DM, Mikulska M, Viscoli C, Gyssens IC, Kern WV, Klyasova G, Marchetti O, Engelhard D, Akova M. 2013. European guidelines for empirical antibacterial therapy for febrile neutropenic patients in the era of growing resistance: summary of the 2011 4th European Conference on Infections in Leukemia. Haematologica 98:1826–1835. doi: 10.3324/haematol.2013.091025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beumier M, Casu GS, Hites M, Wolff F, Cotton F, Vincent JL, Jacobs F, Taccone FS. 2015. Elevated beta-lactam concentrations associated with neurological deterioration in ICU septic patients. Minerva Anestesiol 81:497–506. [PubMed] [Google Scholar]
- 38.Wong G, Brinkman A, Benefield RJ, Carlier M, De Waele JJ, El Helali N, Frey O, Harbarth S, Huttner A, McWhinney B, Misset B, Pea F, Preisenberger J, Roberts MS, Robertson TA, Roehr A, Sime FB, Taccone FS, Ungerer JP, Lipman J, Roberts JA. 2014. An international, multicentre survey of beta-lactam antibiotic therapeutic drug monitoring practice in intensive care units. J Antimicrob Chemother 69:1416–1423. doi: 10.1093/jac/dkt523. [DOI] [PubMed] [Google Scholar]
- 39.Wong G, Briscoe S, Adnan S, McWhinney B, Ungerer J, Lipman J, Roberts JA. 2013. Protein binding of beta-lactam antibiotics in critically ill patients: can we successfully predict unbound concentrations? Antimicrob Agents Chemother 57:6165–6170. doi: 10.1128/AAC.00951-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sime FB, Udy AA, Roberts JA. 2015. Augmented renal clearance in critically ill patients: etiology, definition and implications for beta-lactam dose optimization. Curr Opin Pharmacol 24:1–6. doi: 10.1016/j.coph.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 41.Hughes WT, Armstrong D, Bodey GP, Bow EJ, Brown AE, Calandra T, Feld R, Pizzo PA, Rolston KV, Shenep JL, Young LS. 2002. 2002 guidelines for the use of antimicrobial agents in neutropenic patients with cancer. Clin Infect Dis 34:730–751. doi: 10.1086/339215. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.