

ABSTRACT
Dabigatran etexilate (DE) is a P-glycoprotein (P-gp) probe substrate, and its active anticoagulant moiety, dabigatran, is a substrate of the multidrug and toxin extrusion protein-1 (MATE-1) transporter. The antiretroviral pharmacokinetic enhancers, ritonavir and cobicistat, inhibit both these transporters. Healthy volunteers received single doses of DE at 150 mg alone, followed by ritonavir at 100 mg or cobicistat at 150 mg daily for 2 weeks. DE was then given 2 h before ritonavir or cobicistat. One week later, DE was given simultaneously with ritonavir or cobicistat. No significant increases in dabigatran pharmacokinetic (PK) exposure or thrombin time (TT) measures were observed with the simultaneous administration of ritonavir. Separated administration of ritonavir resulted in a mean decrease in dabigatran PK exposure of 29% (90% confidence interval [CI], 18 to 40%) but did not significantly change TT measures. However, cobicistat increased dabigatran PK exposure (area under the concentration-versus-time curve from time zero to infinity and maximum plasma concentration) by 127% each (90% CI, 81 to 173% and 59 to 196%, respectively) and increased TT measures (33% for the area-under-the-effect curve from time zero to 24 h [90% CI, 22 to 44%] and 51% for TT at 24 h [90% CI, 22 to 78%]) when given simultaneously with dabigatran. Similar increases were observed when cobicistat was administered separately by 2 h from the administration of dabigatran. In all comparisons, no significant increase in the dabigatran elimination half-life was observed. Therefore, it is likely safe to coadminister ritonavir with DE, while there is a potential need for reduced dosing and prudent clinical monitoring with the coadministration of cobicistat due to the greater net inhibition of intestinal P-gp transport and increased bioavailability. (This study has been registered at ClinicalTrials.gov under identifier NCT01896622.)
KEYWORDS: cobicistat, ritonavir, dabigatran, P-gp, MATE-1, transporters, pharmacokinetics, pharmacodynamics, antiretroviral, anticoagulants, boosting agents, pharmacokinetic enhancers, DOACs, thrombin time, drug-drug interaction, ABC transporters, antiretroviral agents, drug transport
INTRODUCTION
With the introduction of potent combination antiretroviral therapy, human immunodeficiency virus (HIV) infection has transformed from a terminal illness to a chronic, treatable condition. Hence, 50% of the current HIV-infected population is estimated to be older than 50 years of age (1–4). Drug interactions between antiretroviral agents and medications commonly used to treat comorbid conditions associated with aging are in need of further investigation. Potential interactions with anticoagulant medications are of particular concern, considering that many patients require short-term or chronic anticoagulation to prevent systemic embolism. Moreover, HIV infection itself has become recognized as a condition characterized by a hypercoagulable state and premature immunologic aging. Studies have shown that thromboembolic events may be as much as 10 times more prevalent in HIV-infected patients than in the general population, and even nonelderly HIV-infected individuals remain at a higher risk (5–11).
Until recently, the only available oral anticoagulants were vitamin K antagonists (VKAs), such as warfarin. Warfarin presents various clinical challenges in attainment and maintenance of an appropriate therapeutic index. Warfarin is metabolized by a variety of cytochrome P450 enzymes and is subject to extensive drug-drug interactions with several common antiretroviral medications (12, 13). Dabigatran, administered as the inactive prodrug dabigatran etexilate (Pradaxa), was the first of the direct oral anticoagulants (DOACs) approved by the Food and Drug Administration (FDA), which was in 2010, and is indicated for the prevention of stroke and systemic embolism (14). Dabigatran is also the first DOAC to have an FDA-approved reversal agent, idarucizumab (Praxbind), which is a humanized antibody fragment that potently and selectively binds to dabigatran and its metabolites to neutralize their anticoagulant effect.
Following oral absorption, dabigatran etexilate is rapidly and completely hydrolyzed by esterases via 2 intermediate metabolites to its active dabigatran moiety. Dabigatran then competitively binds to thrombin, inhibiting the conversion of fibrinogen into fibrin during the coagulation cascade (14). As an advantage over warfarin and many other DOACs, neither dabigatran etexilate nor dabigatran is a substrate, inhibitor, or inducer of cytochrome P450 (CYP450) enzymes (14). However, dabigatran etexilate is a highly sensitive probe substrate of the efflux transporter P-glycoprotein (P-gp), defined as a drug that experiences a ≥2-fold increase in exposure upon coadministration with the P-gp inhibitors verapamil or quinidine, which are commonly used in drug interaction studies (15). P-gp is highly expressed on the apical surface of enterocytes and functions to limit the intestinal permeability of several drugs through active extrusion into the intestinal lumen enterocytes (14).
Ritonavir and cobicistat are pharmacokinetic (PK) enhancers used in combination with certain antiretroviral agents to increase their exposure. Historically, ritonavir has been used as a mainstay in combination antiretroviral therapy as a PK enhancer for protease inhibitors due to its inhibitory effects on CYP3A4, which allows less frequent dosing and improved adherence (16, 17). Cobicistat, a newer PK enhancer without antiviral activity, is now being increasingly used in a number of FDA-approved coformulated fixed-dose antiretroviral products. When it is dosed at 150 mg once daily, cobicistat provides levels of exposure of various antiretroviral substrates bioequivalent to those of ritonavir dosed at 100 mg once daily, resulting in similar rates of HIV virologic suppression (18–21). Cobicistat also exhibits key properties thought to reduce the potential for collateral drug interactions, including more selective inhibition of CYP3A and a lack of induction of hepatic metabolizing enzymes, in contrast to ritonavir's more promiscuous mixed inhibitory and inductive effects on various CYP enzymes (1A2, 2B6, 2C8, 2C9, and 2C19) and UDP-glucuronosyltransferase (UGT) metabolizing enzymes (20, 22).
Inhibition of P-gp-mediated efflux can serve as a secondary mechanism by which PK enhancers can increase the intestinal absorption of these antiretroviral substrates. However, this may also result in potentially significant drug-drug interactions with other P-gp substrates, such as dabigatran etexilate (23–28). Additionally, dabigatran is primarily renally eliminated by glomerular filtration (∼80%). Thus, coadministration with ritonavir or cobicistat may further serve to increase dabigatran concentrations due to their inhibition of the multidrug and toxin extrusion protein-1 (MATE-1) transporter, which is involved in the tubular secretion of creatinine (18, 29). Whereas ritonavir and cobicistat share similar 50% inhibitory concentrations (IC50) for P-gp (∼36 ± 10 μM) and MATE-1 (∼1.34 to 1.87 μM), cobicistat is actively transported into tubular cells by organic cation transporter 2 (OCT2) (18, 29). This active transport may lead to greater accumulation of cobicistat into tubular cells and increased MATE-1 inhibition in vivo compared with the results achieved with ritonavir (29). Therefore, we undertook the current investigation to determine whether coadministration of ritonavir or cobicistat with dabigatran etexilate increases the PK exposure and pharmacodynamic (PD) effects of dabigatran in healthy volunteers and, if so, whether separating the administration of the drugs would circumvent or significantly mitigate this interaction, as has been previously demonstrated with verapamil (28). The results from the cobicistat arm of the study were previously reported in abbreviated form (30). Here, the full PK and PD results and comparison of the results obtained with both of these PK enhancers are reported in further detail.
(Some of the data contained in this report were previously presented in abstract form at the 16th International Workshop on Clinical Pharmacology of HIV & Hepatitis Therapy, May 2014, Washington, DC; at the annual Conference on Retroviruses and Opportunistic Infections in February 2016 in Boston, MA, and February 2017 in Seattle, WA; and in a publication in the journal Circulation.)
RESULTS
Study population.
Thirty-four subjects (n = 16 in arm A and n = 18 in arm B) completed the study at least through phase 2. In arm B (cobicistat), two participants did not complete phase 3 of the study due to an unrelated illness (viral conjunctivitis, sore throat), resulting in the use of concomitant medications in one subject and a failure to follow medication administration and protocol instructions in the other subject. All data from phase 1 and phase 2 for these participants were included in the analysis. The baseline characteristics of the participants in the two study arms did not differ significantly, except that males comprised 56% of the participants in arm A and 72% of the participants in arm B. The average age of the participants was 39 ± 11 years in arm A and 37 ± 9 years in arm B. The average weight and body mass index for participants in both arms were 83.9 ± 30.5 kg and 28.4 ± 9.2 kg/m2, respectively.
Pharmacokinetics.
Dabigatran PK parameter values and geometric mean ratios (GMRs) are displayed in Tables 1 and 2, and the concentration-versus-time profiles for dabigatran in arm A (ritonavir) and arm B (cobicistat) are shown in Fig. 1. In arm A, there were no statistically significant changes in any of the dabigatran PK parameter values when dabigatran and ritonavir were administered simultaneously compared to the values obtained when dabigatran was administered alone. However, there were statistically significant and similar decreases in the dabigatran area under the concentration-versus-time curve (AUC) from time zero to infinity (AUC0–∞) and the maximum plasma concentration (Cmax) by 29% (P < 0.05) and 27% (P < 0.05), respectively, when dabigatran and ritonavir were administered separately (by 2 h) compared to the values obtained when dabigatran was administered alone (phase 2 versus phase 1, respectively). The dabigatran half-life (t1/2) during phase 2 was not significantly decreased from that during phase 1. However, the time to reach Cmax (Tmax) was decreased by 27% (P < 0.05) and the apparent oral clearance (CL/F) and the volume of distribution (V/F) were increased by 37% and 48%, respectively (P < 0.05) (Table 1).
TABLE 1.
Geometric mean values of dabigatran pharmacokinetic parameters in arm A (RTV)a
| Pharmacokinetic parameter | AUC0–24 (ng · h/ml) | AUC0–∞ (ng · h/ml) | Cmax (ng/ml) | Tmax (h) | CL/F (liters/hr) | V/F (liters) | t1/2 (h) | kel (1/h) |
|---|---|---|---|---|---|---|---|---|
| Geometric mean value (% CV) | ||||||||
| Phase 1 (DE alone) | 747.84 (48.77) | 814.87 (48.24) | 93.41 (44.22) | 3.35 (35.46) | 190.08 (39.13) | 159.11 (39.60) | 6.31 (9.20) | 0.11 (8.70) |
| Phase 2 (DE + RTV separated) | 528.13 (36.84) | 576.12 (37.60) | 68.08 (34.71) | 2.43 (25.30) | 260.36 (32.61) | 235.90 (36.76) | 6.56 (13.54) | 0.11 (13.96) |
| Phase 3 (DE + RTV simultaneously) | 832.10 (43.91) | 938.63 (41.60) | 105.65 (36.07) | 3.53 (39.84) | 159.81 (42.31) | 135.55 (40.13) | 6.14 (12.10) | 0.11 (14.01) |
| GMR (90% CI) for comparison of phases | ||||||||
| Phase 2 vs phase 1 | 0.71 (0.60–0.81)b | 0.71 (0.60–0.82)b | 0.73 (0.61–0.85)b | 0.73 (0.62–0.84)b | 1.37 (1.16–1.58)b | 1.48 (1.25–1.71)b | 1.04 (0.99–1.09) | 0.95 (0.90–1.01) |
| Phase 3 vs phase 1 | 1.11 (0.89–1.33) | 1.15 (0.93–1.37) | 1.13 (0.90–1.36) | 1.05 (0.80–1.30) | 0.84 (0.64–1.04) | 0.85 (0.68–1.03) | 0.97 (0.91–1.03) | 1.02 (0.95–1.09) |
AUC0—24, area under the concentration-versus-time curve from time zero to 24 h; AUC0–∞, area under the concentration-versus-time curve from time zero to infinity; Cmax, maximum plasma concentration; Tmax, time to reach the maximum plasma concentration; CL/F, apparent oral clearance; V/F, apparent oral volume of distribution; t1/2, plasma half-life; kel, elimination rate constant; DE, dabigatran etexilate; RTV, ritonavir; CV, coefficient of variation; CI, confidence interval.
P < 0.05, based on a paired Student t test.
TABLE 2.
Geometric mean values of dabigatran pharmacokinetic parameters in arm B (COBI)a
| Pharmacokinetic parameter | AUC0–24 (ng · h/ml) | AUC0–∞ (ng · h/ml) | Cmax (ng/ml) | Tmax (h) | CL/F (liters/hr) | V/F (liters) | t1/2 (h) | kel (1/h) |
|---|---|---|---|---|---|---|---|---|
| Geometric mean value (% CV) | ||||||||
| Phase 1 (DE alone) | 959.49 (63.71) | 1,046.94 (66.55) | 129.74 (61.49) | 3.09 (32.51) | 143.28 (50.61) | 130.00 (45.69) | 6.29 (18.68) | 0.11 (15.95) |
| Phase 2 (DE + COBI separated) | 2051.98 (61.46) | 2,193.93 (60.87) | 258.14 (51.66) | 3.74 (30.25) | 68.37 (48.51) | 57.45 (47.77) | 5.82 (11.76) | 0.12 (11.26) |
| Phase 3 (DE + COBI simultaneously) | 2303.39 (40.37) | 2,483.26 (40.83) | 302.40 (44.47) | 3.20 (18.04) | 60.40 (57.69) | 51.61 (43.71) | 5.92 (15.54) | 0.12 (15.69) |
| GMR (90% CI) for comparison of phases | ||||||||
| Phase 2 vs phase 1 | 2.14 (1.65–2.63)b | 2.10 (1.65–2.54)b | 1.99 (1.42–2.56)b | 1.21 (1.00–1.42) | 0.48 (0.39–0.56)b | 0.44 (0.35–0.53)b | 0.93 (0.88–0.98)c | 1.08 (1.01–1.14)c |
| Phase 3 vs phase 1 | 2.31 (1.83–2.78)b | 2.27 (1.81–2.73)b | 2.27 (1.59–2.96)b | 1.03 (0.88–1.18) | 0.44 (0.37–0.51)b | 0.41 (0.34–0.48)b | 0.92 (0.88–0.97)c | 1.08 (1.03–1.16)c |
AUC0–24, area under the concentration-versus-time curve from time zero to 24 h; AUC0-∞, area under the concentration-versus-time curve from time zero to infinity; Cmax, maximum plasma concentration; Tmax, time to reach the maximum plasma concentration; CL/F, apparent oral clearance; V/F, apparent oral volume of distribution; t1/2, plasma half-life; kel, elimination rate constant; DE, dabigatran etexilate; COBI, cobicistat; CV, coefficient of variation; CI, confidence interval.
P < 0.001, based on a paired Student t test.
P < 0.05, based on a paired Student t test.
FIG 1.
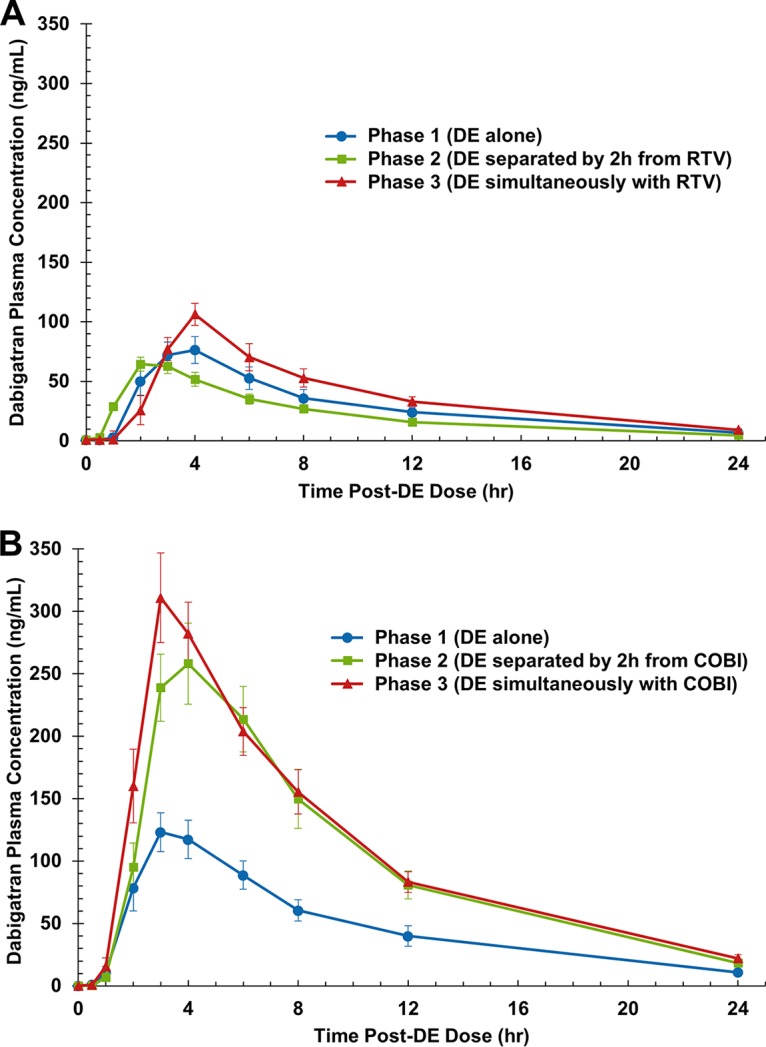
Mean ± SEM dabigatran concentration-versus-time curves after administration of dabigatran alone, administration of dabigatran separated by 2 h from administration of ritonavir (RTV) (A) or cobicistat (COBI) (B), and simultaneous administration of dabigatran with ritonavir (A) and cobicistat (B). DE, dabigatran etexilate.
In arm B, the dabigatran AUC0–∞ and Cmax were significantly increased by 127% each when dabigatran and cobicistat were administered simultaneously (phase 3 versus phase 1; P < 0.001 for both comparisons). When dabigatran and cobicistat administration was separated by 2 h, the increase in dabigatran PK values (phase 2 versus phase 1) was minimally mitigated; the dabigatran AUC0–∞ and Cmax values were increased by 110% and 99%, respectively (P < 0.001 for both comparisons). The elimination rate constant (kel), t1/2, and Tmax during administration in both phases 2 and 3 were not markedly altered compared to the values obtained after dabigatran administration alone (phase 1). Instead, CL/F and V/F were significantly decreased by 52% and 56%, respectively, with separated administration in phase 2 and by 56% and 59%, respectively, with simultaneous administration in phase 3 (P < 0.001 for all comparisons) (Table 2).
Thrombin time.
The PD of dabigatran were evaluated by measuring its anticoagulant effect on thrombin time (TT), and the effect-versus-time curves for ritonavir and cobicistat are presented in Fig. 2. In arm A (ritonavir), regardless of whether the dabigatran and ritonavir doses were separated by 2 h or given simultaneously, there was no significant decrease in the area-under-the-effect curve from time zero to 24 h (AUEC0–24) compared to the value obtained after administration of dabigatran alone (Table 3). There was also no significant difference in the AUEC0–24 between phase 3 and phase 2. TT at 24 h postdose (TT24) was not significantly altered with separated administration in phase 2 but was increased by 31% with simultaneous administration in phase 3 (P < 0.05). Conversely, in arm B (cobicistat), there were statistically significant and similar increases in the AUEC0–24 of 30% and 33% when cobicistat doses were separated or given simultaneously, respectively, compared to the values obtained after administration of dabigatran alone (P < 0.001 for both comparisons). Significant increases in TT24 of 46% and 51% were also observed with separated administration in phase 2 (P < 0.001) and simultaneous administration in phase 3 (P < 0.001), respectively, compared to the values obtained after administration of dabigatran alone (Table 4). Again, no significant difference in TT was noted between simultaneous (phase 3) and separated (phase 2) administration of cobicistat and dabigatran (data not shown).
FIG 2.
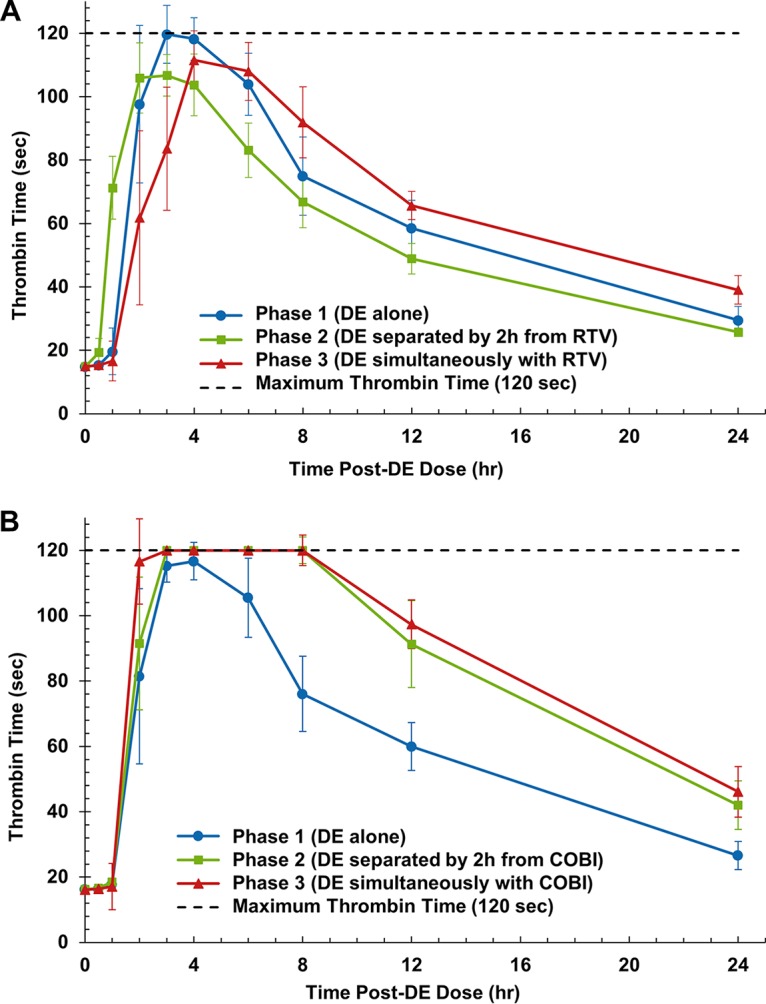
Median ± 90% confidence interval thrombin time-versus-time curves after administration of dabigatran alone, administration of dabigatran separated by 2 h from administration of ritonavir (RTV) (A) or cobicistat (COBI) (B), and simultaneous administration of ritonavir (A) and cobicistat (B). DE, dabigatran etexilate.
TABLE 3.
Geometric mean TT anticoagulation parameters in arm A (RTV)a
| Parameter | AUEC0–24 (s · h) | TT24 (s) |
|---|---|---|
| Geometric mean value (% CV) | ||
| Phase 1 (DE alone) | 1,479 (20.7) | 28.6 (27.3) |
| Phase 2 (DE + RTV separated) | 1,320 (18.7) | 25.3 (17.8) |
| Phase 3 (DE + RTV simultaneously) | 1,424 (26.6) | 37.4 (27.6) |
| GMR (90% CI) for comparison of phases | ||
| Phase 2 vs phase 1 | 0.89 (0.79–1.00) | 0.88 (0.76–1.01) |
| Phase 3 vs phase 1 | 0.96 (0.82–1.11) | 1.31 (1.09–1.52)b |
AUEC0–24, area-under-the-time-effect curve from time zero to 24 h; DE, dabigatran etexilate; RTV, ritonavir; TT24, thrombin time at 24 h; CV, coefficient of variation; CI, confidence interval.
P < 0.05, based on a paired Student t test.
TABLE 4.
Geometric mean TT anticoagulation parameters in arm B (COBI)a
| Parameter | AUEC0–24 (s · h) | TT24 (s) |
|---|---|---|
| Geometric mean value (% CV) | ||
| Phase 1 (DE alone) | 1,508 (16.9) | 29.2 (25.4) |
| Phase 2 (DE + COBI separated) | 1,964 (12.2) | 42.5 (27.8) |
| Phase 3 (DE + COBI simultaneously) | 2,038 (10.9) | 45.4 (28.8) |
| GMR (90% CI) for comparison of phases | ||
| Phase 2 vs phase 1 | 1.30 (1.21–1.39)b | 1.46 (1.30–1.61)b |
| Phase 3 vs phase 1 | 1.33 (1.22–1.44)b | 1.51 (1.24–1.78)b |
AUEC0–24, area-under-the-time-effect curve from time zero to 24 h; TT24, thrombin time at 24 h; COBI, cobicistat; CV, coefficient of variation; CI, confidence interval.
P < 0.001, based on a paired Student t test.
Safety and tolerability.
All study drugs were generally well tolerated. No subject experienced an event related to bleeding or anticoagulation. Adverse events (AEs) were mostly mild (grade 1) and included gastrointestinal symptoms (n = 20), headache (n = 6), genitourinary AEs (n = 5, arm B), bruising (n = 1, arm A), and elevated serum creatinine levels (n = 1, arm B). Two participants in arm A experienced elevations in serum cholesterol levels, and one subject experienced an elevation in alanine aminotransferase (ALT) levels. The only grade 2 AE determined to be possibly related to a study intervention (dabigatran or ritonavir) was heartburn; this resolved without medical intervention, and no further follow-up was warranted.
DISCUSSION
HIV-infected individuals across all age spectra are at an increased risk of thrombosis (31). As patients living with HIV continue to experience increased longevity in the modern era of effective antiretroviral therapy, the incidence of common chronic comorbidities seen in elderly populations is also expected to increase in this population. The treatment and prevention of venous thromboembolic events among HIV-infected individuals receiving antiretroviral therapy represent challenges for clinicians due to the risk of many potential and significant drug interactions. This study aimed to evaluate the effect of simultaneous and separated administration (by 2 h) of the antiretroviral PK enhancers ritonavir and cobicistat on the PK and PD of dabigatran.
The values of the dabigatran PK parameters obtained in this study were generally consistent with those previously reported. The mean area under the concentration-versus-time curve (AUC) from time zero to 24 h (AUC0–24) of dabigatran when administered alone was 747.84 ng · h/ml and 959.49 ng · h/ml in the ritonavir and cobicistat arms, respectively, which are similar to the values reported in previous PK studies with healthy volunteers (28). The mean t1/2 across all study phases in both arms was ∼6 h, which is shorter than the reported 12 to 14 h observed with repeated dosing but similar to the 7 to 8 h reported with single doses (14, 28). The Tmax was ∼2.5 h in a fasted state, which is a slight increase compared with the ∼1 h reported in the prescribing information, and was ∼3.5 h in the presence of a standardized breakfast, which is similar to what has been previously reported (14).
Previous investigations have shown that dabigatran coadministration with recognized intestinal P-gp transporter inhibitors (e.g., amiodarone, clarithromycin, dronedarone, ketoconazole, quinidine, and verapamil) results in increased dabigatran exposure by 49 to 153% (14). Moreover, the maximum anticoagulation effect directly corresponded with plasma concentrations in a linear dose-response fashion (14, 28, 32). Härtter et al. illustrated that this interaction could potentially be obviated by separated administration of the P-gp probe substrate and inhibitor medications by 2 h, with minimal alterations in dabigatran PK exposure and the PD markers of TT AUEC and maximum TT (28). As an in vitro P-gp inhibitor, ritonavir was also expected to result in a similar perturbation in dabigatran PK and PD outcomes. Contrary to our hypothesis, we observed no significant interaction between ritonavir and dabigatran when they were administered simultaneously. Curiously, the separated administration of ritonavir and dabigatran etexilate by 2 h in phase 2 resulted in an unexpected statistically significant decrease in dabigatran PK exposure by 29% and a corresponding statistically insignificant decrease in the TT AUEC by 11%. There was no significant change in t1/2, but there was a significant increase in CL/F and V/F, suggesting that this observation was due principally to the decreased bioavailability of dabigatran. It is plausible that this observation was a result of mixed P-gp induction and inhibition by ritonavir, the former of which may have prevailed with separated administration in phase 2 but was effectively negated by inhibition of P-gp with simultaneous administration in phase 3. Such mixed inhibitory and inductive properties of ritonavir on various metabolizing enzymes and transporters (including P-gp) have been previously observed (29, 33–35). However, the proposed mechanism behind this observation needs to be further validated.
We expected to see a similar result when administering cobicistat with dabigatran etexilate, as ritonavir and cobicistat share similar intestinal drug concentration/IC50 ratio values (∼16.7) for inhibiting P-gp in vitro (22). However, we observed a significant interaction that increased dabigatran exposure by 99 to 127% and increased its anticoagulant effect by at least 30 to 51%. The true clinical impact of cobicistat on the anticoagulant effects of dabigatran may be greater than that measured in this study due to utilization of the thrombin time assay, STA-thrombin, which only reports a maximum TT value of up to 120 s. To account for this, this study evaluated TT24 as a secondary PD outcome. Unfortunately, no clinical guideline to date has provided information or recommendations regarding the goal TT parameters that should be monitored while patients are treated with dabigatran or other DOACs. Furthermore, patients commonly receive twice-daily dosing with dabigatran rather than the single doses utilized in this study. Notably, TT at 12 h postdose (TT12) showed increases nearly identical to those of TT24 with cobicistat coadministration; TT12 increased by 51% and 53% in phases 2 and 3 (data not shown), respectively, whereas TT24 increased by 46% and 51% in phases 2 and 3, respectively. Despite these limitations, TT was selected as a direct PD marker of the dabigatran drug action as TT correlates linearly with plasma dabigatran concentrations and is a more a sensitive PD measure of dabigatran activity than other anticoagulation tests available clinically at most hospital laboratories (e.g., activated partial thromboplastin time and the international normalized ratio [INR]) (36). In addition to TT, the study initially planned to collect and analyze the ecarin clotting time (ECT). However, during the conduct of the study, the reagent for ECT analysis became unavailable commercially, and thus, ECT could no longer be analyzed.
Interestingly, the observed increase in dabigatran exposure and the prolonged effects on thrombin time persisted, despite the separation of cobicistat and dabigatran etexilate administration by 2 h. No significant difference between separate and simultaneous administration of cobicistat with dabigatran etexilate was found. These data demonstrate that separation of administration by 2 h was not an adequate method for circumventing this interaction. Since the participants took their doses with food and the dabigatran Cmax was typically observed to occur 3 to 4 h after dosing, it is possible that separation of cobicistat and dabigatran administration by ≥4 h may have circumvented the interaction. However, a regimen employing such a schedule may present a considerable challenge for adherence, particularly in patients receiving typical twice-daily dosing of dabigatran. Again, while there was no alteration of drug elimination, significant increases in oral bioavailability, as exemplified by decreases in both CL/F and V/F, were observed. These results suggest that cobicistat-mediated intestinal P-gp inhibition and not renal MATE-1 transporter inhibition was primarily responsible for the increases in dabigatran PK exposure and the PD effects.
This study was conducted in healthy HIV-negative volunteers receiving single doses of dabigatran, as is standard in most drug interaction studies. The clinical application of these results will require further investigation in HIV-infected patients chronically receiving antiretrovirals and dabigatran. Numerous reports in the literature suggest that antiretroviral therapy itself can induce changes to the expression of drug transporters. For example, protease inhibitors have been shown to increase the cell surface expression of P-gp in the peripheral blood mononuclear cells of healthy volunteers (37). Furthermore, a recent study by Kis et al. showed that HIV-infected individuals receiving long-term antiretroviral therapy have increased P-gp gene expression (3.2-fold) compared to treatment-naive HIV-infected patients and thus may have altered susceptibility to these drug interactions (38). Nonetheless, the results reported herein suggest that dabigatran etexilate likely can be safely coadministered with ritonavir-boosted regimens without the risk of a clinically significant interaction. This was recently demonstrated in a trial described in a case report by Perram et al., in which initiation of treatment with dabigatran etexilate at a low dose of 110 mg daily with subsequent titration based on monitoring of the dabigatran trough concentration was successfully employed in an HIV-infected patient receiving ritonavir-boosted antiretroviral therapy (39). In contrast, when treating HIV-infected individuals receiving cobicistat-boosted therapy, clinicians may need to consider prescribing an alternative anticoagulant agent in order to avoid this potentially clinically significant interaction. However, currently available alternative DOACs (rivaroxaban, apixaban, and edoxaban) are expected to interact with cobicistat due to CYP3A4 inhibition, leaving the remaining chronic anticoagulant options of enoxaparin, which is administered via subcutaneous injection, or warfarin, which requires periodic monitoring of the INR, among other limitations.
The use of dabigatran etexilate with strong P-gp inhibitors is contraindicated by the European Medicines Agency, and the FDA recommends the use of a reduced dose of 75 mg twice daily in patients with moderate renal impairment (creatinine clearance of 30 to 50 ml/min) who are concomitantly receiving strong P-gp inhibitors. In the FDA clinical pharmacology review of the dabigatran etexilate (Pradaxa) new drug application, any interaction resulting in dabigatran exposure increases of >150% required a dose or regimen adjustment (40). Thus, the magnitude of the interaction observed in this study (a 127% increase in exposure) suggests the potential need for reduced dabigatran etexilate dosing (e.g., 75 mg twice daily rather than 150 mg twice daily) and/or prudent clinical monitoring of patients for signs and symptoms of bleeding when it is administered with cobicistat. Given that there was no detectable change in drug elimination, it is likely inappropriate to reduce the dabigatran dosing interval frequency (i.e., switching from twice-daily dosing to once-daily dosing). Regardless of the dose administered, patients receiving concomitant dabigatran and cobicistat may actually benefit from the monitoring of anticoagulant laboratory parameters, as was employed in the aforementioned case report. This strategy, to date, has not been recommended clinically for the routine use of dabigatran etexilate but has already been suggested in certain situations, such as in patients with renal impairment and patients over 75 years of age. This approach, while cumbersome, may be warranted, given that both elevated TT24 and dabigatran concentrations have been associated with an increased risk of bleeding in patients receiving dabigatran etexilate therapy (41, 42). However, the limited availability of assays for the monitoring of dabigatran concentrations at most clinical institutions, the lack of clear guidance on the plasma target concentrations, and the interpatient variability in drug exposure are potential impediments to this approach. Fortunately, the availability of idarucizumab (Praxbind) may be considered a layer of safety when dabigatran concentrations cannot be monitored, insofar as binding of dabigatran and rapid restoration of the thrombin clotting pathway can be accomplished in cases of life-threatening or uncontrolled bleeding or if there is a need for emergent surgery.
Ultimately, our study highlights the potential differences that can be observed between these similar pharmacokinetic enhancers and the potential risk in the broad application of conclusions derived from ritonavir drug-drug interaction studies to cobicistat, and vice versa. Specifically, these results demonstrate that cobicistat provides a significantly greater net inhibition of P-gp transport than ritonavir in vivo. Given these results, studies with agents with a narrow therapeutic index or drugs that are known substrates of P-gp should be performed with both cobicistat and ritonavir. Furthermore, clinicians should be very cautious in switching patients from ritonavir-boosted therapy to cobicistat-boosted therapy when patients are concomitantly receiving P-gp substrates, as it cannot be assumed that the risk of clinically relevant drug interactions will be similar.
MATERIALS AND METHODS
Study design.
This was a single-center, two-arm, single-sequence, open-label study (ClinicalTrials.gov under identifier NCT01896622) to evaluate the influence of ritonavir (arm A) or cobicistat (arm B) on the PK/PD of dabigatran in healthy volunteers (Fig. 3). This study was conducted at the Clinical Research Center at the National Institutes of Health (Bethesda, MD, USA). All participants gave written informed consent, and the clinical research was conducted according to guidelines for human experimentation specified by the U.S. Department of Health and Human Services. This study was approved by the National Institute of Allergy and Infectious Diseases Institutional Review Board.
FIG 3.

Study design. Abbreviations: COBI, cobicistat; DE, dabigatran etexilate; PD, pharmacodynamics; PK, pharmacokinetics; RTV, ritonavir.
Each arm of the study was conducted similarly. On study day 0 during phase 1, following an overnight fast, the participants were administered a single oral 150-mg dose of dabigatran etexilate (Pradaxa; Boehringer Ingleheim Pharmaceuticals Inc., Ridgefield, CT). Phase 1 was followed by a 5-day washout period. During phase 2, participants in arm A took ritonavir (Norvir; AbbVie Inc., North Chicago, IL) at 100 mg once daily in the morning for 22 days (study days 5 through 26), while participants in arm B took cobicistat (Tybost; Gilead, Foster City, CA) at 150 mg once daily in the morning for 22 days. In arm A, on day 15 of ritonavir dosing (study day 19), participants took their morning ritonavir dose 2 h after they took a single 150-mg dose of dabigatran etexilate; similarly, on study day 19 in arm B, participants took their morning cobicistat dose 2 h after they took a single 150-mg dose of dabigatran etexilate. During phase 3, participants continued taking ritonavir or cobicistat, and on study day 26, the participants were administered their morning ritonavir or cobicistat dose at the same time as the single 150-mg dose of dabigatran etexilate.
During each study phase, blood samples were collected into tubes containing EDTA at time zero (predose) and 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h following each single dose of dabigatran etexilate for determination of dabigatran plasma concentrations. Blood was centrifuged after collection, and plasma was harvested and frozen at −70°C until the time of analysis. Blood samples for determination of the thrombin time (TT) were collected at time points identical to those listed above in tubes containing 0.105 M (3.2%) sodium citrate. Additional blood was collected throughout the study and 28 days following the final day of PK sampling (study day 54) for laboratory safety monitoring. Additionally, participants were assessed via routine questioning for adherence and adverse events throughout the course of the investigation.
Study participants.
The study population consisted of HIV-negative individuals receiving no other concomitant prescription or over-the-counter or herbal medications. To be included in the study, participants were required to be 18 to 70 years of age, HIV seronegative (determined by enzyme-linked immunosorbent assay), and free of concurrent illnesses per medical history, physical examination, and screening laboratory values, including liver function tests and serum creatinine, total and direct bilirubin, and hemoglobin concentration determination. Screening laboratory values were required to be within institutional normal ranges, except for fasting total cholesterol and triglyceride levels, which were required to be below 270 mg/dl. Participants were excluded if they had a history of or a present increased risk of bleeding or any planned invasive or surgical procedures within 28 days of study participation. Females of childbearing potential were required to have a negative urine or serum pregnancy test before beginning to receive study drugs and to practice abstinence or use effective nonhormonal methods of birth control throughout the investigation. The use of tobacco products was not permitted.
Dabigatran and thrombin time assays.
Dabigatran and the dabigatran-d3 ethyl ester HCl (DBG-d3 ethyl ester) internal standard were separated using a newly developed ultraperformance liquid chromatography (UPLC) method with detection by tandem mass spectrometry (UPLC-MS-MS) using multiple-reaction monitoring (MRM). The UPLC-MS-MS analysis was performed using an Acquity UPLC liquid handling system and a Quattro Premier XE triple quadrupole mass spectrometer (Waters Corp., Milford, MA, USA) controlled by MassLynx (version 4.1) mass spectrometry and chromatography manager software. The separation was performed on an Acquity BEH analytical C18 column (2.1 by 50 mm; particle size, 1.7 μm) preceded by a Vanguard BEH C18 precolumn (2.1 by 5 mm; particle size, 1.7 μm) (Waters Corp., Milford, MA, USA) using a mobile-phase gradient starting with a 10:90 (vol/vol) mixture of acetonitrile (ACN) and 20 mM formic acid (FA) buffer solution (pH 3.0) at a flow rate of 0.400 ml/min. Calibration curves for dabigatran were linear from 0.50 ng/ml to 300.0 ng/ml, and the R2 value was >0.996. Percent errors, as a measure of accuracy, were <15%, and the inter- and intra-assay coefficients of variation for dabigatran were 2.85 to 10.38% and 3.25 to 9.68%, respectively, at three different drug concentrations. The limit of quantitation for dabigatran was 0.50 ng/ml, and the limit of detection was 0.10 ng/ml. The overall recovery of dabigatran and DBG-d3 ethyl ester (internal standard) was >90%.
Thrombin time (TT) was determined using the STA-thrombin reagent (Diagnostica Stago, Asnières-sur-Seine, France) at the NIH Clinical Center Department of Laboratory Medicine. Sodium citrate plasma samples were incubated for 2 min at 37°C. STA-thrombin (200 μl) was then added to the sample, and the resulting time (in seconds) for each sample to completely clot was reported and tabulated. The maximum limit of the TT assay was 120 s.
Pharmacokinetic and thrombin time analysis.
Dabigatran PK and TT parameter values were determined by noncompartmental analysis using Phoenix WinNonlin software (version 6.4; Certara, St. Louis, MO). Maximum plasma concentrations (Cmax), the time to reach Cmax (Tmax), and TT at 24 h postdosing (TT24) were determined from direct observation of the data. The elimination rate constant (kel) was estimated as the absolute value of the slope of a linear regression of a natural logarithm of concentration-versus-time plots using at least three points, excluding Cmax, during the elimination phase. The half-life (t1/2) was calculated as (ln 2)/kel. The PK parameters area under the concentration-time curve from time zero to 24 h (AUC0–24) and the TT area-under-the-effect curve from time zero to 24 h (AUEC0–24) were determined using the “linear-up, log-down” trapezoidal rule. The AUC from time zero to infinity (AUC0–∞) was determined by dividing the last measured concentration by kel and adding this value to AUC0–24. Apparent oral clearance (CL/F) was estimated as the dose divided by AUC0–∞. The apparent volume of distribution (V/F) was estimated as CL/F divided by kel.
Statistical analysis.
Sample size was calculated with regard to the reported variability in the dabigatran AUC in healthy volunteers (854 ng · h/ml with a geometric coefficient of variation of 61.8%) (27). On the basis of these data and an α value of 0.05, a sample size of 16 yielded a greater than 80% power to detect a change of 70% in the dabigatran AUC0–∞ with concomitant ritonavir or cobicistat administration. This change was selected, as it was thought to be clinically significant. Dabigatran PK parameter values derived before and after ritonavir or cobicistat exposure were compared using a paired Student's t test. Statistical significance was defined a priori as a P value of <0.05 (Systat software, version 11; Systat Software, Inc., Richmond, CA, USA). Adjustments were not implemented for multiple comparisons. Geometric mean ratios (GMRs) and 90% confidence intervals (CI) were computed for separated ritonavir or cobicistat and dabigatran administration (phase 2 versus phase 1) and simultaneous ritonavir or cobicistat and dabigatran administration (phase 3 versus phase 1) (Microsoft Excel 2011 software; Microsoft Corp., Redmond, WA, USA).
ACKNOWLEDGMENTS
We acknowledge the healthy volunteer participants of the study, the National Institutes of Health (NIH) Clinical Center 5-SWS Day Hospital nursing staff, the National Institute of Allergy and Infectious Diseases (NIAID) OP-8 HIV Clinic case managers, Mike Proschan of NIAID for providing sample size calculations, and Paul Wakim of NIH CC Biostatistics for advice with statistical analysis and review.
P.K., L.A.G., K.M.B., and J.M.G. wrote the manuscript; L.A.G., S.R.P., J.L., and C.H. designed the research; P.K., L.A.G., K.M.B., A.K., M.M., R.M.A., K.N., S.R.P., and C.H. performed the research; and P.K., L.A.G., K.M.B., R.M.A., J.M.G., K.N., and J.L. analyzed the data.
We have no relevant conflicts of interest to disclose.
Funding for this study was provided by the NIH Clinical Center Pharmacy Department and the NIAID intramural research program.
REFERENCES
- 1.Centers for Disease Control and Prevention. 2016. HIV surveillance report, 2015, vol. 27. http://www.cdc.gov/hiv/library/reports/hiv-surveillance.html.
- 2.Prejean J, Song R, Hernandez A, Ziebell R, Green T, Walker F, Lin LS, An Q, Mermin J, Lansky A, Hall HI, HIV Incidence Surveillance Group. 2011. Estimated HIV incidence in the United States, 2006-2009. PLoS One 6:e17502. doi: 10.1371/journal.pone.0017502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Collaboration of Observational HIV Epidemiological Research Europe (COHERE) in EuroCoord, Lewden C, Bouteloup V, De Wit S, Sabin C, Mocroft A, Wasmuth JC, van Sighem A, Kirk O, Obel N, Panos G, Ghosn J, Dabis F, Mary-Krause M, Leport C, Perez-Hoyos S, Sobrino-Vegas P, Stephan C, Castagna A, Antinori A, d'Arminio Monforte A, Torti C, Mussini C, Isern V, Calmy A, Teira R, Egger M, Grarup J, Chêne G. 2012. All-cause mortality in treated HIV-infected adults with CD4 500/mm3 compared with the general population: evidence from large European observational cohort collaboration. Int J Epidemiol 41:433–445. doi: 10.1093/ije/dyr164. [DOI] [PubMed] [Google Scholar]
- 4.New Jersey Department of Health. 2015. New Jersey HIV/AIDS report. New Jersey Department of Health, Newark, NJ: http://www.nj.gov/health/hivstdtb/documents/newsletter/semi0615.pdf Accessed 28 July 2016. [Google Scholar]
- 5.Malek J, Rogers R, Kufera J, Hirshon JM. 2011. Venous thromboembolic disease in the HIV-infected patient. Am J Emerg Med 29:278–282. doi: 10.1016/j.ajem.2009.09.034. [DOI] [PubMed] [Google Scholar]
- 6.Bibas M, Biava G, Antinori A. 2011. HIV-associated venous thromboembolism. Mediterr J Hematol Infect Dis 3:e2011030. doi: 10.4084/mjhid.2011.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klein SK, Slim EJ, de Kruif MD, Keller TT, ten Cate H, van Gorp EC, Brandjes DP. 2005. Is chronic HIV infection associated with venous thrombotic disease? A systematic review. Neth J Med 63:129–136. [PubMed] [Google Scholar]
- 8.Fultz SL, McGinnis KA, Skanderson M, Ragni MV, Justice AC. 2004. Association of venous thromboembolism with human immunodeficiency virus and mortality in veterans. Am J Med 116:420–423. doi: 10.1016/j.amjmed.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 9.Copur AS, Smith PR, Gomez V, Bergman M, Homel P. 2002. HIV infection is a risk factor for venous thromboembolism. AIDS Patient Care STDs 16:205–209. doi: 10.1089/10872910252972258. [DOI] [PubMed] [Google Scholar]
- 10.Ahonkhai AA, Gebo KA, Streiff MB, Moore RD, Segal JB.. 2008. Venous thromboembolism in patients with HIV/AIDS: a case-control study. J Acquir Immune Defic Syndr 48:310–314. doi: 10.1097/QAI.0b013e318163bd70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saber AA, Aboolian A, LaRaja RD, Baron H, Hanna K. 2001. HIV/AIDS and the risk of deep vein thrombosis: a study of 45 patients with lower extremity involvement. Am Surg 67:645–647. [PubMed] [Google Scholar]
- 12.Ageno W, Gallus AS, Wittkowsky A, Crowther M, Hylek EM, Palareti G. 2012. Oral anticoagulant therapy: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest 141(Suppl):e44S–e88S. doi: 10.1378/chest.11-2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bristol-Myers Squibb Pharmaceutical Company. 2016. Coumadin package insert. Bristol-Myers Squibb Pharmaceutical Company, Princeton, NJ. [Google Scholar]
- 14.Boehringer Ingelheim Pharmaceuticals, Inc. 2015. Pradaxa package insert. Boehringer Ingelheim Pharmaceuticals, Inc, Ridgefield, CT. [Google Scholar]
- 15.Prueksaritanont T, Tatosian D, Chu X, Railkar R, Evers R, Chavez-Eng C, Lutz R, Zeng W, Yabut J, Chan GH, Cai X, Latham AH, Hehman J, Stypinski D, Brejda J, Zhou C, Thornton B, Bateman KP, Fraser I, Stoch SA. 2017, Validation of a microdose probe drug cocktail for clinical drug interaction assessments for drug transporters and CYP3A. Clin Pharmacol Ther 101:519–530. [DOI] [PubMed] [Google Scholar]
- 16.Abbott Laboratories. 2010. Norvir package insert. Abbott Laboratories, North Chicago, IL. [Google Scholar]
- 17.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. U.S. Department of Health and Human Services, Washington, DC: http://aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf Accessed 3 June 2014. [Google Scholar]
- 18.Mathias AA, German P, Murray BP, Wei L, Jain A, West S, Warren D, Hui J, Kearney BP. 2010. Pharmacokinetics and pharmacodynamics of GS-9350: a novel pharmacokinetic enhancer without anti-HIV activity. Clin Pharmacol Ther 87:322–329. doi: 10.1038/clpt.2009.228. [DOI] [PubMed] [Google Scholar]
- 19.Kakuda TN, Opsomer M, Timmers M, Iterbeke K, Van De Casteele T, Hillewaert V, Petrovic R, Hoetelmans RMW. 2014. Pharmacokinetics of darunavir in fixed-dose combination with cobicistat compared with coadministration of darunavir and ritonavir as single agents in healthy volunteers. J Clin Pharmacol 54:949–957. doi: 10.1002/jcph.290. [DOI] [PubMed] [Google Scholar]
- 20.German P, Warren D, West S, Hui J, Kearney BP. 2010. Pharmacokinetics and bioavailability of an integrase and novel pharmacoenhancer-containing single-tablet fixed-dose combination regimen for the treatment of HIV. J Acquir Immune Defic Syndr 55:323–329. doi: 10.1097/QAI.0b013e3181eb376b. [DOI] [PubMed] [Google Scholar]
- 21.Elion R, Cohen C, Gathe J, Shalit P, Hawkins T, Liu HC, Mathias AA, Chuck SL, Kearney BP, Warren DR, GS-US-216-0105 Study Team. 2011. Phase 2 study of cobicistat versus ritonavir each with once-daily atazanavir and fixed-dose emtricitabine/tenofovir DF in the initial treatment of HIV infection. AIDS 25:1881–1886. doi: 10.1097/QAD.0b013e32834b4d48. [DOI] [PubMed] [Google Scholar]
- 22.Lepist EI, Phan TK, Roy A, Tong L, Maclennan K, Murray B, Ray AS. 2012. Cobicistat boosts the intestinal absorption of transport substrates, including HIV protease inhibitors and GS-7340, in vitro. Antimicrob Agents Chemother 56:5409–5413. doi: 10.1128/AAC.01089-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robertson S, Penzak SR, Huang S-M. 2012. Drug interactions, p 239–257. In Atkinson AJ, Huang S-M, Lertora JJL, Markey SP (ed), Principles of clinical pharmacology, 3rd ed Elsevier, San Diego, CA. [Google Scholar]
- 24.Penzak SR, Shen JM, Alfaro RM, Remaley AT, Natarajan V, Falloon J. 2004. Ritonavir decreases the nonrenal clearance of digoxin in healthy volunteers with known MDR1 genotypes. Ther Drug Monit 26:322–329. doi: 10.1097/00007691-200406000-00018. [DOI] [PubMed] [Google Scholar]
- 25.Food and Drug Administration. 2012. Food and Drug Administration guidance for industry: drug interaction studies. Study design, data analysis, implications for dosing and labeling recommendations. Food and Drug Administration, Rockville, MD: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM292362.pdf. [Google Scholar]
- 26.European Medicines Agency. 2012. European Medicines Agency guidelines on the investigation of drug interactions. European Medicines Agency, London, England: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf. [Google Scholar]
- 27.Ding R, Tayrouz Y, Riedel K-D, Burhenne J, Weiss J, Mikus G, Haefeli WE. 2004. Substantial pharmacokinetic interaction between digoxin and ritonavir. Clin Pharmacol Ther 76:73–84. doi: 10.1016/j.clpt.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 28.Härtter S, Sennewald R, Nehmiz G, Reilly P. 2013. Oral bioavailability of dabigatran etexilate (Pradaxa®) after co-medication with verapamil in healthy participants. Br J Clin Pharmacol 75:1053–1062. doi: 10.1111/j.1365-2125.2012.04453.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marzolini C, Gibbons S, Khoo S, Back D. 2016. Cobicistat versus ritonavir boosting and differences in the drug-drug interaction profiles with co-medications. J Antimicrob Chemother 71:1755–1758. doi: 10.1093/jac/dkw032. [DOI] [PubMed] [Google Scholar]
- 30.Gordon LA, Kumar P, Brooks KM, Kellogg A, McManus M, Alfaro RM, Nghiem K, George JM, Lozier J, Penzak SR, Hadigan C. 2016. Antiretroviral boosting agent cobicistat increases the pharmacokinetic exposure and anticoagulant effect of dabigatran in HIV-negative healthy volunteers. Circulation 134:1909–1911. doi: 10.1161/CIRCULATIONAHA.116.025257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacobson MC, Dezube BJ, Aboulafia DM. 2004. Thrombotic complications in patients infected with HIV in the era of highly active antiretroviral therapy: a case series. Clin Infect Dis 39:1214–1222. doi: 10.1086/424664. [DOI] [PubMed] [Google Scholar]
- 32.Delavenne X, Ollier E, Basset T, Bertoletti L, Accassat S, Garcin A, Laporte S, Zufferey P, Mismetti P. 2013. A semi-mechanistic absorption model to evaluate drug-drug interaction with dabigatran: application with clarithromycin. Br J Clin Pharmacol 76:107–113. doi: 10.1111/bcp.12055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tseng AL, Luetkehoelter J, Walmsley SL. 2017. Increase in international normalized ratio after switching from atazanavir/ritonavir to darunavir/cobicistat in a patient on warfarin: boosters are not always equal. AIDS 31:175–176. doi: 10.1097/QAD.0000000000001275. [DOI] [PubMed] [Google Scholar]
- 34.Gervasoni C, Riva A, Cozzi V, Capetti A, Rizzardini G, Clementi E, Cattaneo D. 2017. Effects of ritonavir and cobicistat on dolutegravir exposure: when the booster can make the difference. J Antimicrob Chemother 72:1842–1844. doi: 10.1093/jac/dkx055. [DOI] [PubMed] [Google Scholar]
- 35.Perloff MD, Von Moltke LL, Marchand JE, Greenblatt DJ. 2001. Ritonavir induces P-glycoprotein expression, multidrug resistance-associated protein (MRP1) expression, and drug transporter-mediated activity in a human intestinal cell line. J Pharm Sci 90:1829–1837. [DOI] [PubMed] [Google Scholar]
- 36.Stangier J, Rathgen K, Stähle H, Gansser D, Roth W. 2007. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol 64:292–303. doi: 10.1111/j.1365-2125.2007.02899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alam C, Whyte-Allman SK, Omeragic A, Bendayan R. 2016. Role and modulation of drug transporters in HIV-1 therapy. Adv Drug Deliv Rev 103:121–143. doi: 10.1016/j.addr.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 38.Kis O, Sankaran-Walters S, Hoque MT, Walmsley SL, Dandekar S, Bendayan R. 2016. HIV-1 alters intestinal expression of drug transporters and metabolic enzymes: implications for antiretroviral drug disposition. Antimicrob Agents Chemother 60:2771–2781. doi: 10.1128/AAC.02278-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perram J, Joseph J, Holloway C. 2015. Novel oral anticoagulants and HIV: dabigatran use with antiretrovirals. BMJ Case Rep 2015:bcr2015211651. doi: 10.1136/bcr-2015-211651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Food and Drug Administration. 2010. Clinical pharmacology review NDA 22-512, dabigatran. Food and Drug Administration, Rockville, MD: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2010/022512Orig1s000ClinPharmR_Corrrected%203.11.2011.pdf Accessed 28 March 2017. [Google Scholar]
- 41.Huisman MV, Lip GY, Diener HC, Brueckmann M, van Ryn J, Clemens A. 2012. Dabigatran etexilate for stroke prevention in patients with atrial fibrillation: resolving uncertainties in routine practice. Thromb Haemost 107:838–847. doi: 10.1160/TH11-10-0718. [DOI] [PubMed] [Google Scholar]
- 42.Reilly PA, Lehr T, Haertter S, Connolly SJ, Yusuf S, Eikelboom JW, Ezekowitz MD, Nehmiz G, Wang S, Wallentin L, RE-LY Investigators. 2014. The effect of dabigatran plasma concentrations and patient characteristics on the frequency of ischemic stroke and major bleeding in atrial fibrillation patients: the RE-LY Trial (Randomized Evaluation of Long-Term Anticoagulation Therapy). J Am Coll Cardiol 63:321–328. doi: 10.1016/j.jacc.2013.07.104. [DOI] [PubMed] [Google Scholar]