

ABSTRACT
Despite recent successful control efforts, malaria remains a leading global health burden. Alarmingly, resistance to current antimalarials is increasing and the development of new drug families is needed to maintain malaria control. Current antimalarials target the intraerythrocytic developmental stage of the Plasmodium falciparum life cycle. However, the invasive extracellular parasite form, the merozoite, is also an attractive target for drug development. We have previously demonstrated that heparin-like molecules, including those with low molecular weights and low anticoagulant activities, are potent and specific inhibitors of merozoite invasion and blood-stage replication. Here we tested a large panel of heparin-like molecules and sulfated polysaccharides together with various modified chemical forms for their inhibitory activity against P. falciparum merozoite invasion. We identified chemical modifications that improve inhibitory activity and identified several additional sulfated polysaccharides with strong inhibitory activity. These studies have important implications for the further development of heparin-like molecules as antimalarial drugs and for understanding merozoite invasion.
KEYWORDS: Plasmodium falciparum, heparin, invasion, merozoite, polysaccharide
INTRODUCTION
Despite gains in malaria control and a push to elimination in some areas, malaria remains a significant disease globally, with Plasmodium falciparum being the leading cause of malaria (1). Recent evidence of the emergence and spread of artemisinin resistance in several countries raises concerns that current therapies will lose their clinical value (2), making continued drug discovery and development high priorities. Malaria disease occurs during blood-stage infection by P. falciparum, in which the merozoite form of the parasite invades and replicates within red blood cells (RBCs). All current drugs, including those of the artemisinin class, target the intra-RBC stage of development (3). However, targeting and blocking of merozoite invasion also present attractive approaches for therapeutics to prevent parasite invasion of RBCs, reducing parasite burden and disease (4, 5). The use of compounds that block invasion in combination with current drugs providing activity at different stages of the blood-stage life cycle may be valuable, and drug combinations are increasingly used to treat various infections to maximize efficacy and reduce the risk of drug resistance development.
Merozoite invasion involves numerous receptor-ligand interactions, with multiple, redundant invasion pathways having been identified (6). Nevertheless, sulfated carbohydrates and heparin-like molecules (HLMs) have been identified to be a group of compounds that block essential invasion events and are able to inhibit multiple invasion pathways (7). Inhibitory HLMs include heparin (7, 8), curdlan sulfate (9, 10), polyvinyl sulfonate sodium salt (11), suramin (12), carrageenans (13), sulfated cyclodextrins (14), fucosylated chondroitin sulfate (15), and K5 polysaccharides (7). The ability of HLMs to disrupt invasion may be due to the targeting by HLMs of multiple essential or important merozoite ligands. Merozoite invasion into RBCs proceeds through a number of steps: (i) initial contact and weak deformation of the RBC, involving merozoite surface antigens; (ii) strong deformation of the RBC, involving microneme and rhoptry proteins and the actin-myosin motor of the parasite; (iii) pore opening between the parasite and the RBC; (iv) tight junction formation between the parasite and RBC; and (v) internalization (16). HLMs bind proteins involved in the preinvasion stage and the initial stage of attachment to the RBC, such as merozoite surface protein 1 (MSP1) (7), along with rhoptry and microneme proteins, which are involved in the reorientation and signaling steps of invasion, which trigger strong deformation of the RBCs (17–19). Although the precise mechanisms of action are not known, it is possible that these merozoite proteins interact with sulfate groups on the RBC surface, and HLMs may inhibit invasion by disrupting essential receptor-ligand interactions. While heparin has the capacity to inhibit at multiple invasion steps, it appears that the dominant inhibitory activity of HLMs is mediated at the early invasion stages, as it has been demonstrated by live video microscopy of merozoite invasion that heparin blocks preinvasion steps (7, 16). However, the capacity of HLMs to also bind proteins involved in downstream invasion steps may also contribute to effective inhibition and the observed inability to select for heparin-resistant parasites lines (7). Of further potential therapeutic benefit, HLMs are also known to disrupt both the rosetting and sequestration of infected RBCs (15, 20–26), which are important mediators of pathogenesis. The ability of HLMs to inhibit both merozoite invasion and sequestration/rosetting highlights the potential of these molecules to reduce parasitemia and disease severity. Owing to the anticoagulant activity of heparin, it cannot be used as an antimalarial agent. However, it may be possible to reduce the anticoagulant activity of HLMs while maintaining their ability to inhibit P. falciparum (7). Indeed, curdlan sulfate, which has a 10-fold reduced anticoagulation activity compared to heparin, has been tested in a small human trial which suggested that treatment with curdlan sulfate reduced malaria disease severity (27). Further, HLMs, such as K5 polysaccharides, as well as other polyanions that lack anticoagulant activity, have been proposed to be potential therapeutics for viral diseases (reviewed in reference 28) and can inhibit merozoite invasion (7).
In previous work, we identified a number of key structural features of HLMs for invasion-inhibitory activity by testing chemically modified K5 polysaccharides and heparins together with their oligosaccharides (7). Our findings suggest the importance of N- and O-sulfate residues, the presence of ≥2 sulfate units per disaccharide, specific spatial arrangements of sulfation requiring sulfate groups positioned together on a single saccharide unit, and a minimum chain length of 6 monosaccharide residues for optimal inhibitory activity (7). Structure/function studies have also successfully been used to develop small-drug HLMs, such as the pentasaccharide anticoagulant fondaparinux, for other clinical applications (29). Here we build on this knowledge by testing HLMs with specific modifications to further investigate structural features that mediate high levels of inhibitory activity and identify chemical modifications that increase activity. Further, we tested a large panel of sulfated polysaccharides prepared from a wide range of sources to identify inhibitory compounds. We aimed to identify compounds with strong invasion-inhibitory activity that may have potential for therapeutic development.
RESULTS
Heparin can be modified to increase inhibitory activity and remove anticoagulant activity.
Due to the high anticoagulant activity of heparin, it cannot be used directly as an antimalarial agent. Different modifications of heparin compounds can reduce the off-target effects of compounds, such as anticoagulation activity, and increase their bioavailability and half-life. We investigated a panel of compounds comprising modified heparin and HLMs for their inhibitory activity. These included HLMs with the nonsulfated uronic acid ring opened and cleaved at the diol site after periodate oxidation treatment, HLMs with the carboxyl groups of hexuronic acid residues reduced, and HLMs with the hydroxyl groups acylated (for the full list of compounds, see Table S1 in the supplemental material). Periodate oxidation of nonsulfated uronic acid residues, which has been reported to abolish anticoagulation activity (30), increased the activity of some but not all compounds (compounds with improved inhibition following treatment included mucosal heparin [MH] desulfated at position 2 [MH de-2-S], mucosal heparin lacking 6-O-sulfate [MH de-6-S], and mucosal heparin 3 kDa in length, and compounds with decreased inhibition following treatment included bemiparin and fondaparinux; P = 0.195 for overall impact of periodate treatment) (Table 1). The molecular basis for the increased activity is currently unknown, but one possibility is that the increased conformational flexibility of these modified compounds may allow a higher capacity to bind merozoite target antigens. We also assessed the impact of esterification of hydroxyl groups by testing the inhibitory activity of mucosal heparin (porcine) that was both periodate treated and esterified (i.e., glycol-split [gc] mucosal heparin [MH gc butyrate]). Esterification of the hydroxyl groups resulted in a 32% increase in inhibitory activity compared to that of the nonesterified parent compound (MH gc) (mean ± standard error of the mean [SEM] percent inhibition at 20 μg/ml, 65% ± 4.7% for MH gc and 97% ± 0.1% for MH gc butyrate; P < 0.001). Indeed, periodate-treated and esterified heparin was one of the most highly inhibitory compounds tested. These results demonstrate the potential for the development of compounds with increased inhibitory activity based on heparin and modified molecules.
TABLE 1.
Effect of glycol splitting by periodate treatment on inhibitory activity of heparin against merozoite invasion in growth inhibition assaysa
| Parent compound | % inhibition (SEM) |
Gain of inhibition (%) | |
|---|---|---|---|
| Parent compound | Modified compound | ||
| MH | 68 (7) | 77 (9) | 9 |
| MH de-2-S | 27 (2) | 54 (6) | 27b |
| MH de-6-S | 5 (1) | 61 (5.8) | 56b |
| MH of 3 kDa | 37 (2) | 70 (8) | 33b |
| MH H2O2 | 57 (4) | 62 (4) | 5 |
| Enoxaparin | 64 (5) | 80 (3) | 14 |
| Bemiparin | 45 (2) | 31 (5) | −14 |
| Fondaparinux | 34 (0) | 11 (4) | −23b |
A panel of modified heparin compounds at 100 μg/ml each was tested for inhibition of P. falciparum in standard growth inhibition assays. Comparisons of the parent and periodate-treated compounds were made. Gain of inhibition was calculated as (percent inhibition of the modified compound − percent inhibition of parent compound). Positive values indicate increased inhibitory activity of the modified compound compared to that of the parent compound. Negative values indicate reduced inhibitory activity of the modified compound compared to that of the parent compound. Data are the mean percent inhibition ± SEM from two assays performed in duplicate. Abbreviation: MH, mucosal heparin (porcine).
Significant differences in inhibitory activity between parent and modified compounds (P > 0.05, corrected for multiple comparisons by the Holm-Sidak method).
The inhibitory activity of HLMs requires sulfation, and activity occurs across a range of sizes.
We next compared the parent and modified compounds to identify features important in inhibitory activity. Consistent with our prior reports (7), sulfation was a key feature of inhibitory compounds, with the de-O- and de-N-sulfated compounds having reduced activity compared to the parent compounds (P < 0.0001 for overall impact of desulfation) (Table 2). Further, overall longer-chain heparin molecules (molecular mass, >3 to 25 kDa) showed a trend toward having higher inhibitory activity than shorter-chain compounds (molecular mass, 3 kDa) (Mann-Whitney test, P = 0.06) (Table 3). However, inhibition was not strictly size dependent; low-molecular-mass heparin, such as enoxaparin (molecular mass, ∼3 kDa), had activity comparable to that of full-length heparin (molecular mass, 3 to 25 kDa) (Table 3). Further, a number of size-fractionated highly sulfated small HLMs (di-, tetra-, and hexasaccharides), including heparan sulfate (HS)-derived hexasaccharides, had substantial growth-inhibitory activity (Fig. 1). However, it should be noted that heparin oligosaccharides that are less than 6-mers in general have little inhibitory activity (7), suggesting that the inhibition by the hexasaccharides tested here may have been due to the specific sulfation conformation or patterns of these compounds that convey higher than usual inhibitory activity.
TABLE 2.
Effect of de-2, -6, or -N sulfation (with and without re-N-Ac) on inhibitory activity of heparin against merozoite invasion in growth inhibition assaysa
| Modification | Parent compound | % inhibition (SEM) |
Loss of inhibition (%) | |
|---|---|---|---|---|
| Parent compound | Modified compound | |||
| De-2-Sb | MH | 68 (7) | 26 (2) | 42c |
| MH gc | 77 (9) | 54 (6) | 23 | |
| LH | 74 (7) | 49 (0) | 25 | |
| MH of 5 kDa | 75 (6) | 45 (4) | 30 | |
| MH of 5 kDa gc | 73 (8) | 41 (1) | 32 | |
| MH H2O2 gc | 62 (4) | 7 (1) | 55c | |
| MH of 3 kDa | 37 (0) | 15 (4) | 22c | |
| MH of 3 kDa gc | 70 (8) | 11 (5) | 59c | |
| De-6-S | MH | 68 (7) | 5 (1) | 63c |
| MH gc | 77 (9) | 61 (6) | 16 | |
| MH gc de-2-S | 54 (6) | 21 (1) | 33c | |
| MH of 5 kDa | 75 (6) | 41 (4) | 34c | |
| De-NS (NH)b | MH gc | 77 (9) | 17 (4) | 60c |
| Partially N-Acb | MH gc | 77 (9) | 62 (6) | 15 |
| MH of 5 kDa gc | 73 (8) | 64 (2) | 9 | |
| MH H2O2 | 57 (4) | 15 (3) | 42c | |
| MH H2O2 gc de-2-S | 7 (1) | 3 (1) | 4 | |
| MH gc de-2-s | 54 (6) | 24 (8) | 30 | |
| MH de-2-S | 26 (2) | 28 (3) | −2 | |
| LH de-2-S | 49 (0) | 8 (6) | 41c | |
| Totally N-Ac | MH | 68 (7) | 47 (4) | 21 |
| MH de-2-S | 26 (2) | 23 (2) | 3 | |
| MH gc | 77 (9) | 8 (3) | 69c | |
| MH gc de-2-S | 54 (6) | 0 (3) | 54c | |
| LH de-2-S | 49 (0) | −8 (6) | 57c | |
Modified heparin compounds at 100 μg/ml each were tested for inhibition of P. falciparum in growth inhibition assays. Comparisons of parent and desulfated modified compounds were made. In the majority of cases, desulfation resulted in a reduction of inhibitory activity. The loss of inhibitory activity was calculated as (percent inhibition of the parent compound − percent inhibition of the modified compound). Positive values indicate reduced inhibitory activity of the modified compound compared to that of the parent compound. Negative values indicate increased inhibitory activity of the modified compound compared to that of the parent compound. Data are mean inhibition ± SEM from two assays performed in duplicate. Abbreviations: MH, mucosal heparin (porcine); LH, lung heparin (bovine); gc, glycol splitting; NS, nonsulfated; N-Ac, N-acetylated.
Significant difference of modification across groups of modified compounds.
Significant differences in inhibitory activity between the parent and modified compounds (P > 0.05, corrected for multiple comparisons by the Holm-Sidak method).
TABLE 3.
Inhibitory activity of heparin compounds of different sizes against merozoite invasion in growth inhibition assaysa
| Size group | Compound (size estimate [kDa]) | % inhibition (SEM) |
|---|---|---|
| Long chain | Mucosal heparin (>3–25) | 68 (7) |
| Lung heparin (>3–25) | 74 (5) | |
| Mucosal heparin of 5 kDa (>3–8) | 75 (4) | |
| Short chain | Mucosal heparin (3) | 37 (0) |
| Enoxaparin (3) | 64 (6) | |
| Bemiparin (3) | 45 (3) | |
| Fondaparinux (pentasaccharide) (3) | 34 (0) |
Heparin compounds of different oligosaccharide chain lengths at 100 μg/ml each were tested for inhibition of P. falciparum in growth inhibition assays. Data are the mean percent inhibition ± SEM from two assays performed in duplicate. Mucosal heparin is from a porcine source, and lung heparin is from a bovine source.
FIG 1.
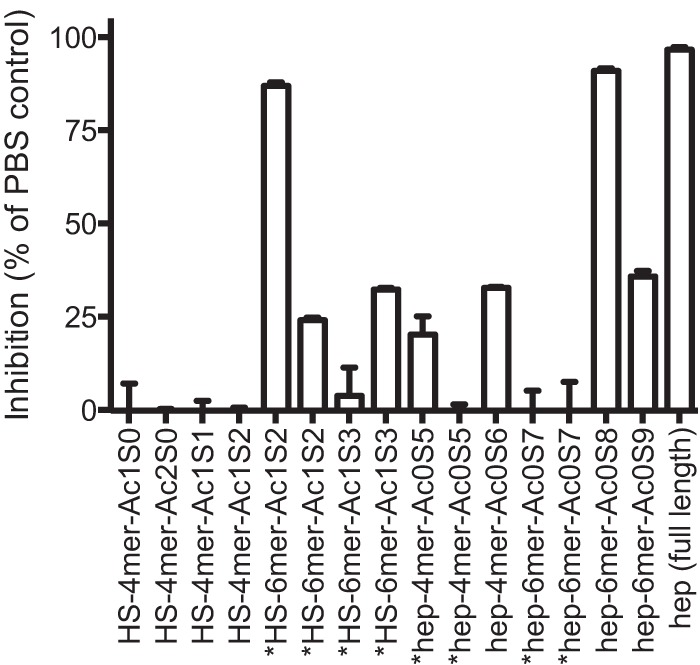
Growth-inhibitory activity of fractionated heparin and heparan sulfate compounds. Fractionated heparin and heparan sulfate tetra- and hexasaccharides were tested for growth-inhibitory activity in two-cycle assays. Data are means ± SEMs from two assays performed in duplicate. Abbreviations: HS, heparan sulfate; hep, heparin; 4-mer, tetrasaccharide; 6-mer, hexasaccharide; Ac1 and Ac2, N-sulfation at positions 1 and 2, respectively; Ac0, a lack of N-sulfation; S0 to S9, O-sulfation at the indicated position; *, different preparations of the same fraction.
Highly sulfated CSs inhibit P. falciparum growth.
Having shown that a key feature of inhibitory HLMs is a high level of sulfation, we investigated whether highly sulfated chondroitin sulfate (CS) compounds inhibited merozoite invasion. We have previously shown that chondroitin sulfate C (CSC) and CSA, which have low degrees of sulfation, are noninhibitory (7). Here we tested CSD (which has a low level of sulfation) and highly sulfated CSE and CSB. CSD has sulfation at uronate 2-S and galactosamine 6-S, CSE has sulfation at 4-S and 6-S of galactosamine, and CSB has sulfation at 2-S of uronate and either 4-S or 6-S of galactosamine. CSE had substantial inhibitory activity (50% inhibitory concentration [IC50], approximately 25 μg/ml) (Fig. 2A). Highly sulfated CSB-2,6-OS also had modest inhibitory activity at high concentrations, whereas CSD was not inhibitory. This suggests that, as for the K5 heparin-like molecules tested previously (7), the location of at least 2 sulfates together on a single oligosaccharide enhances inhibitory activity. To further investigate the inhibitory activity of CSE, a number of CSE hexasaccharides with different sulfation levels were tested in invasion inhibition assays with purified merozoites. Invasion inhibition assays differ from standard growth inhibition assays, in that in invasion inhibition assays compounds are incubated with merozoites and RBCs for only 30 min while invasion is occurring, whereas in growth inhibition assays drugs are incubated with the cultures over the course of the entire 48 h of the blood-stage development cycle, and invasion inhibition assays are more sensitive than standard growth inhibition assays (31). Inhibition appeared to increase with the sulfation level; 50% inhibition was observed with CSE hexasaccharides having 7 sulfate groups when they were tested at 100 μg/ml (Fig. 2B). However, CSE hexasaccharides had no activity at this concentration in standard growth inhibition assays, possibly because short-chain CS oligosaccharides have too few sulfate groups to mediate complete inhibition. The discrepancy between standard growth and invasion inhibition assays is likely due to the increased sensitivity of invasion assays, which use purified merozoites (31), suggesting that the inhibitory activity of compounds is at the threshold of that detected by standard growth inhibition assays.
FIG 2.

Identification of chondroitin sulfates with inhibitory activity against P. falciparum merozoite invasion. (A) CSD, CSE, and highly sulfated CSB polysaccharides were tested in growth inhibition assays at concentrations of 0 to 100 μg/ml. Data are means ± SEMs from three assays performed in duplicate. (B) Fractionated CSE hexasaccharides were tested in invasion inhibition assays. The degree of sulfation is 5, 6, or 7 sulfate groups per molecule. Data are means ± ranges from one assay performed in duplicate.
To characterize the functional mechanism of CSE inhibition, schizont rupture and merozoite invasion in the presence of CSE were analyzed via flow cytometry, with parasite stages being differentiated by ethidium bromide staining (7) and live video imaging (16, 32). As with the inhibitory mechanism of heparin (7), cultures incubated with CSE showed evidence of a slight delay in the time to schizont rupture compared to that for uninhibited cultures (Fig. 3A), but the predominant mechanism of inhibition appeared to be merozoite invasion inhibition, resulting in very low rates of ring formation (Fig. 3B). Live video imaging demonstrated that CSE prevented the invasion of merozoites into RBCs at early invasion steps; we observed schizont rupture, merozoite dispersal, and initial contact of the merozoites with RBCs. However, no oscillatory deformation was observed, and merozoites dissociated from the RBC surface without a clear reorientation of merozoites and echinocytosis of the RBC (Fig. 3C). A single invasion event was observed in over 9 schizont rupture events in 6,000 s of observation for inhibited cultures, whereas 21 invasion events in 13 schizont rupture events were observed in 5,442 s of filming for uninhibited cultures. Having observed that the CSE inhibitory mechanism appeared to be similar to that of heparin, we tested the ability of CSE to disrupt the binding of MSP1, a target of heparin inhibition, to heparin (7). CSE was unable to disrupt the binding of native or recombinant MSP1-42 to heparin (Fig. 3D and E). This suggests that CSE may inhibit initial steps of merozoite invasion via targeting alternative merozoite surface proteins or invasion ligands.
FIG 3.

CSE disrupts initial contact of the merozoite with the RBC but not heparin binding to MSP1-42. (A, B) Flow cytometry of late-stage parasite cultures, in which parasite stages were differentiated on the basis of ethidium bromide staining, was used to track parasite rupture (in which the results are given as percent schizonts) (A) and merozoite invasion (in which the results are given as percent ring forms) (B) in cultures treated with CSE at 100 μg/ml and uninhibited cultures treated with PBS. After 3 h of incubation, there were increased frequencies of schizonts and decreased ring forms in cultures incubated with CSE. Data are the means ± SEMs from two assays performed in duplicate. *, P < 0.05. (C) Live video microscopy of merozoite invasion in the presence of CSE. Merozoites were able to make initial contact with the RBC, but the contact was not sustained and the merozoites disassociated from the RBC surface. Times (in seconds) are indicated in the lower right corner, and the white arrows highlight a single merozoite that attached to and then disassociated from the RBC. (D) Heparin bead binding assays with P. falciparum protein extract. The protein extract was incubated with heparin beads along with soluble inhibitors, as indicated. Unbound and bead-bound fractions were probed for MSP1-42 binding via Western blotting. MSP1-42 was found in the unbound fraction when it was incubated with heparin as a soluble inhibitor, indicating that soluble heparin was able to outcompete MSP1-42 for binding. However, MSP1-42 was found in the bound fraction when it was incubated with soluble de-6-OS-heparin, CSE, or CSC, indicating that these compounds were not able to compete with heparin binding. (E) MSP1-42 coated on ELISA plates was incubated with heparin-BSA along with soluble heparin, CSC, and CSE at increasing concentrations. The binding of heparin-BSA was detected with anti-BSA antibodies. Soluble heparin, but not CSE or CSC, inhibited the binding of heparin-BSA to MSP1-42.
Identification of inhibitory sulfated carbohydrates.
Having shown that inhibitory activity appears to be reliant on the sulfation level and that longer chain lengths are needed for substantial activity, we tested a large panel of polysaccharides to test the impact of sulfation and resulfation on inhibitory activity and to attempt to identify polysaccharides that have the potential to be used as base compounds for novel drug development (for the full list of compounds and sources, see Table S2). Polysaccharides were tested in standard growth inhibition assays at 2, 10, 20, and 100 μg/ml. We tested 87 compounds prepared from a variety of sources with different levels and patterns of sulfation. Initial testing identified 50 compounds with inhibitory activity of greater than 20% when tested at a concentration of 20 μg/ml or lower, with 14 compounds being highly inhibitory at concentrations of 2 μg/ml (Table 4). The other 37 compounds showed weak or no inhibitory activity and were not further studied (Table 5). The stage specificity of inhibition of merozoite invasion by the inhibitory compounds with an IC50 of <10 μg/ml in growth inhibition assays was confirmed in direct invasion inhibition assays using purified merozoites (Fig. 4) (4, 31). The most highly inhibitory compounds, which were those with an IC50 of <2 μg/ml, as determined in standard growth inhibition assays, and those that had confirmed invasion-inhibitory activity, were chemically oversulfated ι-carrageenan, inulin sulfate, propylene glycol alginic sulfate, psyllium sulfate, scleroglucan sulfate, tragacanth sulfate, xylan sulfate (also known as pentosan polysulfate), chemically oversulfated λ-carrageenan, pullulan sulfate, and chemically oversulfated dextran. De-N-acetylated dermatan sulfate and de-N-acetylated heparin (bovine) also had inhibitory activity in standard 48-h growth inhibition assays; however, this was not confirmed in direct invasion inhibition assays, suggesting that the inhibition seen in the growth inhibition assays was not specific to merozoite invasion and may have been due to the nonspecific activity of the sample or other mechanisms of inhibition. The anticoagulation activity of highly inhibitory compounds was tested by assessing the activated partial thromboplastin time. All compounds had reduced anticoagulation activity compared to heparin (Table S3), indicating that these compounds may be more suitable base compounds for future drug development.
TABLE 4.
HLMs and sulfated polysaccharides with inhibitory activity against merozoite invasion in growth inhibition assaysa
| Inhibitor [reference(s)] | % inhibition (SEM) |
|---|---|
| Very strong inhibitors | |
| Inulin sulfate | 93 (4.5) |
| De-N-Ac Hep (bovine)b | 92 (2.5) |
| Dextran sulfatec (40, 41) | 90 (0.3) |
| Xylan sulfated | 86 (6.4) |
| Propylene glycol alginic sulfate | 77 (3.6) |
| Chemically oversulfated N-Ac Hep | 72 (1.3) |
| De-N-Ac Hepb (porcine) | 72 (1.3) |
| De-N-Ac dermatan Sb | 67 (13.7) |
| Chemically oversulfated λ-carrageenanc (13, 42) | 65 (10.4) |
| Tragacanth sulfate | 64 (11.7) |
| Chemically oversulfated ι-carrageenan (13) | 64 (6.4) |
| Scleroglucan sulfate | 55 (8) |
| Strong inhibitors | |
| Cyclodextrin sulfate | 96 (2) |
| Welan sulfate | 96 (1.3) |
| Agarose sulfate | 95 (1.4) |
| Arabic sulfatee | 95 (1.6) |
| Glycogen sulfate | 95 (2.5) |
| Phenoxyacetyl cellulose sulfate | 95 (2.3) |
| Chemically oversulfated free amino Hep | 95 (2.5) |
| Konjac glucomannan sulfate | 95 (2.8) |
| Levan sulfate | 94 (3.2) |
| Pullulan sulfatec | 94 (1.2) |
| Taramind sulfate | 94 (1.5) |
| Ghatti sulfate | 92 (2.8) |
| λ-Carrageenane (13, 42) | 90 (2) |
| Paramylon sulfate | 90 (1.9) |
| Psyllium seed gum sulfate | 89 (2) |
| Stachyose sulfate | 89 (4.6) |
| Agarose sulfatec | 87 (2.6) |
| Chemically oversulfated κ-carrageenane (13, 42) | 85 (2.9) |
| Gellan sulfate (82) | 82 (9.6) |
| Amylopectin sulfate | 74 (1) |
| Tara sulfateb | 71 (3.7) |
| Heparin (bovine lung) (7) | 70 (8.1) |
| Guar sulfatec | 68 (7.1) |
| Alginic sulfate | 62 (11) |
| Psyllium sulfate | 56 (10) |
| Moderate inhibitors | |
| Dextrin sulfate | 56 (9.1) |
| Karaya sulfate | 97 (0.1) |
| Dextrin sulfate | 96 (0) |
| Fucogalactan sulfate | 96 (1) |
| Arabic sulfate | 94 (1) |
| Carboxymethyl cellulose sulfate | 94 (1.1) |
| Amylose sulfate | 86 (2.1) |
| Pectin sulfatec | 84 (2) |
| Locust bean gum sulfate | 79 (3.7) |
| Chitosan sulfate | 61 (2.7) |
| Guar sulfate | 59 (3.6) |
The compounds at 2, 10, and 20 μg/ml each were tested in growth inhibitory assays. Inhibitory compounds are listed according to their estimated IC50 and ordered on the basis of their inhibitory activity. Very strong inhibitors had estimated IC50s of <2 μg/ml, strong inhibitors had estimated IC50s of 2 to 10 μg/ml, and moderate inhibitors had estimated IC50s of 10 to 20 μg/ml. Inhibitory activity at 2 for very strong inhibitors, 10 for strong inhibitors, and 20 for moderate inhibitors (in μg/ml) is as indicated. Data are means ± SEMs from two assays performed in duplicate. Highly inhibitory compounds were additionally screened in invasion inhibition assays with purified merozoites to confirm targeting of merozoite invasion. Where published previously, the reference numbers are listed beside the compounds. Abbreviations: de-N-Ac, de-N-acetylated; Hep, heparin; N-Ac, N-acetylated.
The compounds were noninhibitory in invasion inhibition assays, suggesting that these samples may contain a nonspecific growth inhibitory substance or act through a mechanism separate to invasion inhibition.
Prepared using pyridine sulfur trioxide complex.
Xylan sulfate is also known as pentosan polysulfate.
Prepared using piperidine-N-sulfonic acid.
TABLE 5.
Carbohydrate compounds with weak or no inhibitory activity against merozoite invasion in growth inhibition assaysa
| Inhibitor (reference) | % inhibition (SD) |
|---|---|
| Weak inhibitors | |
| Alginic sulfateb | 35 (0) |
| Ardeparin | 62 (1) |
| Certoparin | 65 (4) |
| Curdlan sulfate (10) | 30 (0) |
| Dalteparin (LMM heparin) | 70 (5) |
| De-N-sulfated enoxiparin | 66 (0) |
| Enoxiparin (LMM heparin) | 63 (5) |
| Ghatti sulfatec | 73 (3) |
| Hypromellose sulfate | 20 (1) |
| Locust bean gum sulfatec | 46 (3) |
| N-Ac enoxaparin | 21 (1) |
| Pectin sulfateb | 28 (3) |
| Pullulan sulfate | 41 (6) |
| Reviparin | 60 (0) |
| Sulodexide | 37 (2) |
| Tinzaparin | 70 (6) |
| Tylose sulfate | 23 (1) |
| Noninhibitors | |
| Chitosan sulfatec | 19 (2) |
| CSA | 1.2 (3) |
| CSC (7) | 0.8 (2) |
| Curdlan sulfateb (10) | 11 (0) |
| Danaparoid | 4 (1) |
| De-N-Ac chitosan | 0 (1) |
| De-N-Ac CSC | 0 (1) |
| Ethyl cellulose sulfate | 4 (8) |
| Gum rosin sulfate | 3 (2) |
| Hyaluronic acid | 0 (3) |
| Heparan sulfate | 6 (6) |
| Hydroxyethyl cellulose sulfate | 0 (0) |
| Methylcellulose sulfate | 0 (0) |
| N-Propylated heparin (porcine) | 3 (1) |
| Propylmethyl sulfate | 0 (3) |
| Scleroglucan sulfatec | 0 (0) |
| Starch sulfate | 13.3 (1) |
| Storax sulfate | 10 (1) |
| Xanthan sulfate | 6 (3) |
The compounds were tested in growth inhibition assays at 2, 10, 20, and 100 μg/ml. Weakly inhibitors had estimated IC50s of between 20 and 100 μg/ml, and noninhibitors showed <20% growth inhibition at 100 μg/ml. The inhibitory activity at 100 μg/ml is indicated. Data are means ± SEMs from two assays performed in duplicate. Abbreviations: CS, chondroitin sulfate; LMM, low molecular mass; N-Ac, N-acetylated; de-N-Ac, de-N-acetylated.
Prepared using piperidine-N-sulfonic acid.
Prepared using pyridine sulfur trioxide complex.
FIG 4.

Invasion-inhibitory activity of sulfated polysaccharides. Highly active HLMs and sulfated carbohydrates were tested in invasion inhibition assays to confirm their activity against merozoite invasion. All compounds were tested at 10 μg/ml. Data are expressed as the percent inhibition from one assay performed in duplicate relative to the inhibition caused by PBS as a reference control. Three CSC-negative controls were included in the assay, and all were noninhibitory (data not shown). †, prepared using pyridine sulfur trioxide complex; $, prepared using piperidine-N-sulfonic acid.
DISCUSSION
Merozoite invasion of the RBC is a critical step during parasite infection and an attractive target for therapeutics that may have potential for use in combination with current antimalarials (4, 5, 7). While heparin has been used as adjunctive treatment for malaria complications, its use as a therapeutic in malaria is no longer recommended due to the risk of serious bleeding-related side effects from its high anticoagulant activity (33). In the study described here, we identified chemical modifications of heparin that increase the inhibitory activity of merozoite invasion, such as periodate oxidation of nonsulfated uronic acid residues and treatment to esterify hydroxyl groups. Importantly, periodate treatment has been reported to greatly reduce the anticoagulation activity of heparin (30), suggesting that the development of HLMs that have high antimalarial activity and reduced anticoagulation activity may be possible. Similarly treated HLMs have recently been tested for inhibition of lung cancer growth in mice and have no anticoagulation activity or toxicity in heart, liver, kidney, or lung tissue (34). On the other hand, desulfation and de-N-acetylation significantly decreased thei inhibitory activity, consistent with sulfation mediating inhibition and the findings of our prior published studies (7). We demonstrated that highly sulfated heparin and chondroitin sulfate compounds have substantial inhibitory activity against P. falciparum merozoite invasion. Short-chain heparins and heparin and HS tetra- and hexasaccharides, along with chemically oversulfated CSB polysaccharides and naturally sulfated CSE polysaccharides and oligosaccharides, were identified to have inhibitory activity.
As with heparin-like compounds, chondroitin sulfate compounds have been proposed to be the basis for the development of drugs with a number of applications, suggesting that these compounds may have use as base molecules for antimalarial drug development (35). CSE demonstrated significant inhibitory activity that appeared to target initial contact events of merozoite invasion and to have a low level of inhibitory activity against schizont rupture, similar to the previously reported mechanisms of inhibition of heparin (7). However, CSE was unable to inhibit the binding of MSP1-42, which was previously identified to be a target of heparin inhibition (7), suggesting that CSE may target another merozoite surface protein. Multiple merozoite microneme and rhoptry proteins have been reported to bind heparin (17–19). However, the timing of CSE inhibitory activity is at the initial contact/preinvasion steps, prior to the substantial deformation of the RBC that is triggered by these proteins (16), which suggests that the CSE inhibitory function targets merozoite surface proteins that are thought to be involved in these initial stages of invasion.
The targeting of inhibitory HLMs to prevent the early stages of invasion suggests that HLMs inhibit the binding of the merozoite to the RBC by disruption of the receptor-ligand interaction to sulfated receptors. These initial contact events are thought to be mediated by multiple merozoite surface proteins via low-affinity interactions with the RBC surface. As many of these interactions are likely to be with sulfated surface receptors, the ability of HLMs to disrupt multiple interactions across multiple invasion steps is likely to ensure the efficacy of HLMs across all parasite strains and limit the emergence of drug resistance. Indeed, previous attempts to induce heparin resistance in vitro have failed (7). It is possible that HLMs are active at different stages of the parasite life cycle, as suggested by the weak inhibitory effect on schizont rupture. It is possible that HLMs may also function by coating the RBC surface rather than the merozoite, further contributing to the inability to induce resistant parasites.
A major priority for the future development of drugs on the basis of this approach is the generation of compounds with much greater potency. This might be achieved through the chemical modification of compounds or through the synthesis of mimetics with a mechanism of action similar to that of the compounds evaluated in this study. Further, inhibitory HLMs and the CS oligosaccharide compounds identified to have inhibitory activity may be the basis for the development in the future of modified HLMs with increased bioavailability and improved inhibitory activity. Modifications may include those identified above, including periodate treatment and esterification. The activity of 4-mer and 6-mer oligosaccharides suggests that it may be possible to identify and optimize short saccharides with high invasion-inhibitory activity; while our data suggest that a 6-mer oligosaccharide is needed for the inhibitory activity of heparin, stachyose sulfate (a tetramer) was also identified to be a strong inhibitor of invasion. At this time, there is little information available concerning the oral availability of the active compounds reported here. However, there have been efforts to improve the oral availability of heparin derivatives (reviewed in reference 36), and the expectation is that such approaches would also prove effective for these compounds, if required. Among the successful methods that have been reported are the use of conjugates with polycarbophil-cysteine (37) and deoxycholic acid (38). Further, the use of nanoparticles has been reported both to improve oral availability and to prolong HLM drug activity (39), which may allow heparin-based compounds to remain active for multiple parasite life cycles.
Alternatively, the inhibitory sulfated polysaccharides identified here from the testing of a large panel of sulfated polysaccharides prepared from a wide range of sources may be used as base molecules for future drug development. We identified a number of highly inhibitory compounds targeting merozoite invasion with estimated IC50s of <10 μg/ml: agarose sulfate, alginic sulfate, amylopectin sulfate, arabic sulfate, cyclodextrin sulfate, chemically oversulfated ι-carrageenan, λ-carrageenan, chemically oversulfated λ-carrageenan, dextran sulfate, dextrin sulfate, gellan sulfate, ghatti sulfate, glycogen sulfate, guar sulfate, inulin sulfate, Konjac glucomannan sulfate, levan sulfate, paramylon sulfate, phenoxyacetyl cellulose sulfate, pullulan sulfate, propylene glycol alginic sulfate, psyllium sulfate, scleroglucan sulfate, tragacanth sulfate, taramind sulfate, welan sulfate, and xylan sulfate. While dextran sulfates, carrageenans, gellan sulfates, and xylan sulfate have previously been reported to inhibit P. falciparum growth in vitro (13, 40–42), the remaining compounds have not previously been identified to be P. falciparum inhibitors. All of these compounds had greater inhibitory activity than heparin, with IC50s of less than 10 μg/ml. Importantly, these compounds have a reduced anticoagulation potential compared with that of heparin, suggesting that these compounds are more suitable for future drug development, as bleeding-related complications would be avoided with these compounds. For future drug development, base compounds may be extracted from natural sources. Indeed, the extraction of sulfated seaweed polysaccharides, such as carrageenans from algae, has become routine due to their broad application (reviewed in reference 43). The compounds identified here may also be used as the basis for future structure/functional studies and the development of small-molecule inhibitors that can be synthetically developed. The synthetic and chemically modified non-glycosaminoglycan-based compounds investigated in this study have the additional advantages of being isolated from nonmammalian sources, circumventing possible concerns with prion diseases or the provenance of supplies of mammalian origin. This is of particular relevance following recent reports regarding the contamination of pharmaceutical-grade heparin (44, 45). Further, while this work has focused on the inhibition of merozoite invasion, the compounds identified here may also have further therapeutic benefit by disrupting parasite sequestration and rosette formation (15, 20–26). The combined ability to disrupt two separate stages of the parasite life cycle increases the time window of activity of any dual-acting compounds. Further studies are needed to assess whether the structural features required for merozoite invasion inhibition are also important in sequestration and rosette inhibition. However, the pursuit of sulfate HLMs as base compounds for novel drug development is supported by several reports of the capacity of similar compounds to inhibit both parasite invasion and parasite sequestration in in vivo models (11, 14, 25, 40, 46, 47).
In conclusion, this work has identified a number of carbohydrate compounds with high inhibitory activity against merozoite invasion of RBCs, and a number of modifications that enhance the inhibitory activity were identified. The optimization of highly inhibitory compounds on the basis of these observations may provide opportunities for the development of novel therapeutics useful in combating malarial disease.
MATERIALS AND METHODS
Parasite culture.
P. falciparum 3D7 or D10-PfGFP isolates (48) were cultured as described previously (49, 50) in culture medium consisting of RPMI-HEPES (pH 7.4) supplemented with 50 μg/ml hypoxanthine, 20 μg/ml gentamicin, 25 mM sodium bicarbonate (NaHCO3), and 0.5% AlbuMAX II (Gibco). RBCs from group O-positive blood donors were used to culture the parasites. Ethical approval for use of RBCs for parasite culture was obtained from the Alfred Human Research and Ethics Committee for the Burnet Institute. Cultures were gassed with 1% O2, 4% CO2, 95% N2 and incubated at 37°C. The parasites were initially synchronized using 5% d-sorbitol treatment for 5 min, as described previously (51). For invasion inhibition assays, sorbitol treatment cultures were further synchronized using heparin synchronization; heparin cannot be used for the selection of heparin-resistant cultures; therefore, it is unlikely that heparin synchronization affected the testing of the HLMs (7). Live video filming of merozoite invasion was performed as described previously (7, 32).
Growth inhibition assays and invasion inhibition assays.
High-throughput growth inhibition assays were performed as described previously (48, 50, 52, 53). Duplicate suspensions of synchronized parasites at 2% parasitemia and 1% hematocrit were incubated with compounds in 96-well sterile U-bottom plates (Falcon) for 44 h for one-cycle assays or 72 h for two-cycle assays and analyzed by flow cytometry with staining of parasites with 10 μg/ml ethidium bromide (Bio-Rad) for 1 h in darkness. The level of parasitemia was measured using a BD FACSCalibur or BD FACSCanto II flow cytometer. Samples were analyzed using FlowJo software (TreeStar) with gating on intact RBCs and then determining the level of parasitemia by the use of ethidium bromide-positive RBCs. The inhibitory effects of the compounds were normalized by comparison of the growth with that of the growth controls, and for each assay the results are presented as a percentage of the growth of the controls.
Invasion inhibition assays with isolated merozoites were conducted as described previously (4, 31) (for detailed methods, see Methods in Malaria Research [54]). Highly synchronized late-stage schizonts were magnet purified by use of a magnetically activated cell sorting (MACS) magnet separation column (Miltenyi Biotec) and treated with trans-epoxysuccinyl-l-leucylamido(4-guanidino)butane E64 until mature merozoites were formed. Merozoites were isolated by membrane filtration and incubated with uninfected RBCs at 0.5% hematocrit and the test compounds at the concentrations indicated above in 50-μl volumes. The estimated number of merozoites per test was approximately 7 × 106. Invasion occurred under agitated conditions for 10 min and then under static conditions for a further 20 min. Following invasion, the cultures were washed twice and returned to the culture medium. The parasites were analyzed by flow cytometry at 40 h postinvasion as described above for the growth inhibition assays.
Modification of heparin-like-molecules.
Porcine mucosal heparan sulfates (HS) (catalog number HO-10595, a 12- to 15-kDa highly sulfated HS, and catalog number HS1098, a 15-kDa HS with a low level of sulfation) and 12.5-kDa mucosal heparin (MH) were purchased from Celsus Laboratories, Inc. (Cincinnati, OH, USA). Bovine lung heparin (LH) was from Calbiochem (Melbourne, Australia); sulodexide (consisting of heparin with a low level of sulfation and low-molecular-mass dermatan sulfate at an 80:20 ratio) was purchased as Vessel, manufactured by Alfa Wasserman, Bologna, Italy; Arixtra (a synthetic heparin pentasaccharide) was from GlaxoSmithKline; enoxaparin (a 3-kDa low-molecular-mass heparin) was purchased as Clexane from Sanofi-Adventis; and bemiparin (a 3-kDa low-molecular-mass heparin) was purchased as Hibro, manufactured by Laboratorios Farmaceuticos Rovi SA.
The heparin compounds were modified by published methods, as follows. Glycol-split heparins and partially (50%) glycol-split heparin were prepared by periodate oxidation followed by NaBH4 reduction as described previously (30, 55, 56). Fully desulfated heparins were prepared by treatment of their pyridinium salts with dimethyl sulfoxide containing 10% water at 100°C by the method of Nagasawa et al. (57). De-N-sulfated and partially de-N-sulfated heparins were prepared by treatment of their pyridinium salts with dimethyl sulfoxide containing 5% methanol for up to 1.5 h at 50°C (58). De-N-sulfated heparins were N-acetylated by treatment of heparin with acetic anhydride in 0.5 M NaHCO3 at 4°C (59). 2-O-Desulfated heparins and glycol-split heparins were prepared as described previously by dissolving the heparin in 0.2 M NaOH followed by lyophilization (60), using an adaptation of a previously described method (61). Mucosal heparin (porcine) lacking 6-O-sulfate (MH de-6-S) was prepared by treatment of the pyridinium salt of heparin with N,O-bis(trimethylsilyl)acetamide in pyridine for 2 h at 60°C (62). Heparin was decarboxylated by treatment with 1-ethyl-3-(3-dimethyl-aminopropyl)carbodiimide and subsequent sodium borohydride reduction as described previously (63; adapted from reference 64).
Mucosal heparin (porcine) 5 kDa in length (obtained by Smith degradation) was prepared by treatment of periodate-oxidized mucosal heparin (porcine) with sodium hydroxide, followed by reduction with sodium borohydride and acid hydrolysis (65). O-acylated derivatives (butyl and hexyl) of heparin fragments were prepared from their tributylammonium salts in N,N-dimethylformamide using carboxylic acid anhydrides and 4-(dimethylamino)pyridine as the catalyst (66, 67). The 3- to 4-kDa MH peroxide (MH H2O2) was prepared by hydrogen peroxide-induced free radical degradation (68). The 3-kDa MH gc (glycol-split mucosal heparin [porcine]) was prepared by limited (10-min) nitrous oxide degradation of glycol-split heparin at pH 4 (reaction A, which cleaves both at glucosamine [Glc]NS and at GlcNH) using the method of Lindahl et al. (69), followed by sodium borohydride reduction. The 3-kDa MH gc-CHO was prepared by the above-described method without subsequent borohydride reduction to leave a terminal reactive aldehyde moiety. Hydrazone derivatives of the 3-kDa MH gc-CHO with 4-phenylsemicarbazide or benzhydrazide were prepared with a 5-fold molar excess of 4-phenylsemicarbazide or benzhydrazide in 100 mM sodium acetate, pH 6, overnight at room temperature (20°C). Reductive amination of the 3-kDa MH gc-CHO with anthranilic acid or 1,3,6-tri-SO3-aminonaphthaline (ANTS) was performed with a 5-fold molar excess of anthranilic acid or ANTS and a 25-fold molar excess of sodium cyanoborahydride (NaBH3CN) in 100 mM sodium acetate, pH 6, overnight at room temperature (20°C).
Confirmation of chemical modifications.
Following de-N-sulfation of the glucosamine residues in the heparin derivatives and their subsequent re-N-acetylation, the presence or absence of unsubstituted glucosamine GlcNH was determined by degradation of the derivative by nitrous acid at pH 4 using reaction B (69), which cleaves only adjacent to unsubstituted glucosamine residues, and analysis on polyacrylamide gels to determine the reduction of the size. The reaction was also quantified by colorimetric analysis of the resultant anhydromannose residues by reaction with 3-methyl-2-benzothiazolinone hydrazone (70). Size analysis by polyacrylamide gel electrophoresis (PAGE) was used to demonstrate that no degradation of the modified heparins had occurred following glycol splitting. The apparent sizes of the heparin fragments cleaved by peroxide (periodate of nitrous acid cleavage) were determined by PAGE analysis using a mini-gel apparatus (Bio-Rad, Hercules, CA). The fragments were run on 15% resolving gels or 30% Tris-glycine gels (71, 72), and known heparin-derived molecular mass standards of 16.7, 10.6, 6.7, and 3.1 kDa, which were a generous gift from Nova Nordisk (Gentofte, Denmark), were used to determine the size of the fragments (73). In addition, the structures of carboxyl-reduced heparins, the de-2-O-sulfate heparins, and the glycol-split derivatives were analyzed by polyacrylamide gel electrophoresis, and the chemical structures were determined by 1H nuclear magnetic resonance spectroscopy, as previously published (60, 63).
Preparation of oligosaccharide fractions from heparin, heparan sulfate, and chondroitin sulfate E.
Heparin and HS oligosaccharide fragments were prepared as described previously (74). Briefly, heparin (200 mg; from porcine intestinal mucosa; Sigma-Aldrich) was incubated with heparin lyase I (100 U; Sigma-Aldrich) and HS (200 mg; from porcine intestinal mucosa; Celsus Laboratories, Inc.) with heparinase III (650 mU; Ibex Technologies, Montreal, Canada) (75) in 5 mM sodium phosphate buffer (pH 7.1) containing 0.2 M NaCl. Digestion was carried out at 30°C and stopped when the reaction was 40% complete. After desalting on a short Sephadex G10 column, the oligosaccharides were fractionated on a Bio-Gel P-4 column (1.6 by 90 cm) with elution by 0.1 M NH4Cl (pH 3.5).
CSE (2 mg; from squid cartilage; AMS Biotechnology, Abingdon, England) was digested with 20 mU of chondroitinase ABC (Sigma) in the same phosphate buffer described above (400 μl) under conditions identical to those described above. Oligosaccharide fractionation was carried out on a Superdex peptide column (Amersham Biosciences, Little Chalfont, England) with elution with 0.05 M ammonium acetate.
The tetra- and hexasaccharide fractions were subfractionated by strong-anion exchange on a short cartridge column (1 ml; HiTrap Q-Sepharose HP; Amersham Biosciences) with UV detection at 232 nm. Elution was carried out with a linear gradient of NaCl (solvent A, 0.1 M NaCl; solvent B, 1.5 M NaCl, pH 3.5) as described previously (76). Oligosaccharide subfractions were collected, desalted, and freeze-dried before quantitation by carbazole assay for hexuronic acid content (77).
Heparin binding assays.
For heparin-agarose bead binding assays, proteins were extracted from P. falciparum schizonts and placed into 1% Triton X-100 in phosphate-buffered saline (PBS) as described previously (78). Proteins from the culture supernatants were collected by allowing highly synchronous schizonts to rupture into protein-free culture medium, and cells were removed by centrifugation. Binding of solubilized proteins to heparin-agarose beads was performed as described previously (7); heparin-agarose beads were washed twice in PBS and then blocked with 1% casein in PBS overnight at 4°C. Schizont protein extracts were incubated overnight at 4°C with beads containing 0.1% casein and 200 μg/ml of test inhibitor (heparin from Sigma-Aldrich; de-6-OS-heparin from Iduron, Alderley Edgy, United Kingdom; CSE from Sikagaku, Tokyo, Japan; and CSC from Sigma-Aldrich) or PBS as a control (50 μl of packed beads plus 100 μl of protein supernatant). Unbound proteins in the supernatant were collected through micro-Bio-Spin chromatography columns (Bio-Rad), and the beads were washed 5 times with PBS containing 0.1% casein, 1% Triton X-100, and protease inhibitors. Bound proteins were eluted from the beads with 50 μl of warmed reducing sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) sample buffer. Bound and unbound proteins were separated by SDS-PAGE under reducing conditions and transferred onto polyvinylidene difluoride membranes for probing with antibodies to detect MSP1-19 by Western blotting. MSP1-19 antibodies were raised in rabbits and purified as described previously (79). Use of rabbits for antibody generation was approved by the University of Melbourne Animal Ethics and Welfare Committee.
Heparin binding to recombinant MSP1-42 was determined by enzyme-linked immunosorbent assay (ELISA) as described previously (7). Recombinant MSP1-42 (expressed as His-tagged proteins in Escherichia coli [80]; from Carole Long, National Institutes of Health) was coated (1 μg/ml) onto 96-well plates (Nunc MaxiSorp) in PBS overnight at 4°C. The plates were washed, blocked with 1% casein, and then incubated with heparin-bovine serum albumin (BSA) or BSA, along with increasing concentrations of soluble inhibitors of heparin, CSC, and CSE. The plates were washed, and bound heparin-BSA or BSA was detected with anti-BSA antibodies (rabbit; Sigma-Aldrich), followed by anti-rabbit immunoglobulin-conjugated horseradish peroxidase and 2,2′-azino-bis(3-ethylbenzthiazoline-6-sulfonic acid) (Sigma-Aldrich). All incubations were performed in PBS with 0.1% casein and 0.05% Tween 20 for 1 h at room temperature.
Chemical sulfation of polysaccharides.
The sulfation of all non-glycosaminoglycan-based carbohydrates was achieved utilizing chlorosulfonic acid, unless indicated otherwise in the tables footnotes and figure legends, where sulfation was carried out essentially as described by Yoshida et al. using pyridine sulfur trioxide complex and piperidine-N-sulfonic acid (81). Carbohydrates were purchased from Sigma-Aldrich, Dextra Laboratories, Celsus Laboratories, Novartis, Wako Chemicals, and EDQM (Conseil de l'Europe), as indicated in Table S2 in the supplemental material. Precursor carbohydrates requiring sulfation (500 mg) were added to prechilled dry pyridine (VWR) in advance of the addition of chlorosulfonic acid (1:16, vol/vol; VWR). The mixture was incubated at 95°C for 2 h prior to cooling with the assistance of an ice bath. Sodium hydroxide (10 M; Thermo Fisher) was added to the mixture with stirring until precipitation occurred. The contents were subsequently transferred to ice-cold ethanol (VWR) presaturated with sodium acetate (VWR). The precipitate was washed extensively before dissolution in and dialysis (3.5-kDa cutoff; Medicell Membranes) against double-distilled H2O. The dialyzed solution was frozen and lyophilized before size exclusion chromatography was performed using high-performance liquid chromatography (HPLC)-grade H2O (Fisher) and a prepacked PD-10 column (GE Healthcare) per the manufacturers' instructions. The sulfation of highly inhibitory compounds was confirmed by recording the attenuated total reflectance Fourier transform infrared spectroscopy (FTIR) spectra using a Nicolet iS5 IR-TF spectrometer (Thermo Fisher) at the Institute of Science and Technology for Medicine Facility of Keele University, scanning in the 4,000- to 400-cm−1 region with a spectral resolution of 2 cm−1 over 32 scans (Fig. S1). The spectrum of background air was obtained and subtracted from all spectra. All carbohydrate spectra were recorded using Thermo Fisher Omnic software. In order to further improve the comparison between samples, the mean for 5 FTIR spectra per sample was normalized to the relative absorbance (i.e., dividing the absorbance value of each point of the spectrum by the ratio of a mutually common and identical spectral region for each precursor and modified polysaccharide pair). First derivatives of all spectral data for precursor/modified polysaccharide pairs were plotted and overlaid using Prism software (GraphPad Software, Inc.).
Assessing anticoagulation activity by activated partial thromboplastin time.
The anticoagulation activity of a subset of sulfated polysaccharides which had high merozoite-inhibitory activity was assessed by measuring the activated partial thromboplastin time. Cuvettes, ball bearings, 50 mM calcium chloride, and test compounds (or controls) were all prewarmed to 37°C using a Thrombotrack Solo coagulation analyzer (Axis-Shield). Fifty microliters of normal human citrated plasma, 25 μl of aqueous test sample or an HPLC-grade water control, and 50 μl of the Pathromtin SL reagent (Siemens) were incubated in a cuvette for 2 min at 37°C. The time for clot formation to occur was ascertained immediately following the addition of 25 μl of a 50 mM calcium chloride solution to the cuvette.
Statistical analysis.
Statistical analysis was performed in GraphPad Prism (version 6) software. Comparison of the activity of the individual parent compound and its modified HLM in growth inhibition assays was performed using paired t tests for each combination of compounds. For each individual modification, P values were adjusted using the Holm-Sidak method to decrease the risk of determination of false discovery rates due to the performance of multiple comparisons. The overall impact of a specific modification (for example, de-2-sulfation) was assessed by the Wilcoxon matched-pair sign-rank test for all pairs of parent and modified compounds. For all comparisons, a P value of <0.05 was considered statistically significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Carole Long for providing the recombinant MSP1-42 protein and the Red Cross Blood Bank (Melbourne, Australia) for providing RBCs for parasite culture.
This work was supported by the National Health and Medical Research Council of Australia (a project grant to J.G.B. and an early career fellowship to M.J.B.), the Australian Government (an Australia postgraduate award to M.J.B.), the University of Melbourne (a top-up award to M.J.B.), the Wellcome Trust of the United Kingdom (to W.C., M.S., and E.Y.), the Royal Society (to M.S. and P.H.), the Medical Research Council of the United Kingdom (to M.S., L.C., and E.Y.), the Engineering and Physical Sciences Research Council (to M.S.), and the Biotechnology and Biological Sciences Research Council of the United Kingdom (to M.S. and E.Y.). The Burnet Institute is supported by the National Health and Medical Research Council Australia Infrastructure for Research Institutes Support Scheme and by Victorian State Government Operational Infrastructure Support.
The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.00709-17.
REFERENCES
- 1.World Health Organization. 2015. World malaria report 2015. WHO Press, Geneva, Switzerland. [Google Scholar]
- 2.Amaratunga C, Lim P, Suon S, Sreng S, Mao S, Sopha C, Sam B, Dek D, Try V, Amato R, Blessborn D, Song L, Tullo GS, Fay MP, Anderson JM, Tarning J, Fairhurst RM. 2016. Dihydroartemisinin-piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect Dis 16:357–365. doi: 10.1016/S1473-3099(15)00487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilson DW, Langer C, Goodman CD, McFadden GI, Beeson JG. 2013. Defining the timing of action of antimalarial drugs against Plasmodium falciparum. Antimicrob Agents Chemother 57:1455–1467. doi: 10.1128/AAC.01881-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boyle MJ, Wilson DW, Beeson JG. 2013. New approaches to studying Plasmodium falciparum merozoite invasion and insights into invasion biology. Int J Parasitol 43:1–10. doi: 10.1016/j.ijpara.2012.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Wilson DW, Goodman CD, Sleebs BE, Weiss GE, de Jong NW, Angrisano F, Langer C, Baum J, Crabb BS, Gilson PR, McFadden GI, Beeson JG. 2015. Macrolides rapidly inhibit red blood cell invasion by the human malaria parasite, Plasmodium falciparum. BMC Biol 13:52. doi: 10.1186/s12915-015-0162-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beeson JG, Drew DR, Boyle MJ, Feng G, Fowkes FJI, Richards JS. 2016. Merozoite surface proteins in red blood cell invasion, immunity and vaccines against malaria. FEMS Microbiol Rev 40:343–372. doi: 10.1093/femsre/fuw001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boyle MJ, Richards JS, Gilson PR, Chai W, Beeson JG. 2010. Interactions with heparin-like molecules during erythrocyte invasion by Plasmodium falciparum merozoites. Blood 115:4559–4568. doi: 10.1182/blood-2009-09-243725. [DOI] [PubMed] [Google Scholar]
- 8.Butcher GA, Parish CR, Cowden WB. 1988. Inhibition of growth in vitro of Plasmodium falciparum by complex polysaccharides. Trans R Soc Trop Med Hyg 82:558–559. doi: 10.1016/0035-9203(88)90504-4. [DOI] [PubMed] [Google Scholar]
- 9.Havlik I, Rovelli S, Kaneko Y. 1994. The effect of curdlan sulphate on in vitro growth of Plasmodium falciparum. Trans R Soc Trop Med Hyg 88:686–687. doi: 10.1016/0035-9203(94)90230-5. [DOI] [PubMed] [Google Scholar]
- 10.Evans SG, Morrison D, Kaneko Y, Havlik I. 1998. The effect of curdlan sulphate on development in vitro of Plasmodium falciparum. Trans R Soc Trop Med Hyg 92:87–89. doi: 10.1016/S0035-9203(98)90969-5. [DOI] [PubMed] [Google Scholar]
- 11.Kisilevsky R, Crandall I, Szarek WA, Bhat S, Tan C, Boudreau L, Kain KC. 2002. Short-chain aliphatic polysulfonates inhibit the entry of Plasmodium into red blood cells. Antimicrob Agents Chemother 46:2619–2626. doi: 10.1128/AAC.46.8.2619-2626.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fleck SL, Birdsall B, Babon J, Dluzewski AR, Martin SR, Morgan WD, Angov E, Kettleborough CA, Feeney J, Blackman MJ, Holder AA. 2003. Suramin and suramin analogues inhibit merozoite surface protein-1 secondary processing and erythrocyte invasion by the malaria parasite Plasmodium falciparum. J Biol Chem 278:47670–47677. doi: 10.1074/jbc.M306603200. [DOI] [PubMed] [Google Scholar]
- 13.Adams Y, Smith SL, Schwartz-Albiez R, Andrews KT. 2005. Carrageenans inhibit the in vitro growth of Plasmodium falciparum and cytoadhesion to CD36. Parasitol Res 97:290–294. doi: 10.1007/s00436-005-1426-3. [DOI] [PubMed] [Google Scholar]
- 14.Crandall IE, Szarek WA, Vlahakis JZ, Xu Y, Vohra R, Sui J, Kisilevsky R. 2007. Sulfated cyclodextrins inhibit the entry of Plasmodium into red blood cells. Implications for malarial therapy. Biochem Pharmacol 73:632–642. [DOI] [PubMed] [Google Scholar]
- 15.Bastos MF, Albrecht L, Kozlowski EO, Lopes SCP, Blanco YC, Carlos BC, Castiñeiras C, Vicente CP, Werneck CC, Wunderlich G, Ferreira MU, Marinho CRF, Mourão PAS, Pavão MSG, Costa FTM. 2014. Fucosylated chondroitin sulfate inhibits Plasmodium falciparum cytoadhesion and merozoite invasion. Antimicrob Agents Chemother 58:1862–1871. doi: 10.1128/AAC.00686-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss GE, Gilson PR, Taechalertpaisarn T, Tham W-H, de Jong NWM, Harvey KL, Fowkes FJI, Barlow PN, Rayner JC, Wright GJ, Cowman AF, Crabb BS. 2015. Revealing the sequence and resulting cellular morphology of receptor-ligand interactions during Plasmodium falciparum invasion of erythrocytes. PLoS Pathog 11:e1004670. doi: 10.1371/journal.ppat.1004670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi K, Takano R, Takemae H, Sugi T, Ishiwa A, Gong H, Recuenco FC, Iwanaga T, Horimoto T, Akashi H, Kato K. 2013. Analyses of interactions between heparin and the apical surface proteins of Plasmodium falciparum. Sci Rep 3:3178. doi: 10.1038/srep03178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baum J, Chen L, Healer J, Lopaticki S, Boyle M, Triglia T, Ehlgen F, Ralph SA, Beeson JG, Cowman AF. 2009. Reticulocyte-binding protein homologue 5—an essential adhesin involved in invasion of human erythrocytes by Plasmodium falciparum. Int J Parasitol 39:371–380. doi: 10.1016/j.ijpara.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi K, Kato K, Sugi T, Takemae H, Pandey K, Gong H, Tohya Y, Akashi H. 2010. Plasmodium falciparum BAEBL binds to heparan sulfate proteoglycans on the human erythrocyte surface. J Biol Chem 285:1716–1725. doi: 10.1074/jbc.M109.021576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skidmore MA, Dumax-Vorzet AF, Guimond SE, Rudd TR, Edwards EA, Turnbull JE, Craig AG, Yates EA. 2008. Disruption of rosetting in Plasmodium falciparum malaria with chemically modified heparin and low molecular weight derivatives possessing reduced anticoagulant and other serine protease inhibition activities. J Med Chem 51:1453–1458. doi: 10.1021/jm701337t. [DOI] [PubMed] [Google Scholar]
- 21.Udomsangpetch R, Wåhlin B, Carlson J, Berzins K, Torii M, Aikawa M, Perlmann P, Wahlgren M. 1989. Plasmodium falciparum-infected erythrocytes form spontaneous erythrocyte rosettes. J Exp Med 169:1835–1840. doi: 10.1084/jem.169.5.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rowe A, Berendt AR, Marsh K, Newbold CI. 1994. Plasmodium falciparum: a family of sulphated glycoconjugates disrupts erythrocyte rosettes. Exp Parasitol 79:506–516. doi: 10.1006/expr.1994.1111. [DOI] [PubMed] [Google Scholar]
- 23.Barragan A, Spillmann D, Kremsner PG, Wahlgren M, Carlson J. 1999. Plasmodium falciparum: molecular background to strain-specific rosette disruption by glycosaminoglycans and sulfated glycoconjugates. Exp Parasitol 91:133–143. doi: 10.1006/expr.1998.4349. [DOI] [PubMed] [Google Scholar]
- 24.Carlson J, Ekre HP, Helmby H, Gysin J, Greenwood BM, Wahlgren M. 1992. Disruption of Plasmodium falciparum erythrocyte rosettes by standard heparin and heparin devoid of anticoagulant activity. Am J Trop Med Hyg 46:595–602. doi: 10.4269/ajtmh.1992.46.595. [DOI] [PubMed] [Google Scholar]
- 25.Vogt AM, Pettersson F, Moll K, Jonsson C, Normark J, Ribacke U, Egwang TG, Ekre H-P, Spillmann D, Chen Q, Wahlgren M. 2006. Release of sequestered malaria parasites upon injection of a glycosaminoglycan. PLoS Pathog 2:e100. doi: 10.1371/journal.ppat.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kyriacou HM, Steen KE, Raza A, Arman M, Warimwe G, Bull PC, Havlik I, Rowe JA. 2007. In vitro inhibition of Plasmodium falciparum rosette formation by curdlan sulfate. Antimicrob Agents Chemother 51:1321–1326. doi: 10.1128/AAC.01216-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Havlik I, Looareesuwan S, Vannaphan S, Wilairatana P, Krudsood S, Thuma PE, Kozbor D, Watanabe N, Kaneko Y. 2005. Curdlan sulphate in human severe/cerebral Plasmodium falciparum malaria. Trans R Soc Trop Med Hyg 99:333–340. doi: 10.1016/j.trstmh.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 28.Rusnati M, Vicenzi E, Donalisio M, Oreste P, Landolfo S, Lembo D. 2009. Sulfated K5 Escherichia coli polysaccharide derivatives: a novel class of candidate antiviral microbicides. Pharmacol Ther 123:310–322. doi: 10.1016/j.pharmthera.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 29.Petitou M, van Boeckel CAA. 2004. A synthetic antithrombin III binding pentasaccharide is now a drug! What comes next? Angew Chem Int Ed Engl 43:3118–3133. doi: 10.1002/anie.200300640. [DOI] [PubMed] [Google Scholar]
- 30.Pisano C, Aulicino C, Vesci L, Casu B, Naggi A, Torri G, Ribatti D, Belleri M, Rusnati M, Presta M. 2005. Undersulfated, low-molecular-weight glycol-split heparin as an antiangiogenic VEGF antagonist. Glycobiology 15:1C–6C. [DOI] [PubMed] [Google Scholar]
- 31.Boyle MJ, Wilson DW, Richards JS, Riglar DT, Tetteh KKA, Conway DJ, Ralph SA, Baum J, Beeson JG. 2010. Isolation of viable Plasmodium falciparum merozoites to define erythrocyte invasion events and advance vaccine and drug development. Proc Natl Acad Sci U S A 107:14378–14383. doi: 10.1073/pnas.1009198107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilson PR, Crabb BS. 2009. Morphology and kinetics of the three distinct phases of red blood cell invasion by Plasmodium falciparum merozoites. Int J Parasitol 39:91–96. doi: 10.1016/j.ijpara.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 33.World Health Organization Malaria Action Programme. 1986. Severe and complicated malaria. Trans R Soc Trop Med Hyg 80(Suppl):3–50. doi: 10.1016/0035-9203(86)90407-4. [DOI] [PubMed] [Google Scholar]
- 34.Yu L, Garg HG, Li B, Linhardt RJ, Hales CA. 2010. Antitumor effect of butanoylated heparin with low anticoagulant activity on lung cancer growth in mice and rats. Curr Cancer Drug Targets 10:229–241. doi: 10.2174/156800910791054176. [DOI] [PubMed] [Google Scholar]
- 35.Yamada S, Sugahara K. 2008. Potential therapeutic application of chondroitin sulfate/dermatan sulfate. Curr Drug Discov Technol 5:289–301. doi: 10.2174/157016308786733564. [DOI] [PubMed] [Google Scholar]
- 36.Neves AR, Correia-da-Silva M, Sousa E, Pinto M. 2016. Strategies to overcome heparins' low oral bioavailability. Pharmaceuticals (Basel) 9:E37. doi: 10.3390/ph9030037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kast CE, Guggi D, Langoth N, Bernkop-Schnürch A. 2003. Development and in vivo evaluation of an oral delivery system for low molecular weight heparin based on thiolated polycarbophil. Pharm Res 20:931–936. doi: 10.1023/A:1023803706746. [DOI] [PubMed] [Google Scholar]
- 38.Park JW, Jeon OC, Kim SK, Al-Hilal TA, Moon HT, Kim CY, Byun Y. 2010. Anticoagulant efficacy of solid oral formulations containing a new heparin derivative. Mol Pharm 7:836–843. doi: 10.1021/mp900319k. [DOI] [PubMed] [Google Scholar]
- 39.Hoffart V, Lamprecht A, Maincent P, Lecompte T, Vigneron C, Ubrich N. 2006. Oral bioavailability of a low molecular weight heparin using a polymeric delivery system. J Control Release 113:38–42. doi: 10.1016/j.jconrel.2006.03.020. [DOI] [PubMed] [Google Scholar]
- 40.Xiao L, Yang C, Patterson PS, Udhayakumar V, Lal AA. 1996. Sulfated polyanions inhibit invasion of erythrocytes by plasmodial merozoites and cytoadherence of endothelial cells to parasitized erythrocytes. Infect Immun 64:1373–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clark DL, Su S, Davidson EA. 1997. Saccharide anions as inhibitors of the malaria parasite. Glycoconj J 14:473–479. doi: 10.1023/A:1018551518610. [DOI] [PubMed] [Google Scholar]
- 42.Recuenco FC, Takano R, Chiba S, Sugi T, Takemae H, Murakoshi F, Ishiwa A, Inomata A, Horimoto T, Kobayashi Y, Horiuchi N, Kato K. 2014. Lambda-carrageenan treatment exacerbates the severity of cerebral malaria caused by Plasmodium berghei ANKA in BALB/c mice. Malar J 13:487. doi: 10.1186/1475-2875-13-487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cunha L, Grenha A. 2016. Sulfated seaweed polysaccharides as multifunctional materials in drug delivery applications. Mar Drugs 14:E42. doi: 10.3390/md14030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guerrini M, Beccati D, Shriver Z, Naggi A, Viswanathan K, Bisio A, Capila I, Lansing JC, Guglieri S, Fraser B, Al-Hakim A, Gunay NS, Zhang Z, Robinson L, Buhse L, Nasr M, Woodcock J, Langer R, Venkataraman G, Linhardt RJ, Casu B, Torri G, Sasisekharan R. 2008. Oversulfated chondroitin sulfate is a contaminant in heparin associated with adverse clinical events. Nat Biotechnol 26:669–675. doi: 10.1038/nbt1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kishimoto TK, Viswanathan K, Ganguly T, Elankumaran S, Smith S, Pelzer K, Lansing JC, Sriranganathan N, Zhao G, Galcheva-Gargova Z, Al-Hakim A, Bailey GS, Fraser B, Roy S, Rogers-Cotrone T, Buhse L, Whary M, Fox J, Nasr M, Dal Pan GJ, Shriver Z, Langer RS, Venkataraman G, Austen KF, Woodcock J, Sasisekharan R. 2008. Contaminated heparin associated with adverse clinical events and activation of the contact system. N Engl J Med 358:2457–2467. doi: 10.1056/NEJMoa0803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Marques J, Vilanova E, Mourão PAS, Fernàndez-Busquets X. 2016. Marine organism sulfated polysaccharides exhibiting significant antimalarial activity and inhibition of red blood cell invasion by Plasmodium. Sci Rep 6:24368. doi: 10.1038/srep24368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen J-H, Lim J-D, Sohn E-H, Choi Y-S, Han E-T. 2009. Growth-inhibitory effect of a fucoidan from brown seaweed Undaria pinnatifida on Plasmodium parasites. Parasitol Res 104:245–250. doi: 10.1007/s00436-008-1182-2. [DOI] [PubMed] [Google Scholar]
- 48.Wilson DW, Crabb BS, Beeson JG. 2010. Development of fluorescent Plasmodium falciparum for in vitro growth inhibition assays. Malar J 9:152. doi: 10.1186/1475-2875-9-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beeson JG, Brown GV, Molyneux ME, Mhango C, Dzinjalamala F, Rogerson SJ. 1999. Plasmodium falciparum isolates from infected pregnant women and children are associated with distinct adhesive and antigenic properties. J Infect Dis 180:464–472. doi: 10.1086/314899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Persson KEM, Lee CT, Marsh K, Beeson JG. 2006. Development and optimization of high-throughput methods to measure Plasmodium falciparum-specific growth inhibitory antibodies. J Clin Microbiol 44:1665–1673. doi: 10.1128/JCM.44.5.1665-1673.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lambros C, Vanderberg JP. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65:418–420. doi: 10.2307/3280287. [DOI] [PubMed] [Google Scholar]
- 52.McCallum FJ, Persson KEM, Mugyenyi CK, Fowkes FJI, Simpson JA, Richards JS, Williams TN, Marsh K, Beeson JG. 2008. Acquisition of growth-inhibitory antibodies against blood-stage Plasmodium falciparum. PLoS One 3:e3571. doi: 10.1371/journal.pone.0003571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Drew DR, Hodder AN, Wilson DW, Foley M, Mueller I, Siba PM, Dent AE, Cowman AF, Beeson JG. 2012. Defining the antigenic diversity of Plasmodium falciparum apical membrane antigen 1 and the requirements for a multi-allele vaccine against malaria. PLoS One 7:e51023. doi: 10.1371/journal.pone.0051023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moll K, Ljungström I, Perlmann H, Scherf A, Wahlgren M. 2013. Methods in malaria research. EVIMalaR, Glasgow, United Kingdom, and MR4/ATCC, Manassas, VA https://www.beiresources.org/Publications/MethodsinMalariaResearch.aspx. [Google Scholar]
- 55.Casu B, Guerrini M, Naggi A, Perez M, Torri G, Ribatti D, Carminati P, Giannini G, Penco S, Pisano C, Belleri M, Rusnati M, Presta M. 2002. Short heparin sequences spaced by glycol-split uronate residues are antagonists of fibroblast growth factor 2 and angiogenesis inhibitors. Biochemistry 41:10519–10528. doi: 10.1021/bi020118n. [DOI] [PubMed] [Google Scholar]
- 56.Naggi A, Casu B, Perez M, Torri G, Cassinelli G, Penco S, Pisano C, Giannini G, Ishai-Michaeli R, Vlodavsky I. 2005. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J Biol Chem 280:12103–12113. doi: 10.1074/jbc.M414217200. [DOI] [PubMed] [Google Scholar]
- 57.Nagasawa K, Inoue Y, Kamata T. 1977. Solvolytic desulfation of glycosaminoglycuronan sulfates with dimethyl sulfoxide containing water or methanol. Carbohydr Res 58:47–55. doi: 10.1016/S0008-6215(00)83402-3. [DOI] [PubMed] [Google Scholar]
- 58.Inoue Y, Nagasawa K. 1976. Selective N-desulfation of heparin with dimethyl sulfoxide containing water or methanol. Carbohydr Res 46:87–95. doi: 10.1016/S0008-6215(00)83533-8. [DOI] [PubMed] [Google Scholar]
- 59.Hopwood JJ, Elliott H. 1981. Selective depolymerisation of heparin to produce radio-labelled substrates for sulfamidase, 2-acetamido-2-deoxy-alpha-d-glucosidase, acetyl-CoA:2-amino-2-deoxy-alpha-d-glucoside N-acetyltransferase, and 2-acetamido-2-deoxy-d-glucose 6-sulfate sulfatase. Carbohydr Res 91:165–190. doi: 10.1016/S0008-6215(00)86029-2. [DOI] [PubMed] [Google Scholar]
- 60.Garg HG, Mrabat H, Yu L, Freeman C, Li B, Zhang F, Linhardt RJ, Hales CA. 2008. Significance of the 2-O-sulfo group of l-iduronic acid residues in heparin on the growth inhibition of bovine pulmonary artery smooth muscle cells. Carbohydr Res 343:2406–2410. doi: 10.1016/j.carres.2008.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jaseja M, Rej RN, Sauriol F, Perlin AS. 1989. Novel regio-stereoselective modifications of heparin in alkaline solution: nuclear magnetic resonance spectroscopic evidence. Can J Chem 67:1449–1456. doi: 10.1139/v89-221. [DOI] [Google Scholar]
- 62.Matsuo M, Takano R, Kamei-Hayashi K, Hara S. 1993. A novel regioselective desulfation of polysaccharide sulfates: specific 6-O-desulfation with N,O-bis(trimethylsilyl)acetamide. Carbohydr Res 241:209–215. doi: 10.1016/0008-6215(93)80107-P. [DOI] [PubMed] [Google Scholar]
- 63.Garg HG, Mrabat H, Yu L, Freeman C, Li B, Zhang F, Linhardt RJ, Hales CA. 2010. Effect of carboxyl-reduced heparin on the growth inhibition of bovine pulmonary artery smooth muscle cells. Carbohydr Res 345:1084–1087. doi: 10.1016/j.carres.2010.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karamanos NK, Hjerpe A, Tsegenidis T, Engfeldt B, Antonopoulos CA. 1988. Determination of iduronic acid and glucuronic acid in glycosaminoglycans after stoichiometric reduction and depolymerization using high-performance liquid chromatography and ultraviolet detection. Anal Biochem 172:410–419. doi: 10.1016/0003-2697(88)90463-0. [DOI] [PubMed] [Google Scholar]
- 65.Islam T, Butler M, Sikkander SA, Toida T, Linhardt RJ. 2002. Further evidence that periodate cleavage of heparin occurs primarily through the antithrombin binding site. Carbohydr Res 337:2239–2243. doi: 10.1016/S0008-6215(02)00229-X. [DOI] [PubMed] [Google Scholar]
- 66.Bârzu T, Desmoulière A, Herbert JM, Level M, Herault JP, Petitou M, Lormeau JC, Gabbiani G, Pascal M. 1992. O-acylated heparin derivatives with low anticoagulant activity decrease proliferation and increase alpha-smooth muscle actin expression in cultured arterial smooth muscle cells. Eur J Pharmacol 219:225–233. doi: 10.1016/0014-2999(92)90300-S. [DOI] [PubMed] [Google Scholar]
- 67.Bârzu T, Level M, Petitou M, Lormeau JC, Choay J, Schols D, Baba M, Pauwels R, Witvrouw M, De Clercq E. 1993. Preparation and anti-HIV activity of O-acylated heparin and dermatan sulfate derivatives with low anticoagulant effect. J Med Chem 36:3546–3555. doi: 10.1021/jm00075a009. [DOI] [PubMed] [Google Scholar]
- 68.Volpi N, Mascellani G, Bianchini P. 1992. Low molecular weight heparins (5 kDa) and oligoheparins (2 kDa) produced by gel permeation enrichment or radical process: comparison of structures and physicochemical and biological properties. Anal Biochem 200:100–107. doi: 10.1016/0003-2697(92)90283-D. [DOI] [PubMed] [Google Scholar]
- 69.Lindahl U, Bäckström G, Jansson L, Hallén A. 1973. Biosynthesis of heparin. II. Formation of sulfamino groups. J Biol Chem 248:7234–7241. [PubMed] [Google Scholar]
- 70.Riesenfeld J, Rodén L. 1990. Quantitative analysis of N-sulfated, N-acetylated, and unsubstituted glucosamine amino groups in heparin and related polysaccharides. Anal Biochem 188:383–389. doi: 10.1016/0003-2697(90)90624-I. [DOI] [PubMed] [Google Scholar]
- 71.Turnbull JE, Hopwood JJ, Gallagher JT. 1999. A strategy for rapid sequencing of heparan sulfate and heparin saccharides. Proc Natl Acad Sci U S A 96:2698–2703. doi: 10.1073/pnas.96.6.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Turnbull JE. 1993. Oligosaccharide mapping and sequence analysis of glycosaminoglycans. Methods Mol Biol 19:253–267. [DOI] [PubMed] [Google Scholar]
- 73.Kristensen HI, Tromborg EM, Nielsen JR, Nielsen JI, Johansen KB, Ostergaard PB. 1991. Development and validation of a size exclusion chromatography method for determination of molecular masses and molecular mass distribution in low molecular weight heparin. Thromb Res 64:131–141. doi: 10.1016/0049-3848(91)90113-B. [DOI] [PubMed] [Google Scholar]
- 74.Chai W, Luo J, Lim CK, Lawson AM. 1998. Characterization of heparin oligosaccharide mixtures as ammonium salts using electrospray mass spectrometry. Anal Chem 70:2060–2066. doi: 10.1021/ac9712761. [DOI] [PubMed] [Google Scholar]
- 75.Leteux C, Chai W, Nagai K, Herbert CG, Lawson AM, Feizi T. 2001. 10E4 antigen of scrapie lesions contains an unusual nonsulfated heparan motif. J Biol Chem 276:12539–12545. doi: 10.1074/jbc.M010291200. [DOI] [PubMed] [Google Scholar]
- 76.Chai W, Beeson JG, Lawson AM. 2002. The structural motif in chondroitin sulfate for adhesion of Plasmodium falciparum-infected erythrocytes comprises disaccharide units of 4-O-sulfated and non-sulfated N-acetylgalactosamine linked to glucuronic acid. J Biol Chem 277:22438–22446. doi: 10.1074/jbc.M111401200. [DOI] [PubMed] [Google Scholar]
- 77.Bitter T, Muir HM. 1962. A modified uronic acid carbazole reaction. Anal Biochem 4:330–334. doi: 10.1016/0003-2697(62)90095-7. [DOI] [PubMed] [Google Scholar]
- 78.Sanders PR, Gilson PR, Cantin GT, Greenbaum DC, Nebl T, Carucci DJ, McConville MJ, Schofield L, Hodder AN, Yates JR, Crabb BS. 2005. Distinct protein classes including novel merozoite surface antigens in raft-like membranes of Plasmodium falciparum. J Biol Chem 280:40169–40176. doi: 10.1074/jbc.M509631200. [DOI] [PubMed] [Google Scholar]
- 79.Stanisic DI, Richards JS, McCallum FJ, Michon P, King CL, Schoepflin S, Gilson PR, Murphy VJ, Anders RF, Mueller I, Beeson JG. 2009. Immunoglobulin G subclass-specific responses against Plasmodium falciparum merozoite antigens are associated with control of parasitemia and protection from symptomatic illness. Infect Immun 77:1165–1174. doi: 10.1128/IAI.01129-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Singh S, Miura K, Zhou H, Muratova O, Keegan B, Miles A, Martin LB, Saul AJ, Miller LH, Long CA. 2006. Immunity to recombinant Plasmodium falciparum merozoite surface protein 1 (MSP1): protection in Aotus nancymai monkeys strongly correlates with anti-MSP1 antibody titer and in vitro parasite-inhibitory activity. Infect Immun 74:4573–4580. doi: 10.1128/IAI.01679-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yoshida T, Yasuda Y, Mimura T, Kaneko Y, Nakashima H, Yamamoto N, Uryu T. 1995. Synthesis of curdlan sulfates having inhibitory effects in vitro against AIDS viruses HIV-1 and HIV-2. Carbohydr Res 276:425–436. doi: 10.1016/0008-6215(95)00186-W. [DOI] [PubMed] [Google Scholar]
- 82.Recuenco FC, Kobayashi K, Ishiwa A, Enomoto-Rogers Y, Fundador NGV, Sugi T, Takemae H, Iwanaga T, Murakoshi F, Gong H, Inomata A, Horimoto T, Iwata T, Kato K. 2014. Gellan sulfate inhibits Plasmodium falciparum growth and invasion of red blood cells in vitro. Sci Rep 4:4723. doi: 10.1038/srep04723. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.