

ABSTRACT
This study assessed the molecular epidemiology, resistance mechanisms, and susceptibility profiles of a collection of 150 extensively drug-resistant (XDR) Pseudomonas aeruginosa clinical isolates obtained from a 2015 Spanish multicenter study, with a particular focus on resistome analysis in relation to ceftolozane-tazobactam susceptibility. Broth microdilution MICs revealed that nearly all (>95%) of the isolates were nonsusceptible to piperacillin-tazobactam, ceftazidime, cefepime, aztreonam, imipenem, meropenem, and ciprofloxacin. Most of them were also resistant to tobramycin (77%), whereas nonsusceptibility rates were lower for ceftolozane-tazobactam (31%), amikacin (7%), and colistin (2%). Pulsed-field gel electrophoresis–multilocus sequence typing (PFGE-MLST) analysis revealed that nearly all of the isolates belonged to previously described high-risk clones. Sequence type 175 (ST175) was detected in all 9 participating hospitals and accounted for 68% (n = 101) of the XDR isolates, distantly followed by ST244 (n = 16), ST253 (n = 12), ST235 (n = 8), and ST111 (n = 2), which were detected only in 1 to 2 hospitals. Through phenotypic and molecular methods, the presence of horizontally acquired carbapenemases was detected in 21% of the isolates, mostly VIM (17%) and GES enzymes (4%). At least two representative isolates from each clone and hospital (n = 44) were fully sequenced on an Illumina MiSeq. Classical mutational mechanisms, such as those leading to the overexpression of the β-lactamase AmpC or efflux pumps, OprD inactivation, and/or quinolone resistance-determining regions (QRDR) mutations, were confirmed in most isolates and correlated well with the resistance phenotypes in the absence of horizontally acquired determinants. Ceftolozane-tazobactam resistance was not detected in carbapenemase-negative isolates, in agreement with sequencing data showing the absence of ampC mutations. The unique set of mutations responsible for the XDR phenotype of ST175 clone documented 7 years earlier were found to be conserved, denoting the long-term persistence of this specific XDR lineage in Spanish hospitals. Finally, other potentially relevant mutations were evidenced, including those in penicillin-binding protein 3 (PBP3), which is involved in β-lactam (including ceftolozane-tazobactam) resistance, and FusA1, which is linked to aminoglycoside resistance.
KEYWORDS: Pseudomonas aeruginosa, whole-genome sequencing, extensively drug resistant, high-risk clones
INTRODUCTION
The increasing prevalence of nosocomial infections produced by multidrug-resistant (MDR), and particularly, extensively drug-resistant (XDR) Pseudomonas aeruginosa strains severely compromises the selection of appropriate treatments and is therefore associated with significant morbidity and mortality (1–3). This growing threat results from the extraordinary capacity of this pathogen for developing resistance to nearly all available antibiotics by the selection of mutations in chromosomal genes and from the increasing prevalence of transferable resistance determinants, particularly those encoding class B carbapenemases (metallo-β-lactamases [MBLs]) or extended-spectrum β-lactamases (ESBLs), frequently cotransferred with genes encoding aminoglycoside-modifying enzymes (4, 5). The emergence of MDR/XDR global clones disseminated in several hospitals worldwide, the high-risk clones, adds further concern (6, 7). Beyond classical molecular epidemiology and phenotypically targeted resistance mechanism assessment, recent whole-genome sequencing (WGS) studies are providing relevant information for building up the complex resistome of MDR/XDR high-risk clones (8–13).
On the other hand, the recent introduction of novel β-lactam–β-lactamase inhibitor combinations, such as ceftolozane-tazobactam, which are stable against AmpC hydrolysis, partially alleviates the urgent clinical need for new agents to combat infections by MDR/XDR P. aeruginosa (14–18). However, the emergence of resistance to these antibiotics is of particular concern and should therefore be closely monitored (17, 19).
Thus, the objective of this work was to determine the molecular epidemiology, resistance mechanisms, and susceptibility profiles of a large collection of recent XDR P. aeruginosa clinical isolates obtained from a multicenter study in Spain, with a particular focus on WGS resistome analysis in relation to ceftolozane-tazobactam susceptibility.
RESULTS AND DISCUSSION
Clonal epidemiology of XDR P. aeruginosa from Spain.
Through SpeI–pulsed-field gel electrophoresis (SpeI-PFGE), 14 unique restriction patterns were documented (not shown). One representative isolate from each pattern and hospital was further genotyped by multilocus sequence typing (MLST), and the results are presented in Fig. 1. As shown, most of the XDR P. aeruginosa isolates studied belonged to well-established P. aeruginosa high-risk clones (7). Among them, sequence type 175 (ST175) was detected in all 9 participating hospitals and was found to be by far the most frequent sequence type, accounting for 68% (n = 101) of the studied collection of XDR P. aeruginosa isolates (Fig. 1A and B), with a frequency ranging from 58% to 100% for each hospital. ST175 was distantly followed by ST244 (n = 16), ST253 (n = 12), ST235 (n = 8), and ST111 (n = 2) (Fig. 1A); indeed, these well-established high-risk clones were not only less common but also less disseminated, each being detected in just 1 to 2 of the participating hospitals (Fig. 1B). With the single exception of ST313 detected in 2 isolates from the same hospital, all the other STs (7%) were detected just in single isolates, including ST179, ST274, ST395, ST455, ST2221, and 4 STs not previously described (ST2533, ST2534, ST2535, and ST2536). Interestingly, one of the isolates, related to ST244, could not be typed due to an indel mutation in mutL, resulting in a mutator phenotype, as confirmed through the determination of the rifampin resistance spontaneous mutation rates (20). This phenomenon has been previously described in the cystic fibrosis (CF) setting (20–24), in which mismatch repair system-deficient mutators (mutS and mutL) are positively selected (25).
FIG 1.

(A) Prevalence of MLST genotypes in the studied collection. (B) Distribution of high-risk clones in participating hospitals. (C) β-Lactam resistance mechanisms detected. AmpC↑ + OprD−, AmpC hyperproduction plus OprD deficiency.
Overall, the obtained results correlated well with those from previous molecular epidemiological surveys performed in Spain, as well as in other countries worldwide, indicating that MDR/XDR profiles are usually conferred by a very limited number of genotypes recognized as high-risk clones (26–34). It is particularly noteworthy that ST175 was already the predominant XDR clone in Spain in 2008, according to the results of a multicenter study on bloodstream infections, indicating the long-term persistence of this clone in Spanish hospitals. In contrast, the prevalences of ST235 and ST111, the high-risk clones showing a wider geographical distribution worldwide, are found to be relatively low in Spain.
Antimicrobial susceptibility profiles and β-lactam resistance mechanisms.
Broth microdilution panels were performed for the whole XDR P. aeruginosa collection. As shown in Table 1, nearly all of the isolates (>95%) were determined to be nonsusceptible to ticarcillin, piperacillin-tazobactam, ceftazidime, cefepime, aztreonam, imipenem, meropenem, and ciprofloxacin, applying both CLSI and EUCAST clinical breakpoints. Moreover, most of the isolates were also nonsusceptible to tobramycin. On the contrary, the highest susceptibility rates were recorded for colistin, amikacin, and the recently introduced antipseudomonal β-lactam–β-lactamase inhibitor combination ceftolozane-tazobactam. Nearly all isolates (97.2%) were susceptible to colistin according to both CLSI and EUCAST breakpoints. However, slight discrepancies in the susceptibility rates for amikacin (92.7% versus 86%) were documented when applying CLSI or EUCAST clinical breakpoints, respectively (Table 1). Likewise, while the susceptibility percentages of ceftolozane-tazobactam were identical according to CLSI and EUCAST (68.7%), slight differences were documented for resistance rates (20.7% versus 31.3%, respectively).
TABLE 1.
Antibiotic susceptibility rates, MIC50, and MIC90 for the 150 XDR P. aeruginosa isolates studied
| Antibiotica | MIC50 (mg/liter) | MIC90 (mg/liter) | CLSI guidelinesb |
EUCAST guidelinesb |
||
|---|---|---|---|---|---|---|
| % S | % R | % S | % R | |||
| TIC | 256 | >512 | 0.0 | 94.0 | 0.0 | 100.0 |
| PIP/TZ | 128 | 256 | 1.3 | 80.0 | 1.3 | 98.7 |
| CAZ | 32 | >64 | 2.0 | 80.7 | 2.0 | 98.0 |
| FEP | 32 | >64 | 3.3 | 60.0 | 3.3 | 96.7 |
| TOL/TZ | 4 | >64 | 68.7 | 20.7 | 68.7 | 31.3 |
| ATM | 16 | 64 | 16.7 | 41.3 | 0.0 | 41.3 |
| IMI | 32 | >64 | 0.0 | 100.0 | 0.0 | 98.7 |
| MER | 16 | >64 | 0.0 | 99.3 | 0.0 | 86.0 |
| CIP | >16 | >16 | 1.3 | 97.3 | 1.3 | 98.7 |
| TOB | 32 | >32 | 22.7 | 74.7 | 22.7 | 77.3 |
| AMI | 4 | 16 | 92.7 | 4.0 | 86.0 | 7.3 |
| COL | 2 | 2 | 97.3 | 2.7 | 97.3 | 2.7 |
TIC, ticarcillin; PIP/TZ, piperacillin-tazobactam; CAZ, ceftazidime; FEP, cefepime; TOL/TZ, ceftolozane-tazobactam; ATM, aztreonam; IMI, imipenem; MER, meropenem; CIP, ciprofloxacin; TOB, tobramycin; AMI, amikacin; COL, colistin.
S, susceptible; R, resistant.
Through the use of phenotypic (cloxacillin inhibition test) and molecular methods (PCR), AmpC hyperproduction plus OprD deficiency (79%) was found to be the main cause of antipseudomonal penicillin, cephalosporin, and carbapenem resistance, whereas the presence of horizontally acquired carbapenemases was detected in 21% of the isolates, mostly including VIM MBLs and GES class A carbapenemases (Fig. 1C).
Interestingly, isolates harboring horizontally acquired carbapenemases accounted for nearly all the ceftolozane-tazobactam resistance documented and belonged to the previously described high-risk clones ST111, ST175, ST235, and ST253 (Data Set S1 in the supplemental material).
The distribution of ceftolozane-tazobactam MICs for the studied collection is shown in Fig. 2. A bimodal distribution was clearly identified, with the second wave, including MICs above 16 μg/ml, accounting for all carbapenemase-producing strains. However, it should be noted that an important proportion of isolates showed MICs of 4 (32%) and 8 (11%) μg/ml. The clinical susceptibility breakpoint for this combination has been set at ≤4 μg/ml by both CLSI and EUCAST. Additionally, only CLSI has defined an intermediate category (MIC = 8 μg/ml). Therefore, the frequent ceftolozane-tazobactam peribreakpoint MICs of XDR isolates argue for the need for clinical studies evaluating optimal dosing and/or the use of combined therapy.
FIG 2.
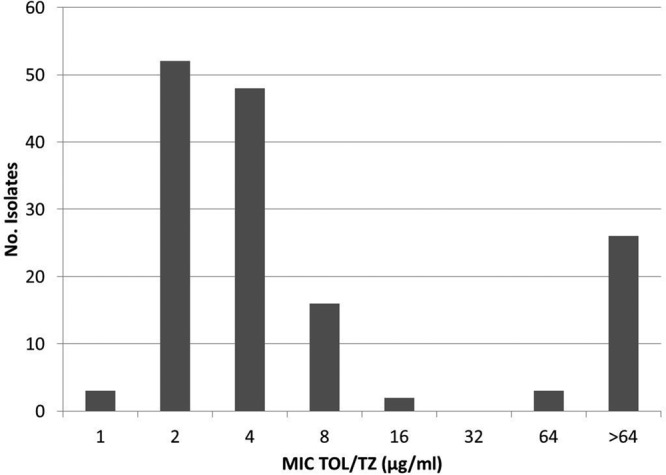
Distribution of ceftolozane-tazobactam (TOL/TZ) MICs for the studied collection of XDR P. aeruginosa isolates.
Deciphering the resistome of XDR P. aeruginosa from Spain.
To define the resistome of XDR P. aeruginosa strains from Spain, at least two (when available) representative isolates from each different clone and hospital were fully sequenced (n = 44). The detected horizontally acquired resistance determinants and sequence variations in a set of 164 chromosomal genes related with P. aeruginosa mutational resistance are shown in Data Set S1. Figure 3 summarizes the main resistance mechanisms detected, including the horizontally acquired β-lactamases and aminoglycoside-modifying enzymes, as well as a summary of the most relevant mutations likely contributing to the resistance profiles after integrating single-nucleotide polymorphism (SNP) refining, as described in Materials and Methods.
FIG 3.

Main antibiotic resistance-related mutations and horizontally acquired resistance determinants of the 44 XDR P. aeruginosa isolates that were fully sequenced. 1, Hospital code, isolate identification (ID). 2, ST244 mutator variant showing a 6-bp insertion in mutL.
β-Lactam resistome.
β-Lactam resistance in widespread ST175 was mainly caused by AmpC hyperproduction plus OprD inactivation, detected in 14 of the 18 isolates sequenced. Moreover, in most of the cases, it was due to the previously described combination of mutations in the AmpC regulator ampR (G154R) and oprD (Q142X), detected in isolates from 7 of the 9 hospitals (Fig. 3 and Data Set S1) (8, 26). Remarkably, this combination of mutations was already common among ST175 isolates recovered 7 years earlier, denoting the long-term persistence of this specific XDR lineage in Spanish hospitals. In contrast, the 4 remaining ST175 isolates not overexpressing AmpC showed diverse acquired β-lactamases (VIM-2, VIM-20, GES-5, and/or OXA-2) and different OprD-inactivating mutations, suggesting a parallel independent evolution of β-lactam resistance in these ST175 lineages. Moreover, these 4 ST175 isolates were the only ones showing ceftolozane-tazobactam resistance according to the CLSI breakpoint (Fig. 3 and Data Set S1). AmpC overexpression was also common in isolates belonging to other STs, as demonstrated by phenotypic methods and reverse transcriptase reverse transcription-PCR (RT-PCR) assays, but mostly due to the presence of missense mutations within ampC regulators ampD (n = 16) and/or dacB (n = 10), with just 2 isolates harboring a mutated ampR (Fig. 3 and Data Set S1). These results are therefore in agreement with previous data indicating that ampD and dacB mutations are the most frequent drivers of AmpC hyperproduction in P. aeruginosa clinical strains (35). Interestingly, mutations leading to ampC overexpression were documented in nearly all isolates from all clones except ST235, in which the documented β-lactam resistance, including that against ceftolozane-tazobactam, was exclusively linked with the presence of an outstanding variety of acquired class I integron-encoded β-lactamases (VIM-47, GES-1, GES-19, OXA-2, and/or OXA-10) (Fig. 3 and Data Set S1), illustrating the huge capacity of ST235 for acquiring exogenous resistance determinants (7, 10). Acquired class A, B, and D β-lactamases were also detected among ST111, ST253, and ST179 ceftolozane-tazobactam-resistant AmpC-hyperproducing P. aeruginosa isolates (Fig. 3 and Data Set S1).
Besides ampC overexpression, recent studies have revealed that β-lactam resistance development, including the novel β-lactam–β-lactamase inhibitor combinations ceftolozane-tazobactam and ceftazidime-avibactam, may result from mutations leading to the structural modification of AmpC or other β-lactamases (17, 35–39). Moreover, a recent work by Berrazeg et al. (40) identified diverse AmpC variants associated with high-level cephalosporin resistance, including ceftolozane-tazobactam and ceftazidime-avibactam, found at a low prevalence (around 1%) among clinical P. aeruginosa isolates. The AmpC sequence variations (Pseudomonas-derived cephalosporinase [PDC] variants) detected among the studied isolates are shown in Table 2. Intraclonal variation of AmpC sequences was not detected. Indeed, all isolates belonging to the widespread ST175, as well as those from ST244, showed the wild-type P. aeruginosa PAO1 AmpC sequence (PDC-1). Moreover, none of the PDC variants documented have been previously associated with ceftolozane-tazobactam resistance, and they are most likely natural polymorphisms not involved in resistance. These findings are thus consistent with the fact that none of the isolates not producing an acquired β-lactamase showed a ceftolozane-tazobactam MIC above 8 μg/ml, although the minor effects of some of these AmpC substitutions should be fully ruled out in future works.
TABLE 2.
Amino acid variations in the sequences of PDC enzymes from the XDR P. aeruginosa isolates studied
| Wild type or ST | No. of isolates | PDC | Residue at amino acid positiona: |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 7 | 21 | 27 (1) | 55 (29) | 79 (53) | 97 (71) | 105 (79) | 155 (129) | 176 (150) | 205 (179) | 356 (330) | 391 (365) | |||
| PAO1 | PDC-1b | P | T | G | A | R | A | T | Q | L | V | V | G | |
| ST175 | 18 | PDC-1 | ||||||||||||
| ST244 | 6 | PDC-1 | ||||||||||||
| ST235 | 5 | PDC-35 | D | V | A | L | A | |||||||
| ST111 | 2 | PDC-3 | A | |||||||||||
| ST253 | 2 | PDC-34 | D | T | A | R | L | A | ||||||
| ST313 | 2 | PDC-37 | S | D | A | L | I | A | ||||||
| ST455 | 1 | PDC-3 | A | |||||||||||
| ST179 | 1 | PDC-8 | A | R | ||||||||||
| ST274 | 1 | PDC-10 | A | A | ||||||||||
| ST395 | 1 | PDC-8 | A | R | ||||||||||
| ST2221 | 1 | PDC-41c | L | T | K | A | P | Q | L | I | A | |||
| ST2533 | 1 | PDC-5 | Q | A | ||||||||||
| ST2534 | 1 | PDC-31 | A | L | ||||||||||
| ST2535 | 1 | PDC-3 | A | |||||||||||
| ST2536 | 1 | PDC-12 | A | A | L | |||||||||
Number of amino acid residues changed in each PCD. Number in parentheses refers to the mature protein of PAO1, after cleavage of the 26 N-terminal amino acid residues of the signal peptide.
Amino acid residues of P. aeruginosa reference strain PAO1 (http://v2.pseudomonas.com).
This strain has the following additional mutations: D3H, T4A, R5T, F6I, C8N, I12L, L18F, P23S, I25A, D32E (6), S59T (33), L71V (45), G77A (51), Q117R (91), A136G (110), S150A (124), Q174H (148), L200S (174), Q203R (177), Q213H (187), H215F (189), D217R (191), E220A (194), A222Q (196), L223Q (197), A224G (198), Q225L (199), R235H (209), G248A (222), V251L (225), T253S (227), D263E (237), D272E (246), R273K (247), S306D (280), T316A (290), P322A (296), R324K (298), I325V (299), E335D (309), V353L (327), L361V (335), and K396V (370).
Beyond β-lactamases, there is growing evidence on the role of target (essential penicillin-binding proteins [PBPs]) modification in P. aeruginosa β-lactam resistance, particularly when involving mutations in ftsI, encoding PBP3, an essential high-molecular-weight molecular class B PBP with transpeptidase activity (41). Indeed, data from CF patients (21, 42) and from in vitro studies (43) have recently demonstrated that PBP3 is under strong mutational pressure, with specific mutations contributing to β-lactam resistance development. Four of the isolates showed unique mutations in PBP3, including two (R504C and F533L) located within the protein domains implicated in the formation and stabilization of the inactivating complex β-lactam–PBP3 (44). Moreover, these two specific mutations have been documented to be selected in vivo during the course of chronic respiratory infection in CF patients (21, 42) and upon meropenem exposure in vitro (39). Thus, in addition to the documented OprD deficiency and AmpC hyperproduction, these PBP3 mutations likely contribute to the β-lactam resistance phenotype of these isolates, perhaps explaining the increased ceftolozane-tazobactam MICs (8 μg/ml) in the absence of any acquired β-lactamase or AmpC mutation (Data Set S1). Although unique polymorphisms were also detected in some strains for other PBPs, including high-molecular-weight (PBP1a, PBP1b, and PBP2) and low-molecular-weight (PBP5 and PBP7) enzymes (Data Set S1), their roles in β-lactam resistance, if any, still need to be experimentally addressed.
Finally, other relevant contributing factors to β-lactam resistance are the resistance-nodulation-division (RND) efflux pumps. The analysis of efflux pump gene expression coupled to the sequencing of their regulatory components revealed frequent mutations leading to overexpression. Particularly frequent were mexZ mutations, leading to the overexpression of the efflux pump MexXY-OprM (involved in cefepime resistance); these were detected in most (73%) of the strains analyzed (Fig. 3). Moreover, around 25% of the strains overexpressed the efflux pump MexAB-OprM (affecting all antipseudomonal β-lactams except imipenem) due to mexR (nalB), nalC, or nalD mutations (Fig. 3). mexT mutations coupled to MexEF-OprN overexpression and OprD downregulation (and thus involved in carbapenem resistance) were also detected in a few strains (Fig. 3). Additionally, several sequence variations in unique residues were detected in the efflux pump components (Data Set S1); however, their contributions to the XDR resistance profile, if any, still need to be further explored.
Fluoroquinolone resistome.
As shown in Fig. 3 and Data Set S1, ciprofloxacin resistance was linked to the presence of missense mutations in gyrA, gyrB, parC, and/or parE quinolone resistance-determining regions (QRDRs). Up to 89% of the sequenced isolates showed missense mutations within gyrA QRDR; specifically, all of them harbored the classical GyrA-T83I mutation, and around half of them, including all ST175 isolates, showed an additional GyrA-D87N mutation (8, 12, 45). Furthermore, nearly all of the isolates showing very high-level ciprofloxacin resistance (MICs > 16 μg/ml) were additionally mutated in the parC QRDR. While the ParC-S87W mutation was encountered in all ST175 and ST253 isolates, most of the ST111, ST235, and ST244 isolates harbored the ParC-S87L mutation (Fig. 3 and Data Set S1). On the other hand, gyrB or parE QRDR mutations were only occasionally detected. Thus, the combination of GyrA and ParC QRDR mutations appears to be responsible for the high-level fluoroquinolone resistance documented in most high-risk clones. As commented above, many of the studied isolates overexpressed one or several RND efflux pumps known to be involved in fluoroquinolone resistance. However, their contribution to the fluoroquinolone resistance profiles in the presence of multiple QRDR mutations is expected to be marginal (41). Nevertheless, two isolates showed MexCD-OprJ overexpression due to nfxB mutations, likely contributing to ciprofloxacin resistance together with the GyrB (and ParE in one case) mutations detected (Fig. 3 and Data Set S1). Indeed, nfxB mutation-driven MexCD-OprJ overexpression has been shown to be a frequent mechanisms selected upon fluoroquinolone exposure in vitro and in vivo (43, 46, 47).
Aminoglycoside resistome.
Up to 27 (61.4%) of the sequenced XDR P. aeruginosa isolates harbored aminoglycoside-modifying enzymes (Fig. 3 and Data Set S1). The presence of aminoglycoside-modifying enzymes correlated well with the documented aminoglycoside resistance profiles. All detected genes [aac(6′)-33, aacA4, aacA29a, aadA1, aadA13, aadA6, aadB, and aacC1] were contained within class I integrons and were accompanied by the presence of genes encoding β-lactamases in 12 of the sequenced isolates (44.4%). As previously described (8), all ST175 isolates harbored an aadB gene.
Moreover, a high proportion of isolates overexpressed the MexXY efflux pump (72.7%); accordingly, the major MexXY expression regulator mexZ was very often mutated (70.5%), including the characteristic G195D mutation in all ST175 isolates (Fig. 3 and Data Set S1). These results point out the underlying strong evolutionary pressure onto mexZ and the relevance of MexXY overexpression for aminoglycoside resistance development; these results are in agreement with those of other recently published works (21, 48, 49).
In addition to these well-known P. aeruginosa contributors to aminoglycoside resistance, there is recent growing evidence that high-level aminoglycoside resistance can rise in the absence of aminoglycoside-modifying enzymes as the result of a stepwise process in which novel genetic determinants are apparently involved (21, 50–52). Among them, fusA1, which codes for elongation factor G, is particularly noteworthy. Indeed, 4 of the studied isolates from different genetic backgrounds showed unique mutations in this gene (Data Set S1); their implication in aminoglycoside resistance profiles is under investigation.
Polymyxins and fosfomycin resistome.
In recent years, old antibiotics, such as the polymyxins, have been increasingly used in the clinical setting to combat infections by MDR/XDR Gram-negative pathogens (53). Resistance development to this class of antibiotics has been related with the presence of mutations within P. aeruginosa two component-regulatory systems (such as PmrAB, PhoPQ, and ParRS) involved in lipopolysaccharide (LPS) biosynthesis (54–57). However, individual alterations within the two-component regulatory systems have been proven to be insufficient for the acquisition of high-level polymyxin resistance (56–60). In agreement with this, although the presence of unique mutations within these genes was frequent, only 3 of the isolates were documented to show colistin resistance, according to established clinical breakpoints (Data Set S1).
Fosfomycin might also be a useful alternative in combined treatments of XDR P. aeruginosa infections, provided that high-level resistance is not evidenced (61). Up to 23 (52%) of the sequenced isolates showed MICs below the EUCAST epidemiological cutoff (ECOFF) (128 μg/ml), whereas the remaining 21 (48%) isolates showed high-level resistance (>1,024 μg/ml) (Data Set S1). Fosfomycin resistance in P. aeruginosa is known to be caused mainly by the mutation of glpT, a gene coding for a glycerol-3-phosphate permease (62). As previously documented (8), fosfomycin resistance among ST175 isolates correlated well with the presence of a T211P mutation in GlpT. While all other strains showing fosfomycin resistance showed polymorphisms in GlpT, some of them were also detected among susceptible isolates (Data Set S1).
Concluding remarks.
Our results, in agreement with those obtained in other countries, indicate that P. aeruginosa XDR profiles are conferred by a very limited number of genotypes recognized as high-risk clones. However, while ST111 and especially ST235 are the predominant high-risk clones worldwide (7), ST175 is by far the most frequent one in Spain. Beyond epidemiological implications, the differential distribution of high-risk clones among XDR isolates significantly impacts the prevalence of the involved resistance mechanisms, since ST111 and ST235 are strongly linked to the production of acquired β-lactamases. Indeed, chromosomal mutation was found to be the main cause of β-lactam and carbapenem resistance among XDR P. aeruginosa strains from Spain (79%), whereas the presence of horizontally acquired carbapenemases was detected in only 21% of the isolates, contrasting with the data from countries showing a high prevalence of ST111 or ST235, in which the vast majority of XDR isolates are carbapenemase producers (10, 13, 63).
P. aeruginosa XDR isolates from Spain were generally nonsusceptible to all antipseudomonal agents except colistin, amikacin, and ceftolozane-tazobactam, which found to be susceptible in 2/3 of the isolates. Moreover, ceftolozane-tazobactam resistance (MIC, >8 μg/ml) correlated smoothly with the presence of horizontally acquired β-lactamases, in agreement with previous evidence (64). Thus, the prevalence of such enzymes is a good marker of the prevalence of ceftolozane-tazobactam resistance and the other way around.
The assessment of WGS resistomes allowed us to perform a deeper analysis of genotype-phenotype correlations. However, the complex repertoire of P. aeruginosa mutation-driven resistance mechanisms and the difficulty of differentiating relevant mutations from natural polymorphisms or random drift mutations determine this to be an exigent task. To minimize this limitation, the full list of mutations in the 164 genes studied was refined to include only those more likely to be involved in the resistance phenotypes, including the input from resistance gene expression. The presence of classical mutational mechanisms, such as those leading to overexpression of the β-lactamase AmpC or efflux pumps, the inactivation of the carbapenem porin OprD, and/or QRDR mutations, was confirmed in most isolates and correlated well with the resistance phenotypes in the absence of horizontally acquired determinants. Ceftolozane-tazobactam resistance (MIC, >8 μg/ml) in the absence of horizontally acquired β-lactamases was not detected, consistently with ampC sequencing data, not evidencing previously described resistance mutations. However, given the increasing use of this novel combination, the potential emergence and fixation of such AmpC variants should be closely monitored (17).
Regarding clone-specific mutations, the previously documented unique set of mutations responsible for the XDR phenotype of the widespread ST175 clone (8) were found to be conserved 7 years later, denoting the long-term persistence of this specific XDR lineage in Spanish hospitals. Our results also provided evidence for the existence and important role of less expected resistance mutations, such as those in PBP3, involved in β-lactam (including ceftolozane-tazobactam) resistance, and FusA1, involved in aminoglycoside resistance.
MATERIALS AND METHODS
XDR P. aeruginosa collection and susceptibility testing.
The P. aeruginosa collection studied included 150 XDR clinical isolates, each recovered in 2015 from a different infected patient (bacteremia, pneumonia, urinary tract, intra-abdominal, or skin and soft tissue infections) from 9 hospitals located in 6 different Spanish regions in the context of a multicenter clinical study (EudraCT 2013-005583-25, PI JP Horcajada). The MICs of ticarcillin, piperacillin-tazobactam, ceftazidime, cefepime, ceftolozane-tazobactam, aztreonam, imipenem, meropenem, ciprofloxacin, tobramycin, amikacin, and colistin were determined by broth microdilution according to EUCAST guidelines (http://www.eucast.org/clinical_breakpoints/). EUCAST version 7.1 and CLSI 2017 (65) clinical breakpoints were used for interpretation. According to the established recommendations, the XDR profile was defined as nonsusceptibility to at least one agent in all but 1 or 2 antibiotic classes (66). P. aeruginosa reference strain PAO1 was used as a control.
Molecular typing.
Clonal relatedness among isolates was first evaluated by pulsed-field gel electrophoresis (PFGE). For this purpose, bacterial DNA embedded in agarose plugs, prepared as described previously (67), was digested with the restriction enzyme SpeI. DNA separation was then performed in a contour-clamped homogeneous-electric-field DRIII apparatus (Bio-Rad, La Jolla, CA) under the following conditions: 6 V/cm2 for 26 h, with pulse times of 5 to 40 s. Finally, the obtained DNA macrorestriction patterns were interpreted according to the criteria established by Tenover et al. (68). Representative isolates from each unique macrorestriction pattern and hospital were further analyzed by multilocus sequence typing (MLST) using available protocols and databases (http://pubmlst.org/paeruginosa/).
Characterization of resistance mechanisms.
AmpC hyperproduction, OprD deficiency, and the presence of horizontally acquired β-lactamases were first explored through previously established phenotypic and molecular (PCR) methods (26, 69). The levels of expression, in comparison with the P. aeruginosa reference strain PAO1, of the genes encoding the chromosomal β-lactamase AmpC (ampC) and four P. aeruginosa efflux pump components (mexB [MexAB-OprM], mexD [MexCD-OprJ], mexF [MexEF-OprN], and mexY [MexXY-OprM]), were determined in selected isolates by real-time reverse transcription-PCR (RT-PCR) with an Eco real-time PCR system (Illumina), according to previously described protocols (70). Additionally, when needed, the nucleotide sequences of genes involved in antibiotic resistance were confirmed through PCR amplification, followed by Sanger sequencing, using previously described protocols (71).
Library preparation and WGS.
Genomic DNA for selected representative isolates was obtained by using a commercially available extraction kit (High Pure PCR template preparation kit; Roche Diagnostics). Indexed paired-end libraries were prepared with the Nextera XT DNA library preparation kit (Illumina, Inc., USA) and then sequenced on an Illumina MiSeq benchtop sequencer with the MiSeq reagent kit version 2 (Illumina, Inc.), resulting in 250-bp paired-end reads.
Variant calling.
Previously defined and validated protocols were used with slight modifications (48, 72). Briefly, paired-ended reads were aligned to the P. aeruginosa PAO1 reference genome (GenBank accession no. NC_002516.2) with Bowtie 2 version 2.2.4 (http://bowtie-bio.sourceforge.net/bowtie2/index.shtml) (73); eventually, pileup and raw files were obtained by using SAMtools version 0.1.16 (https://sourceforge.net/projects/samtools/files/samtools/) (74) and PicardTools version 1.140 (https://github.com/broadinstitute/picard). The Genome Analysis Toolkit (GATK) version 3.4-46 (https://www.broadinstitute.org/gatk/) was used for realignment around indels (75). Median PAO1 coverage ranged from 77 to 97%. SNPs were extracted from the raw files if they met the following criteria: a quality score (Phred-scaled probability of the samples reads being homozygous reference) of at least 50, a root-mean-square (RMS) mapping quality of at least 25, and a coverage depth of at least 3 reads, excluding all ambiguous variants. Microindels were extracted from the total pileup files applying the following criteria: a quality score of at least 500, an RMS mapping quality of at least 25, and support of at least one-fifth of the covering reads. Finally, all positions in which most isolates belonging to a defined clone showed some variation were manually and individually checked without applying any filtering in all other isolates of the same clone.
De novo assembly.
Sequence reads from each isolate were de novo assembled using Velvet version 1.2.10 (https://www.ebi.ac.uk/~zerbino/velvet/) (76), with a k-mer length of 31 and the following parameters: scaffolding = no, ins_length = 500, cov_cutoff = 3, and min_contig_lgth = 500.
Profiling of antibiotic resistance genes.
SNPs and indels for each isolate were annotated by using SnpEff software version 4.2 (http://snpeff.sourceforge.net/index.html) (77), with default options. A previously described (8, 21) set of 164 chromosomal genes known to be related to chromosomal antibiotic resistance in P. aeruginosa were analyzed (Data Set S1). Indels and premature stop codons were considered to result in the inactivation of the corresponding product. In order to refine the potential contributions of the documented SNPs to the phenotypes, several points were considered: (i) SNPs commonly distributed among wild-type strains, such as P. aeruginosa PA14, were considered natural polymorphisms, (ii) SNPs previously demonstrated to cause a phenotype were considered as such, (iii) SNPs in resistance gene regulators which correlated with resistance gene expression data (i.e., AmpC β-lactamase or efflux pumps) were also considered to be involved in the corresponding phenotypes, and (iv) all other SNPs were considered of uncertain effect. In addition, to identify possible horizontally acquired antimicrobial resistance genes, we used the online tool ResFinder version 2.1 (https://cge.cbs.dtu.dk//services/ResFinder/) (78).
Accession number(s).
Sequence files have been deposited in the European Nucleotide Archive under study number PRJEB21341 and accession numbers ERS1792085 to ERS1792128.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Plan Nacional de I+D+i 2013-2016 and Instituto de Salud Carlos III, Subdirección General de Redes y Centros de Investigación Cooperativa, Ministerio de Economía, Industria y Competitividad, Spanish Network for Research in Infectious Diseases (grants REIPI RD16/0016/0002, RD16/0016/0004 RD16/0016/0005, RD16/0016/0007, RD16/0016/0008, RD16/0016/0010, and RD16/0016/0015), and grants PI15/00088 (PI AO) and PI13/00984 (PI JPH) cofinanced by European Development Regional Fund (ERDF) “A way to achieve Europe,” Operative program Intelligent Growth 2014-2020.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01589-17.
REFERENCES
- 1.Livermore MD. 2009. Has the era of untreatable infections arrived? J Antimicrob Chemother 64(Suppl 1):29–36. doi: 10.1093/jac/dkp255. [DOI] [PubMed] [Google Scholar]
- 2.Mesaros N, Nordmann P, Plésiat P, Roussel-Delvallez M, Van Eldere J, Glupczynski Y, Van Laethem Y, Jacobs F, Lebecque P, Malfroot A, Tulkens PM, Van Bambeke F. 2007. Pseudomonas aeruginosa: resistance and therapeutic options at the turn of the new millennium. Clin Microbiol Infect 13:560–578. doi: 10.1111/j.1469-0691.2007.01681.x. [DOI] [PubMed] [Google Scholar]
- 3.Peña C, Suarez C, Gozalo M, Murillas J, Almirante B, Pomar V, Aguilar M, Granados A, Calbo E, Rodríguez-Baño J, Rodríguez F, Tubau F, Martínez-Martínez L, Oliver A, Spanish Network for Research in Infectious Diseases REIPI. 2012. Prospective multicenter study of the impact of carbapenem resistance on mortality in Pseudomonas aeruginosa bloodstream infections. Antimicrob Agents Chemother 56:1265–1272. doi: 10.1128/AAC.05991-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lister PD, Wolter DJ, Hanson ND. 2009. Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin Microbiol Rev 22:582–610. doi: 10.1128/CMR.00040-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poole K. 2011. Pseudomonas aeruginosa: resistance to the max. Front Microbiol 5:2–65. doi: 10.3389/fmicb.2011.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woodford N, Turton JF, Livermore DM. 2011. Multiresistant Gram-negative bacteria: the role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol Rev 35:736–755. doi: 10.1111/j.1574-6976.2011.00268.x. [DOI] [PubMed] [Google Scholar]
- 7.Oliver A, Mulet X, López-Causapé C, Juan C. 2015. The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist Updat 21-22:41–59. [DOI] [PubMed] [Google Scholar]
- 8.Cabot G, López-Causapé C, Ocampo-Sosa AA, Sommer LM, Domínguez MÁ Zamorano L, Juan C, Tubau F, Rodríguez C, Moyá B, Peña C, Martínez-Martínez L, Plesiat P, Oliver A. 2016. Deciphering the resistome of the widespread Pseudomonas aeruginosa sequence type 175 international high-risk clone through whole-genome sequencing. Antimicrob Agents Chemother 60:7415–7423. doi: 10.1128/AAC.02676-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jeukens J, Freschi L, Kukavica-Ibrulj I, Emond-Rheault JG, Tucker NP, Levesque RC. 2 June 2017. Genomics of antibiotic-resistance prediction in Pseudomonas aeruginosa. Ann N Y Acad Sci doi: 10.1111/nyas.13358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Treepong P, Kos VN, Guyeux C, Blanc DS, Bertrand X, Valot B, Hocquet D. 22 June 2017. Global emergence of the widespread Pseudomonas aeruginosa ST235 clone. Clin Microbiol Infect doi: 10.1016/j.cmi.2017.06.018. [DOI] [PubMed] [Google Scholar]
- 11.Jaillard M, van Belkum A, Cady KC, Creely D, Shortridge D, Blanc B, Barbu EM, Dunne WM Jr, Zambardi G, Enright M, Mugnier N, Le Priol C, Schicklin S, Guigon G, Veyrieras JB. 2017. Correlation between phenotypic antibiotic susceptibility and the resistome in Pseudomonas aeruginosa. Int J Antimicrob Agents 50:210–218. doi: 10.1016/j.ijantimicag.2017.02.026. [DOI] [PubMed] [Google Scholar]
- 12.Kos VN, Déraspe M, McLaughlin RE, Whiteaker JD, Roy PH, Alm RA, Corbeil J, Gardner H. 2015. The resistome of Pseudomonas aeruginosa in relationship to phenotypic susceptibility. Antimicrob Agents Chemother 59:427–436. doi: 10.1128/AAC.03954-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Turton JF, Wright L, Underwood A, Witney AA, Chan YT, Al-Shahib A, Arnold C, Doumith M, Patel B, Planche TD, Green J, Holliman R, Woodford N. 2015. High-resolution analysis by whole-genome sequencing of an international lineage (sequence type 111) of Pseudomonas aeruginosa associated with metallo-carbapenemases in the United Kingdom. J Clin Microbiol 53:2622–2631. doi: 10.1128/JCM.00505-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moyá B, Zamorano L, Juan C, Pérez JL, Ge Y, Oliver A. 2010. Activity of a new cephalosporin, CXA-101 (FR264205), against beta-lactam-resistant Pseudomonas aeruginosa mutants selected in vitro and after antipseudomonal treatment of intensive care unit patients. Antimicrob Agents Chemother 54:1213–1217. doi: 10.1128/AAC.01104-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moyá B, Beceiro A, Cabot G, Juan C, Zamorano L, Alberti S, Oliver A. 2016. Pan-β-lactam resistance development in Pseudomonas aeruginosa clinical strains: molecular mechanisms, penicillin-binding protein profiles, and binding affinities. Antimicrob Agents Chemother 56:4771–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Duin D, Bonomo RA. 2016. Ceftazidime/avibactam and ceftolozane/tazobactam: second-generation β-lactam/β-lactamase inhibitor combination. Clin Infect Dis 63:234–241. doi: 10.1093/cid/ciw243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haidar G, Philips NJ, Shields RK, Snyder D, Cheng S, Potoski BA, Doi Y, Hao B, Press EG, Cooper VS, Clancy CJ, Nguyen MH. 2017. Ceftolozane-tazobactam for the treatment of multidrug-resistant Pseudomonas aeruginosa infections: clinical effectiveness and evolution of resistance. Clin Infect Dis 65:110–120. doi: 10.1093/cid/cix182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munita JM, Aitken SL, Miller WR, Perez F, Rosa R, Shimose LA, Lichtenberger PN, Abbo LM, Jain R, Nigo M, Wanger A, Araos R, Tran TT, Adachi J, Rakita R, Shelburne S, Bonomo RA, Arias CA. 2017. Multicenter evaluation of ceftolozane/tazobactam for serious infections caused by carbapenem-resistant Pseudomonas aeruginosa. Clin Infect Dis 65:158–161. doi: 10.1093/cid/cix014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Castanheira M, Mills JC, Farrell DJ, Jones RN. 2014. Mutation-driven β-lactam resistance mechanisms among contemporary ceftazidime-nonsusceptible Pseudomonas aeruginosa isolates from U.S. hospitals. Antimicrob Agents Chemother 58:6844–6850. doi: 10.1128/AAC.03681-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oliver A, Cantón R, Campo P, Baquero F, Blázquez J. 2000. High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288:1251–1254. doi: 10.1126/science.288.5469.1251. [DOI] [PubMed] [Google Scholar]
- 21.López-Causapé C, Sommer LM, Cabot G, Rubio R, Ocampo-Sosa AA, Johansen HK, Figuerola J, Cantón R, Kidd TJ, Molin S, Oliver A. 2017. Evolution of the Pseudomonas aeruginosa mutational resistome in an international cystic fibrosis clone. Sci Rep 7:5555. doi: 10.1038/s41598-017-05621-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kidd TJ, Grimwood K, Ramsay KA, Rainey P, Bell SC. 2011. Comparison of three molecular techniques for typing Pseudomonas aeruginosa isolates in sputum samples. J Clin Microbiol 49:263–268. doi: 10.1128/JCM.01421-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.García-Castillo M, Máiz L, Morosini MI, Rodríguez-Baños M, Suarez L, Fernández-Olmos A, Baquero F, Cantón R, del Campo R. 2012. Emergence of a mutL mutation causing multilocus sequence typing-pulsed-field gel electrophoresis discrepancy among Pseudomonas aeruginosa isolates from a cystic fibrosis patient. J Clin Microbiol 50:1777–1778. doi: 10.1128/JCM.05478-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.López-Causapé C, Rojo-Molinero E, Mulet X, Cabot G, Moyá B, Figuerola J, Togores B, Pérez JL, Oliver A. 2013. Clonal dissemination, emergence of mutator lineages and antibiotic resistance evolution in Pseudomonas aeruginosa cystic fibrosis chronic lung infection. PLoS One 8:e71001. doi: 10.1371/journal.pone.0071001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mena A, Smith EE, Burns JL, Speert DP, Moskowitz SM, Perez JL, Oliver A. 2008. Genetic adaptation of Pseudomonas aeruginosa to the airways of cystic fibrosis patients is catalyzed by hypermutation. J Bacteriol 190:7910–7917. doi: 10.1128/JB.01147-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cabot G, Ocampo-Sosa AA, Domínguez MA, Gago JF, Juan C, Tubau F, Rodríguez C, Moyá B, Peña C, Martínez-Martínez L, Oliver A, Spanish Network for Research in Infectious Diseases (REIPI). 2012. Genetic markers of widespread extensively drug-resistant Pseudomonas aeruginosa high-risk clones. Antimicrob Agents Chemother 56:6349–6357. doi: 10.1128/AAC.01388-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulet X, Cabot G, Ocampo-Sosa AA, Domínguez MA, Zamorano L, Juan C, Tubau F, Rodríguez C, Moyá B, Peña C, Martínez-Martínez L, Oliver A, Spanish Network for Research in Infectious Diseases (REIPI). 2013. Biological markers of Pseudomonas aeruginosa epidemic high-risk clones. Antimicrob Agents Chemother 57:5527–5535. doi: 10.1128/AAC.01481-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peña C, Cabot G, Gómez-Zorrilla S, Zamorano L, Ocampo-Sosa A, Murillas J, Almirante B, Pomar V, Aguilar M, Granados A, Calbo E, Rodríguez-Baño J, Rodríguez-López F, Tubau F, Martínez-Martínez L, Oliver A, Spanish Network for Research in Infectious Diseases (REIPI). 2015. Influence of virulence genotype and resistance profile in the mortality of Pseudomonas aeruginosa bloodstream infections. Clin Infect Dis 60:539–548. doi: 10.1093/cid/ciu866. [DOI] [PubMed] [Google Scholar]
- 29.Castanheira M, Deshpande LM, Costello A, Davies TA, Jones RN. 2014. Epidemiology and carbapenem resistance mechanisms of carbapenem-non-susceptible Pseudomonas aeruginosa collected during 2009–11 in 14 European and Mediterranean countries. J Antimicrob Chemother 69:1804–1814. doi: 10.1093/jac/dku048. [DOI] [PubMed] [Google Scholar]
- 30.Cholley P, Thouverez M, Hocquet D, van der Mee-Marquet N, Talon D, Bertrand X. 2011. Most multidrug-resistant Pseudomonas aeruginosa isolates from hospitals in eastern France belong to a few clonal types. J Clin Microbiol 49:2578–2583. doi: 10.1128/JCM.00102-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guzvinec M, Izdebski R, Butic I, Jelic M, Abram M, Koscak I, Baraniak A, Hryniewicz W, Gniadkowski M, Tambic Andrasevic A. 2014. Sequence types 235, 111, and 132 predominate among multidrug-resistant Pseudomonas aeruginosa clinical isolates in Croatia. Antimicrob Agents Chemother 58:6277–6283. doi: 10.1128/AAC.03116-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mano Y, Saga T, Ishii Y, Yoshizumi A, Bonomo RA, Yamaguchi K, Tateda K. 2015. Molecular analysis of the integrons of metallo- β-lactamase-producing Pseudomonas aeruginosa isolates collected by nationwide surveillance programs across Japan. BMC Microbiol 15:41. doi: 10.1186/s12866-015-0378-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wright LL, Turton JF, Livermore DM, Hopkins KL, Woodford N. 2015. Dominance of international ‘high-risk clones’ among metallo-β-lactamase-producing Pseudomonas aeruginosa in the UK. J Antimicrob Chemother 70:103–110. doi: 10.1093/jac/dku339. [DOI] [PubMed] [Google Scholar]
- 34.Feng W, Sun F, Wang Q, Xiong W, Qiu X, Dai X, Xia P. 2017. Epidemiology and resistance characteristics of Pseudomonas aeruginosa isolates from the respiratory department of a hospital in China. J Glob Antimicrob Resist 8:142–147. doi: 10.1016/j.jgar.2016.11.012. [DOI] [PubMed] [Google Scholar]
- 35.Moya B, Dötsch A, Juan C, Blázquez J, Zamorano L, Haussler S, Oliver A. 2009. Beta-lactam resistance response triggered by inactivation of a nonessential penicillin-binding protein. PLoS Pathog 5:e1000353. doi: 10.1371/journal.ppat.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cabot G, Bruchmann S, Mulet X, Zamorano L, Moyà B, Juan C, Haussler S, Oliver A. 2014. Pseudomonas aeruginosa ceftolozane-tazobactam resistance development requires multiple mutations leading to overexpression and structural modification of AmpC. Antimicrob Agents Chemother 58:3091–3099. doi: 10.1128/AAC.02462-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lahiri SD, Johnstone MR, Ross PL, McLaughlin RE, Olivier NB, Alm RA. 2014. Avibactam and class C β-lactamases: mechanism of inhibition, conservation of the binding pocket, and implications for resistance. Antimicrob Agents Chemother 58:5704–5713. doi: 10.1128/AAC.03057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haidar G, Clancy CJ, Chen L, Samanta P, Shields RK, Kreiswirth BN, Nguyen MH. 2017. Identifying spectra of activity and therapeutic niches for ceftazidime-avibactam and imipenem-relebactam against carbapenem-resistant Enterobacteriaceae. Antimicrob Agents Chemother 61:e00642-17. doi: 10.1128/AAC.00642-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fraile-Ribot PA, Mulet X, Cabot G, Del Barrio-Tofiño E, Juan C, Pérez JL, Oliver A. 2017. In vivo emergence of resistance to novel cephalosporin-β-lactamase inhibitor combinations through the duplication of the amino acid D149 from OXA-2 β-lactamase (OXA-539) in ST235 Pseudomonas aeruginosa. Antimicrob Agents Chemother 61:e01117-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berrazeg M, Jeannot K, Ntsogo Enguéné VY, Broutin I, Loeffert S, Fournier D, Plésiat P. 2015. Mutations in β-lactamase AmpC increase resistance of Pseudomonas aeruginosa isolates to antipseudomonal cephalosporins. Antimicrob Agents Chemother 59:6248–6255. doi: 10.1128/AAC.00825-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen W, Zhang YM, Davies C. 2016. Penicillin-binding protein 3 is essential for growth of Pseudomonas aeruginosa. Antimicrob Agents Chemother 61:e01651-16. doi: 10.1128/AAC.01651-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diaz Caballero J, Clark ST, Coburn B, Zhang Y, Wang PW, Donaldson SL, Tullis DE, Yau YC, Waters VJ, Hwang DM, Guttman DS. 2015. Selective sweeps and parallel pathoadaptation drive Pseudomonas aeruginosa evolution in the cystic fibrosis lung. mBio 6:e00981-15. doi: 10.1128/mBio.00981-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cabot G, Zamorano L, Moyá B, Juan C, Navas A, Blázquez J, Oliver A. 2016. Evolution of Pseudomonas aeruginosa antimicrobial resistance and fitness under low and high mutation rates. Antimicrob Agents Chemother 60:1767–1778. doi: 10.1128/AAC.02676-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Han S, Zaniewski RP, Marr ES, Lacey BM, Tomaras AP, Evdokimov A, Miller JR, Shanmugasundaram V. 2010. Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 107:22002–22007. doi: 10.1073/pnas.1013092107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bruchmann S, Dötsch A, Nouri B, Chaberny IF, Häussler S. 2013. Quantitative contributions of target alteration and decreased drug accumulation to Pseudomonas aeruginosa fluoroquinolone resistance. Antimicrob Agents Chemother 57:1361–1368. doi: 10.1128/AAC.01581-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maciá MD, Borrell N, Segura M, Gómez C, Pérez JL, Oliver A. 2006. Efficacy and potential for resistance selection of antipseudomonal treatments in a mouse model of lung infection by hypermutable Pseudomonas aeruginosa. Antimicrob Agents Chemother 50:975–983. doi: 10.1128/AAC.50.3.975-983.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reinhardt A, Köhler T, Wood P, Rohner P, Dumas JL, Ricou B, van Delden C. 2007. Development and persistence of antimicrobial resistance in Pseudomonas aeruginosa: a longitudinal observation in mechanically ventilated patients. Antimicrob Agents Chemother 51:1341–1350. doi: 10.1128/AAC.01278-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marvig RL, Sommer LM, Molin S, Johansen HK. 2015. Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat Genet 47:57–64. doi: 10.1038/ng.3148. [DOI] [PubMed] [Google Scholar]
- 49.Prickett MH, Hauser AR, McColley SA, Cullina J, Potter E, Powers C, Jain M. 2017. Aminoglycoside resistance of Pseudomonas aeruginosa in cystic fibrosis results from convergent evolution in the mexZ gene. Thorax 72:40–47. doi: 10.1136/thoraxjnl-2015-208027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feng Y, Jonker MJ, Moustakas I, Brul S, Ter Kuile BH. 2016. Dynamics of mutations during development of resistance by Pseudomonas aeruginosa against five antibiotics. Antimicrob Agents Chemother 60:4229–4236. doi: 10.1128/AAC.00434-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schurek KN, Marr AK, Taylor PK, Wiegand I, Semenec L, Khaira BK, Hancock RE. 2008. Novel genetic determinants of low-level aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob Agents Chemother 52:4213–4219. doi: 10.1128/AAC.00507-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.El'Garch F, Jeannot K, Hocquet D, Llanes-Barakat C, Plésiat P. 2007. Cumulative effects of several nonenzymatic mechanisms on the resistance of Pseudomonas aeruginosa to aminoglycosides. Antimicrob Agents Chemother 51:1016–1021. doi: 10.1128/AAC.00704-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bergen PJ, Landersdorfer CB, Lee HJ, Li J, Nation RL. 2012. ‘Old’ antibiotics for emerging multidrug-resistant bacteria. Curr Opin Infect Dis 25:626–633. doi: 10.1097/QCO.0b013e328358afe5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morita Y, Tomida J, Kawamura Y. 2014. Responses of Pseudomonas aeruginosa to antimicrobials. Front Microbiol 4:422. doi: 10.3389/fmicb.2013.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tomaras AP, McPherson CJ, Kuhn M, Carifa A, Mullins L, George D, Desbonnet C, Eidem TM, Montgomery JI, Brown MF, Reilly U, Miller AA, O'Donnell JP. 2014. LpxC inhibitors as new antibacterial agents and tools for studying regulation of lipid biosynthesis in Gram-negative pathogens. mBio 5(5):e01551-14. doi: 10.1128/mBio.01551-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gutu AD, Sgambati N, Strasbourger P, Brannon MK, Jacobs MA, Haugen E, Kaul RK, Johansen HK, Høiby N, Moskowitz SM. 2013. Polymyxin resistance of Pseudomonas aeruginosa phoQ mutants is dependent on additional two-component regulatory systems. Antimicrob Agents Chemother 57:2204–2215. doi: 10.1128/AAC.02353-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fernández L, Jenssen H, Bains M, Wiegand I, Gooderham WJ, Hancock RE. 2012. The two-component system CprRS senses cationic peptides and triggers adaptive resistance in Pseudomonas aeruginosa independently of ParRS. Antimicrob Agents Chemother 56:6212–6222. doi: 10.1128/AAC.01530-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moskowitz SM, Brannon MK, Dasgupta N, Pier M, Sgambati N, Miller AK, Selgrade SE, Miller SI, Denton M, Conway SP, Johansen HK, Høiby N. 2012. PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob Agents Chemother 56:1019–1030. doi: 10.1128/AAC.05829-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miller AK, Brannon MK, Stevens L, Johansen HK, Selgrade SE, Miller SI, Høiby N, Moskowitz SM. 2011. PhoQ mutations promote lipid A modification and polymyxin resistance of Pseudomonas aeruginosa found in colistin-treated cystic fibrosis patients. Antimicrob Agents Chemother 55:5761–5769. doi: 10.1128/AAC.05391-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee JY, Chung ES, Na IY, Kim H, Shin D, Ko KS. 2014. Development of colistin resistance in pmrA-, phoP-, parR- and cprR-inactivated mutants of Pseudomonas aeruginosa. J Antimicrob Chemother 69:2966–2971. doi: 10.1093/jac/dku238. [DOI] [PubMed] [Google Scholar]
- 61.Asuphon O, Montakantikul P, Houngsaitong J, Kiratisin P, Sonthisombat P. 2016. Optimizing intravenous fosfomycin dosing in combination with carbapenems for treatment of Pseudomonas aeruginosa infections in critically ill patients based on pharmacokinetic/pharmacodynamic (PK/PD) simulation. Int J Infect Dis 50:23–29. doi: 10.1016/j.ijid.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 62.Castañeda-García A, Rodríguez-Rojas A, Guelfo JR, Blázquez J. 2009. The glycerol-3-phosphate permease GlpT is the only fosfomycin transporter in Pseudomonas aeruginosa. J Bacteriol 191:6968–6974. doi: 10.1128/JB.00748-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Edelstein MV, Skleenova EN, Shevchenko OV, D'souza JW, Tapalski DV, Azizov IS, Sukhorukova MV, Pavlukov RA, Kozlov RS, Toleman MA, Walsh TR. 2013. Spread of extensively resistant VIM-2-positive ST235 Pseudomonas aeruginosa in Belarus, Kazakhstan, and Russia: a longitudinal epidemiological and clinical study. Lancet Infect Dis 13:867–876. doi: 10.1016/S1473-3099(13)70168-3. [DOI] [PubMed] [Google Scholar]
- 64.Juan C, Zamorano L, Pérez JL, Ge Y, Oliver A. 2010. Activity of a new antipseudomonal cephalosporin, CXA-101 (FR264205), against carbapenem-resistant and multidrug-resistant Pseudomonas aeruginosa clinical strains. Antimicrob Agents Chemother 54:846–851. doi: 10.1128/AAC.00834-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.CLSI. 2017. Performance standards for antimicrobial susceptibility testing, 27th ed, vol 37 CLSI document M100. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 66.Magiorakos AP, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, Monnet DL. 2012. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 18:268–281. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- 67.Kaufmann ME. 1998. Pulsed-field gel electrophoresis. Methods Mol Med 15:33–50. doi: 10.1385/0-89603-498-4:33. [DOI] [PubMed] [Google Scholar]
- 68.Tenover FC, Arbeit RD, Goering RV, Mickelsen A, Murray BE, Persing DH, Swaminathan B. 1995. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol 33:2233–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mikucionyte G, Zamorano L, Vitkauskiene A, López-Causapé C, Juan C, Mulet X, Oliver A. 2016. Nosocomial dissemination of VIM-2-producing ST235 Pseudomonas aeruginosa in Lithuania. Eur J Clin Microbiol Infect Dis 35:195–200. doi: 10.1007/s10096-015-2529-0. [DOI] [PubMed] [Google Scholar]
- 70.Juan C, Moyá B, Pérez JL, Oliver A. 2006. Stepwise upregulation of the Pseudomonas aeruginosa chromosomal cephalosporinase conferring high-level beta-lactam resistance involves three AmpD homologues. Antimicrob Agents Chemother 50:1780–1787. doi: 10.1128/AAC.50.5.1780-1787.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cabot G, Ocampo-Sosa AA, Tubau F, Macia MD, Rodríguez C, Moya B, Zamorano L, Suárez C, Peña C, Martínez-Martínez L, Oliver A, Spanish Network for Research in Infectious Diseases (REIPI). 2011. Overexpression of AmpC and efflux pumps in Pseudomonas aeruginosa isolates from bloodstream infections: prevalence and impact on resistance in a Spanish multicenter study. Antimicrob Agents Chemother 55:1906–1911. doi: 10.1128/AAC.01645-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marvig RL, Johansen HK, Molin S, Jelsbak L. 2013. Genome analysis of a transmissible lineage of Pseudomonas aeruginosa reveals pathoadaptive mutations and distinct evolutionary paths of hypermutators. PLoS Genet 9:e1003741. doi: 10.1371/journal.pgen.1003741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Phillippakis AA, del Angel G, Rivas MA, Hanna M, McKenna A, Fennell TJ, Kernytsky AM, Sivachenko AY, Cibulskis K, Gabriel SB, Altshuler D, Daly MJ. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. 2012. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.