

ABSTRACT
Combinations of antibiotics, each individually effective against Mycobacterium abscessus, are routinely coadministered based on the concept that this minimizes the spread of antibiotic resistance. However, our in vitro data contradict this assumption and instead document antagonistic interactions between two antibiotics (clarithromycin and amikacin) used to treat M. abscessus infections. Clinically relevant concentrations of clarithromycin induced increased resistance to both amikacin and itself. The induction of resistance was dependent on whiB7, a transcriptional activator of intrinsic antibiotic resistance that is induced by exposure to many different antibiotics. In M. abscessus, the deletion of whiB7 (MAB_3508c) resulted in increased sensitivity to a broad range of antibiotics. WhiB7 was required for transcriptional activation of genes that confer resistance to three commonly used anti-M. abscessus drugs: clarithromycin, amikacin, and tigecycline. The whiB7-dependent gene that conferred macrolide resistance was identified as erm(41) (MAB_2297), which encodes a ribosomal methyltransferase. The whiB7-dependent gene contributing to amikacin resistance was eis2 (MAB_4532c), which encodes a Gcn5-related N-acetyltransferase (GNAT). Transcription of whiB7 and the resistance genes in its regulon was inducible by subinhibitory concentrations of clarithromycin but not by amikacin. Thus, exposure to clarithromycin, or likely any whiB7-inducing antibiotic, may antagonize the activities of amikacin and other drugs. This has important implications for the management of M. abscessus infections, both in cystic fibrosis (CF) and non-CF patients.
KEYWORDS: Mycobacterium abscessus, drug resistance, antagonize, synergize, whiB7, macrolide, aminoglycoside, clarithromycin, amikacin, cystic fibrosis, antibiotic resistance, synergy
INTRODUCTION
Mycobacterium abscessus is a rapidly growing, intrinsically drug-resistant, nontuberculous mycobacterium that has become a global health problem. A recent epidemiological study documented its prevalence and transmission between hospital settings throughout the world, suggesting that M. abscessus has become a resident hospital pathogen rather than an opportunistic pathogen (1). Numerous local outbreaks of M. abscessus infections were described in 2016 (1–5). In addition to invading patients with inflammatory lung diseases, such as cystic fibrosis (6), M. abscessus causes serious cutaneous, joint, soft tissue, surgical site, and disseminated infections (7).
The fact that M. abscessus is among the most difficult to treat mycobacterial infections has accelerated its spread and evolution as a dangerous pathogen (8, 9). It is resistant to most antibiotics (10), including those commonly used to treat Mycobacterium tuberculosis infections (rifampin, isoniazid, and ethambutol). The only clinically available antibiotics known to have significant in vitro activity against M. abscessus are clarithromycin (a macrolide), amikacin (an aminoglycoside), cefoxitin (a cephalosporin), and imipenem (a carbapenem) (7). Clarithromycin is the cornerstone for M. abscessus therapy and is routinely coadministered in combinations with amikacin, cefoxitin, imipenem, or tigecycline for 6 to 12 months. However, treatment outcome is unpredictable, and many cases are untreatable (11–14).
This serious problem has motivated studies that have revealed a complex overlapping network of resistance mechanisms (15). In both M. abscessus and M. tuberculosis, high levels of intrinsic antibiotic resistance are provided, in part, by their low-permeability cell envelopes (10, 16, 17). In addition, M. tuberculosis upregulates the expression of resistance genes (15, 18–20) able to combat internalized antibiotics. These genes include eis (encoding an acetyltransferase that modifies aminoglycosides [18, 19, 21]), erm (encoding a ribosomal methyltransferase that prevents macrolide binding [22]), and tap (encoding an efflux pump able to export drugs, including aminoglycosides, tetracyclines, and para-aminosalicylic acid [23]). Although these genes all encode proteins that can provide resistance to certain antibiotics, increased expression is needed to optimize their activities. Importantly, all of these resistance genes are upregulated by WhiB7, a transcriptional activator that is conserved across mycobacteria and related actinomycetes (18–20, 24).
The WhiB7-dependent network of resistance genes is activated by environmental stress signals and a broad range of antibiotics, including macrolides, lincosamides, aminoglycosides, tetracyclines, and pleuromutilins (18–20, 25). Importantly, the induction of WhiB7-mediated resistance by one type of inducer can promote cross-resistance to other types of antibiotics (18). Recently published M. abscessus data revealed that an orthologous WhiB7 regulon (including erm and tap identified in Mycobacterium smegmatis and M. tuberculosis) was upregulated in response to the macrolide erythromycin (26), a known inducer of WhiB7-mediated resistance in other Mycobacterium species (18). In fact, an erm ortholog [erm(41)] has been identified as an inducible macrolide resistance gene in M. abscessus (27, 28).
Since M. abscessus therapy typically includes the macrolide clarithromycin, a whiB7 inducer, we speculated that it might affect the M. abscessus antibiotic resistance spectra (specifically, aminoglycoside resistance). Exposure to clarithromycin should increase the expression of the macrolide resistance gene erm(41) and of potential aminoglycoside resistance genes (including the M. abscessus ortholog of tap). Studies reported here show that exposure to clarithromycin induces intrinsic aminoglycoside resistance in a WhiB7-dependent manner. The activation of cross-resistance to amikacin by clarithromycin has important clinical implications for treating M. abscessus infections, including those in patients with cystic fibrosis.
RESULTS
Clarithromycin induces resistance to amikacin.
Clarithromycin is known to induce expression of the M. abscessus erm(41) gene, which results in increased macrolide resistance (28). To determine whether clarithromycin induced resistance to other antibiotics, M. abscessus ATCC 19977 cultures were incubated in ½ or ¼ the MIC of clarithromycin for 24 h, followed by the addition of antibiotics used to treat M. abscessus infections (amikacin, imipenem, cefoxitin, or tigecycline). Growth inhibition was determined using the resazurin colorimetric assay to establish the MIC. Preincubation with clarithromycin resulted in a 4-fold increase in amikacin MIC (from 3.1 to 12.5 mg/liter) (Table 1) but caused no change in the MICs of imipenem, cefoxitin, or tigecycline (data not shown). Corresponding studies were done using six independently isolated clinical strains of M. abscessus (Table 1). Five of the six strains (strains no. 2 to 6) displayed a 4-fold increase in amikacin MIC when preincubated with clarithromycin; strain no. 1 had high levels of constitutive amikacin resistance without preincubation, likely due to mutation(s) in its 16S rRNA (29). Increased amikacin resistance resulting from clarithromycin exposure occurred rapidly; a 4-fold increase in amikacin MIC took place with 1 h of preincubation (data not shown).
TABLE 1.
Amikacin resistance of M. abscessus strains preincubated with subinhibitory concentrations of clarithromycin
| Clarithromycin concn (mg/liter) | Amikacin MIC (mg/liter) by straina |
||||||
|---|---|---|---|---|---|---|---|
| ATCC 19977 | No. 1 | No. 2 | No. 3 | No. 4 | No. 5 | No. 6 | |
| 0 | 3.1 | >50 | 3.1 | 3.1 | 3.1 | 3.1 | 3.1 |
| 0.05 | 12.5 | >50 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 |
| 0.1 | 12.5 | >50 | 12.5 | 12.5 | 12.5 | 12.5 | 12.5 |
Values are the median values of 3 experiments.
Unlike amikacin, which is bactericidal, clarithromycin is bacteriostatic at the clinically relevant concentrations used in our studies. To test the effect of clarithromycin preincubation on the bactericidal effect of amikacin, cultures were exposed for 2.5 h to clarithromycin concentrations that were subinhibitory for growth (0.1 mg/liter; ½ the MIC) (Fig. 1A and B) or to therapeutic concentrations found in the lung (20 mg/liter; 100× the MIC) (Fig. 1C and D). The cultures were then challenged using a range of amikacin concentrations, including those found in the lungs during amikacin therapy (3 to 12 mg/liter; Fig. 1A and C), as well as those used to define sensitivity (<16 mg/liter) or intermediate resistance (32 mg/liter [30]) (Fig. 1B and D). Amikacin bactericidal activity was recorded as the change in the number of CFU over 4 to 6 days. Without clarithromycin preincubation, low concentrations of amikacin (3 to 6 mg/liter) prevented increases in the number of viable cells, and higher concentrations (9 to 32 mg/liter) reduced the number of CFU in a time- and concentration-dependent manner (Fig. 1A and B). Even at the lowest amikacin concentration tested (3 mg/liter), no increase in CFU occurred over the time period (0 to 120 h). However, after preincubation with ½ the MIC of clarithromycin, bacterial growth occurred at amikacin concentrations up to 16 mg/liter (Fig. 1A and B), and the bactericidal activity of 32 mg/liter amikacin was noticeably reduced (Fig. 1B). After preincubation in 100× the MIC clarithromycin (Fig. 1C and D), the bactericidal activity of amikacin in the 3 to 12 mg/liter range was completely suppressed (Fig. 1C), and its bactericidal activity at higher concentrations (16 to 32 mg/liter) was markedly reduced (Fig. 1D). Without preincubation, inhibition of amikacin bactericidal activity by clarithromycin occurred to a lesser extent (see Fig. S2 in the supplemental material).
FIG 1.

Effect of clarithromycin preincubation on amikacin bactericidal activity. WT M. abscessus was untreated or preincubated with clarithromycin (Cla) at sub-MIC (0.1 mg/liter) (A and B) or 100× the MIC (20 mg/liter) (C and D) for 2.5 h. Following preincubation, amikacin (Amik) was added to the mixture described in panels A and C at 3 to 12 mg/liter, or in panels B and D at 16 to 32 mg/liter. CFU were determined at 24-h intervals post-amikacin addition. The dashed line represents the limit of detection. Data points are the mean of 3 duplicates, with standard deviation presented as error bars.
whiB7 was essential for resistance to a broad spectrum of antibiotics.
In other mycobacteria, WhiB7 is a central regulator of multidrug resistance, including inducible macrolide resistance (18, 24). We constructed a deletion of the whiB7 homolog in M. abscessus (MAB_3508c) and analyzed its resistance using both MIC and kill curve (CFU) assays. The MIC data showed that resistance to a variety of antibiotics was decreased in the ΔwhiB7 mutant compared to the wild type (Table 2). The ΔwhiB7 mutant was more sensitive to aminoglycosides (tobramycin, gentamicin, sisomicin, and amikacin), phenicols (chloramphenicol), tetracyclines (tigecycline), and clarithromycin than the wild type. Sensitivity to imipenem, cefoxitin, doxycycline, tetracycline, clindamycin, levofloxacin, moxifloxacin, rifabutin, rifamycin, isoniazid, or ethambutol was unaltered (Table S2). Kill curve analyses of tobramycin, amikacin, and clarithromycin against wild-type (WT) and ΔwhiB7 mutant strains demonstrated increased activities of all antibiotics against the mutant ΔwhiB7 (Fig. 2). Resistance was restored when the whiB7 mutant was complemented (ΔwhiB7-C; Fig. 2). For both aminoglycosides (amikacin and tobramycin), all concentrations tested allowed growth of the wild type (at reduced rates) but were bactericidal for the ΔwhiB7 mutant (Fig. 2A and B). Importantly, clarithromycin prevented ΔwhiB7 mutant strain CFU increases at 0.2 or 0.4 mg/liter, whereas WT CFU continuously increased at these concentrations (Fig. 2C). Clinically relevant concentrations of clarithromycin (3 to 20 mg/liter) caused much higher rates of killing in the ΔwhiB7 mutant (Fig. S3).
TABLE 2.
Effect of whiB7 deletion on the resistance profile of M. abscessus
| Antibiotic | MIC (mg/liter)a |
|||
|---|---|---|---|---|
| 0 mg/liter clarithromycin |
½ the MIC of clarithromycin |
|||
| WT | ΔwhiB7 mutant | WT | ΔwhiB7 mutant | |
| Amikacin | 3.1 | 0.8 | 12.5 | 0.8 |
| Tobramycin | 6.3 | 1.6 | 25 | 1.6 |
| Gentamicin | 6.3 | 1.6 | 25 | 1.6 |
| Sisomicin | 3.1 | 0.8 | 12.5 | 0.8 |
| Chloramphenicol | 100 | 25 | 100 | 25 |
| Tigecycline | 0.8 | 0.2 | 0.8 | 0.2 |
| Clarithromycin | 0.2 | 0.05 | 0.2 | 0.05 |
| Clarithromycinb | 3.1 | 0.1 | 25 | 0.4 |
Values are the medians of 3 experiments.
Clarithromycin resistance was also measured after 7 days of incubation, which is standard for macrolide MIC analysis.
FIG 2.

Effect of deleting whiB7 on amikacin, tobramycin, and clarithromycin resistance. WT (left), ΔwhiB7 mutant (middle), and ΔwhiB7-C (right) cultures were incubated in amikacin (A), tobramycin (B), or clarithromycin (C), at the indicated concentrations. CFU were monitored at 24-h intervals. Data points are the means of 3 replicates, with standard deviations shown as error bars. The data represent 3 independent experiments.
Role of whiB7 in clarithromycin-induced activation of amikacin resistance.
Studies were done to determine whether clarithromycin-induced activation of amikacin resistance was dependent on WhiB7. Cultures were untreated or preincubated with clarithromycin at ½ the MIC (relative to their respective MICs) for 24 h and assayed for amikacin resistance. Without clarithromycin preinduction, the ΔwhiB7 mutant had a 4-fold reduction in amikacin MIC compared to the WT (3.1 to 0.8 mg/liter; Table 2). Preincubation with clarithromycin resulted in a 4-fold increase in WT amikacin MIC but had no effect on ΔwhiB7 mutant amikacin resistance. Additionally, induction of amikacin resistance was not observed in cultures of the ΔwhiB7 mutant preincubated with a concentration of clarithromycin that induced resistance in the WT (0.1 mg/liter; data not shown). Preincubation with clarithromycin also generated a whiB7-dependent 4-fold increase in MIC of other aminoglycosides (tobramycin, gentamicin, and sisomicin; Table 2).
To examine whether additional antibiotics induced amikacin resistance in a whiB7-dependent manner, WT and ΔwhiB7 mutant cultures were preincubated for 24 h with ½ the MIC of clarithromycin, chloramphenicol, tigecycline, or amikacin. Clarithromycin and tigecycline induced a 4-fold increase in amikacin resistance, while chloramphenicol induced a 2-fold increase in the WT but not in the ΔwhiB7 mutant (Table 3); amikacin, however, did not induce resistance to itself in either strain (Table 3). Preincubation with cefoxitin or imipenem, two other drugs commonly used to control M. abscessus, did not induce amikacin resistance (data not shown).
TABLE 3.
Amikacin resistance is affected by preincubation with subinhibitory concentrations of various antibiotics in M. abscessus WT and ΔwhiB7 mutant
| Preincubation with ½ the MIC | Amikacin MIC (mg/liter)a |
|
|---|---|---|
| WT | ΔwhiB7 mutant | |
| No drug | 3.1 | 0.8 |
| Clarithromycin | 12.5 | 0.8 |
| Amikacin | 3.1 | 0.8 |
| Chloramphenicol | 6.1 | 0.4 |
| Tigecycline | 12.5 | 0.1 |
Values are the medians of 3 experiments.
Clarithromycin induced a whiB7-dependent increase in clarithromycin resistance.
Previous studies showed that erm(41) is responsible for clarithromycin-induced macrolide resistance (28); erm(41) transcription increases for 24 h, followed by increased resistance to clarithromycin within the first week. Therefore, we exposed cultures to clarithromycin for 24 h and then measured the clarithromycin MIC after 7 days to assess the consequences of whiB7 disruption on the induction of macrolide resistance. Without preincubation, the ΔwhiB7 mutant was 8-fold more sensitive than the WT to clarithromycin (Table 2). Preinduction of the WT culture with ½ the MIC of clarithromycin induced a further 8-fold increase in resistance to clarithromycin in WT but had no effect on the ΔwhiB7 mutant MIC (Table 2). In total, the preincubated WT culture assayed at 7 days had a 64-fold increase in clarithromycin MIC relative to the ΔwhiB7 mutant. In conclusion, whiB7-dependent induction of clarithromycin resistance extends over many days and was a major determinant of clarithromycin resistance in M. abscessus.
WhiB7-dependent induction of macrolide and aminoglycoside resistance genes.
We observed whiB7-dependent upregulation of macrolide and aminoglycoside (including amikacin) resistance in cultures preincubated with clarithromycin (Table 2). Quantitative reverse transcription-PCR (RT-qPCR) verified that WhiB7 was induced by clarithromycin and was needed for erm(41) induction in M. abscessus (Fig. 3). It has been established that erm(41) is responsible for inducible macrolide resistance in M. abscessus (28); studies in other mycobacteria (18) showed that the induction of orthologous erm genes is dependent on WhiB7 and causes inducible macrolide resistance. In summary, our studies showed that erm(41) and whiB7 also provided inducible macrolide resistance in M. abscessus.
FIG 3.
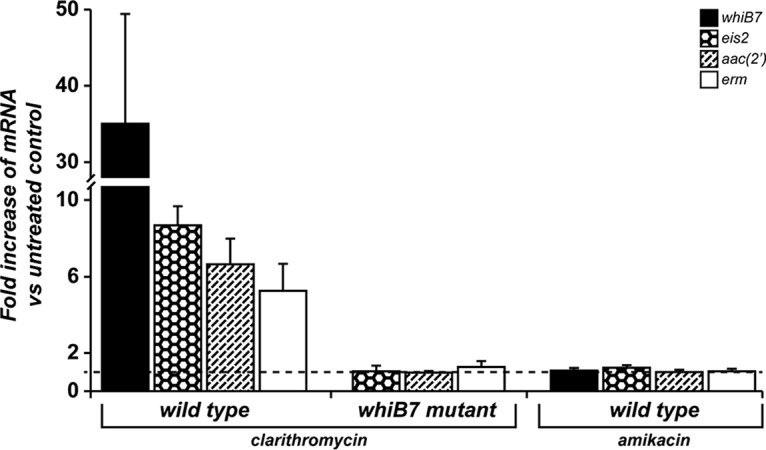
Antibiotic-induced transcription of whiB7, eis2, aac(2′), and erm in M. abscessus WT and a ΔwhiB7 mutant. Increases in whiB7, eis2, aac(2′), and erm mRNAs after 2.5 h of treatment with clarithromycin (0.1 mg/liter WT and 0.025 mg/liter ΔwhiB7 mutant) or amikacin (1.6 mg/liter) relative to untreated controls of the same strain are shown. Data are the mean of three biologically independent samples, with standard deviation as the error bars. The dashed line indicates a value (treated versus untreated ratio of 1) corresponding to no antibiotic-induced change.
To identify the amikacin resistance factors, we initially focused on two possible aminoglycoside resistance genes, MAB_1409c and MAB_4395, induced by the macrolide erythromycin in M. abscessus (26). MAB_1409c is the ortholog of M. tuberculosis tap, encoding an efflux pump known to provide aminoglycoside resistance that is under WhiB7 regulation (23). MAB_4395 [aac(2′)] is annotated in online databases as an aminoglycoside 2′-N-acetyltransferase gene (31). These genes were disrupted and the mutants analyzed for resistance to amikacin and other representative aminoglycosides (tobramycin, sisomicin, and gentamicin). The Δtap mutant displayed no significant change in resistance to any of these antibiotics (Table S3). The Δaac(2′) mutant was more sensitive to several aminoglycosides, including tobramycin (16-fold), sisomicin (4-fold), and gentamicin (2-fold), but there were no changes in its resistance to amikacin (Table S3). Preincubation with clarithromycin increased the MICs to tobramycin, sisomicin, and gentamicin (but not amikacin) in the WT but not the Δaac(2′) mutant (Table S3), implying that upregulation of aac(2′) accounts for resistance to some aminoglycosides but not amikacin. RT-qPCR demonstrated that Δaac(2′) expression was indeed under WhiB7 control (Fig. 3). Additionally, amikacin resistance was induced 4-fold by clarithromycin in the Δaac(2′) mutant, indicating that aac(2′) was not necessary for M. abscessus amikacin resistance. These results indicated that M. abscessus contained additional WhiB7-regulated genes that provide amikacin resistance.
A recently published study confirmed our observations regarding the Δaac(2′) mutant and identified MAB_4532c (eis2) as an amikacin resistance determinant in M. abscessus (21, 32). We used RT-qPCR to determine whether eis2 was in the WhiB7 regulon and could be responsible for clarithromycin-induced amikacin resistance. Indeed, eis2 transcription was induced by clarithromycin in a WhiB7-dependent manner (Fig. 3), establishing its inclusion in the WhiB7 regulon. Consistent with the findings of resistance studies described above, this gene was not induced by amikacin (Fig. 3). These in vitro data provide evidence that eis2 is likely to play an important role in WhiB7-dependent, clarithromycin-induced amikacin resistance in M. abscessus.
DISCUSSION
Although combinations of antibiotics are routinely used to treat bacterial diseases and often have synergistic activities, there can also be antagonistic interactions (33–35). In 1952, Jawetz and Gunnison first reported that bacteriostatic antibiotics can inhibit the activities of bactericidal antibiotics (36) (see review in reference 33). We discovered a mechanism for antagonism between antibiotics currently used for M. abscessus therapy. Our studies revealed that clarithromycin, a bacteriostatic macrolide that is the cornerstone antibiotic for the treatment of M. abscessus infections, antagonized the activity of a partnered bactericidal aminoglycoside (amikacin). Based on previous studies in M. smegmatis and M. tuberculosis, we explored whether the antagonism relied on antibiotic-induced stress signals that triggered genes within the WhiB7 resistance regulon.
Treatment of M. abscessus lung and cystic fibrosis infections involves 2 to 3 antibiotics taken for up to a year. This traditional practice is costly and has numerous undesirable side effects. A recent meta-analysis and systematic review of seroconversion in pulmonary M. abscessus infections found that antibiotic treatment outcomes were successful only 41% of the time with surgical intervention and 35% of the time without surgery, concluding that most patients will retain chronic infection (37). The ineffectiveness of the standard triple therapy (clarithromycin, amikacin, and cefoxitin) in eliminating infection has been confirmed in a hollow fiber M. abscessus lung disease model (38). Following up reports that clarithromycin induced clarithromycin resistance, we explored the concept that it might also alter resistance to amikacin and other coadministered antibiotics. Our data revealed that preincubation with clarithromycin increased the amikacin MIC 4-fold and reduced or eliminated its bactericidal activity throughout a clinically relevant concentration range (3 to 32 mg/liter; Fig. 1). A review of drug concentrations in patients undergoing treatment for pneumonia showed that the amikacin maximum concentration of drug in serum (Cmax) in lungs reached 10 mg/liter after standard intravenous (i.v.) treatments (15 mg/kg of body weight/day dosing) (39). Even at higher i.v. doses used to treat cystic fibrosis patients (35 mg/kg/day amikacin), a Cmax of only 11 mg/liter is achieved (40). This implies that clarithromycin treatment of M. abscessus may allow bacterial growth at concentrations of amikacin higher than those achieved in patients if administered as little as 1 h before amikacin. The antagonistic activities of clarithromycin and amikacin were still present, although at a lower rate, if they were added at the same time (Fig. S2). A range of other antibiotics, including chloramphenicol and tigecycline, also induced amikacin resistance (Table 3). In addition, we found that clarithromycin also increased resistance to three other aminoglycosides (Table 2). These results are clinically important proof that exposure to specific antibiotics can induce a multidrug resistance state in M. abscessus. In other mycobacteria, WhiB7 provides multidrug resistance and is upregulated in response to many different antibiotics, including a range of macrolides. We therefore genetically inactivated whiB7 in M. abscessus to analyze its role in the response to antibiotic exposure and identified several genes in its regulon that may provide drug resistance.
WhiB7 function was required for resistance to aminoglycosides (amikacin, tobramycin, gentamicin, and sisomicin), macrolides (clarithromycin), tigecycline, and chloramphenicol (Table 2), and for resisting the bactericidal effects of clarithromycin at high concentrations (Fig. S3). It was also required for increased levels of resistance induced by preincubation with clarithromycin, tigecycline, or chloramphenicol (Table 3). Curiously, while WhiB7 was needed for resistance to amikacin, preincubation with amikacin did not amplify resistance to itself (Table 3). RT-qPCR analyses demonstrated that amikacin did not induce transcription of whiB7 or other genes under WhiB7 control (Fig. 3). Therefore, while WhiB7 was a key player in amikacin resistance, the inability of amikacin to induce whiB7 expression minimized amikacin's ability to increase aminoglycoside resistance. Knowing that clarithromycin induced amikacin resistance and is an established whiB7 inducer (18), we used it to explore the WhiB7 resistance regulon in M. abscessus.
Resistance spectra, clarithromycin-induced expression, and whiB7 dependence were analyzed for the aminoglycoside resistance genes tap (MAB_1409c) and aac(2′) (MAB_4395), as well as the macrolide resistance gene erm(41) (MAB_2297). RT-qPCR studies showed that transcription of whiB7, tap, aac(2′), and erm(41) was induced by clarithromycin treatment in a whiB7-dependent manner (Fig. 3). However, tap and aac(2′) (a resistance determinant for several aminoglycosides) could not be linked to amikacin resistance (Table S3).
To identify the whiB7-dependent mediator of amikacin resistance, we analyzed the recently reported mycobacterial amikacin resistance determinant eis2 (MAB_ 4532c), an eis paralog (32). The deletion of eis2 causes an 8-fold reduction in amikacin MIC in M. abscessus (32), and eis2 was upregulated by erythromycin (26). Our data demonstrated that in M. abscessus, eis2 was within the WhiB7 regulon. eis2 transcription was induced ∼8-fold by clarithromycin in a WhiB7-dependent manner (Fig. 3), but not by amikacin. Independent studies of M. abscessus cultures growing in a different medium carried out in Ghosh lab at the Wadsworth Center have shown that amikacin can induce whiB7, but the response was much weaker than that of other antibiotics tested; higher concentrations were required, the fold induction was lower, and the response was delayed (41). The M. tuberculosis genome does not contain eis2; therefore, WhiB7 is linked to amikacin resistance in M. abscessus but not in M. tuberculosis.
Divergence of resistance in these two species reflects evolutionary selective pressures in different niches, as human pathogens, or in environmental communities. Eis belongs to a family of ubiquitous acetyltransferases (GNATs) that have remarkably flexible substrate specificities (42). The Eis protein in M. tuberculosis (Rv2416c) acetylates antibiotics having different structures (aminoglycosides and capreomycin), histone-like proteins that fold its chromosome (43), as well as at least one host protein (44). This allows it not only to provide aminoglycoside and capreomycin resistance, but it also allows the enhancement of intracellular survival (eis) in macrophages. However, disruption of eis in M. tuberculosis (21) does not alter amikacin resistance. Disruption of eis2 increases sensitivity of M. abscessus to capreomycin as well as a range of other aminoglycosides (32). Phylogenetic analysis of eis2 indicates that it does not cluster with the mycobacterial eis genes. Interestingly, it clusters with homologs found in Streptomyces species (32, 45), bacteria classified together with mycobacteria as members of the Actinobacteria taxon. Streptomyces spp. inhabit soil environments throughout the world and are best known as nonpathogenic producers of the majority of known antibiotics, including macrolides, aminoglycosides, and capreomycin. The fact that Streptomyces genomes carry functional whiB7 homologs (20) suggests that the eis2 may have served to provide resistance to antibiotics produced by these organisms and was retained in M. abscessus.
Our studies revealed that WhiB7 activates resistance to three of the five commonly used M. abscessus drugs, clarithromycin, amikacin, and tigecycline, but not imipenem or cefoxitin, and suggest that it has a central role in treatment failure. We demonstrated that clarithromycin preexposure increased resistance not only to itself but also to amikacin and other aminoglycosides. A wide variety of compounds inhibiting translation (including lincosamides, tetracyclines, aminoglycosides, and macrolides) or other functions (including fluoroquinolones and acivicin) are whiB7 inducers in M. smegmatis (18). In addition to antibiotics, signals for whiB7 expression in other mycobacteria include palmitic acid, lung surfactant, iron restriction, sputum, and macrophage infection (20, 26, 46–48). Exposure of M. abscessus to any of these conditions during lung infections may induce whiB7 and the antibiotic resistance functions it upregulates. The importance of WhiB7-controlled erm(41) expression can be seen directly by comparing treatment outcomes with M. abscessus subspecies that do not have a functional Erm(41). Erm(41) is responsible for constitutive macrolide and inducible macrolide resistance in M. abscessus (28). However, M. abscessus can be split into subspecies which do (M. abscessus subsp. abscessus T28 and M. abscessus subsp. bolletii) or do not (M. abscessus subsp. abscessus C28 and M. abscessus subsp. massiliense) contain a functional erm(41) (49). Treatment success of M. abscessus subsp. massiliense is 70%, compared to 41% to 35% in M. abscessus with a functional erm(41) gene, emphasizing the importance of WhiB7-induced erm(41) expression on negative M. abscessus treatment outcome.
Our in vitro data suggest that clarithromycin and amikacin, which are front-line coadministered drugs, may have antagonistic effects during treatment. An effective concentration of i.v.-administered amikacin in the lungs may not be achievable in patients whose therapy includes clarithromycin, compounding the issue with Erm(41)-mediated macrolide resistance. This implies that WhiB7-mediated inducible antibiotic resistance decreases the clinical effectiveness of 2 of the 3 antibiotics used in M. abscessus triple therapy. Furthermore, mutations in the M. tuberculosis whiB7 locus can cause constitutive expression of whiB7 and lead to increased expression of its resistance regulon (50). Similar mutations in M. abscessus may provide clinically relevant antibiotic-resistant mutants. Together, our data argue against coadministering clarithromycin and i.v. amikacin, since these drugs can be antagonistic. However, if alternative antibiotics cannot be identified, amikacin should only be given by inhalation, yielding a much higher Cmax (∼970 mg/liter) (51) and thus overcoming induced amikacin resistance. Our studies suggest that inhibitors of WhiB7 might increase killing by macrolides, tigecycline, and aminoglycosides, thereby minimizing the spread of high-level resistance.
MATERIALS AND METHODS
Bacterial strains.
All cloning was done in Escherichia coli DH5α grown in LB broth supplemented with 50 mg/liter kanamycin, 100 mg/liter ampicillin, or 50 μg/ml apramycin where appropriate. All M. abscessus strains used in these studies (described in Table 4) were classified as Mycobacterium abscessus subsp. abscessus based on 16S and hps65 gene sequencing. ATCC 19977 was purchased from the ATCC, and clinical M. abscessus strains were obtained from Patrick Tang at the British Columbia Centre for Disease Control. All M. abscessus strains were grown in Mueller-Hinton II (MHII) broth supplemented with 0.05% tyloxapol at 37°C in rolling test tubes, or in flasks shaking at 200 rpm. MHII was supplemented with 50 mg/liter kanamycin, 50 mg/liter apramycin, or 100 mg/liter zeocin when appropriate.
TABLE 4.
Strain descriptions
| Strain | Descriptiona | Reference or source |
|---|---|---|
| M. abscessus | Mycobacterium abscessus (ATCC 19977) strain containing pJV53-zeo; Zeor | 53 |
| M. abscessus strain 1 | Rough clinical strain isolated from sputum | This study |
| M. abscessus strain 2 | Rough clinical strain isolated from sputum | This study |
| M. abscessus strain 3 | Rough clinical strain isolated from hand ulcer | This study |
| M. abscessus strain 4 | Rough clinical strain isolated from sputum | This study |
| M. abscessus strain 5 | Rough clinical strain isolated from bone joint abscess | This study |
| M. abscessus strain 6 | Rough clinical strain isolated from sputum | This study |
| ΔwhiB7 mutant | M. abscessus with deletion of whiB7 (MAB_3508c) from bp 80–131; Kanr,b | This study |
| Δaac(2′) mutant | M. abscessus with deletion of aac(2′) (MAB_4395) from bp 200–300; Kanr,b | This study |
| Δtap mutant | M. abscessus with deletion of tap (MAB_1409c) from bp 551–560; Kanr,b | This study |
| ΔwhiB7-C mutant | ΔwhiB7 (Kanrb) with pMV261 expressing MAB_3509c (upstream gene in operon) and whiB7 gene from native promoter; Aprrc | This study |
Zeor, zeomycin resistance; Kanr, kanamycin resistance; Aprr, apramycin resistance.
Resistance cassette from pMV261 (52).
Resistance cassette from pT10full.
Cloning.
PCRs were performed using Q5 high-fidelity DNA polymerase (catalog no. M0491S; New England BioLabs), according to the manufacturer's instructions. All restriction enzymes were purchased from New England BioLabs and used according to the manufacturer's instructions. Ligations were done with T4 DNA ligase (catalog no. 15224-041; Invitrogen) overnight at 16°C. The primers used are listed in Table S1. Blunt-end reactions were done with the Klenow fragment (catalog no. M0210S; New England BioLabs), as per the manufacturer's instructions.
Construction of whiB7, tap, and aac(2′) mutants.
whiB7, tap, and aac(2′) genes were PCR amplified with primers whiB7-FW and whiB7-RV, tap-FW and tap-RV, and aac(2′)-FW and aac(2′)-RV, respectively. The resulting products were A-tailed with Taq DNA polymerase, and the fragments were ligated into pGEM-T Easy to create pGem-whiB7, pGem-tap, and pGem-aac(2′). Plasmids were PCR amplified using primers whiB7-iPCR-FW and whiB7-iPCR-RV, tap-iPCR-FW and tap-iPCR-RV, and aac(2′)-iPCR-FW and aac(2′)-iPCR-RV that added terminal unique HindIII (forward [FW] primers) and StuI (reverse [RV] primers) sites. The PCR products were then digested with HindIII and StuI. An aph kanamycin resistance gene, isolated from pMV261 (52) using StuI and HindIII, was then ligated into StuI/HindIII-digested pGem-whiB7, pGem-tap, and pGem-aac(2′) PCR products.
Fragments containing whiB7, tap, or aac(2′) genes disrupted with the kanamycin resistance gene were isolated from the pGEM backbone with SphI and SacI to yield linear DNA. Linear DNA products were electroporated into M. abscessus containing the pJV53-zeo plasmid (53), and the resulting transformants were selected for on MHII-kanamycin agar plates. Gene disruptions were confirmed using primers whiB7-OS-FW and whiB7-OS-RV, tap-OS-FW and tap-OS-RV, and aac(2′)-OS-FW and aac(2′)-OS-RV.
Construction of pWhiB7-C complementing plasmid.
To complement the whiB7 mutant, whiB7, including ∼500 bp of its upstream region (to encompass the native promoter), was PCR amplified from M. abscessus genomic DNA using primers WhiB7-C-FW and WhiB7-C-RV. The resulting PCR product was digested with HindIII and PstI and cloned into a similarly digested modified pMV261 backbone (using the plasmid pFB7 [19]) to construct pMV261-whiB7.
To remove the pMV261 kanamycin resistance gene and introduce the apramycin resistance gene, the plasmid pMV261-whiB7 was digested with SpeI and blunted, followed by HindIII digestion. From pT10full (J. Burian, unpublished data), the apramycin resistance gene was removed by digestion with NcoI, whose end was blunted, followed by HindIII digestion. The resulting HindIII/blunt digestion products were ligated to construct pWhiB7-C (plasmid map and sequence are shown in Fig. S1).
MIC determination.
M. abscessus was inoculated into MHII medium and grown for 48 h in rolling test tubes at 37°C to a final optical density at 600 nm (OD600) of 2 to 5. For preinduction, cultures were diluted to an OD600 of 0.01 in 3 ml of MHII medium containing 0.05 to 0.1 mg/liter clarithromycin, 1.6 mg/liter or 0.4 mg/liter amikacin, 0.4 mg/liter or 0.1 mg/liter tigecycline, or 50 mg/liter or 12.5 mg/liter chloramphenicol for 24 h at 37°C in rolling test tubes. After 24 h, cultures were diluted to an OD600 of 0.005, and 100 μl was added to 100 μl of MHII medium containing serial 2-fold dilutions of antibiotics in 96-well plates (product no. 3370; Costar). Plates were then incubated for 48 h or 7 days, followed by the addition of 30 μl of 10 mg/100 ml resazurin/water. Plates were incubated for an additional 24 h, and wells that remained blue were deemed negative for growth. Wells that turned pink were assigned as growth.
CFU analysis.
M. abscessus was inoculated into MHII medium and grown in rolling test tubes at 37°C for 48 h to a final OD600 of 2 to 5. Cultures were then diluted to an OD600 of 0.005 in 3 ml of MHII medium in test tubes. For preinduction experiments, M. abscessus was incubated for 2.5 h with 0.1 mg/liter or 20 mg/liter clarithromycin before supplementing cultures with a range of amikacin concentrations (3 to 32 mg/liter). For CFU kill curves, a log-phase (to OD600 0.7 to 1.5) culture was diluted to an OD600 of 0.005, and 3-ml volumes were added to test tubes with appropriate concentrations of amikacin, tobramycin, or clarithromycin. At specified times, 100 μl of culture was removed from each tube, and serial 10-fold dilutions were done. Ten microliters of each dilution was spotted onto MHII agar plates, which were then incubated at 37°C. Colonies were counted after 5 days.
RNA extraction.
M. abscessus was inoculated into MHII medium and grown in shaking flasks at 37°C for 48 h to an OD600 of 0.5 to 0.6. Cultures were split, and ½ the MIC clarithromycin or amikacin was added (WT, 0.1 mg/liter clarithromycin or 1.6 mg/liter amikacin; ΔwhiB7 mutant, 0.05 mg/liter clarithromycin) for comparison with an untreated control. The cultures were then incubated for 3 h shaking at 37°C. RNA was extracted as previously described (18).
Quantitative PCR.
Synthesis of cDNA and quantitative PCR (qPCR) analysis were previously described (18). Generation of cDNA was done with qScript cDNA synthesis kit (catalog no. 95047-100; Quanta), as per the manufacturer's instructions, with a total of 100 ng of isolated RNA. The Bioline SensiFAST SYBR No-ROX kit (BIO-98005) was used for qPCR analysis using a Bio-Rad Opticon 2. The primers used for whiB7 were whiB7-qPCR-FW and whiB7-qPCR-RV; for erm(41), the primers were erm-qPCR-FW and erm-qPCR-RV; for aac(2′), the primers were aac(2′)-qPCR-FW and aac(2′)-qPCR-RV; and for eis2, the primers were eis2-qPCR-FW and eis2-qPCR-RV. Concentrations were calculated against a standard curve of genomic DNA dilutions paired with the same primers. Values were standardized to an internal control, sigA, which was measured using the primers sigA-qPCR-FW and sigA-qPCR-RV. Fold increase was calculated by comparison to a nontreated control run in parallel.
Supplementary Material
ACKNOWLEDGMENTS
We are very grateful to Gaye Sweet for her guidance in the laboratory, thoughts, and comments on the manuscript, Patrick Tang for providing clinical M. abscessus strains, and Pallavi Ghosh for communicating unpublished data and her comments on the manuscript.
This work was supported by grants from The Canadian Institute of Health Research (MOP-82855 to C.J.T.) and the British Columbia Lung Association (to C.J.T.).
Footnotes
For a companion article on this topic, see https://doi.org/10.1128/AAC.01347-17.
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.01353-17.
REFERENCES
- 1.Bryant JM, Grogono DM, Rodriguez-Rincon D, Everall I, Brown KP, Moreno P, Verma D, Hill E, Drijkoningen J, Gilligan P, Esther CR, Noone PG, Giddings O, Bell SC, Thomson R, Wainwright CE, Coulter C, Pandey S, Wood ME, Stockwell RE, Ramsay KA, Sherrard LJ, Kidd TJ, Jabbour N, Johnson GR, Knibbs LD, Morawska L, Sly PD, Jones A, Bilton D, Laurenson I, Ruddy M, Bourke S, Bowler IC, Chapman SJ, Clayton A, Cullen M, Dempsey O, Denton M, Desai M, Drew RJ, Edenborough F, Evans J, Folb J, Daniels T, Humphrey H, Isalska B, Jensen-Fangel S, Jonsson B, Jones AM, et al. . 2016. Emergence and spread of a human-transmissible multidrug-resistant nontuberculous mycobacterium. Science 354:751–757. doi: 10.1126/science.aaf8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kapnadak SG, Hisert KB, Pottinger PS, Limaye AP, Aitken ML. 2016. Infection control strategies that successfully controlled an outbreak of Mycobacterium abscessus at a cystic fibrosis center. Am J Infect Control 44:154–159. doi: 10.1016/j.ajic.2015.08.023. [DOI] [PubMed] [Google Scholar]
- 3.Johnston DI, Chisty Z, Gross JE, Park SY. 2016. Investigation of Mycobacterium abscessus outbreak among cystic fibrosis patients, Hawaii 2012. J Hosp Infect 94:198–200. doi: 10.1016/j.jhin.2016.04.015. [DOI] [PubMed] [Google Scholar]
- 4.Cheng A, Sheng WH, Huang YC, Sun HY, Tsai YT, Chen ML, Liu YC, Chuang YC, Huang SC, Chang CI, Chang LY, Huang WC, Hsueh PR, Hung CC, Chen YC, Chang SC. 2016. Prolonged postprocedural outbreak of Mycobacterium massiliense infections associated with ultrasound transmission gel. Clin Microbiol Infect 22:382.e1–382.e11. doi: 10.1016/j.cmi.2015.11.021. [DOI] [PubMed] [Google Scholar]
- 5.Guimarães T, Chimara E, do Prado GV, Ferrazoli L, Carvalho NG, Simeao FC, de Souza AR, Costa CA, Viana Niero C, Brianesi UA, di Gioia TR, Gomes LM, Spadao FS, Silva MD, de Moura EG, Levin AS. 2016. Pseudooutbreak of rapidly growing mycobacteria due to Mycobacterium abscessus subsp bolletii in a digestive and respiratory endoscopy unit caused by the same clone as that of a countrywide outbreak. Am J Infect Control 44:e221–e226. doi: 10.1016/j.ajic.2016.06.019. [DOI] [PubMed] [Google Scholar]
- 6.Floto RA, Olivier KN, Saiman L, Daley CL, Herrmann JL, Nick JA, Noone PG, Bilton D, Corris P, Gibson RL, Hempstead SE, Koetz K, Sabadosa KA, Sermet-Gaudelus I, Smyth AR, van Ingen J, Wallace RJ, Winthrop KL, Marshall BC, Haworth CS. 2016. US Cystic Fibrosis Foundation and European Cystic Fibrosis Society consensus recommendations for the management of non-tuberculous mycobacteria in individuals with cystic fibrosis. Thorax 71(Suppl 1):i1–i22. doi: 10.1136/thoraxjnl-2015-207360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Groote MA, Huitt G. 2006. Infections due to rapidly growing mycobacteria. Clin Infect Dis 42:1756–1763. doi: 10.1086/504381. [DOI] [PubMed] [Google Scholar]
- 8.Kasperbauer SH, De Groote MA. 2015. The treatment of rapidly growing mycobacterial infections. Clin Chest Med 36:67–78. doi: 10.1016/j.ccm.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 9.Brown-Elliott BA, Wallace RJ Jr. 2002. Clinical and taxonomic status of pathogenic nonpigmented or late-pigmenting rapidly growing mycobacteria. Clin Microbiol Rev 15:716–746. doi: 10.1128/CMR.15.4.716-746.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nessar R, Cambau E, Reyrat JM, Murray A, Gicquel B. 2012. Mycobacterium abscessus: a new antibiotic nightmare. J Antimicrob Chemother 67:810–818. doi: 10.1093/jac/dkr578. [DOI] [PubMed] [Google Scholar]
- 11.Jeon K, Kwon OJ, Lee NY, Kim BJ, Kook YH, Lee SH, Park YK, Kim CK, Koh WJ. 2009. Antibiotic treatment of Mycobacterium abscessus lung disease: a retrospective analysis of 65 patients. Am J Respir Crit Care Med 180:896–902. doi: 10.1164/rccm.200905-0704OC. [DOI] [PubMed] [Google Scholar]
- 12.Huang YC, Liu MF, Shen GH, Lin CF, Kao CC, Liu PY, Shi ZY. 2010. Clinical outcome of Mycobacterium abscessus infection and antimicrobial susceptibility testing. J Microbiol Immunol Infect 43:401–406. doi: 10.1016/S1684-1182(10)60063-1. [DOI] [PubMed] [Google Scholar]
- 13.Tung YJ, Bittaye SO, Tsai JR, Lin CY, Huang CH, Chen TC, Lin WR, Chang K, Lai CC, Lu PL, Chen YH. 2015. Risk factors for microbiologic failure among Taiwanese adults with Mycobacterium abscessus complex pulmonary disease. J Microbiol Immunol Infect 48:437–445. doi: 10.1016/j.jmii.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 14.Koh WJ, Jeong BH, Kim SY, Jeon K, Park KU, Jhun BW, Lee H, Park HY, Kim DH, Huh HJ, Ki CS, Lee NY, Kim HK, Choi YS, Kim J, Lee SH, Kim CK, Shin SJ, Daley CL, Kim H, Kwon OJ. 2017. Mycobacterial characteristics and treatment outcomes in Mycobacterium abscessus. Lung Dis Clin Infect Dis 64:309–316. doi: 10.1093/cid/ciw724. [DOI] [PubMed] [Google Scholar]
- 15.Smith T, Wolff KA, Nguyen L. 2013. Molecular biology of drug resistance in Mycobacterium tuberculosis. Curr Top Microbiol Immunol 374:53–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jarlier V, Nikaido H. 1990. Permeability barrier to hydrophilic solutes in Mycobacterium chelonei. J Bacteriol 172:1418–1423. doi: 10.1128/jb.172.3.1418-1423.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen L, Chinnapapagari S, Thompson CJ. 2005. FbpA-dependent biosynthesis of trehalose dimycolate is required for the intrinsic multidrug resistance, cell wall structure, and colonial morphology of Mycobacterium smegmatis. J Bacteriol 187:6603–6611. doi: 10.1128/JB.187.19.6603-6611.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burian J, Ramon-Garcia S, Sweet G, Gomez-Velasco A, Av-Gay Y, Thompson CJ. 2012. The mycobacterial transcriptional regulator whiB7 gene links redox homeostasis and intrinsic antibiotic resistance. J Biol Chem 287:299–310. doi: 10.1074/jbc.M111.302588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burian J, Yim G, Hsing M, Axerio-Cilies P, Cherkasov A, Spiegelman GB, Thompson CJ. 2013. The mycobacterial antibiotic resistance determinant WhiB7 acts as a transcriptional activator by binding the primary sigma factor SigA (RpoV). Nucleic Acids Res 41:10062–10076. doi: 10.1093/nar/gkt751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morris RP, Nguyen L, Gatfield J, Visconti K, Nguyen K, Schnappinger D, Ehrt S, Liu Y, Heifets L, Pieters J, Schoolnik G, Thompson CJ. 2005. Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 102:12200–12205. doi: 10.1073/pnas.0505446102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zaunbrecher MA, Sikes RD Jr, Metchock B, Shinnick TM, Posey JE. 2009. Overexpression of the chromosomally encoded aminoglycoside acetyltransferase eis confers kanamycin resistance in Mycobacterium tuberculosis. Proc Natl Acad Sci U S A 106:20004–20009. doi: 10.1073/pnas.0907925106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buriánková K, Doucet-Populaire F, Dorson O, Gondran A, Ghnassia JC, Weiser J, Pernodet JL. 2004. Molecular basis of intrinsic macrolide resistance in the Mycobacterium tuberculosis complex. Antimicrob Agents Chemother 48:143–150. doi: 10.1128/AAC.48.1.143-150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramón-García S, Mick V, Dainese E, Martin C, Thompson CJ, De Rossi E, Manganelli R, Ainsa JA. 2012. Functional and genetic characterization of the Tap efflux pump in Mycobacterium bovis BCG. Antimicrob Agents Chemother 56:2074–2083. doi: 10.1128/AAC.05946-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramón-García S, Ng C, Jensen PR, Dosanjh M, Burian J, Morris RP, Folcher M, Eltis LD, Grzesiek S, Nguyen L, Thompson CJ. 2013. WhiB7, an Fe-S-dependent transcription factor that activates species-specific repertoires of drug resistance determinants in actinobacteria. J Biol Chem 288:34514–34528. doi: 10.1074/jbc.M113.516385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Geiman DE, Raghunand TR, Agarwal N, Bishai WR. 2006. Differential gene expression in response to exposure to antimycobacterial agents and other stress conditions among seven Mycobacterium tuberculosis whiB-like genes. Antimicrob Agents Chemother 50:2836–2841. doi: 10.1128/AAC.00295-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miranda-CasoLuengo AA, Staunton PM, Dinan AM, Lohan AJ, Loftus BJ. 2016. Functional characterization of the Mycobacterium abscessus genome coupled with condition specific transcriptomics reveals conserved molecular strategies for host adaptation and persistence. BMC Genomics 17:553. doi: 10.1186/s12864-016-2868-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maurer FP, Castelberg C, Quiblier C, Bottger EC, Somoskovi A. 2014. Erm(41)-dependent inducible resistance to azithromycin and clarithromycin in clinical isolates of Mycobacterium abscessus. J Antimicrob Chemother 69:1559–1563. doi: 10.1093/jac/dku007. [DOI] [PubMed] [Google Scholar]
- 28.Nash KA, Brown-Elliott BA, Wallace RJ Jr. 2009. A novel gene, erm(41), confers inducible macrolide resistance to clinical isolates of Mycobacterium abscessus but is absent from Mycobacterium chelonae. Antimicrob Agents Chemother 53:1367–1376. doi: 10.1128/AAC.01275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prammananan T, Sander P, Brown BA, Frischkorn K, Onyi GO, Zhang Y, Bottger EC, Wallace RJ Jr. 1998. A single 16S ribosomal RNA substitution is responsible for resistance to amikacin and other 2-deoxystreptamine aminoglycosides in Mycobacterium abscessus and Mycobacterium chelonae. J Infect Dis 177:1573–1581. doi: 10.1086/515328. [DOI] [PubMed] [Google Scholar]
- 30.Woods GL. 2000. Susceptibility testing for mycobacteria. Clin Infect Dis 31:1209–1215. doi: 10.1086/317441. [DOI] [PubMed] [Google Scholar]
- 31.Ripoll F, Pasek S, Schenowitz C, Dossat C, Barbe V, Rottman M, Macheras E, Heym B, Herrmann JL, Daffe M, Brosch R, Risler JL, Gaillard JL. 2009. Non mycobacterial virulence genes in the genome of the emerging pathogen Mycobacterium abscessus. PLoS One 4:e5660. doi: 10.1371/journal.pone.0005660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rominski A, Selchow P, Becker K, Brulle JK, Dal Molin M, Sander P. 2017. Elucidation of Mycobacterium abscessus aminoglycoside and capreomycin resistance by targeted deletion of three putative resistance genes. J Antimicrob Chemother 72:2191–2200. doi: 10.1093/jac/dkw466. [DOI] [PubMed] [Google Scholar]
- 33.Rahal JJ., Jr 1978. Antibiotic combinations: the clinical relevance of synergy and antagonism. Medicine (Baltimore) 57:179–195. doi: 10.1097/00005792-197803000-00005. [DOI] [PubMed] [Google Scholar]
- 34.Aaron SD, Ferris W, Henry DA, Speert DP, Macdonald NE. 2000. Multiple combination bactericidal antibiotic testing for patients with cystic fibrosis infected with Burkholderia cepacia. Am J Respir Crit Care Med 161:1206–1212. doi: 10.1164/ajrccm.161.4.9907147. [DOI] [PubMed] [Google Scholar]
- 35.Schimpff SC. 1986. Empiric antibiotic therapy for granulocytopenic cancer patients. Am J Med 80:13–20. [PubMed] [Google Scholar]
- 36.Jawetz E, Gunnison JB. 1952. An experimental basis of combined antibiotic action. JAMA 150:693–695. doi: 10.1001/jama.1952.63680070011015. [DOI] [PubMed] [Google Scholar]
- 37.Diel R, Ringshausen F, Richter E, Welker L, Schmitz J, Nienhaus A. 2017. Microbiological and clinical outcomes of treating non-Mycobacterium avium complex nontuberculous mycobacterial pulmonary disease: a systematic review and meta-analysis. Chest 152:120–142. doi: 10.1016/j.chest.2017.04.166. [DOI] [PubMed] [Google Scholar]
- 38.Ferro BE, Srivastava S, Deshpande D, Pasipanodya JG, van Soolingen D, Mouton JW, van Ingen J, Gumbo T. 2016. Failure of the amikacin, cefoxitin, and clarithromycin combination regimen for treating pulmonary Mycobacterium abscessus infection. Antimicrob Agents Chemother 60:6374–6376. doi: 10.1128/AAC.00990-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cruciani M, Gatti G, Cazzadori A, Concia E. 1996. Pharmacokinetics of antimicrobial agents in the respiratory tract. Zentralbl Bakteriol 284:1–31. doi: 10.1016/S0934-8840(96)80150-2. [DOI] [PubMed] [Google Scholar]
- 40.Canis F, Husson MO, Turck D, Vic P, Launay V, Ategbo S, Vincent A, Courcol RJ. 1997. Pharmacokinetics and bronchial diffusion of single daily dose amikacin in cystic fibrosis patients. J Antimicrob Chemother 39:431–433. doi: 10.1093/jac/39.3.431. [DOI] [PubMed] [Google Scholar]
- 41.Hurst-Hess K, Rudra P, Ghosh P. 2017. Mycobacterium abscessus WhiB7 regulates a species-specific repertoire of genes to confer extreme antibiotic resistance. Antimicrob Agents Chemother 61:e01347-17. doi: 10.1128/AAC.01347-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vetting MW, S de Carvalho LP, Yu M, Hegde SS, Magnet S, Roderick SL, Blanchard JS. 2005. Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys 433:212–226. doi: 10.1016/j.abb.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 43.Ghosh S, Padmanabhan B, Anand C, Nagaraja V. 2016. Lysine acetylation of the Mycobacterium tuberculosis HU protein modulates its DNA binding and genome organization. Mol Microbiol 100:577–588. doi: 10.1111/mmi.13339. [DOI] [PubMed] [Google Scholar]
- 44.Kim KH, An DR, Song J, Yoon JY, Kim HS, Yoon HJ, Im HN, Kim J, Kim do J, Lee SJ, Kim KH, Lee HM, Kim HJ, Jo EK, Lee JY, Suh SW. 2012. Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc Natl Acad Sci U S A 109:7729–7734. doi: 10.1073/pnas.1120251109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pricer RE, Houghton JL, Green KD, Mayhoub AS, Garneau-Tsodikova S. 2012. Biochemical and structural analysis of aminoglycoside acetyltransferase Eis from Anabaena variabilis. Mol Biosyst 8:3305–3313. doi: 10.1039/c2mb25341k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rohde KH, Veiga DF, Caldwell S, Balazsi G, Russell DG. 2012. Linking the transcriptional profiles and the physiological states of Mycobacterium tuberculosis during an extended intracellular infection. PLoS Pathog 8:e1002769. doi: 10.1371/journal.ppat.1002769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gold B, Rodriguez GM, Marras SA, Pentecost M, Smith I. 2001. The Mycobacterium tuberculosis IdeR is a dual functional regulator that controls transcription of genes involved in iron acquisition, iron storage and survival in macrophages. Mol Microbiol 42:851–865. doi: 10.1046/j.1365-2958.2001.02684.x. [DOI] [PubMed] [Google Scholar]
- 48.Schwab U, Rohde KH, Wang Z, Chess PR, Notter RH, Russell DG. 2009. Transcriptional responses of Mycobacterium tuberculosis to lung surfactant. Microb Pathog 46:185–193. doi: 10.1016/j.micpath.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mougari F, Bouziane F, Crockett F, Nessar R, Chau F, Veziris N, Sapriel G, Raskine L, Cambau E. 2017. Selection of resistance to clarithromycin in Mycobacterium abscessus subspecies. Antimicrob Agents Chemother 61:e00943-16. doi: 10.1128/AAC.00943-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reeves AZ, Campbell PJ, Sultana R, Malik S, Murray M, Plikaytis BB, Shinnick TM, Posey JE. 2013. Aminoglycoside cross-resistance in Mycobacterium tuberculosis due to mutations in the 5′ untranslated region of whiB7. Antimicrob Agents Chemother 57:1857–1865. doi: 10.1128/AAC.02191-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luyt CE, Clavel M, Guntupalli K, Johannigman J, Kennedy JI, Wood C, Corkery K, Gribben D, Chastre J. 2009. Pharmacokinetics and lung delivery of PDDS-aerosolized amikacin (NKTR-061) in intubated and mechanically ventilated patients with nosocomial pneumonia. Crit Care 13:R200. doi: 10.1186/cc8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR, Bloom BR. 1991. New use of BCG for recombinant vaccines. Nature 351:456–460. doi: 10.1038/351456a0. [DOI] [PubMed] [Google Scholar]
- 53.Medjahed H, Reyrat JM. 2009. Construction of Mycobacterium abscessus defined glycopeptidolipid mutants: comparison of genetic tools. Appl Environ Microbiol 75:1331–1338. doi: 10.1128/AEM.01914-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.