

Abstract
ALS is characterised by a focal onset of motor neuron loss, followed by contiguous outward spreading of pathology throughout the nervous system, resulting in paralysis and death generally within a few years after diagnosis. The aberrant release and uptake of toxic proteins including SOD1 and TDP-43 and their subsequent propagation, accumulation and deposition in motor neurons may explain such a pattern of pathology. Previous work has suggested that the internalization of aggregates triggers stress granule formation. Given the close association of stress granules and TDP-43, we wondered whether internalisation of SOD1 aggregates stimulated TDP-43 cytosolic aggregate structures. Addition of recombinant mutant G93A SOD1 aggregates to NSC-34 cells was found to trigger a rapid shift of TDP-43 to the cytoplasm where it was still accumulated after 48 h. In addition, SOD1 aggregates also triggered cleavage of TDP-43 into fragments including a 25 kDa fragment. Collectively, this study suggests a role for protein aggregate uptake in TDP-43 pathology.
Keywords: Protein aggregation, Propagation, Prion, TDP-43, SOD1, ALS
Introduction
Amyotrophic lateral sclerosis (ALS) is an incurable neurodegenerative disorder characterised by the loss of both the upper and lower motor neurons in the brain and spinal cord, respectively, resulting in the progressive paralysis of the muscles of speech, limbs, swallowing and respiration, due to the progressive degeneration of innervating motor neurons (Cleveland and Rothstein 2001). The neuropathology of all cases of ALS are characterised by disease-specific proteins, mutant and wild type alike; mislocalised and abnormally accumulated as misfolded, insoluble aggregates in the cytoplasm of afflicted motor neurons (Leigh et al. 1991; Bosco et al. 2010; Ross and Poirier 2004). However, aggregation in ALS is not restricted to disease-specific proteins as more than 70 proteins, which have in common the biophysical property of being supersaturated, can be found in ALS deposits suggesting a collapse of protein homeostasis (Ciryam et al. 2017). Proteinaceous inclusions, containing misfolded aggregated proteins, peptides and fragments, also occur in many other neurodegenerative disorders including Alzheimer’s disease (AD), Parkinson’s disease (PD), frontotemporal dementia (FTD), Huntington’s disease (HD) (Chiti and Dobson 2006); and prion diseases, such as Creutzfeldt-Jakob disease, Kuru and fatal familial insomnia diseases (Prusiner 1982; Prusiner 1984). How misfolded proteins cause cellular dysfunction is unknown; one hypothesis is that exposed hydrophobic surfaces interact inappropriately with cellular components (Bolognesi et al. 2010).
Mutations in several genes cause familial ALS (fALS) which account for 5–10% of total ALS cases and contribute to the development of sporadic ALS (sALS, 90% of ALS cases) (Andersen and Al-Chalabi 2011). In fALS, mutations in the gene encoding the Cu/Zn superoxide dismutase (SOD1), a ubiquitously expressed homodimeric enzyme, results in the monomerisation (McAlary et al. 2013; Polling et al. 2014), misfolding and aggregation of this normally stable protein (Banci et al. 2009). However, recently misfolded SOD1 species have been increasingly identified in non-SOD1 fALS and sALS cases (Bosco et al. 2010; Forsberg et al. 2010), suggesting that misfolded SOD1 may play a pathological role in all types of ALS (Pokrishevsky et al. 2012).
In addition to SOD1, TAR DNA binding protein (TDP-43) has been identified as a major component of cytoplasmic inclusions in sALS, SOD1-negative fALS and ALS with dementia; as well as the most common pathological subtype of frontotemporal dementia (FTD) with ubiquitinated inclusions (Arai et al. 2006; Cairns et al. 2007; Davidson et al. 2007; Mackenzie et al. 2010; Neumann et al. 2006, 2007). However, aggregate formation pathways and final structures are likely different between SOD1 and TDP-43 (Farrawell et al. 2015). Clinical and pathological overlap between the different forms of ALS and FTD has raised the possibility that there is a pathogenic mechanistic-link between these disorders and has prompted their reclassification as TDP-43-proteinopathies. The shared pathology includes neuronal cytoplasmic inclusions (NCIs), loss of the normal nuclear TDP-43, ubiquitination and hyperphosphorylation of TDP-43 and lastly formation of abnormal fragments of TDP-43 in post-mortem tissue (Neumann et al. 2007; Nonaka et al. 2009; Zhang et al. 2009). However, TDP-43 is also found in inclusions in Machado-Joseph disease (Tan et al. 2009), spinocerebellar ataxia (Toyoshima et al. 2011), Huntington’s disease (Schwab et al. 2008), Alzheimer’s disease (Davidson et al. 2011), inclusion body myositis (Weihl et al. 2008) and Parkinson’s disease (Nakashima-Yasuda et al. 2007), suggesting that TDP-43 accumulation is not ALS or FTD specific. Of interest, AD-associated Aβ has been implicated in triggering the phosphorylation and cytosolic accumulation of pathogenic TDP-43 in rodent models and in brain autopsies from AD patients (Herman et al. 2011). This may explain the presence of TDP-43 pathology in a proportion of AD cases (Wilson et al. 2011). Interestingly, the process of internalisation of tau aggregates triggers stress granule formation and accumulation (Brunello et al. 2016), and given that TDP-43 is associated with stress granule formation, this may explain the accumulation of TDP-43 with other neurodegenerative diseases where aggregate propagation is involved in pathology.
In the current study, we examined whether exogenous recombinant SOD1 protein aggregates can induce and/or contribute to TDP-43 pathology, specifically its mislocalisation and aggregation. Upon incubation with large recombinantly formed SOD1 aggregates with NSC-34 cells, WT TDP-43 was found mislocalised to the cytoplasm of both mouse neuronal-like cells and human embryonic kidney cells. We also demonstrate that, upon addition of the SOD1 aggregates, fragments of TDP-43 can be observed in the cytoplasm of NSC-34 cells. Thus, we conclude that addition of recombinant SOD1 aggregates to the extracellular environment of neuron-like cells in culture results in TDP-43 mislocalisation, aggregation and fragmentation.
Materials and methods
Cell lines
The mouse neuroblastoma × spinal cord hybrid cell line (NSC-34 cells) (Cashman et al. 1992) were routinely cultured in DMEM/F-12 supplemented with 10% (v/v) FBS and 2 mM GlutaMAX. Cells were maintained in an incubator at 37 °C under a humidified atmosphere containing 5% (v/v) CO2. The pCMV6-AC-tGFP expression vector containing TDP-43WT cDNA was obtained from Origene. TDP-tomato red (TdTomato) constructs were created by replacing the tGFP sequences in the TDP-43-tGFP plasmids with tdTomato (by Genscript, USA). Cells were then incubated in DMEM-F-12 serum-free culture medium containing 2 μg WT TDP-tomato red (TR) plasmid DNA and Lipofectamine 2000 for 5 h at 37 °C under a humidified atmosphere containing 5% (v/v) CO2. Cells were then washed once with serum-free media and replenished with complete culture medium. Cells were then incubated for a longer period of time either for 19, 43 or 67 h (24, 48 and 72 h in total, respectively).
Aggregation of WT and mutant G93A SOD1
WT and G93A SOD1 were expressed and purified from Escheria coli as previously outlined (Roberts et al. 2013; Lindberg et al. 2002). SOD1 aggregation was performed in vitro as previously described (Roberts et al. 2013). Briefly, solutions of purified WT or G93A mutant SOD1 protein (1 mg/mL) in PBS were co-incubated with 20 mM dithiothreitol (DTT) and 5 mM ethylenediaminetetraacetic acid (EDTA) for 72 h at 37 °C with shaking using a digital shaker (universal IKA® MS 3, 230 V) (Sigma, St. Louis, MO).
Treatment of transfected cells with SOD1
Cells were visualised prior to experimentation using an Eclipse TE2000 inverted microscope (Nikon, Tokyo, Japan) to confirm transfection and determine transfection efficiency. NSC-34 cells were incubated with labelled biotinylated aggregates, or soluble (non-aggregated) WT and mutant G93A SOD1 proteins (20 μg/mL) or no protein as a control at 37 °C/CO2, for either 2 or 72 h. Post-incubation, cells were analysed either by confocal microscopy or FloIT. In the case where cells were analysed by confocal microscopy, inclusions were counted manually and expressed as a proportion of transfected cells. To account for toxicity of the addition of aggregates, the proportion of cells containing cytoplasmic TDP-43WT-tdtomato was normalised to the amount of cell loss over 72 h. To assess the toxicity of SOD1 to cells expressing TDP-43WT-tdtomato constructs in NSC-34 cells over a time course, an IncuCyte® automated fluorescent microscope (Essen BioScience, USA) was used as previously described (McAlary et al. 2016). Briefly, NSC-34 cells were plated into 12-wells plates at a confluency of 60% and were transfected 24 h post-plating. Cells were dissociated 24 h post-transfection and plated into 96-well plates at a confluency of 20% in phenol-red-free DMEM-F12 supplemented with 10% FBS. At least three images were acquired per-well at 2-h intervals for 72 h in both phase and red channels. The number of TDP-43WT-tdtomato-positive cells at each time point for each transfection was normalised to the initial value determined in the first scan after plating. Then, the normalised values of the SOD1 treatments at each time point were divided by the normalised TDP-43WT-tdtomato alone data at the same time points to determine the relative cell viability. The proportion of cells containing cytoplasmic TDP-43WT-tdtomato for each treatment was then normalised to the relative viability at 72 h.
FloIT assay
The FloIT assay was performed as in previously published work (Whiten et al. 2016). Briefly, NSC-34 cells were transiently transfected with TDP-43 (TDP-43WT-tdtomato). Following transient transfection at 24, 48 and 72 h in total, cells were harvested and washed using centrifugation. An aliquot of cells (2 × 105cells/0.15 mL) were collected then analysed for transfection efficiency. The remaining cells (4 × 105cells/0.35 mL) were washed as above and lysed prior to analysis in lysis buffer. Cell lysates were then incubated with RedDot2 (1:1000) at RT for 2 min. Events were collected using a LSRFortessa X-20 Cell Analyzer (BD Biosciences) (excitation 561 nm, emission collected with 586/15 band-pass filter and excitation 633, emission collected with 675/15 nm band-pass filter for tomato red and RedDot2). Lysates were firstly gated on forward and side scatter, and then, the fluorescence from RedDot2 was determined by flow cytometry. All parameters were set to log10 during acquisition from cell lysates. The forward scatter threshold was set to the minimum value (200 AU) to minimise the exclusion of small protein inclusions. Nuclei were identified and enumerated based on RedDot2 fluorescence and forward scatter and then excluded from further analysis. The remaining particles were analysed for the presence of inclusions based on GFP/tdTomato fluorescence, forward scatter and comparison lysates prepared from cells expressing only the corresponding fluorescent protein. The number of inclusions in the population can be normalised to the number of nuclei and reported as inclusions/100 transfected cells (iFloIT) according to the equation outlined in (Whiten et al. 2016). Analysis of all events was determined using FlowJo software (Tree Star, Ashland, OR).
Subcellular fractionation assay of NSC34 cells
NSC-34 cells were incubated with WT SOD1 in a soluble or aggregated form (20 μg/mL) in PBS for 2 h at 37 °C/5% CO2. Post-incubation, the cells were washed three times with PBS (300×g for 5 min) harvested using 0.5% trypsin and 5 mM EDTA and washed (500×g for 5 min). The cells were washed three times with ice cold PBS (500×g for 3 min) for fractionation using a Subcellular Protein Fractionation Kit for Cultured Cells as per manufacturer’s instructions (Thermo Fisher Scientific).
Protein concentration was determined using the BCA method. Supernatants (20 μg protein/lane) were separated under reducing conditions (5% β-mercaptoethanol) using Any kD Mini-PROTEAN TGX Stain-Free™ Precast Gels. Proteins were then transferred to nitrocellulose membranes using a Trans-Blot Turbo Transfer System (Bio-Rad). Total protein per lane was then imaged and measured with a Bio-Rad Criterion Stain Free Imager and Image Lab software. Membranes were blocked with heat denatured casein (HDC) for 1 h at 37 °C. To test the purity of the separation, rabbit anti-EEA1 pAb (1:500), rabbit anti-Vimentin pAb (1:500) and mouse anti-actin mAb (1:5000), diluted in HDC/PBS for 1 h at 37 °C were used to probe the ME, NE and PE fractions. Membranes were visualised using chemiluminescent substrate and Amersham Hyperfilm ECL (GE Healthcare). Images of films were collected using a GS-800 Calibrated Densitometer (Bio-Rad). The processing of films was achieved using GBX Developer and Replenisher and GBX Fixer and Replenisher (Kodak Australiasia, Collingwood, Victoria Australia). Images of the films were collected using a GS-800 Calibrated Densitometer (Bio-Rad).
Presentation of data and statistical analyses
Data is presented as the mean ± SD. ANOVA paired with Tukey’s HSD multiple comparison post-test tests were used to analyse and compare differences between multiple treatments. Unpaired Student’s t tests were performed for single treatment comparisons. Prism 5 for Windows (Version 5.01) (GraphPad Software, San Diego, CA) was used to generate these statistical analyses. Differences were defined as significant for P < 0.05.
Results
Transiently expressed TDP-43WT-tdTomato is observed in the cytosol of NSC-34 cells upon treatment with recombinant SOD1 protein aggregates
TDP-43 redistribution and aggregation is observed in the cytoplasm of affected cells in a diverse set of neurodegenerative diseases. However, the factors responsible for TDP-43 mislocalisation and aggregation remain ambiguous. Both SOD1 and TDP-43 have been implicated in fALS and sALS pathogenesis; although, whether SOD1 misfolding or aggregation contribute to TDP-43 pathology is yet to be established. Given that Aβ aggregates can affect TDP-43 deposition (Herman et al. 2011) and uptake of tau aggregates causes stress granule formation (Brunello et al. 2016), we tested whether exogenously added SOD1 aggregates have an effect on TDP-43 cytosolic mislocalisation and aggregation. Initially, we followed cells transiently expressing WT TDP-43-tomato red (TDP-43WT TR) in live cells. Post-transfection, cells were incubated with either WT or mutant G93A SOD1 in both their aggregated and soluble state (Fig. 1a). The percentage of cells containing cytosolic TDP-43 in foci that measured >1 μm per (this definition would include stress granules and inclusions) was determined by confocal microscopy. Cytosolic TDP-43 by this definition did not distinguish between cells where TDP-43WT was cleared from the nucleus from TDP-43 that had accumulated in the cytosol even though some nuclear TDP-43WT remained (Fig. 1b). Examples of TDP-43 morphology scored in this work are given in Fig. 1b. This included a granular appearance, large inclusions, with or without clearance of TDP-43 from the nucleus.
Fig. 1.

Treatment of NSC-34 cells with large recombinant SOD1 protein aggregates. a Schematic diagram of the experimental outline. Recombinant human SOD1 was aggregated (followed by ThT fluorescence), or not, and then added to NSC-34 cells expressing TDP-43WT-TR. TEM bar represents 50 nm. b TDP-43 mislocalisation was initially determined by manually counting fluorescent foci larger than 1 μm; various examples of TDP-43 structures scored as mislocalised are shown here. Bars represent 10 μm
The addition of SOD1 protein aggregates to NSC-34 cells resulted in a rapid and significant increase in percentage of cells that contained cytosolic TDP-43WT when treated with SOD1 aggregates (15.6 ± 2%) compared to the controls (absence of protein treatment; Fig. 2a). However, there was also a significant increase in cells displaying mislocalised TDP-43WT -TR when incubated with soluble G93A SOD1 (10.4 ± 2%) and soluble WT SOD1 (8.7 ± 2%). Furthermore, little to no cells were observed to contain TDP-43WT-positive cytoplasmic inclusions without protein treatment after the 2 h incubation period. While cells incubated with aggregated SOD1 aggregates had more TDP-43WT -TR inclusions than those incubated with soluble G93A, no significant differences were detected between them. In contrast, cells treated with SOD1 aggregates had significantly more TDP-43WT -TR aggregates when compared to cells treated with soluble WT SOD1 (Fig. 2a).
Fig. 2.
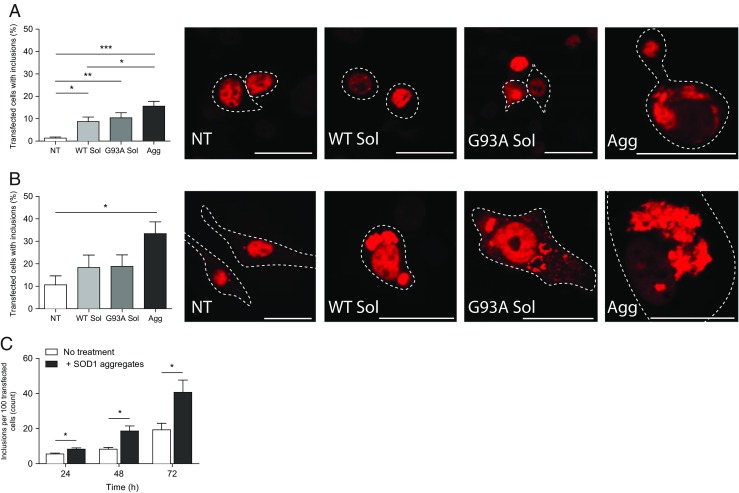
Exogenous recombinant SOD1 aggregates induce TDP-43-TR mislocalisation and aggregation. The percentage of NSC-34 cells containing TDP43WT-positive aggregates was assessed by the number of cells containing mislocalised TDP43 into foci that measured >1 μm per treatment including both TDP-43WT cleared from the nucleus and TDP-43WT that had accumulated in the cytosol even though some nuclear TDP43WT remained, as determined by Image J. Results shown for 2 h (a) and 72 h (b) as means ± SD, n = 3; ∗ P < 0.05, ∗∗ P < 0.01, ∗∗∗ P < 0.001. Example confocal images of TDP-43 pathology from respective time points. Bars represent 25 μm. c FloIT analysis of cell lysates. Following transfection (24, 48 and 72 h), adherent NSC-34 cells transiently transfected with TDP-43WT were co-cultured with 20 μg/ml and aggregated G93A SOD1 for indicated time intervals at 37 °C. Cells were harvested for supernatant analysis and then lysed. Cell lysates were incubated with RedDot2 for 2 min at RT. Transfection efficiencies, the number of inclusions and nuclei were determined by flow cytometry and results means ± SD, n = 3, *P < 0.05 compared to corresponding control (no protein treatment)
Given TDP-43 translocation can be a rapid response to stress (Zhang et al. 2014) that can be reversible (Liu-Yesucevitz et al. 2010), we next tested whether the TDP-43WT mislocalisation associated with SOD1 aggregate treatment persisted for 72 h. After the extended incubation with SOD1 aggregates, a large number of cells still exhibited cytosolic TDP-43WT, which appeared as larger clusters of aggregates or inclusion bodies, when assessed by confocal microscopy (Fig. 2b).
Incubation of aggregated SOD1 resulted in a significantly higher percentage of transfected cells with miscolcalised TDP-43WT compared to the control cells (Fig. 2b). In contrast to the 2 h experiment, there was no significant increase in cells displaying mislocalised TDP-43WT when incubated with soluble G93A SOD1 and soluble WT SOD1. However, it is important to note that complete loss of normal nuclear staining was still not observed and that addition of SOD1 aggregates leads to a variety of aggregation states (examples shown in Fig. 1b).
Given that the above data relied on manual counting which can be biased by the images taken and used for counting, an unbiased and novel flow cytometric (FloIT) method was next employed to quantify TDP-43 aggregation after addition of SOD1 aggregates. Cells were transiently transfected with TDP-43WT-tdTomato and incubated with human recombinant G93A mutant SOD1 aggregates for either 24, 48 or 72 h (Fig. 2c). At each time point, a significant increase in the number of TDP-43WT inclusions were identified in cells incubated in the presence of mutant SOD1 aggregates compared to the corresponding cells in the absence of mutant SOD1 aggregates (Fig. 2c).
Exogenous recombinant SOD1 aggregates induce the cytosolic mislocalisation and fragmentation of endogenous mouse TDP-43 in naive NSC-34 cells
Previous work suggests that the accumulation of the truncated TDP-43 fragment CTF25 is a pathological feature of TDP-43 proteinopathies (Neumann et al. 2007). Since this fragment has been suggested to play a critical role in ALS pathogenesis, we investigated whether adding aggregated SOD1 to NSC-34 cells had any effect on the formation of CTF25. We have previously shown that exogenously added recombinant SOD1 is capable of entering into NSC-34 cells (Grad et al. 2014), escaping into the cytosol (Zeineddine et al. 2015) and causes ER stress (Sundaramoorthy et al. 2013). To begin to investigate the effect of SOD1 aggregates on endogenous TDP-43 location and truncation, a subcellular fractionation assay and western blot was carried out on NSC-34 cell lysates (Fig. 3).
Fig. 3.

Exogenous recombinant SOD1 proteins induce TDP43 fragmentation and cytosolic mislocalisation in NSC34 cells. Cytoplasmic extract (C), membrane extract (ER/Golgi) (M), nuclear extract (N) and pellet extract (cytoskeleton) (P) fractions by centrifugation from NSC-34 cells treated with either a SOD1 aggregates or b WT SOD1 soluble (20 μg/mL) or c–d no added protein were separated by SDS PAGE under reducing conditions, transferred to nitrocellulose membrane and incubated with anti-TDP-43, anti-actin, anti-EEA1 or anti-vimentin Abs (as indicated)
TDP-43 location was then investigated by immunoblotting of the cytosolic, membrane (ER/Golgi), nuclear and cytoskeletal supernatant fractions (Fig. 3). In the SOD1 aggregate-treated fractions (Fig. 3a), TDP-43 was detected as a range of bands but predominantly as distinct bands at 43 and ~25 kDa in all fractions to some degree. In contrast, in untreated controls and cells treated with soluble SOD1, TDP-43 was found predominantly in the nuclear fraction (Fig. 3b, c). In samples treated with aggregates, there were also bands found in the membrane fraction at approximately 40 and 18 kDa. The additional 40 kDa band was also detected in the nuclear (N) and cytoskeletal (P) fractions at much lower levels. Interestingly, after treatment with SOD1 aggregates, the full-length mouse TDP-43 was predominantly cytosolic and associated with the membrane fraction suggesting a rapid movement from the nucleus after SOD1 aggregate addition. This was in contrast to the work presented above which shows that only a proportion of TDP-43WT-tdTomato becomes cytosolic after SOD1 incubation. This is likely to be due to the fact that we are overexpressing human TDP-43 in that system. To further confirm the purity of the fractions, control immunoblotting using an anti-actin, anti-EEA1 and anti-vimentin antibody was performed (Fig. 3d). These bands were detected in the membrane fraction for EEA1 (180 kDa), and cytoskeleton fraction for vimentin (54 kDa), while actin (42 kDa) acted as a loading control and could be found in all fractions.
Discussion
Mutations in the TDP-43 gene are found in sporadic and familial ALS, implicating TDP-43 as a contributing factor to disease (Kabashi et al. 2008; Sreedharan et al. 2008). TDP-43 has previously been reported to spontaneously form aggregates that resemble TDP-43 deposits in degenerating neurons in ALS FTD-U patients (Johnson et al. 2009). Furthermore, previous studies have reported that in the TDP-43 protein sequence, the C-terminal domain is critical for spontaneous aggregation and that TDP-43 is intrinsically aggregation prone (Johnson et al. 2009).
Consistent with misfolded SOD1 detected in sALS, previous work has demonstrated that WT SOD1 misfolding can be propagated cell to cell (Grad et al. 2014), at least partly due to protein aggregate uptake via macropinocytosis, subsequent stress and protein aggregation (Zeineddine et al. 2015; Sundaramoorthy et al. 2013; Zeineddine and Yerbury 2015). Further, internalisation of tau aggregates was found to stimulate stress granule formation and accumulation (Brunello et al. 2016). Here we have shown that exogenously added large SOD1 protein aggregates are capable of inducing the cytoplasmic mislocalisation and accumulation of TDP-43 in the motor neuron-like cell line (NSC-34). Furthermore, the present study demonstrates that the addition of large SOD1 aggregates induces rapid fragmentation of TDP-43 in the cytoplasmic fraction of NSC-34 cells. Collectively, this suggests that exogenous protein aggregates are capable of stimulating TDP-43 pathology through an unknown mechanism(s), resulting in their redistribution, fragmentation and aggregation, similar to what has been observed in most cases of ALS (Pokrishevsky et al. 2012; Nonaka et al. 2009; Arai et al. 2010; Correia et al. 2015).
An association between misfolded and/or aggregated SOD1 and TDP-43 mislocalisation has been previously suggested. For example, a study has reported that TDP-43 cytoplasmic mislocalisation and deposition into ubiquitin immunoreactive inclusions were observed in lower motor neurons of end-stage mutant SOD1 transgenic mice (Shan et al. 2009). In addition to this, misfolded human WT SOD1 has been observed in association with a cytosolic accumulation of mutant TDP-43 in TDP-43-fALS and WT TDP-43 in sALS (Pokrishevsky et al. 2012). It has been suggested that mutant TDP-43 may indirectly induce the propagation of WT SOD1 misfolding in fALS and sALS (Pokrishevsky et al. 2012). Given that the aggregation of TDP-43 and SOD1 appear to be distinct processes with structurally and morphologically discrete aggregates (Farrawell et al. 2015), it is unlikely that one will seed aggregation of the other in a conventional manner and therefore must be explained by an alternate mechanism.
Cytoplasmic accumulation of WT TDP-43 as a local response to injury or cell stress has been previously described in sALS (Liu-Yesucevitz et al. 2010). In addition to this, misfolded and aggregated SOD1 can induce ER stress and dysfunction and stress granule formation in cell culture (Nishitoh et al. 2008), rodent ALS models at symptom onset and disease end stage and human ALS patients (Atkin et al. 2006; Atkin et al. 2008; Ilieva et al. 2007; Kaus and Sareen 2015; Saxena et al. 2009). There is evidence that the uptake of extracellular misfolded WT and mutant G93A SOD1 into human and mouse neuronal cells causes disruptions to protein transport between the ER and Golgi apparatus, resulting in ER stress, Golgi fragmentation, and subsequent apoptotic cell death (Sundaramoorthy et al. 2013). ER stress is activated when proteins accumulate within the ER lumen, thus triggering the unfolded protein response (UPR), which in turn may lead to cellular apoptosis if unresolved (Atkin et al. 2014). There is a strong link between ALS pathology and ER stress (Walker and Atkin 2011), and it has been proposed that prolonged ER stress leads to motor neuron death in ALS (Walker and Atkin 2011). While generally the unfolded protein response activated by ER stress is a protective response able to rescue cells from proteotoxicity, in the ALS context this can lead to further aggregation of ALS-associated proteins such as TDP-43 (Suzuki et al. 2011). Taken together, these data are consistent with the idea that ER stress caused by uptake and maintenance of SOD1 aggregates may trigger TDP-43 mislocalisation and accumulation. In the case that uptake of protein aggregates into neurons causes TDP-43 mislocalisation and inclusion formation, it may be possible to inhibit this process by suppressing the uptake of aggregates. This may be possible by utilising molecular chaperones or manipulating macropinocytosis pathways.
TDP-43 mislocalisation is a common response to a variety of stressors including oxidative stress (Colombrita et al. 2009), heat shock stress (Chang et al. 2013), and proteosomal stress (Scotter et al. 2014). Indeed, extranuclear accumulation of WT TDP-43 has been shown to be a common pathology in sALS, FTD and Alzheimer’s disease, and therefore may be a general consequence of cellular stress (Wilson et al. 2011). Interestingly, exogenously applied Aβ amyloid aggregates associated with Alzheimer’s disease have also been shown to induce TDP-43 mislocalisation (Herman et al. 2011) suggesting that uptake of any aggregate regardless of which protein they are made from might be enough to trigger TDP-43 accumulation. This widespread predisposition of various types of neurons to undergo cytosolic accumulation of TDP-43 may be a consequence of the generally low protesostasis capacity of neurons (Yerbury et al. 2016).
Conclusion
In conclusion, the work presented here presents findings to suggest that aggregates made from SOD1 are able to induce TDP-43 mislocalisation and rapid fragmentation consistent with TDP pathology observed in sporadic disease. However, the exact mechanism(s) for this observation were not determined. It is likely that the stress induced by aggregate uptake was enough to trigger the mislocalisation of TDP-43. However, it should be noted that a high concentration (20 μg/ml) of large SOD1 protein aggregates were used in the current study to induce TDP-43 pathology in cell lines. This concentration is higher than that used in a number of similar studies (Nonaka et al. 2013; Münch et al. 2011). Future investigations into the mechanism(s)/role of SOD1 aggregates on the formation of pathological TDP-43 are therefore warranted.
Acknowledgments
The authors would like to thank Prof. Neil Cashman for insightful comments and review of the material presented here.
Author’s contributions
R. Zeineddine performed the experiments, interpreted and analysed data, as well as wrote the initial manuscript. N.E. Farrawell and I. A. Lambert-Smith performed the experiments and analysed the data. J.J. Yerbury designed the experiments, interpreted and analysed the data, and wrote and edited the manuscript.
Compliance with ethical standards
Funding
The project was funded by NHRMC project grant (1003032). JJY is supported by an NHMRC Career Development Fellowship (1084144) and a NHRMC Dementia Teams Grant (1095215).
References
- Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7(11):603–615. doi: 10.1038/nrneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Akiyama H. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Arai T, Hasegawa M, Nonoka T, Kametani F, Yamashita M, Hosokawa M, Niizato K, Tsuchiya K, Kobayashi Z, Ikeda K, Yoshida M, Onaya M, Fujishiro H, Akiyama H. Phosphorylated and cleaved TDP-43 in ALS, FTLD and other neurodegenerative disorders and in cellular models of TDP-43 proteinopathy. Neuropathology. 2010;30(2):170–181. doi: 10.1111/j.1440-1789.2009.01089.x. [DOI] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Turner BJ, Tomas D, Lysaght JA, Nunan J, Rembach A, Nagley P, Beart PM, Cheema SS, Horne MK. Induction of the unfolded protein response in familial amyotrophic lateral sclerosis and association of protein-disulfide isomerase with superoxide dismutase 1. J Biol Chem. 2006;281(40):30152–30165. doi: 10.1074/jbc.M603393200. [DOI] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Walker AK, McLean C, Tomas D, Horne MK. Endoplasmic reticulum stress and induction of the unfolded protein response in human sporadic amyotrophic lateral sclerosis. Neurobiol Dis. 2008;30(3):400–407. doi: 10.1016/j.nbd.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Atkin JD, Farg MA, Soo KY, Walker AK, Halloran M, Turner BJ, Nagley P, Horne MK. Mutant SOD1 inhibits ER-Golgi transport in amyotrophic lateral sclerosis. J Neurochem. 2014;129(1):190–204. doi: 10.1111/jnc.12493. [DOI] [PubMed] [Google Scholar]
- Banci L, Bertini I, Boca M, Calderone V, Cantini F, Girotto S, Vieru M. Structural and dynamic aspects related to oligomerization of apo SOD1 and its mutants. Proc Natl Acad Sci U S A. 2009;106(17):6980–6985. doi: 10.1073/pnas.0809845106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognesi B, Kumita JR, Barros TP, Esbjorner EK, Luheshi LM, Crowther DC, Wilson MR, Dobson CM, Favrin G, Yerbury JJ. ANS binding reveals common features of cytotoxic amyloid species. ACS Chem Biol. 2010;5(8):735–740. doi: 10.1021/cb1001203. [DOI] [PubMed] [Google Scholar]
- Bosco DA, Morfini G, Karabacak M, Song Y, Gros-Louis F, Pasinelli P, Goolsby H, Fontaine BA, Lemay N, McKenna-Yasek D, Frosch MP, Agar JN, Julien J-P, Brady ST, Brown RH. Wild-type and mutant SOD1 share an aberrant conformation and a common pathogenic pathway in ALS. Nat Neurosci. 2010;13:1396–1403. doi: 10.1038/nn.2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunello CA, Yan X, Huttunen HJ. Internalized tau sensitizes cells to stress by promoting formation and stability of stress granules. Sci Rep. 2016;6:30498. doi: 10.1038/srep30498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns NJ, Neumann M, Bigio EH, Holm IE, Troost D, Hatanpaa KJ, Foong C, White CL, 3rd, Schneider JA, Kretzschmar HA, Carter D, Taylor-Reinwald L, Paulsmeyer K, Strider J, Gitcho M, Goate AM, Morris JC, Mishra M, Kwong LK, Stieber A, Xu Y, Forman MS, Trojanowski JQ, Lee VM, Mackenzie IR. TDP-43 in familial and sporadic frontotemporal lobar degeneration with ubiquitin inclusions. Am J Pathol. 2007;171(1):227–240. doi: 10.2353/ajpath.2007.070182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman NR, Durham HD, Blusztajn JK, Oda K, Tabira T, Shaw IT, Dahrouge S, Antel JP. Neuroblastoma x spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev Dynam. 1992;194:209–221. doi: 10.1002/aja.1001940306. [DOI] [PubMed] [Google Scholar]
- Chang HY, Hou SC, Way TD, Wong CH, Wang IF. Heat-shock protein dysregulation is associated with functional and pathological TDP-43 aggregation. Nat Commun. 2013;4:2757. doi: 10.1038/ncomms3757. [DOI] [PubMed] [Google Scholar]
- Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- Ciryam P, Lambert-Smith IA, Bean DM, Freer R, Cid F, Tartaglia GG, Saunders DN, Wilson MR, Oliver SG, Morimoto RI, Dobson CM, Vendruscolo M, Favrin G, Yerbury JJ (2017) Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc Natl Acad Sci U S A [DOI] [PMC free article] [PubMed]
- Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nature Reviews. 2001;2:806–819. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- Colombrita C, Zennaro E, Fallini C, Weber M, Sommacal A, Buratti E, Silani V, Ratti A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J Neurochem. 2009;111(4):1051–1061. doi: 10.1111/j.1471-4159.2009.06383.x. [DOI] [PubMed] [Google Scholar]
- Correia AS, Patel P, Dutta K, Julien JP. Inflammation induces TDP-43 Mislocalization and aggregation. PLoS One. 2015;10(10):e0140248. doi: 10.1371/journal.pone.0140248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson Y, Kelley T, Mackenzie IR, Pickering-Brown S, Plessis DD, Neary D, Snowden JS, Mann DM. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathol. 2007;113(5):521–533. doi: 10.1007/s00401-006-0189-y. [DOI] [PubMed] [Google Scholar]
- Davidson YS, Raby S, Foulds PG, Robinson A, Thompson JC, Sikkink S, Yusuf I, Amin H, DuPlessis D, Troakes C, Al-Sarraj S, Sloan C, Esiri MM, Prasher VP, Allsop D, Neary D, Pickering-Brown SM, Snowden JS, Mann DM. TDP-43 pathological changes in early onset familial and sporadic Alzheimer’s disease, late onset Alzheimer’s disease and Down’s syndrome: association with age, hippocampal sclerosis and clinical phenotype. Acta Neuropathol. 2011;122(6):703–713. doi: 10.1007/s00401-011-0879-y. [DOI] [PubMed] [Google Scholar]
- Farrawell NE, Lambert-Smith IA, Warraich ST, Blair IP, Saunders DN, Hatters DM, Yerbury JJ. Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci Rep. 2015;5:13416. doi: 10.1038/srep13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsberg K, Jonsson PA, Andersen PM, Bergemalm D, Graffmo KS, Hultdin M, Jacobsson J, Rosquist R, Marklund SL, Brannstrom T. Novel antibodies reveal inclusions containing non-native SOD1 in sporadic ALS patients. PLoS One. 2010;5(7):e11552. doi: 10.1371/journal.pone.0011552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grad LI, Yerbury JJ, Turner BJ, Guest WC, Pokrishevsky E, O'Neill MA, Yanai A, Silverman JM, Zeineddine R, Corcoran L, Kumita JR, Luheshi LM, Yousefi M, Coleman BM, Hill AF, Plotkin SS, Mackenzie IR, Cashman NR (2014) Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc Natl Acad Sci U S A [DOI] [PMC free article] [PubMed]
- Herman AM, Khandelwal PJ, Stanczyk BB, Rebeck GW, Moussa CE. Beta-amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Res. 2011;1386:191–199. doi: 10.1016/j.brainres.2011.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilieva EV, Ayala V, Jove M, Dalfo E, Cacabelos D, Povedano M, Bellmunt MJ, Ferrer I, Pamplona R, Portero-Otin M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130(Pt 12):3111–3123. doi: 10.1093/brain/awm190. [DOI] [PubMed] [Google Scholar]
- Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284(30):20329–20339. doi: 10.1074/jbc.M109.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Velde CV, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, Pradat PF, Camu W, Meininger V, Dupre N, Rouleau GA. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40(5):572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kaus A, Sareen D. ALS patient stem cells for unveiling disease signatures of Motoneuron susceptibility: perspectives on the deadly mitochondria, ER stress and calcium triad. Front Cell Neurosci. 2015;9:448. doi: 10.3389/fncel.2015.00448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh PN, Whitwell H, Garofalo O, Buller J, Swash M, Martin JE, Gallo JM, Weller RO, Anderton BH. Ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis. Morphology, distribution, and specificity. Brain. 1991;114(2):775–788. doi: 10.1093/brain/114.2.775. [DOI] [PubMed] [Google Scholar]
- Lindberg MJ, Tibell L, Oliveberg M. Common denominator of Cu/Zn superoxide dismutase mutants associated with amyotrophic lateral sclerosis: decreased stability of the apo state. Proc Natl Acad Sci U S A. 2002;99(26):16607–16612. doi: 10.1073/pnas.262527099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bilgutay A, Zhang YJ, Vanderweyde T, Citro A, Mehta T, Zaarur N, McKee A, Bowser R, Sherman M, Petrucelli L, Wolozin B. Tar DNA binding protein-43 (TDP-43) associates with stress granules: analysis of cultured cells and pathological brain tissue. PLoS One. 2010;5(10):e13250. doi: 10.1371/journal.pone.0013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- McAlary L, Yerbury JJ, Aquilina JA. Glutathionylation potentiates benign superoxide dismutase 1 variants to the toxic forms associated with amyotrophic lateral sclerosis. Scientific Reports. 2013;3:3275. doi: 10.1038/srep03275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlary L, Aquilina JA, Yerbury JJ. Susceptibility of mutant SOD1 to form a destabilized monomer predicts cellular aggregation and toxicity but not in vitro aggregation propensity. Front Neurosci. 2016;10:499. doi: 10.3389/fnins.2016.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Münch C, O’Brien J, Bertolotti A. Prion-like propagation of mutant superoxide dismutase1 misfolding in neuronal cells. Proc Natl Acad Sci U S A. 2011;108(9):3548–3553. doi: 10.1073/pnas.1017275108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima-Yasuda H, Uryu K, Robinson J, Xie SX, Hurtig H, Duda JE, Arnold SE, Siderowf A, Grossman M, Leverenz JB, Woltjer R, Lopez OL, Hamilton R, Tsuang DW, Galasko D, Masliah E, Kaye J, Clark CM, Montine TJ, Lee VM, Trojanowski JQ. Co-morbidity of TDP-43 proteinopathy in Lewy body related diseases. Acta Neuropathol. 2007;114(3):221–229. doi: 10.1007/s00401-007-0261-2. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VMY. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. J Cell Sci. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Neumann M, Kwong LK, Truax AC, Vanmassenhove B, Kretzschmar HA, Van Deerlin VM, Clark CM, Grossman M, Miller BL, Trojanowski JQ, Lee VM. TDP-43-positive white matter pathology in frontotemporal lobar degeneration with ubiquitin-positive inclusions. J Neuropathol Exp Neurol. 2007;66(3):177–183. doi: 10.1097/01.jnen.0000248554.45456.58. [DOI] [PubMed] [Google Scholar]
- Nishitoh H, Kadowaki H, Nagai A, Maruyama T, Yokota T, Fukutomi H, Noguchi T, Matsuzawa A, Takeda K, Ichijo H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008;22(11):1451–1464. doi: 10.1101/gad.1640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonaka T, Arai T, Buratti E, Baralle FE, Akiyama H, Hasegawa M. Phosphorylated and ubiquitinated TDP-43 pathological inclusions in ALS and FTLD-U are recapitulated in SH-SY5Y cells. FEBS Lett. 2009;583(2):394–400. doi: 10.1016/j.febslet.2008.12.031. [DOI] [PubMed] [Google Scholar]
- Nonaka T, Masuda-Suzukake M, Arai T, Hasegawa Y, Akatsu H, Obi T, Yoshida M, Murayama S, Mann DM, Akiyama H, Hasegawa M. Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 2013;4(1):124–134. doi: 10.1016/j.celrep.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Pokrishevsky E, Grad LI, Yousefi M, Wang J, Mackenzie IR, Cashman NR. Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis. PLoS One. 2012;7(4):e35050. doi: 10.1371/journal.pone.0035050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polling S, Mok YF, Ramdzan YM, Turner BJ, Yerbury JJ, Hill AF, Hatters DM. Misfolded polyglutamine, polyalanine, and superoxide dismutase 1 aggregate via distinct pathways in the cell. J Biol Chem. 2014;289(10):6669–6680. doi: 10.1074/jbc.M113.520189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Some speculations about prions, amyloid, and Alzheimer’s disease. N Engl J Med. 1984;310(10):661–663. doi: 10.1056/NEJM198403083101021. [DOI] [PubMed] [Google Scholar]
- Roberts K, Zeineddine R, Corcoran L, Li W, Campbell IL, Yerbury JJ. Extracellular aggregated Cu/Zn superoxide dismutase activates microglia to give a cytotoxic phenotype. Glia. 2013;61(3):409–419. doi: 10.1002/glia.22444. [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10:10–17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Saxena S, Cabuy E, Caroni P. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nat Neurosci. 2009;12(5):627–636. doi: 10.1038/nn.2297. [DOI] [PubMed] [Google Scholar]
- Schwab C, Arai T, Hasegawa M, Yu S, McGeer PL. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J Neuropathol Exp Neurol. 2008;67(12):1159–1165. doi: 10.1097/NEN.0b013e31818e8951. [DOI] [PubMed] [Google Scholar]
- Scotter EL, Vance C, Nishimura AL, Lee YB, Chen HJ, Urwin H, Sardone V, Mitchell JC, Rogelj B, Rubinsztein DC, Shaw CE. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J Cell Sci. 2014;127(Pt 6):1263–1278. doi: 10.1242/jcs.140087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan X, Vocadlo D, Krieger C. Mislocalization of TDP-43 in the G93A mutant SOD1 transgenic mouse model of ALS. Neurosci Lett. 2009;458(2):70–74. doi: 10.1016/j.neulet.2009.04.031. [DOI] [PubMed] [Google Scholar]
- Sreedharan J, Blair LP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, Baralle F, Belleroche Jd, Mitchell D, Leigh N, Chalabi AA, Miller CC, Nicholson G, Shaw CE. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. J Cell Sci. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundaramoorthy V, Walker AK, Yerbury J, Soo KY, Farg MA, Hoang V, Zeineddine R, Spencer D, Atkin JD. Extracellular wildtype and mutant SOD1 induces ER-Golgi pathology characteristic of amyotrophic lateral sclerosis in neuronal cells. Cell Mol Life Sci. 2013;70(21):4181–4195. doi: 10.1007/s00018-013-1385-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Lee K, Matsuoka M. TDP-43-induced death is associated with altered regulation of BIM and Bcl-xL and attenuated by caspase-mediated TDP-43 cleavage. J Biol Chem. 2011;286(15):13171–13183. doi: 10.1074/jbc.M110.197483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CF, Yamada M, Toyoshima Y, Yokoseki A, Miki Y, Hoshi Y, Kaneko H, Ikeuchi T, Onodera O, Kakita A, Takahashi H. Selective occurrence of TDP-43-immunoreactive inclusions in the lower motor neurons in Machado-Joseph disease. Acta Neuropathol. 2009;118(4):553–560. doi: 10.1007/s00401-009-0552-x. [DOI] [PubMed] [Google Scholar]
- Toyoshima Y, Tanaka H, Shimohata M, Kimura K, Morita T, Kakita A, Takahashi H. Spinocerebellar ataxia type 2 (SCA2) is associated with TDP-43 pathology. Acta Neuropathol. 2011;122(3):375–378. doi: 10.1007/s00401-011-0862-7. [DOI] [PubMed] [Google Scholar]
- Walker AK, Atkin JD. Stress signaling from the endoplasmic reticulum: a central player in the pathogenesis of amyotrophic lateral sclerosis. IUBMB Life. 2011;63(9):754–763. doi: 10.1002/iub.520. [DOI] [PubMed] [Google Scholar]
- Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M, Hanson PI, Kimonis V, Pestronk A. TDP-43 accumulation in inclusion body myopathy muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2008;79(10):1186–1189. doi: 10.1136/jnnp.2007.131334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiten DR, San Gil R, McAlary L, Yerbury JJ, Ecroyd H, Wilson MR. Rapid flow cytometric measurement of protein inclusions and nuclear trafficking. Sci Rep. 2016;6:31138. doi: 10.1038/srep31138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson AC, Dugger BN, Dickson DW, Wang DS. TDP-43 in aging and Alzheimer’s disease—a review. Int J Clin Exp Pathol. 2011;4(2):147–155. [PMC free article] [PubMed] [Google Scholar]
- Yerbury JJ, Ooi L, Dillin A, Saunders DN, Hatters DM, Beart PM, Cashman NR, Wilson MR, Ecroyd H (2016) Walking the tightrope: proteostasis and neurodegenerative disease. J Neurochem [DOI] [PubMed]
- Zeineddine R, Yerbury JJ. The role of macropinocytosis in the propagation of protein aggregation associated with neurodegenerative diseases. Front Physiol. 2015;6:277. doi: 10.3389/fphys.2015.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeineddine R, Pundavela JF, Corcoran L, Stewart EM, Do-Ha D, Bax M, Guillemin G, Vine KL, Hatters DM, Ecroyd H, Dobson CM, Turner BJ, Ooi L, Wilson MR, Cashman NR, Yerbury JJ. SOD1 protein aggregates stimulate macropinocytosis in neurons to facilitate their propagation. Mol Neurodegener. 2015;10:57. doi: 10.1186/s13024-015-0053-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Xu YF, Cook C, Gendron TF, Roettges P, Link CD, Lin WL, Tong J, Castanedes-Casey M, Ash P, Gass J, Rangachari V, Buratti E, Baralle F, Golde TE, Dickson DW, Petrucelli L. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc Natl Acad Sci U S A. 2009;106(18):7607–7612. doi: 10.1073/pnas.0900688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Baldie G, Periz G, Wang J. RNA-processing protein TDP-43 regulates FOXO-dependent protein quality control in stress response. PLoS Genet. 2014;10(10):e1004693. doi: 10.1371/journal.pgen.1004693. [DOI] [PMC free article] [PubMed] [Google Scholar]