

Abstract
The novel class of dual-family immunophilins (henceforth abbreviated as DFI) represents naturally occurring chimera of classical FK506-binding protein (FKBP) and cyclophilin (CYN), connected by a flexible linker that may include a three-unit tetratricopeptide (TPR) repeat. Here, I report a comprehensive analysis of all current DFI sequences and their host organisms. DFIs are of two kinds: CFBP (cyclosporin- and FK506-binding protein) and FCBP (FK506- and cyclosporin-binding protein), found in eukaryotes. The CFBP type occurs in select bacteria that are mostly extremophiles, such as psychrophilic, thermophilic, halophilic, and sulfur-reducing. Essentially all DFI organisms are unicellular. I suggest that DFIs are specialized bifunctional chaperones that use their flexible interdomain linker to associate with large polypeptides or multisubunit megacomplexes to promote simultaneous folding or renaturation of two clients in proximity, essential in stressful and denaturing environments. Analysis of sequence homology and predicted 3D structures of the FKBP and CYN domains as well as the TPR linkers upheld the modular nature of the DFIs and revealed the uniqueness of their TPR domain. The CFBP and FCBP genes appear to have evolved in parallel pathways with no obvious single common ancestor. The occurrence of both types of DFI in multiple unrelated phylogenetic clades supported their selection in metabolic and environmental niche roles rather than a traditional taxonomic relationship. Nonetheless, organisms with these rare immunophilins may define an operational taxonomic unit (OTU) bound by the commonality of chaperone function.
Electronic supplementary material
The online version of this article (doi:10.1007/s12192-017-0813-x) contains supplementary material, which is available to authorized users.
Keywords: Chaperone, Immunophilin, Apicomplexa, Extremophiles, Flavobacteria, Spirochetes
Introduction
In Nature, two classes of immunophilins have been recognized for a long time, cyclophilin (CYN) and FK506-binding protein (FKBP) (Galat 2003). [Of note, although the cyclophilins are often given the acronym CYP, I have used the recently accepted root, CYN (Nebert et al. 2004) in this paper]. Members of both CYN and FKBP classes primarily function as chaperones that assist and regulate the folding of client proteins. In 2005, our team reported the founding members of dual-family immunophilins (DFI) that were recognized as natural chimera of the catalytic modules of CYN and FKBP, joined in either order, by a linker region (Adams et al. 2005; Barik 2006). Based on the order of the two modules, the DFIs were later subdivided into two types, with separate acronyms: CFBP (Cyclosporin- and FK506-binding protein, with the architecture CYN-linker-FKBP) and FCBP (FK506- and cyclosporin-binding protein, with the architecture FKBP-linker-CYN) (Adams et al. 2005; Barik 2006; Krücken et al. 2009; this paper). We reported six such immunophilins, one each in Toxoplasma gondii (a protozoan parasite of the Apicomplexa family), Theileria parva (also an Apicomplexan), Tetrahymena thermophilia, and Treponema denticola (a spirochete bacterium) and two in Flavobacterium johnsoni (a bacterium, belonging to Bacteroidetes (formerly Cytophaga-Flavobacterium-Bacteroides or CFB) (Adams et al. 2005; Barik 2006). We also showed that both the CYN and the FKBP modules in representative CFBP and FCBP were functional, possessing chaperone activities (Adams et al. 2005).
Since our first report, complete genome sequences have been available for numerous more microorganisms, many collected from the world’s oceans. Homology search of all GenBank sequences, including these recent ones, revealed over a hundred DFIs (Supplementary Material 1). The large-scale analysis, presented here, confirms that DFIs are absent in metazoa and advanced eukaroyta, such as all vertebrates, including mammals. In contrast, individual CYNs and FKBPs are known to occur in all eukaryotes, even unicellular ones, as also in many prokaryotes; moreover, multiple paralogs of each are also quite common (Galat 2003). For example, the human genome encodes 16 FKBP-like and 15 CYN-like sequences, Saccharomyces cerevisiae (yeast) contains 4 FKBPs and 8 CYNs, and Escherichia coli has 2 CYN and 4 FKBPs (Galat 2003). This relatively rare occurrence of the DFIs has been intriguing for years, but no attempt has been made to understand its significance. In this paper, I have paid special attention to the phylogenetic distribution of the DFI-encoding species, which revealed their extremophilic nature and/or unconventional habitat and nutrient usage. I also analyzed their sequence relationships and bioinformatically predicted structures, which shed light on the nature and phylogeny of this only known bifunctional family of protein chaperones in biology.
Methods
Sequence comparison
Multiple sequence alignments were performed by Clustal Omega (Sievers et al. 2011) at the EMBL-EBI web server (McWilliam et al. 2013) using MBED-like clustering guide tree and HMM profile-profile techniques with default parameters. The output, obtained without residue numbering, was saved in the Newick format and redrawn using the phylogenetic tree printer at the Indiana University web site (http://iubio.bio.indiana.edu/treeapp/treeprint-form.html), from which the phenogram tree with default tree growth and node positions was printed in the rooted form.
Homology-based 3-D structure
3-D modeling was done with I-TASSER (Yang et al. 2015b), which starts from experimentally determined structure templates in the PDB library and uses multiple threading programs by LOMETS, each of which can generate thousands of alignments. I-TASSER only accepts templates of the highest significance in the threading alignments measured by Z-score. One template of the highest Z-score was selected from each threading program, where the threading programs were sorted by the average performance in the large-scale benchmark test experiments. The simulations generated a large ensemble of structural conformations. The final models were selected using the SPICKER program to cluster all structures based on the pair-wise structure similarity, and four models, corresponding to the four largest structure clusters reported, were displayed in Fig. 2. The confidence of each I-TASSER model is quantitatively measured by C-score based on the significance of threading template alignments and the convergence parameters of the structure assembly simulations.
Fig. 2.

Homology-based three-dimensional (3D) structure prediction for T. gondii FCBP. The primary sequence of T. gondii FCBP (Supplementary Material 1) was subjected to homology modeling by TASSER as described in “Methods.” The PDB files of four highest scoring simulations (a–d) were opened in RasMol (Sayle and Milner-White 1995) as “cartoon” display, and in each, the three domains were labeled (FKBP, 3TPR, and CYN). The structures were rotated to keep the FKBP domain (at the N-terminus) to the left so that the structural differences are more easily discernible. The 3D molecular models are also submitted separately as PDB files (Models 1–4)
TPR sequence analysis
The published procedure of the Regan laboratory (Sawyer et al. 2013) was followed, in which the modules of each TPR were aligned to identify repeat-specific features. Since some of the TPR domains had spacer amino acids between the individual repeats, I refrained from analyzing the full 3TPR and compared only the exact TPR motifs. Due to the degeneracy of TPR sequences in general, 34 amino acid residues for each motif were visually demarcated, whereby the most universal residues, also conserved in strategic positions of the FCBP TPRs, served as alignment landmarks (Supplementary Material 1). A total of 30 FCBP 3TPRs were analyzed, and the amino acid frequencies in each position of each motif were plotted by the sequence logo presentation (Fig. 5). The FCBP TPR sequences were also compared head-to-head with the published data of 974 global 3TPRs (Sawyer et al. 2013), which did not include the FCBP TPRs.
Fig. 5.

Sequence consensus among the three TPR motifs of FCBP. The 34-residue sequences of the TPR1, TPR2, and TPR3 motifs of all FCBPs were plotted in sequence logo format using the WebLogo software (Crooks et al. 2004). In each panel, the plot was compared with the corresponding TPR motif of all biological 3TPRs (Sawyer et al. 2013), as described in “Results.” Amino acid numbers (N- to C-terminus) are shown along the X-axis, and the Y-axis represents their relative abundance. The most conserved amino acid residues in the FCBP TPRs are indicated by all arrowheads. Residues that are known to interact with the MEEVD sequence of Hsp90, based on structural studies of CYN and FKBP TPRs (Cliff et al. 2006; Scheufler et al. 2000; Ward et al. 2002; Wu et al. 2004), are specifically indicated with closed arrowheads (detailed in Results); note that they are also among the most highly conserved, supporting the functional importance of a potential FCBP TPR-Hsp90 interaction
Results
DFI genes occur in select microorganisms only
In an effort to gain an ecological and phylogenetic clue to DFI function, I note the following characteristics of the DFI organisms.
Minority bacteria and eukaryotes: DFI is absent in most living species; as mentioned above, none could be found in higher, multicellular eukaryotes (metazoa), including human. Whereas the CFBPs occur in select bacterial species, the FCBPs are found in simple, primitive eukaryotes (Fig. 1; Supplementary Materials 1 and 2). The vast majority of CFBPs are found mainly in Flavobacteria and Spirochetes, and the majority of FCBPs are found in Apicomplexa and the closely related Ciliophora (Lee and Kugrens 1992). No well-characterized archaeal organism contained FCBP/CFBPs (Supplementary Material 2). Overall, the DFI genes are found in complex bacteria or simple protozoa. For example, Spirochetes are bacteria with a distinctive long, helically coiled (spiral) shape, and many have complex life cycles, spanning insect carriers and mammalian hosts (Paster and Dewhirst 2000; Charon et al. 2012; Vedantam and Viswanathan 2012). Essentially, all Flavobacteria switch from rod-shaped cells to long filaments that can intertwine to form clumps (Jooste and Hugo 1999). They also contain a variety of pigments such as yellow pigments (Verma and Rathore 2015) or proteorhodopsin and can be stimulated by light (Gómez-Consarnau et al. 2007); Yoshizawa et al. 2014). In another example, R. biformata forms characteristic rusty red pigmented colonies and, like many Flavobacteria, contains genes for the complex pathway of carotenoid biosynthesis (Oh et al., 2009). The complexity and novelty of the Bacteroidetes Phylum, specially the Flavobacteria family, are also underscored by the fact that many microbes in this family have been phylogenetically reclassified from different initial assignments (Jooste and Hugo 1999; Yoon et al. 2012).
Degradative enzymes: Many DFI organisms are heterotrophs, encoding genes for degradative enzymes that allow them to survive on diverse high molecular weight compounds; such enzymes include protease, sulfatase, pectinase, chitinase, and α-amylases (Jooste and Hugo 1999; Abt et al. 2011; Bauer et al. 2006; Kirchman 2002; Klippel et al. 2011). Some in fact derive their names from the ability to degrade and scavenge. Cellulophaga (neo-Latin for “cellulose-eating”) eponymously excretes hydrolytic enzymes that allow it to metabolize complex biomaterials, such as cellulose and other polysaccharides, proteins, amino acids, and even noncellular chemicals such as Tween, a detergent. Most Flavobacteria are in fact aquatic and major decomposers of high molecular weight organic matter (Klippel et al., 2011; Cottrell and Kirchman 2000; Woyke et al., 2011).
Gliding motility: Many DFI organisms contain flagella or cilia that are used for gliding or swimming motility (Charon et al., 2012; McBride 2004), essential for survival and nutrient scavenging in the wild (see below). Pioneering studies by the McBride laboratory, using Flavobacterium johnsoniae as a model, has identified a large set of genes (“Gld” genes), all of which contributed to optimal gliding (McBride and Braun 2004; Braun and McBride 2005; Hunnicutt et al. 2002). Cells with mutations in these genes grew as well as wild type in laboratory culture but were completely nonmotile. As mentioned earlier, our detailed biochemical studies characterized the GldI gene product as a CFBP (Adams et al. 2005). Interestingly, three of the Gld genes comprised a putative ATP-binding cassette (ABC) transporter, a multisubunit complex (McBride 2004; Braun and McBride 2005). Additionally, these investigators identified a novel protein secretion system, the Por secretion system (PorSS), in this family, and found that the PorSS genes are in fact a subset of the Gld genes, suggesting a relationship between the secretion system and motility (Rhodes et al. 2011; McBride and Zhu 2013). The F. johnsoniae PorSS, for example, is needed for the assembly of the gliding motility apparatus as well as for the secretion of a chitinase. Recently, this group conducted bioinformatic analysis of 37 genomes of members of the Bacteroidetes phylum, which included several FCBP-encoding bacteria in Table 1, and found that the gliding motility genes and PorSS genes are widespread in this phylum (McBride and Zhu 2013). Control genes, associated with other bacterial protein secretion systems, were less common.
Life in nutrient-poor habitats, requiring scavenging: Although phylogenetically diverse (see later), many DFI organisms, particularly Flavobacteria, live in aquatic environments such as marine waters with exceedingly low concentrations of organic nutrient and, therefore, must possess an active nutrient transporter system for scavenging. Not surprisingly also, many DFI organisms can degrade high molecular weight organic matter, as mentioned above. Most Spirochetes and Apicomplexa are pathogens that must scavenge available nutrients in their niche tissues; for instance, T. denticola, a periodontal pathogen, thrives in the anaerobic conditions under the gum (Frederick et al. 2011; Sela 2001). Capnocytophaga spp. are Gram-negative, microaerophilic, and CO2-requiring found in the oropharyngeal tract of mammals and cause infections in animal bite wounds as well as periodontal diseases (Jolivet-Gougeon et al. 2007).
Extremophiles: Many, if not all, of these organisms (Supplementary Material 2) live in various degrees of extreme habitats and often derive energy from the reduction of exotic substrates. For example, Desulfotalea psychrophila is a marine, sulfate-reducing, delta-proteobacterium that is ice-dwelling (sympagic) and can grow at subzero temperatures (Rabus et al. 2004); Flavobacterium psychrophilum is a psychrotroph that infects diverse species of cold water salmon and trout (Duchaud et al. 2007; Nematollahi et al. 2003); Psychroflexus torquis represents a group of Antarctic ice-derived psychrophilic proteobacteria (Feng et al. 2014); Borrelia hermsii, causative agent of tick-borne relapsing fever (Di et al. 2014), must survive at the febrile temperature resulting from the host’s response to the parasite. Lastly, nearly all Flavobacteria and many Spirochetes use a large variety of substrates and are also flexible in this behavior. Treponema spp., the first spirochetes to be isolated from termite hindguts, can use H2 plus CO2 as substrates and are capable of homoacetogenic growth on mono- and disaccharides and (in the case of ZAS-2; Supplementary Material 2) methoxylated benzenoids (Graber and Breznak 2004). They are also capable of mixotrophic growth, i.e., simultaneous utilization of H2 and organic substrates. As its name implies, Desulfobulbus propionicus is able to disproportionate elemental sulfur to sulfate and sulfide and is of great environmental and industrial interest not only due to its impact on the sulfur cycle in aquatic sediments but also its large substrate spectrum and a broad versatility in using various fermentation pathways (Pagani et al. 2011).
Fig. 1.

Phylogenetic location of DFI organisms. Since the DFI organisms (Supplementary Material 2) occupy only a few branches, I have adopted a simplified three-“domain” evolutionary tree (Woese et al. 1990) instead of the more detailed trees proposed more recently (Cavalier-Smith 2010). The major DFI clades are circled, and the number of DFI genes in each clade, counted from Supplementary Material 2, are shown in parentheses. The CFBP-containing organisms (blue) are all prokaryotic, while the FCBP-containing organisms (red) are all eukaryotic. The Dinoflagellates are very similar to other microbes with flagella/cilia, such as Apicomplexa and Ciliophora (also see Fig. 4), and therefore, the single dinoflagellate FCBP was not assigned a separate branch to conserve space. Note that the CFBP and FCBP lineages are well separated, making direct gene transfers implausible, thus suggesting their independent evolution. I propose that the DFI-encoding organisms, although they are phylogenetically diverse, constitute a new Operational Taxonomic Unit (OTU)
Table 1.
Examples of genes in the immediate neighborhood of CFBP genes
| Organism | DFI | Neighboring genes (Ensembl transcript/protein ID) |
|---|---|---|
| Capnocytophaga canimorsus | CFBP | 3′-Phosphoadenosine 5′-phosphate phosphatase (CEN33219); putative phenylacetic acid degradation NADH oxidoreductase paaE (CEN33220); Crotonase (a hexameric protein; CEN33205); Ser HMTase (dimer) |
| Jejuia pallidilutea | CFBP | ABC transporter (GAL90931) |
| Kordia algicida | CFBP | Secreted protein containing N-terminal Zinc-dependent carboxypeptidase related domain (EDP94481); putative transmembrane protein (EDP94488) |
| Leeuwenhoekiella blandensis | CFBP | ABC transporter ATP-binding protein (EAQ50783, EAQ50799); putative integral membrane transport protein; ATPase (EAQ50797); CDP-DAG Ser O-phosphatidyl transferase (EAQ50788) |
| Polaribacter dokdonensis | CFBP | Two-component system sensor His kinase/response regulator, encoded by multiple tandem genes (KOY52959, KOY52960, KOY52961, KOY52962); CYN (KOY52954) |
| Psychroflexus torquis | CFBP | PPIase gliding motility-associated protein GldI (AFU67562); protein with phytase-like active site (AFU67564)a; histidine phosphatase (AFU67565); putative membrane protein (AFU67568) |
Only a few Flavobacterial CFBP are shown as representatives; however, to have a diverse coverage with non-redundant sequences, we avoided closely similar organisms such as two species of the same genus. Neighbors of F. johnsoniae CFBP genes have been described in detail by the McBride group (Braun and McBride 2005; McBride and Braun 2004), and not shown here aPhytases, such as histidine acid phosphatase (HAP), are important for the bioavailability of essential phosphate from phytic acid; this gene is highly conserved in all Flavobacteria. Note that the next gene in this bacterial species is also a histidine phosphatase, as shown
In what follows, I explored the molecular aspects of the DFIs in an attempt to understand the unique features described above.
Modular and unique domain arrangements of the DFIs
The domain arrangements of the DFI family polypeptides exhibited several notable patterns. While the dual-family immunophilins are rare, the single-family double immunophilins, i.e., CYN-CYN or FKBP-FKBP, are actually even rarer (Galat 2003). Specifically, there is currently no example of a chimeric CYN-CYN polypeptide, and the domain structures of the two highly similar double-FKBPs, designated as FKBP51 and FKBP52, comprised two linked FK506-binding domains (of which only one has PPIase activity), followed by a TPR domain at the C-terminus (Galat 2003). The FKBP51/52 are found in the mammals, which do not encode any DFI (Fig. 1). Thus, the FCBP-type DFIs are the only examples of two immunophilin modules of any type connected by a TPR domain. Finally, in TPR - domain CYNs such as human CyP40 and yeast CyP42 and CyP45, the TPR domain is at the C-terminal side of the CYN domain. Thus, the FCBP is the only example of a CYN domain with a TPR to its N-terminal side. The significance of the unique domain structure of DFI chaperones must await an analysis of the relationship between structure and function.
Further examination of the DFI domains and the host organisms revealed several other important features. First, CFBPs are exclusive to prokaryotes (bacteria) (Figs. 1 and 3), whereas FCBPs are exclusive to eukaryotes (Figs. 1 and 4), which is a clear distinction. Second, there is a consistent pattern in the linker sequences as well: (a) All CFBPs (CYN-FKBP) are devoid of TPR (Supplementary Material 1). Since no bacterial CYN or FKBP contains TPR (Galat 2003), this also suggests that the CFBPs may have been created by fusion of a CYN with a TPR-free, small bacterial FKBP. (b) The FCBPs contain TPR, i.e., they are all FKBP-TPR-CYN (Supplementary Material 1). This is reminiscent of the “large” FKBPs in diverse species that contain one, two, or (in rare cases) three FKBP domains at their N-terminal side and a TPR domain at their C-terminal side; besides the FKBP51/52 mentioned above, they include Xap2, FKBP36, FKBP59, FKBP42, and FKBP62. Reciprocally, the “small” FKBPs (e.g., hFKBP12) do not have TPR. Thus, it is possible that FCBPs were created by fusion of a eukaryotic, large FKBP-3TPR with CYN. Indeed, the occurrence of TPR in the FCBPs, but not in the CFBPs, parallels the greater prevalence of the TPR domains in eukaryotes over prokaryotes (Cerveny et al. 2013).
Fig. 3.
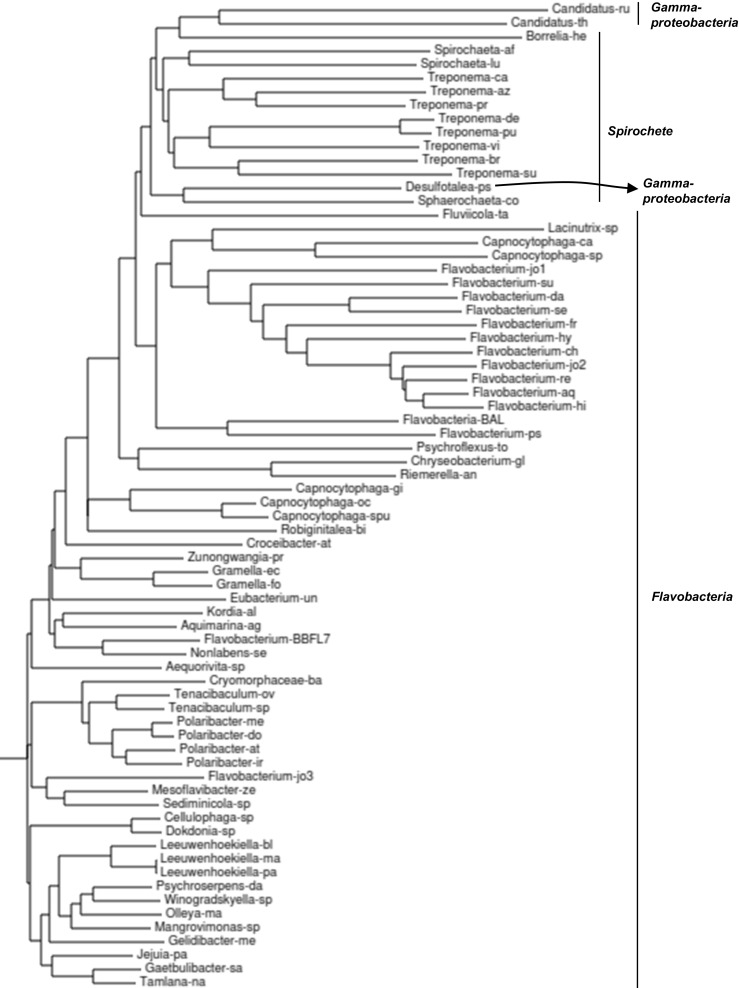
Relationship among CFBP sequences. The phenogram of all major CFBP sequences was derived from multiple sequence alignment as described in “Methods.” The two large bacterial clusters (Spirochetes and Flavobacteria) and the three species of gammaproteobacteria are so marked. Desulfobolus propionicus, also a proteobacteria (Supplementary Material 2), was omitted here to conserve space, but was counted in Fig. 1
Fig. 4.

Relationship among FCBP sequences. The phenogram of all FCBP sequences was derived from multiple sequence alignment as described in “Methods.” These sequences are exclusively eukaryotic, and the clusters are marked
DFI-neighboring genes often code for large and multisubunit proteins that are potential clients for chaperones
There is a large body of evidence in a variety of extant taxa that genes, clustered together in the chromosome, are often regulated as a unit (Ghanbarian and Hurst 2015; Michalak 2008). These genes are often functionally related, and their protein products may also physically interact to form a complex (Michalak 2008; Fraser et al. 2004; Ge et al. 2001; Lee and Sonnhammer 2003). The classic example of transcriptional co-regulation is the bacterial operon, although in eukaryotes the exact molecular mechanism of co-regulation of nuclear genes remains unclear. Co-regulation is most pronounced in the immediate vicinity (<100 kb) but may extend much further. With this premise in mind, I examined the chromosomal location of the DFI genes in a few representative organisms for which the genome sequences are well annotated and found that several of the immediate neighbors on either side of the DFI gene encode proteins that may have a higher need for folding assistance. As shown (Table 1), these proteins are relatively large or constitute multisubunit complexes, and some of them are also membrane-bound, playing a role in transport or motility. As mentioned earlier, two of the first discovered DFI genes were F. johnsoniae CFBPs (Adams et al. 2005) and were located next to the GldI gene, one of several Gld family genes, essential for gliding motility and chitin utilization (McBride and Braun 2004). It is to be noted that the GldI gene itself codes for a membrane lipoprotein (McBride and Braun 2004) with a domain that shows strong homology to FKBP and thus may also act as a chaperone in conjunction with the two CYN-FKBP paralogs that we reported (Adams et al. 2005). One of the two CYN genes of Polaribacter dokdonensis, a flavobacterium (Yoon et al. 2006), is a close neighbor of the CFBP gene (Table 1), perhaps suggesting a co-operative role. Clearly, an extended analysis of DFI gene neighbors in multiple other organisms may reveal the full significance of these observations, but it is tempting to speculate that at least some of the proteins encoded by the neighboring genes could be either clients or accessories of the DFI chaperones and, therefore, have evolved to be expressed together with the DFI gene.
Predicted tertiary structure of a DFI
Our biochemical studies using deletion mutants of T. gondii FCBP documented that the CYN and the FKBP domains can function as independent modules (Adams et al. 2005), and therefore, it was hypothesized that the linker region between them might allow the DFI chaperone the flexibility that it needs to use both modules simultaneously. As no crystal structure of a DFI is yet available, I conducted homology-based tertiary structure modeling of TgFCBP using the I-TASSER program, as described in “Methods,” selecting the top 4 models. As shown (Fig. 2), in all four models the three domains of the structure are clearly discernible: the FKBP domain (with five antiparallel β-strands around a central α-helix, the so-called FK fold) (Sinars et al. 2003), the three-TPR linker (i.e., helix-turn-helix × 3; abbreviated as 3TPR), followed by the CYN domain. While each domain structure is largely constant as expected, it is instructive that they show various degrees of orientation relative to each other, clearly due to both rotational and torsional flexibility of the 3TPR linker (Fig. 2). Thus, the structural ensemble provides support for our simultaneous chaperoning hypothesis, since the FKBP and CYN domains are well separated and may adopt different relative orientations due to the structure and flexibility of TPR.
Origin and phylogeny of the DFI genes
First of all, neither the FCBP nor the CFBP gene is a simple duplication of a CYN/FKBP paralog in the same cell, since a free-standing CYN/FKBP gene does not even exist in many DFI organisms (Supplementary Material 2), and when it does (e.g., in Croceibacter atlanticus), the sequence is nonidentical. To understand the evolutionary history of the DFI genes, I investigated the similarities among the DFI sequences by multiple alignment using Clustal Omega. The FCBP and CFBP sequences were aligned separately because of their different domain structures. These results (Figs. 3 and 4) revealed higher degrees of similarity within each phylogenetic set. For example, all Apicomplexa were clustered together as were all Spirochetes and Flavobacteria. Thus, the relationship among the DFIs conforms to the phylogeny of the organisms in which they occur. It is also notable that DFI genes are not strictly conserved in any given phylogenetic branch; for example, although several Apicomplexa encode DFI, others do not, a notable exception being Plasmodia, such as P. falciparum, causative agent of virulent malaria. I conclude that all DFI genes likely originated by gene fusion (without duplication) sometime after the prokaryotes and the eukaryotes split, and the FCBPs originated after the creation of the Archaea branch (Fig. 1), resulting in independent generation of CFBPs and FCBPs. Thereafter, the DFI sequences within each clade underwent further changes with time to select for the best fit in a particular habitat. Note that these alignments were performed with full-length DFI sequences, including the TPR, but as described below, I also analyzed the TPRs separately to tease out their unique sequence characteristics from the overall sequence.
Unique TPR signature of the FCBP family and its phylogenetic subsets
TPR sequences are roughly 34 amino acid-long, degenerate repeats, in which the properties of key amino acids in specific positions, rather than the exact residues, are conserved (Cerveny et al. 2013; D'Andrea and Regan 2003). Although TPRs occur in various number of repeats, a recent large-scale survey of all TPR-containing proteins in living organisms revealed that three-TPR (3TPR) is the most common, comprising ~40% of all TPRs (Sawyer et al. 2013). The analysis led to the identification of overall amino acid signatures in all 3TPRs as well as distinct signatures of each of the three individual TPR motifs. As described in “Methods,” I adopted this successful strategy to analyze the 3TPR domains of the FCBPs. These plots (Fig. 5) revealed a number of interesting and unique features of the TPRs of the FCBPs. First, as marked by arrows, each FCBP TPR showed major amino acid differences with the corresponding global TPR. In TPR1, for example, the most unique residues were: K5, G8, F12, Y24, E26, G27, Y30 and D/E32. Whereas G8 and Y24 could be seen in a subset of the all 3TPR1 pool, the residues K5, F12, and D/E32 were virtually absent in all 3TPR1s. The residues K5, G8, A20, and Y24 were essentially invariant in all 30 FCBP TPR1 (with the singular exception of L5 in Pseudocohnilembus persalinus) (Supplementary Material 1). Such uniqueness extended to the next two TPR motifs as well. In TPR2, the most unique residues were: L5, N6, C10, and K13, of which L5 and K13 being extremely rare in the global non-FCBP pool. Second, in the TPR3 of FCBPs, multiple unique residues were found that were very rare in the global non-FCBP pool; they were K2, L4, Y5, R11, L17, K21, D23, L24, K29, D31, and N34. Finally, when TPR1, TPR2, and TPR3 within the FCBP family were compared with one another, the consensus signature of the three showed clear differences; in fact, they appeared to be more different than the three TPRs in the global 3TPR pool (Fig. 5). I conclude that the FCBP TPRs have a consensus signature that is unique from the global biological TPRs. It is to be pointed out that these unique residues are conserved among all FCBP organisms even though they span diverse genera and species (Supplementary Material 2). Finally, an invariant tryptophan (W) is present exclusively in the Apicomplexa FCBP TPR domains, located three residues downstream of the first TPR motif (TPR1) (Supplementary Material 1).
Since the FCBP TPRs were clearly distinct from the average sequence of all biological TPRs, I inquired whether they are similar to TPRs found in FKBPs and CYNs. As mentioned before, while most immunophilins are devoid of TPRs, the large FKBPs (such as FKBP51, FKBP52, and FKBP8/38) and large CYNs (such as CYP40) contain 3TPRs. However, as these examples are small in number, I realized that a multiple alignment to find immunophilin-specific TPR consensus will be less useful than searching for specific residues known to function in protein-protein interaction. Among the TPR-interacting proteins that have been studied in detail are the heat shock proteins 70 and 90 (Hsp70 and Hsp90), which also act as chaperones (Mattoo and Goloubinoff, 2014). Both are also ubiquitous proteins with highly conserved sequence. The binding exhibits various degrees of specificity between the two partners; Hsp90, for example, is preferred over Hsp70 to bind to the TPRs of the large FKBPs (Assimon et al. 2015; Guy et al. 2015). In particular, peptide-TPR co-crystal structure and mutational analysis have established that the invariant C-terminal pentapeptide, MEEVD, of Hsp90 (Gupta 1995) interacts with specific residues that are common to the TPRs of long FKBPs and CYNs (Assimon et al. 2015; Cliff et al. 2006; Scheufler et al. 2000; Ward et al. 2002; Wu et al. 2004). The residues, found to be absolutely essential for MEEVD binding, are: K5, N9 in TPR1, N6 in TPR2, and K2, R6 in TPR3. As shown in Fig. 5, these residues are invariant or highly conserved in the FCBP TPRs as well. I have also found Hsp90 homologs in all the DFI organism genomes that I have examined so far, and all of them have C-terminal MEEVD in their conceptually translated sequence. Although originally discovered as a gene induced under heat shock, subsequent studies have shown that proteins of several Hsp families are induced and are protective in cold shock and other stresses as well (García-Descalzo et al. 2011; Hossain and Nakamoto 2002; Kaneko and Kibayashi 2012; Maikova et al. 2016; Sonna et al. 2010). It is, therefore, likely that Hsp90 is one of the proteins that FCBPs interact with, which may further enhance the efficiency of chaperoning of the client proteins under stress conditions, including extreme heat and cold, to which many DFI organisms are subjected to.
Discussion
This communication provides the first theoretical framework of the possible role and evolution of the dual-family immunophilins (DFI) that were reported by our laboratory in 2005 (Adams et al. 2005). Homology-based structure prediction concurred with our hypothesis that the DFIs may act as bifunctional chaperones due to the flexibility of the loop connecting the CYN and FKBP domains. We also showed previously that several DFIs possess protein prolyl isomerase (PPIase) activities (Adams et al. 2005), which play an important role in the proper folding of the client protein by promoting cis-trans isomerization of the Pro residues (Schmidpeter and Schmid 2015). I propose a model (Fig. 6) in which, under the extreme conditions of the DFI organisms, the DFI chaperones play an essential role in the cotranslational folding or renaturation of multiple polypeptides of a large protein complex or the different domains of a large polypeptide. If pre-folded separately by individual immunophilins, these polypeptides may not form a functional multisubunit complex, and such folding may also be kinetically too slow to protect the polypeptides from denaturation in the harsh environment. Proper assembly of complexes such as nutrient transporters may also be essential for their transport to appropriate locations in the membrane. In this scenario, the DFIs themselves may be located in the vicinity of the cell membrane in order to promote cotranslational folding and vectorial transport of membrane proteins and extracellular enzymes.
Fig. 6.

Model for DFI function. In this model, a dual-family immunophilin uses its two chaperone domains (FKBP and CYN, in either order) in cis to assist in proper folding of large polypeptides (A, B) or multisubunit complexes, more rapidly, and efficiently than single chaperones in trans can
Due to the unambiguous placement of CFBPs in prokaryotes and FCBPs in eukaryotes, they do not have a proximal common ancestor, and the lineage-specific ancestors, such as Hydrogenbacter, Archaea, or Diplomonadida (Fig. 1), do not have DFI genes. Thus, independent creation of the ancestral CFBP and FCBP genes, respectively in prokaryotes and eukaryotes, followed by minor sequence changes for each species within a phylum, can explain the current phylogenetic distribution of the DFIs. Even within a phylum, the DFI sequences may have been selected by lifestyle needs of the organism rather than by phylogeny, since as mentioned, Plasmodia is a major Apicomplexan that do not contain a DFI, while several other members of this phylum do.
The CYN and FKBP domains of DFIs offer no surprises, since their primary and predicted tertiary structures are similar to the individual homologs in all living cells (Fig. 2). Our early studies (Adams et al. 2005) revealed that each domain possessed the hallmark PPIase activity and was also inhibited by the class-specific inhibitors, thus proving that the active site and drug-binding residues are intact. A gross examination of several other DFIs revealed that the key enzymatic residues are conserved in them. I presume that the majority of DFIs are functional PPIases and may also have the expected chaperone activity. As a corollary, they may be explored as therapeutic targets (Schiene-Fischer 2015; Ünal and Steinert 2014), perhaps through the designing of selective bispecific drugs (Dunyak et al. 2015; Yang et al. 2015a).
Our studies of TPR domains in the FCBP-type DFIs have revealed unique consensus sequences; they also revealed that the three TPR motifs in these domains are not created equal. Since TPR domains are involved in protein-protein interactions, it is logical to conjecture that the unique residues are involved in selective interactions with FCBP-specific client proteins. This may hold the key to the ability of the FCBP to specifically chaperone the large proteins in extreme environments, as I have suggested. Experimental detection of interacting residues in FCBP-client complexes will shed light on this intriguing possibility.
There are very few published reports of the total microbiome of extreme environments. One study of microbial diversity in a deep-subsurface hydrothermal aquifer in Naica Mine, Mexico (Ragon et al. 2013) employed 18S rRNA sequence as the species identifier and detected only a few organisms in the high-temperature low-nutrient regions, consisting of no eukaryotes, only several Proteobacteria, and a few archaea, as the major OTUs. None of the DFI organisms were found in this particular niche. A more recent and comprehensive study of >30,000 marine microorganisms from global ocean waters allowed a clearer decoupling of function and taxonomy (Louca et al. 2016) and revealed that the environmental conditions, not taxonomy, dictate the metabolic profile of the organisms, thus defining a common “metabolic niche.” Interestingly, the marine microbial population in this study included two CFBP phyla (Bacteroidetes, Spirochetes) and shared several metabolic functions (Louca et al. 2016). This is again consistent with our hypothesis that environment-related metabolic needs, rather than phylogeny per se, resulted in the creation and evolution of the DFI sequences, and therefore, the DFI organisms define a new OTU. Identification of the DFI substrate repertoire from multiple organisms and determination of their roles in natural or simulated conditions will shed light on the physiological function of this unique and rare family of immunophilins.
Electronic supplementary material
(DOCX 43.6 kb)
(DOCX 37.3 kb)
Acknowledgements
I am deeply indebted to two research groups for generously sharing their raw datasets: Professor Lynne Regan (Yale University, New Haven, CT) and her associate, Dr. Jieming Chen (currently in the Institute for Computational Health Sciences, University of California, San Francisco), for sharing the TPR sequence collection used in their analysis (Sawyer et al. 2013), and Dr. Stilianos Louca (University of British Columbia, Canada) for sharing the list of the organisms from their OTU studies (Louca et al. 2016). I thank Professor Andreas Teske (Marine Sciences, University of North Carolina, Chapel Hill, NC) for helpful discussions on the phylogeny and ecology of DFI organisms.
Abbreviations
- CYN
Cyclophilin;
- CFBP
Cyclosporin- and FK506-binding protein
- FCBP
FK506- and cyclosporin-binding protein
- DFI
Dual-family immunophilin
- Hsp
Heat shock protein
- OTU
Operational taxonomic unit
- TPR
Tetratricopeptide repeat
Compliance with ethical standards
Funding
No external funding was used for these studies. The publication cost of this paper was paid by the personal fund of the author.
Footnotes
Electronic supplementary material
The online version of this article (doi:10.1007/s12192-017-0813-x) contains supplementary material, which is available to authorized users.
References
- Abt B, Lu M, Misra M, Han C, Nolan M, Lucas S, et al. Complete genome sequence of Cellulophaga algicola type strain (IC166) Stand Genomic Sci. 2011;4:72–80. doi: 10.4056/sigs.1543845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams B, Musiyenko A, Kumar R, Barik S. A novel class of dual-family immunophilins. J Biol Chem. 2005;280:24308–24314. doi: 10.1074/jbc.M500990200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assimon VA, Southworth DR, Gestwicki JE. Specific binding of tetratricopeptide repeat proteins to heat shock protein 70 (Hsp70) and heat shock protein 90 (Hsp90) is regulated by affinity and phosphorylation. Biochemistry. 2015;54:7120–7131. doi: 10.1021/acs.biochem.5b00801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barik S. Immunophilins: for the love of proteins. Cell Mol Life Sci. 2006;63:2889–2900. doi: 10.1007/s00018-006-6215-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Kube M, Teeling H, Richter M, Lombardot T, Allers E. Whole genome analysis of the marine Bacteroidetes 'Gramella forsetii' reveals adaptations to degradation of polymeric organic matter. Environ Microbiol. 2006;8:2201–2213. doi: 10.1111/j.1462-2920.2006.01152.x. [DOI] [PubMed] [Google Scholar]
- Braun TF, McBride MJ. Flavobacterium johnsoniae GldJ is a lipoprotein that is required for gliding motility. J Bacteriol. 2005;187:2628–2637. doi: 10.1128/JB.187.8.2628-2637.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalier-Smith T. Kingdoms protozoa and Chromista and the eozoan root of the eukaryotic tree. Biol Lett. 2010;23:342–345. doi: 10.1098/rsbl.2009.0948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerveny L, Straskova A, Dankova V, Hartlova A, Ceckova M, Staud F, et al. Tetratricopeptide repeat motifs in the world of bacterial pathogens: role in virulence mechanisms. Infect Immun. 2013;81:629–635. doi: 10.1128/IAI.01035-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charon NW, Cockburn A, Li C, Liu J, Miller KA, Miller MR, et al. The unique paradigm of spirochete motility and chemotaxis. Annu Rev Microbiol. 2012;66:349–370. doi: 10.1146/annurev-micro-092611-150145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliff MJ, Harris R, Barford D, Ladbury JE, Williams MA. Conformational diversity in the TPR domain-mediated interaction of protein phosphatase 5 with Hsp90. Structure. 2006;14:415–426. doi: 10.1016/j.str.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Cottrell MT, Kirchman DL. Natural assemblages of marine proteobacteria and members of the Cytophaga-Flavobacter cluster consuming low- and high-molecular-weight dissolved organic matter. Appl Environ Microbiol. 2000;66:1692–1697. doi: 10.1128/AEM.66.4.1692-1697.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–1190. doi: 10.1101/gr.849004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Andrea LD, Regan L. TPR proteins: the versatile helix. Trends Biochem Sci. 2003;28:655–662. doi: 10.1016/j.tibs.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Di L, Pagan PE, Packer D, Martin CL, Akther S, Ramrattan G, et al. Borrelia Base: a phylogeny-centered browser of Borrelia genomes. BMC Bioinformatics. 2014;15:233. doi: 10.1186/1471-2105-15-233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchaud E, Boussaha M, Loux V, Bernardet JF, Michel C, Kerouault B, et al. Complete genome sequence of the fish pathogen Flavobacterium psychrophilum. Nat Biotechnol. 2007;25:763–769. doi: 10.1038/nbt1313. [DOI] [PubMed] [Google Scholar]
- Dunyak BM, Nakamura RL, Frankel AD, Gestwicki JE. Selective targeting of cells via bispecific molecules that exploit coexpression of two intracellular proteins. ACS Chem Biol. 2015;10:2441–2447. doi: 10.1021/acschembio.5b00426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Powell SM, Wilson R, Bowman JP. Extensive gene acquisition in the extremely psychrophilic bacterial species Psychroflexus torquis and the link to sea-ice ecosystem specialism. Genome Biol Evol. 2014;6:133–148. doi: 10.1093/gbe/evt209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser HB, Hirsh AE, Wall DP, Eisen MB. Coevolution of gene expression among interacting proteins. Proc Natl Acad Sci U S A. 2004;101:9033–9038. doi: 10.1073/pnas.0402591101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederick JR, Sarkar J, McDowell JV, Marconi RT. Molecular signaling mechanisms of the periopathogen, Treponema denticola. J Dent Res. 2011;90:1155–1163. doi: 10.1177/0022034511402994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galat A. Peptidylprolyl cis/trans isomerases (immunophilins): biological diversity–targets–functions. Curr Top Med Chem. 2003;3:1315–1347. doi: 10.2174/1568026033451862. [DOI] [PubMed] [Google Scholar]
- García-Descalzo L, Alcazar A, Baquero F, Cid C. Identification of in vivo HSP90-interacting proteins reveals modularity of HSP90 complexes is dependent on the environment in psychrophilic bacteria. Cell Stress Chaperones. 2011;16:203–218. doi: 10.1007/s12192-010-0233-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H, Liu Z, Church GM, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet. 2001;29:482–486. doi: 10.1038/ng776. [DOI] [PubMed] [Google Scholar]
- Ghanbarian AT, Hurst LD. Neighboring genes show correlated evolution in gene expression. Mol Biol Evol. 2015;32:1748–1766. doi: 10.1093/molbev/msv053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Consarnau L, González JM, Coll-Lladó M, Gourdon P, Pascher T, Neutz R, et al. Light stimulates growth of proteorhodopsin-containing marine Flavobacteria. Nature. 2007;445:210–213. doi: 10.1038/nature05381. [DOI] [PubMed] [Google Scholar]
- Graber JR, Breznak JA. Physiology and nutrition of Treponema primitia, an H2/CO2-acetogenic spirochete from termite hindguts. Appl Environ Microbiol. 2004;70:1307–1314. doi: 10.1128/AEM.70.3.1307-1314.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RS. Phylogenetic analysis of the 90 kD heat shock family of protein sequences and an examination of the relationship among animals, plants, and fungi species. Mol Biol Evol. 1995;12:1063–1073. doi: 10.1093/oxfordjournals.molbev.a040281. [DOI] [PubMed] [Google Scholar]
- Guy NC, Garcia YA, Sivils JC, Galigniana MD, Cox MB. Functions of the Hsp90-binding FKBP immunophilins. Subcell Biochem. 2015;78:35–68. doi: 10.1007/978-3-319-11731-7_2. [DOI] [PubMed] [Google Scholar]
- Hossain MM, Nakamoto H. HtpG plays a role in cold acclimation in cyanobacteria. Curr Microbiol. 2002;44:291–296. doi: 10.1007/s00284-001-0005-9. [DOI] [PubMed] [Google Scholar]
- Hunnicutt DW, Kempf MJ, McBride MJ. Mutations in Flavobacterium johnsoniae gldF and gldG disrupt gliding motility and interfere with membrane localization of GldA. J Bacteriol. 2002;184:2370–2378. doi: 10.1128/JB.184.9.2370-2378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolivet-Gougeon A, Sixou JL, Tamanai-Shacoori Z, Bonnaure-Mallet M. Antimicrobial treatment of Capnocytophaga infections. Int J Antimicrob Agents. 2007;29:367–373. doi: 10.1016/j.ijantimicag.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Jooste PJ, Hugo CJ. The taxonomy, ecology and cultivation of bacterial genera belonging to the family Flavobacteriaceae. Int J Food Microbiol. 1999;53:81–94. doi: 10.1016/S0168-1605(99)00162-2. [DOI] [PubMed] [Google Scholar]
- Kaneko T, Kibayashi K. Mild hypothermia facilitates the expression of cold-inducible RNA-binding protein and heat shock protein 70.1 in mouse brain. Brain Res. 2012;1466:128–136. doi: 10.1016/j.brainres.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Kirchman DL. The ecology of Cytophaga–Flavobacteria in aquatic environments. FEMS Microbiol Ecol. 2002;39:91–100. doi: 10.1111/j.1574-6941.2002.tb00910.x. [DOI] [PubMed] [Google Scholar]
- Klippel B, Lochner A, Bruce DC, Davenport KW, Detter C, Goodwin LA, et al. Complete genome sequences of Krokinobacter sp. strain 4H-3-7-5 and Lacinutrix sp. strain 5H-3-7-4, polysaccharide-degrading members of the family Flavobacteriaceae. J Bacteriol. 2011;193:4545–4546. doi: 10.1128/JB.05518-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krücken J, Greif G, von Samson-Himmelstjerna G. In silico analysis of the cyclophilin repertoire of apicomplexan parasites. Parasit Vectors. 2009;2:27. doi: 10.1186/1756-3305-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Sonnhammer EL. Genomic gene clustering analysis of pathways in eukaryotes. Genome Res. 2003;13:875–882. doi: 10.1101/gr.737703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RE, Kugrens P. Relationship between the flagellates and the ciliates. Microbiol Rev. 1992;56:529–542. doi: 10.1128/mr.56.4.529-542.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louca S, Parfrey LW, Doebeli M. Decoupling function and taxonomy in the global ocean microbiome. Science. 2016;353:1272–1277. doi: 10.1126/science.aaf4507. [DOI] [PubMed] [Google Scholar]
- Maikova A, Zalutskaya Z, Lapina T, Ermilova E. The HSP70 chaperone machines of Chlamydomonas are induced by cold stress. J Plant Physiol. 2016;204:85–91. doi: 10.1016/j.jplph.2016.07.012. [DOI] [PubMed] [Google Scholar]
- Mattoo RU, Goloubinoff P. Molecular chaperones are nanomachines that catalytically unfold misfolded and alternatively folded proteins. Cell Mol Life Sci. 2014;71:3311–3325. doi: 10.1007/s00018-014-1627-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride MJ. Cytophaga-flavobacterium gliding motility. J Mol Microbiol Biotechnol. 2004;7:63–71. doi: 10.1159/000077870. [DOI] [PubMed] [Google Scholar]
- McBride MJ, Braun TF. GldI is a lipoprotein that is required for Flavobacterium johnsoniae gliding motility and chitin utilization. J Bacteriol. 2004;186:2295–2302. doi: 10.1128/JB.186.8.2295-2302.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride MJ, Zhu Y. Gliding motility and Por secretion system genes are widespread among members of the phylum bacteroidetes. J Bacteriol. 2013;195:270–278. doi: 10.1128/JB.01962-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliam H, Li W, Uludag M, Squizzato S, Park YM, Buso N, et al. Analysis tool web services from the EMBL-EBI. Nucleic Acids Res. 2013;41:W597–W600. doi: 10.1093/nar/gkt376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak P. Coexpression, coregulation, and cofunctionality of neighboring genes in eukaryotic genomes. Genomics. 2008;91:243–248. doi: 10.1016/j.ygeno.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Sophos NA, Vasiliou V, Nelson DR. Cyclophilin nomenclature problems, or, 'a visit from the sequence police'. Hum Genomics. 2004;1:381–388. doi: 10.1186/1479-7364-1-5-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nematollahi A, Decostere A, Pasmans F, Haesebrouck F. Flavobacterium psychrophilum infections in salmonid fish. J Fish Dis. 2003;26:563–574. doi: 10.1046/j.1365-2761.2003.00488.x. [DOI] [PubMed] [Google Scholar]
- Oh HM, Giovannoni SJ, Lee K, Ferriera S, Johnson J, Cho JC. Complete genome sequence of Robiginitalea biformata HTCC2501. J Bacteriol. 2009;191:7144–7145. doi: 10.1128/JB.01191-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagani I, Lapidus A, Nolan M, Lucas S, Hammon N, Deshpande S. Complete genome sequence of Desulfobulbus propionicus type strain (1pr3) Stand Genomic Sci. 2011;4:100–110. doi: 10.4056/sigs.1613929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paster BJ, Dewhirst FE. Phylogenetic foundation of spirochetes. J Mol Microbiol Biotechnol. 2000;2:341–344. [PubMed] [Google Scholar]
- Rabus R, Ruepp A, Frickey T, Rattei T, Fartmann B, Stark M, et al. The genome of Desulfotalea psychrophila, a sulfate-reducing bacterium from permanently cold Arctic sediments. Environ Microbiol. 2004;6:887–902. doi: 10.1111/j.1462-2920.2004.00665.x. [DOI] [PubMed] [Google Scholar]
- Ragon M, Van Driessche AE, García-Ruíz JM, Moreira D, López-García P. Microbial diversity in the deep-subsurface hydrothermal aquifer feeding the giant gypsum crystal-bearing Naica Mine, Mexico. Front Microbiol. 2013;4:37. doi: 10.3389/fmicb.2013.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes RG, Nelson SS, Pochiraju S, McBride MJ. Flavobacterium johnsoniae sprB is part of an operon spanning the additional gliding motility genes sprC, sprD, and sprF. J Bacteriol. 2011;193:599–610. doi: 10.1128/JB.01203-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer N, Chen J, Regan L. All repeats are not equal: a module-based approach to guide repeat protein design. J Mol Biol. 2013;425:1826–1838. doi: 10.1016/j.jmb.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayle R, Milner-White EJ. RasMol: Biomolecular graphics for all. Trends Biochem Sci. 1995;20:374–376. doi: 10.1016/S0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, et al. Structure of TPR domain-peptide complexes: critical elements in the assembly of the Hsp70-Hsp90 multichaperone machine. Cell. 2000;101:199–210. doi: 10.1016/S0092-8674(00)80830-2. [DOI] [PubMed] [Google Scholar]
- Schiene-Fischer C. Multidomain peptidyl prolyl cis/trans isomerases. Biochim Biophys Acta. 2015;1850:2005–2016. doi: 10.1016/j.bbagen.2014.11.012. [DOI] [PubMed] [Google Scholar]
- Schmidpeter PA, Schmid FX. Prolyl isomerization and its catalysis in protein folding and protein function. J Mol Biol. 2015;427:1609–1631. doi: 10.1016/j.jmb.2015.01.023. [DOI] [PubMed] [Google Scholar]
- Sela MN. Role of Treponema denticola in periodontal diseases. Crit Rev Oral Biol Med. 2001;12:399–413. doi: 10.1177/10454411010120050301. [DOI] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinars CR, Cheung-Flynn J, Rimerman RA, Scammell JG, Smith DF, Clardy J. Structure of the large FK506-binding protein FKBP51, an Hsp90-binding protein and a component of steroid receptor complexes. Proc Natl Acad Sci U S A. 2003;100:868–873. doi: 10.1073/pnas.0231020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonna LA, Kuhlmeier MM, Khatri P, Chen D, Lilly CM. A microarray analysis of the effects of moderate hypothermia and rewarming on gene expression by human hepatocytes (HepG2) Cell Stress Chaperones. 2010;15:687–702. doi: 10.1007/s12192-010-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ünal CM, Steinert M. Microbial peptidyl-prolyl cis/trans isomerases (PPIases): virulence factors and potential alternative drug targets. Microbiol Mol Biol Rev. 2014;78:544–571. doi: 10.1128/MMBR.00015-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedantam G, Viswanathan VK. Spirochaetes and their twisted ways. Gut Microbes. 2012;3:399–400. doi: 10.4161/gmic.22051. [DOI] [PubMed] [Google Scholar]
- Verma DK, Rathore G. New host record of five Flavobacterium species associated with tropical fresh water farmed fishes from North India. Braz J Microbiol. 2015;46:969–976. doi: 10.1590/S1517-838246420131081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BK, Allan RK, Mok D, Temple SE, Taylor P, Dornan J, et al. A structure-based mutational analysis of cyclophilin 40 identifies key residues in the core tetratricopeptide repeat domain that mediate binding to Hsp90. J Biol Chem. 2002;277:40799–40809. doi: 10.1074/jbc.M207097200. [DOI] [PubMed] [Google Scholar]
- Woese CR, Kandler O, Wheelis ML (1990) Towards a natural system of organisms: proposal for the domains Archaea. Bacteria, and Eucarya, Proc Natl Acad Sci USA 87:4576–4579 [DOI] [PMC free article] [PubMed]
- Woyke T, Chertkov O, Lapidus A, Nolan M, Lucas S, Del Rio TG, et al. Complete genome sequence of the gliding freshwater bacterium Fluviicola taffensis type strain (RW262) Stand Genomic Sci. 2011;5:21–29. doi: 10.4056/sigs.2124912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B, Li P, Liu Y, Lou Z, Ding Y, Shu C. 3D structure of human FK506-binding protein 52: implications for the assembly of the glucocorticoid receptor/Hsp90/immunophilin heterocomplex. Proc Natl Acad Sci U S A. 2004;101:8348–8353. doi: 10.1073/pnas.0305969101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CJ, Takeda M, Terauchi T, Jee J, Kainosho M. Differential large-amplitude breathing motions in the interface of FKBP12-drug complexes. Biochemistry. 2015;54:6983–6995. doi: 10.1021/acs.biochem.5b00820. [DOI] [PubMed] [Google Scholar]
- Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER suite: protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Kang SJ, Oh TK. Polaribacter dokdonensis sp. nov., isolated from seawater. Int J Syst Evol Microbiol. 2006;56:1251–1255. doi: 10.1099/ijs.0.63820-0. [DOI] [PubMed] [Google Scholar]
- Yoon JH, Kang SJ, Park S, Oh TK. Reclassification of the three species of the genus Krokinobacter into the genus Dokdonia as Dokdonia genika comb. nov., Dokdonia diaphoros comb. nov. and Dokdonia eikasta comb. nov., and emended description of the genus Dokdonia Yoon et al. 2005. Int J Syst Evol Microbiol. 2012;62:1896–1901. doi: 10.1099/ijs.0.035253-0. [DOI] [PubMed] [Google Scholar]
- Yoshizawa S, Kumagai Y, Kim H, Ogura Y, Hayashi T, Iwasaki W, et al. Functional characterization of flavobacteria rhodopsins reveals a unique class of light-driven chloride pump in bacteria. Proc Natl Acad Sci U S A. 2014;111:6732–6737. doi: 10.1073/pnas.1403051111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX 43.6 kb)
(DOCX 37.3 kb)