

Abstract
Multinucleated skeletal muscle fibers form through the fusion of myoblasts during development and regeneration. Previous studies identified myomaker (Tmem8c) as a muscle-specific membrane protein essential for fusion. However, the specific function of myomaker and how its function is regulated are unknown. To explore these questions, we first examined the cellular localization of endogenous myomaker. Two independent antibodies showed that whereas myomaker does localize to the plasma membrane in cultured myoblasts, the protein also resides in the Golgi and post-Golgi vesicles. These results raised questions regarding the precise cellular location of myomaker function and mechanisms that govern myomaker trafficking between these cellular compartments. Using a synchronized fusion assay, we demonstrated that myomaker functions at the plasma membrane to drive fusion. Trafficking of myomaker is regulated by palmitoylation of C-terminal cysteine residues that allows Golgi localization. Moreover, dissection of the C terminus revealed that palmitoylation was not sufficient for complete fusogenic activity suggesting a function for other amino acids within this C-terminal region. Indeed, C-terminal mutagenesis analysis highlighted the importance of a C-terminal leucine for function. These data reveal that myoblast fusion requires myomaker activity at the plasma membrane and is potentially regulated by proper myomaker trafficking.
Keywords: intracellular trafficking, membrane fusion, myogenesis, protein palmitoylation, protein trafficking (Golgi), site-directed mutagenesis
Introduction
Myoblast fusion is a fundamental process necessary for muscle formation during development and regeneration (1). We previously identified myomaker, a muscle-specific membrane protein, as one of the central components of the muscle fusion machinery (2, 3). Specifically, myomaker is necessary for skeletal muscle formation and myoblast fusion in mouse, chicken, and zebrafish (2, 4–6). Moreover, ectopic expression of myomaker in cell types that normally do not fuse, such as fibroblasts, induces their fusion with muscle cells (2). Myomaker is, however, not sufficient for fibroblast-fibroblast fusion indicating the necessity of additional factors to fully reconstitute fusion in non-fusogenic cells. Recently, we and others identified myomerger as a muscle-specific factor that functions with myomaker to drive fusion of non-fusogenic cells (7–9). Given that myomaker and myomerger are necessary for myoblast fusion in mice and sufficient to drive fusion in heterologous cells (7–9), understanding the regulation and function of each protein will be necessary for complete elucidation of myoblast fusion mechanisms.
Structural characteristics of myomaker include spanning the bilayer seven times, with a cytosolic C-terminal tail and extracellular N-terminal region, with no known conserved functional domains (10). Given that myomaker is a robust activator of fusion and that fusion fates must be precisely controlled to avoid inappropriate cellular mixing, it is plausible that expression and localization of myomaker is temporally and spatially regulated. Transcription of myomaker is controlled through activity of muscle-specific transcription factors, MyoD and myogenin. Indeed, multiple E-boxes within the myomaker promoter are necessary for expression of myomaker mRNA in muscle (3). In contrast to its transcriptional control, it is not known whether myomaker protein localization is also a mechanism by which myoblasts regulate fusion. Acylation of proteins is a highly conserved post-translational modification that directs membrane proteins to traffic between various cellular membrane compartments, localize proteins to specific membrane domains, or facilitate complex protein-protein interactions (11, 12). We previously showed that the C-terminal region of myomaker is necessary for function and harbors three cysteine residues, which are predicted to be palmitoylated (10). Moreover, myomaker exhibits a N-terminal glycine predicted to undergo myristoylation (13). Although myomaker contains canonical amino acid residues for lipidation, it is not known if myomaker is indeed lipid modified and the associated consequence of potential lipidation on this multipass transmembrane protein.
Cell-cell fusion encompasses numerous pathways including recognition, migration, adhesion, signaling, actin cytoskeletal dynamics, and membrane merger (14). Neither the biochemical function of myomaker, nor its role within these cellular processes, are understood. Elucidation of the precise localization of myomaker is necessary to understand its function. FLAG-tagged myomaker is present on the plasma membrane and in intracellular vesicles (10). The presence of myomaker-FLAG on the plasma membrane of myoblasts suggests that myomaker functions at the cell membrane to induce fusion, however, no direct evidence for this function exists. Moreover, myomaker-FLAG does not exhibit the same level of fusogenic activity as wild-type (WT) myomaker questioning the utility of myomaker-FLAG to faithfully reveal mechanisms of myomaker regulation (10). Development of reagents to investigate the localization and activity of WT myomaker will aid in the ultimate delineation of myomaker function.
Here, using a pair of myomaker antibodies we show that myomaker localizes to the Golgi, intracellular vesicles, and plasma membrane in myoblasts. We demonstrate that whereas myomaker localizes to multiple cellular compartments, inhibition of myomaker at the plasma membrane with an antibody blocks fusion. Investigation of the potential for myomaker lipidation to govern trafficking revealed that palmitoylation of myomaker C-terminal cysteines significantly alters trafficking and function to induce fusion. Further mutational analysis revealed that palmitoylation and a hydrophobic leucine residue within the C-terminal region cooperate for full activity. Taken together, these studies demonstrate that myoblast fusion is partially controlled through regulatory mechanisms that govern myomaker trafficking to the plasma membrane for proper fusion.
Results
Analysis of myomaker antibodies
We sought to develop tools to reveal the expression pattern of endogenous myomaker. We initially generated a rabbit polyclonal antibody against amino acids 137–152 of myomaker (custom antibody) (Fig. 1A). Based on our previous topology analysis, this is an intracellular stretch of amino acids located between transmembrane domains 5 and 6 (10). To assess the utility of the custom myomaker antibody, we performed Western blot analysis on lysates from C2C12 cells retrovirally infected with either an empty virus or myomaker virus, and myomaker knock-out (KO) C2C12 cells. C2C12 cell lysates were collected at multiple days after differentiation and we observed no detectable myomaker expression of empty-infected C2C12 cells at day 0 of differentiation (Fig. 1B). Myomaker has a calculated molecular mass of 24.8 kDa, however, we detected a band close to 20 kDa in empty-infected samples after differentiation and this signal was increased in myomaker-infected samples (Fig. 1B). Moreover, FLAG-myomaker exhibited an expected slower migration (Fig. 1B). The myomaker band was not present in lysates from myomaker KO C2C12 cells further indicating that the antibody indeed recognizes myomaker (Fig. 1C). To test whether the antibody could be used to determine myomaker localization, we immunostained WT and myomaker KO C2C12 cells, and observed staining in WT but not in myomaker KO cultures (Fig. 1D). The custom antibody does not yield a positive signal through live immunofluorescence (not shown), which aligns with our topology model that this is an intracellular epitope.
Figure 1.

Validation of myomaker antibodies. A, schematic of myomaker protein topology with the epitope used for custom antibody generation noted in red. Green labels the potential epitope range for the Santa Cruz G12 antibody. B, the utility of the custom antibody was tested through Western blot analysis of C2C12 lysates infected with either empty or myomaker retrovirus. Myomaker was not detected at day 0 in empty-infected C2C12 cells but is up-regulated upon differentiation. Myomaker was robustly detected in myomaker-infected samples at all stages of myogenesis. A FLAG tagged version of myomaker (SF1) exhibits an upward shift due to the presence of the FLAG epitope. C, empty-C2C12 cells, myomaker-infected C2C12 cells, and myomaker KO cells were differentiated for 2 days and lysates were subjected to myomaker immunoblotting demonstrating the specificity of the custom myomaker antibody. D, immunostaining with the custom myomaker antibody on WT and myomaker KO C2C12 cells in culture was performed and shows lack of staining in myomaker KO samples further demonstrating specificity. Phalloidin was used to identify cells. E, immunofluorescence microscopy and phase-contrast images of live (non-permeabilized) WT C2C12 cells and myomaker KO C2C12 cells stained with myomaker antibody G12. The staining was performed 2 days after differentiation. Scale bars, 20 μm. Immunofluorescent images were obtained using identical microscope parameters and all brightness/contrast adjustments were applied equally to all images.
A limitation of the custom antibody is that it is unable to detect myomaker at the cell surface, therefore we assessed a commercially available myomaker (TMEM8C) antibody from Santa Cruz Biotechnology (G12). Manufacturer information indicates the epitope ranges from amino acid 110 to 160, however, they were not able to reveal the specific epitope. The potential G12 antibody epitope could overlap with our custom antibody epitope (Fig. 1A). We first tested whether the G12 antibody could detect myomaker on the cell surface through live immunofluorescence. Indeed, we observed labeling for WT C2C12 cells expressing myomaker but not myomaker KO myoblasts (Fig. 1E). This finding confirmed specificity of the G12 antibody and provides direct evidence for the presence of WT myomaker at the surface of fusion-committed cells. Moreover, these data suggest that the G12 antibody epitope potentially corresponds to amino acids 110–130, upstream of our custom antibody epitope. In the work described below, we have used the custom myomaker antibody for all Western blot analysis and the G12 antibody for detection of myomaker at the cell surface. For detection of intracellular myomaker after permeabilization, we have used the custom antibody except where indicated.
Localization of myomaker in the Golgi and intracellular vesicles
To identify which compartments within the cell contain myomaker, we performed co-localization studies with several proteins known to label specific cellular regions. We infected C2C12 cells with myomaker and after 2 days of differentiation, the cells were immunostained with both myomaker and organelle-specific antibodies. Myomaker was enriched in structures that are positive for two Golgi markers, which target cis and trans Golgi regions of the cell (GM130 and Golgin 97) (Fig. 2A). However, we did not detect co-localization with either endoplasmic reticulum or with two vesicular markers, lysosomes and early endosomes (Fig. 2A). To further assess the overlap of myomaker with GM130 and Golgin 97 we performed super-resolution microscopy. Myomaker exhibits a Golgi-localized pattern, residing juxtaposed to both GM130 and Golgin 97, and displaying areas of overlap with Golgin 97 (Fig. 2B). These results indicate that myomaker resides in the Golgi compartment but may not directly interact with GM130 and Golgin 97. Nonetheless, GM130 can serve as a general indicator of myomaker localization in the Golgi region.
Figure 2.

Localization of myomaker in the Golgi and intracellular vesicles. A, C2C12 cells infected with myomaker, differentiated for 2 days, and immunostained with a myomaker antibody and antibodies against GM130 (cis Golgi), Golgin 97 (trans Golgi), PDI (endoplasmic reticulum), Lamp1 (lysosomes), or EEA1 (endosomes). The G12 Santa Cruz antibody was used for co-staining with Golgin 97, whereas all other co-staining utilized our custom myomaker antibody. B, super-resolution microscopy showing localization of myomaker with either GM130 or Golgin 97. Arrows indicate areas of co-localization. Scale bars: A, 10 μm; B, 5 μm.
We previously showed the presence of endogenous myomaker on the plasma membrane of cultured myoblasts (Fig. 1E), suggesting that vesicular myomaker could be transiting to or from the plasma membrane. To further investigate the identity of myomaker vesicles, we analyzed co-localization of myomaker with multiple Rab GPTases, a family of proteins that function as canonical regulators of trafficking proteins between donor and target membranes (15, 16). We did not detect co-localization of myomaker with Rab antibodies that label early endosomes (Rab5), and observed minimal myomaker in late endosomes (Rab7 and -9) or recycling endosomes (Rab11) (Fig. 3). We also assessed localization with Rab8, a marker for vesicles trafficking from the trans Golgi network (17), and Rab10, which labels secretory vesicles (18), however, we also did not observe myomaker in these structures (Fig. 3). Taken together, these data indicate that myomaker is not solely expressed on the plasma membrane and instead this protein is also localized in the Golgi and intracellular vesicles.
Figure 3.
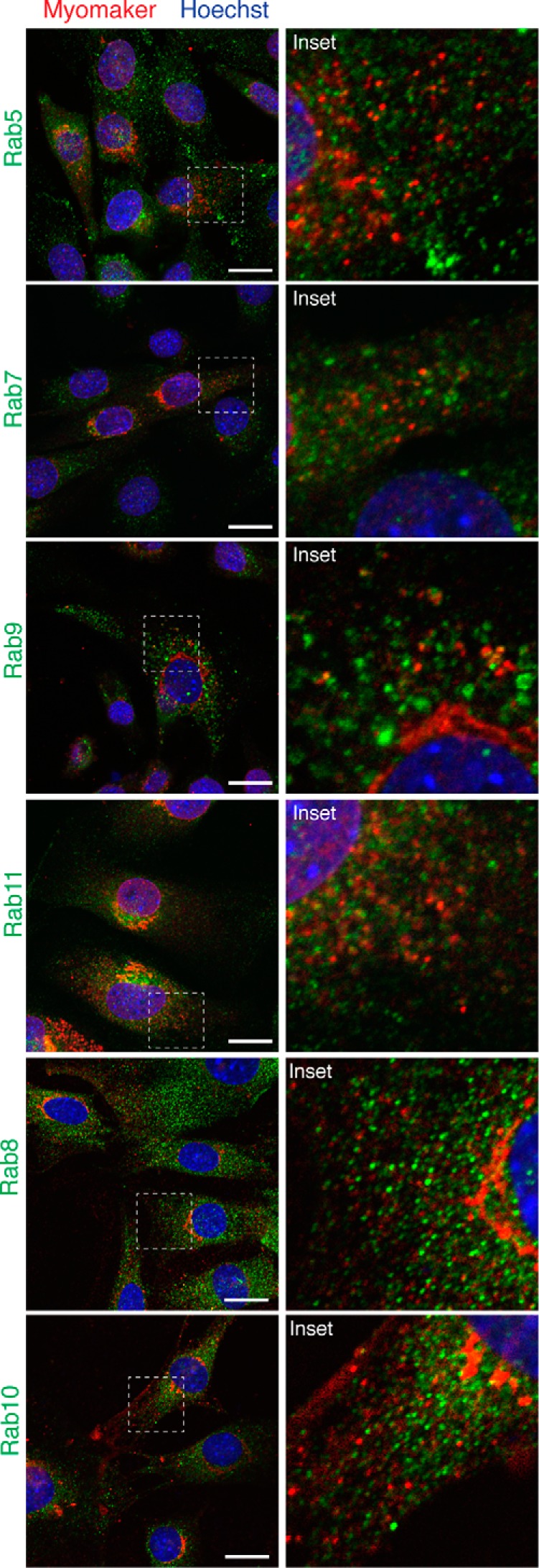
Vesicular myomaker does not co-localize with Rab proteins. Myomaker-infected C2C12 cells were stained to determine whether Rab proteins co-localized with myomaker. Rab immunostaining with antibodies against Rab5 (early endosomes), Rab7 (late endosomes), Rab9 (late endosomes), Rab11 (recycling endosomes), Rab8 (trans Golgi-derived transport vesicles), and Rab10 (Glut4 vesicles of the secretory pathway). Each culture was co-stained with the myomaker goat polyclonal antibody from Santa Cruz (G12). Scale bars, 10 μm.
Our current assays to measure the fusogenic activity of myomaker require retroviral overexpression in either fibroblasts or myomaker null myoblasts. Thus, we next assessed whether localization of retrovirally expressed myomaker was different compared with endogenously expressed myomaker. Empty-infected and myomaker-infected C2C12 myoblasts, with and without differentiation, were immunostained with the custom myomaker antibody. During myogenic differentiation myomaker was consistently expressed in perinuclear and vesicular regions in both myoblasts and myotubes indicating that intracellular localization is not appreciably altered during differentiation (Fig. 4A). Moreover, both cell types displayed myomaker staining in the Golgi and within intracellular vesicles indicating that overexpression of myomaker does not cause dramatic altered localization (Fig. 4B). Quantification of the relative amounts of myomaker in the Golgi using Mander's overlap coefficient (18) further revealed a similar distribution between endogenous and overexpressed myomaker (Fig. 4C). Finally, biochemical fractionation shows that overexpressed myomaker tracks with multiple cellular membranes, similar to the endogenous system (Fig. 4D). We conclude that overexpression of myomaker does not dramatically alter localization validating this system to reveal mechanisms of myomaker regulation.
Figure 4.

Localization of myomaker during myogenic differentiation and after overexpression. A, C2C12 cells were infected with either empty or myomaker retrovirus and immunostained with a custom myomaker antibody after the indicated days of differentiation. GM130 was used as a marker to assess the relative myomaker localization. B, the same cells in A were imaged at high magnification on day 2 of differentiation for quantification of relative localization. C, five independent images were taken for each sample. The amount of myomaker in the Golgi relative to total myomaker was used to determine the Mander's overlap coefficient using NIS elements software. D, empty-infected or myomaker-infected C2C12 cells were biochemically fractionated and the various fractions were analyzed by immunoblotting with the custom myomaker antibody. (P5 = 5,000 × g pellet; P17 = 17,000 × g pellet; P100 = 100,000 × g pellet; S100 = supernatant of 100,000 × g spin.) Data are presented as mean ± S.D. Scale bars, 10 μm.
Fusion is dependent on myomaker activity at the surface of myoblasts
The dynamic localization of myomaker within multiple cellular sites complicates elucidation of myomaker function. Myomaker could act within the Golgi, intracellular vesicles, or at the plasma membrane to promote fusion. To examine whether myoblast fusion requires activity of myomaker at the cell surface, we utilized a fusion-synchronization approach developed previously (19). Specifically, we reversibly blocked fusion of C2C12 myoblasts without blocking pre-fusion stages using lysophosphatidylcholine (LPC),2 an inhibitor of membrane merger. We accumulated ready-to-fuse C2C12 cells in the presence of LPC for 16 h and then removed LPC by replacing the LPC-supplemented differentiation medium with LPC-free medium to allow fusion. Myomaker antibody (G12) was applied at the time of LPC removal and fusion was assayed 30 min later. As a control for the myomaker antibody, we used an antibody for human syncytin 1, a protein that is not expressed on murine cells. Through this approach it is possible to monitor both the initial membrane fusion event that merges the membranes of two cells (hemifusion) and the subsequent pore formation and expansion of this nascent membrane connection required to form a multinucleated cell. Specifically, we labeled one cell population with membrane dye and another with a cytosolic probe and assayed for the presence of co-labeled cells after removal of LPC, with and without the addition of the myomaker antibody. In contrast to the control syncytin 1 antibody, myomaker antibody inhibited formation of multinucleated cells (syncytium formation) (Fig. 5A). Myomaker antibodies also inhibited membrane mixing potentially suggesting that myomaker functions at or before the hemifusion stage between C2C12 myoblasts (Fig. 5A). In an independent set of experiments, myomaker antibodies similarly inhibited fusion of primary murine myoblasts in the synchronized fusion assay (Fig. 5A). We also performed synchronized fusion with our custom myomaker antibody, which recognizes an intracellular region of myomaker and therefore should not inhibit myomaker activity. Indeed, the custom myomaker antibody did not alter fusion after LPC removal (Fig. 5B). Overall, our data demonstrate that myomaker is not only present at the plasma membrane of myoblasts at the time and place of fusion, but is directly involved in the late stages of fusion.
Figure 5.

Myomaker on the surface of myoblasts is involved in synchronized fusion. A, quantification of synchronized fusion for C2C12 myoblasts and primary myoblasts was achieved by assessing formation of multinucleated myotubes (syncytial formation) and membrane merger (hemifusion). LPC was added to synchronize cells in a pre-fusion state and then the cells were washed (+LPC/Wash) to allow fusion. Myomaker antibody or a control syncytin antibody was applied when washing LPC (+LPC/Wash + G12 Myomaker Ab., and +LPC/Wash + Syncytin Ab., respectively). Fusion was assayed 30 min after LPC removal or, to establish the background level of fusion, at the same time point for the cells that remained in LPC containing medium. Both membrane merger and syncytium formation were inhibited by antibodies to myomaker but not by antibodies to syncytin 1, which was used as a negative control. B, synchronized fusion is not perturbed with the custom myomaker antibody that recognizes an intracellular epitope. Quantification of LPC-mediated synchronized fusion for primary myoblasts by assessing formation of multinucleated myotubes and membrane merger. Our custom generated antibody that recognizes an intracellular region of myomaker has no effect. Data are presented as mean ± S.D. (compiled from 3 independent experiments). *, p < 0.05.
Post-Golgi vesicle trafficking is necessary for myomaker function
We next sought to disrupt myomaker localization through mutagenesis to determine the relative contribution of myomaker trafficking to overall fusion activity. Membrane proteins typically harbor a short signal sequence at the N terminus that is important to determine the final destination and aids in protein stability (20). Preliminary evidence of myomaker structure suggests it contains seven transmembrane domains (21). However, experimental evidence is lacking for various aspects of this model, such as whether the first 26 amino acids encode a cleavable signal peptide. To determine whether the myomaker signal sequence is cleaved and its importance for trafficking and function, we generated a mutant that lacks the potential signal sequence (Δ1–26 myomaker). In this mutant protein, we also engineered a synthetic cleavable signal sequence from influenza virus hemagglutinin to the N terminus (22). We infected myomaker KO C2C12 cells with empty, WT myomaker, or Δ1–26 myomaker retrovirus and after 2 days of differentiation cell lysates were analyzed by Western blot for myomaker. Δ1–26 myomaker exhibited faster migration compared with WT myomaker, indicating that myomaker does not contain a cleavable signal sequence (Fig. 6A). We further investigated the importance of the non-cleaved signal sequence for myomaker localization and function. Co-staining of these various myomaker-expressing cells with GM130 and myomaker antibodies revealed that the lack of the signal peptide sequestered myomaker in the Golgi, whereas as WT myomaker was localized to Golgi and intracellular vesicles (Fig. 6B, top panel). When these cells were differentiated for 4 days to assay for an ability to rescue fusion defects in myomaker KO myoblasts, empty-infected cells were unable to fuse, whereas expression of myomaker rescued fusion (Fig. 6B, bottom panel). In contrast, Golgi-restricted Δ1–26 myomaker did not rescue fusion suggesting that myomaker must be trafficked out of the Golgi for proper activity (Fig. 6B, bottom panel). Quantification of the fusion index achieved with each construct also shows that Δ1–26 myomaker exhibits no activity (Fig. 6C).
Figure 6.

Analysis of the myomaker N-terminal region. A, Western blot analysis of myomaker KO C2C12 cells retrovirally infected with empty, myomaker, or a myomaker signal peptide mutant (Δ1–26). The faster migration of the Δ1–26 myomaker protein indicates that myomaker does not harbor a cleavable signal sequence. GAPDH was used as a loading control. B, myomaker KO cells were infected with empty, myomaker, or with Δ1–26 myomaker retrovirus and were differentiated for 2 days followed by co-immunostaining with myomaker and GM130 antibodies (top panel). Each cell line was also differentiated for 4 days and immunostained with a myosin antibody to evaluate fusion. C, quantification of the fusion index from B. D, glycine 2 of myomaker, a potential myristoylation site, was mutated to alanine (G2A myomaker). G2A myomaker was localized to Golgi and intracellular vesicles similar to WT myomaker (top panels) and G2A myomaker functioned to fuse myomaker KO myoblasts (bottom panel). E, quantification of the fusion index from D. Data are presented as mean ± S.D. *, p < 0.05. Scale bars, B and D, top panels, 10 μm; bottom panels, 50 μm.
Regulation of myomaker trafficking through lipid modification
Acylation is a well known post-translation modification that contributes to the sorting of proteins between membrane compartments. For example, myristoylation and palmitoylation are two such lipid modifications that enhance proper membrane binding for both soluble and membrane proteins (11). The second amino acid of myomaker is a glycine, which represents a canonical myristoylation site for the addition of the 14-carbon myristic acid. Although our current toplogy model suggests that the N terminus of myomaker is extracellular, and myristoylation usually occurs on N-terminal glycines in the cytosol, there is no direct evidence for the potential role of myomaker myristoylation. To this end, we mutated the N-terminal glycine to an alanine and evaluated localization and function. Myomaker KO C2C12 cells were infected with empty, myomaker, or G2A myomaker viruses and after 2 days of differentiation, the cells were fixed, permeabilized, and immunostained with both GM130 and our myomaker custom antibody. Both myomaker and G2A myomaker exhibited similar localization (in Golgi and intracellular vesicles) indicating that the predicted myristoylation site does not significantly effect myomaker localization (Fig. 6D, top panel). Visual analysis of the ability of G2A myomaker to rescue fusion revealed similar activity as WT myomaker (Fig. 6D, bottom panel). Quantification of the fusion index for each cell line demonstrates a statistically significant reduction in the function of G2A myomaker compared with WT myomaker (Fig. 6E). We interpret the small magnitude of reduction to likely suggest minimal relevance, at least in our overexpression system.
The C-terminal region of myomaker (STLCCTCV) contains three conserved cysteine residues that could represent sites for lipid modification, but are not a consensus CAAX motif that undergoes prenylation. These cysteines are predicted to be palmitoylated, a reversible modification known to control protein sorting, protein-protein interactions, and protein-lipid interactions (12). To determine whether the C-terminal cysteines of myomaker undergo palmitoylation, we generated a series of constructs that contain mutations in various cysteines and analyzed palmitoylation of each mutant using an acyl-RAC (acyl-resin associated capture) assay (23). Acyl-RAC followed by immunoblotting for myomaker revealed that WT myomaker is indeed palmitoylated (Fig. 7A). We did not observe a positive signal in lysates that were incubated in the absence of hydroxylamine, which cleaves palmitate from cysteines allowing these free cysteines to bind to the thiopropyl-Sepharose resin (Fig. 7A). Mutation of the final seven amino acids (SAAAAAAA) and mutation of the three cysteines (217,218,220A, STLAATAV) abrogated their detection by acyl-RAC indicating the C-terminal cysteines are the only myomaker cysteines capable of palmitoylation (Fig. 7A). Moreover, mutation of the potential PDZ binding motif (219–221A, STLCCAAA) was detected by acyl-RAC indicating either Cys217 or Cys218, or both, are palmitoylated. As expected, addition of the three cysteines to the 215–221A mutant (SAACCACA) displayed a positive signal for acyl-RAC (Fig. 7A).
Figure 7.

Post-translational lipidation of myomaker governs protein trafficking. A, myomaker contains multiple cysteine residues in the C-terminal domain (STLCCTCV) and multiple cysteine-deficient myomaker constructs were generated including mutation of the final seven amino acids of myomaker to alanine ((215–221A) SAAAAAAA), mutation of the final TCV to alanines ((219–221A) STLCCAAA), mutation of the three cysteines in the C-terminal region ((217, 218, 220A) STLAATAV), and a construct where the cysteines were re-introduced to the 215–221A mutant (SAACCACA). Myomaker KO C2C12 cells were infected with WT myomaker or each mutant and 2 days after differentiation, cells were lysed and subjected to acyl-RAC assay to evaluate palmitoylation. Proteins were captured using thiol-reactive Sepharose beads and were analyzed by Western blotting for myomaker. B, the localization of the C-terminal mutants was assessed through co-immunostaining with GM130 and myomaker antibodies. WT myomaker exhibits Golgi and vesicle localization, however, disruption of the C-terminal palmitoylation sites results in only vesicle localization. C, mean fluorescence intensity of myomaker within the Golgi and vesicles was quantified and expressed as a vesicle to Golgi ratio. *, p < 0.05 compared with WT (STLCCTCV). D, the C-terminal region of myomaker (amino acids 197–221) was reconstituted onto the C-terminal region of Tmem8b, which also contains a synthetic, cleavable N-terminal signal sequence and FLAG tag to allow for immunodetection. SF1-Tmem8b localizes to Golgi and post-Golgi regions, whereas SF1-Tmem8b-myomaker197–221 is Golgi restricted. Data are presented as mean ± S.D. Scale bars, 10 μm.
Myomaker palmitoylation could be a mechanism that governs myomaker trafficking. To this end, we examined the localization pattern of each C-terminal mutant construct through co-immunofluorescence with myomaker and GM130 antibodies. Although WT myomaker localized to both Golgi and intracellular vesicles, mutation of the final seven amino acids of the C terminus (215–221A, SAAAAAAA) or disruption of the three cysteines (217,218,220A, STLAATAV) caused the majority of myomaker to localize in intracellular vesicles with minimal detection in the Golgi (Fig. 7B). In contrast, mutation of the potential PDZ binding motif (219–221A, STLCCAAA) or re-addition of the cysteines onto the 215–221A mutant (SAACCACA) localized similar to WT myomaker (Fig. 7B). To quantify these observations, we measured mean fluorescence intensity of the myomaker signal within intracellular vesicles and Golgi to determine the ratio of vesicle to Golgi myomaker. Mean intensity ratio of myomaker increased 15-fold for the 215–221A (SAAAAAAA) construct and 10-fold for the cysteine 217,218,220A (STLAATAV) compared with WT myomaker (Fig. 7C). No significant difference was observed in the vesicle to Golgi ratio between WT myomaker and 219–221A (STLCCAAA) or SAACCACA. These results indicate that palmitoylation of the C-terminal cysteines retains myomaker in the Golgi.
We next determined the sufficiency of the myomaker C-terminal region to alter trafficking of a non-myomaker membrane protein. The C terminus of myomaker (amino acids 197–221) was engineered onto an independent membrane protein, Tmem8b, which shares homology with myomaker within the transmembrane regions (10). However, Tmem8b contains a longer N-terminal cytosolic region and does not exhibit fusogenic activity in myoblasts (10). For immunostaining purposes, we incorporated a FLAG epitope along with a synthetic cleavable signal sequence on the N terminus of Tmem8b (SF1-Tmem8b), and then generated another SF1-Tmem8b construct containing amino acids 197–221 of myomaker on the C terminus (Fig. 7D). Myomaker knock-out myoblasts were infected with each construct and after 2 days of differentiation cells were co-stained with FLAG and GM 130 to explore their localization. SF1-Tmem8b was localized to both Golgi and post-Golgi compartments, whereas addition of the C terminus of myomaker was sufficient to confine the chimeric protein (SF1-Tmem8b-myomaker197–221) to the Golgi (Fig. 7D). These data further confirm that palmitoylation of the C-terminal cysteines of myomaker acts as a Golgi-localization signal.
We have previously shown that the C-terminal region of myomaker, in the context of FLAG-myomaker, is essential for its function to fuse fibroblasts to muscle cells (10). Whether this region is also necessary for the function of WT myomaker to rescue fusion of myomaker KO myoblasts, and the importance of palmitoylation for fusion, has not been investigated. Mutation of the final seven amino acids to alanine (215–221A, SAAAAAAA) completely abrogates the fusion ability in myomaker KO cells, and mutation of the final TCV (219–221A, STLCCAAA), a potential PDZ binding motif, displayed similar activity as WT myomaker (Fig. 8, A and B). The palmitoylation mutant 217,218,220A (STLAATAV) exhibited reduced activity compared with WT myomaker, although the lack of palmitoylation did not completely abolish function (Fig. 8, A and B). These data suggest that the non-cysteine amino acids within the C terminus are also important for myomaker function. Indeed, simply adding back the cysteines to the 215–221A mutant to create SAACCACA resulted in an active protein, although activity was also reduced 50% compared with WT (Fig. 8, A and B).
Figure 8.

Function of myomaker C-terminal mutants reveals the importance of non-cysteine amino acids. A, the various C-terminal mutant constructs were expressed in myomaker KO cells and assayed for their ability to rescue fusion. Cultures after 4 days of differentiation were stained with a myosin antibody and Hoechst. B, quantification of the fusion index. C, immunostaining of live cells using the G12 antibody shows that myomaker is able to localize to the plasma membrane after perturbation of the C-terminal region. Data are presented as mean ± S.D. *, p < 0.05 compared with empty and 215–221A (SAAAAAAA). #, p < 0.05 compared with WT myomaker (STLCCTCV). N.S., not significant. Scale bars, A, 50 μm; C, 10 μm.
We also previously showed that both FLAG-myomaker and the C-terminal deletion FLAG-myomaker were present on the cell surface (10). To assess if deletion of the C-terminal region in the WT context disrupts cell surface localization we immunostained live myoblasts with the G12 antibody. Here, we found that myomaker 215–221A (SAAAAAAA) was indeed present on the cell surface (Fig. 8C). That the most dramatic C-terminal mutant (SAAAAAAA) is still present on the plasma membrane suggests that this region does not globally impact trafficking, and thus, we did not assess the plasma membrane localization of the more subtle C-terminal mutations.
Dissection of myomaker palmitoylation
Our data thus far are derived from mutants that contain alanine substitutions, which is the most commonly used amino acid for mutagenesis due to its benign characteristics such as small side chain. However, serine is often chosen to replace cysteines because they exhibit similar size side chains, potentially promoting normal folding while still lacking an ability for palmitoylation (24). To evaluate whether the nature of amino acid substitution impacts myomaker trafficking and function, we mutated the C-terminal cysteines to serine (217,218,220S, STLSSTSV). Co-immunofluorescence studies revealed that both 217,218,220A and 217,218,220S myomaker localized to intracellular vesicles but not to Golgi (Fig. 9A, top panel). Interestingly, functional studies showed that the insertion of the serine residues dramatically reduced the capability to rescue fusion in myomaker KO myoblasts (Fig. 9, A, lower panel, and B). These results may indicate that the myomaker C-terminal region requires a strict structure for function, which could be the physiological purpose for its palmitoylation.
Figure 9.

Evaluation of which C-terminal cysteine is required for palmitoylation, localization, and function. A, a separate cysteine mutant was generated where the cysteines were mutated to serine ((217, 218, 220S) STLSSTSV). Both myomaker 217,218,220A and 217,218,220S localized to intracellular vesicles with minimal expression in the Golgi as assessed by co-immunostaining with myomaker and GM130 antibodies (top panels). Each construct was also assessed for an ability to rescue fusion in myomaker KO myoblasts through differentiation and immunostaining with a myosin antibody (bottom panels). B, quantification of the fusion index revealed that myomaker 217,218,220S exhibited significantly less fusogenic activity. C, single cysteines were added back to the myomaker 217,218,220S construct to generate the following mutants: STLCSTSV, STLSCTSV, and STLSSTCV. The acyl-RAC assay was performed on cell lines expressing each construct and demonstrates that each cysteine is able to undergo palmitoylation. D, co-immunostaining with myomaker and GM130 antibodies revealed that re-addition of one cysteine was not sufficient to drive Golgi retention (top panels). Assessment of function of each of the serine mutants indicates that restoration of one of the C-terminal cysteines improves function but not to WT levels (bottom panels). E, quantification of the fusion index shows that re-addition of cysteine 217 results in significantly more function compared with re-addition of cysteine 218. Dashed lines representing fusion indexes of STLCCTCV, STLAATAV, and STLSSTSV were added to the graph for comparison. Data are presented as mean ± S.D. *, p < 0.05. Scale bars, A and D, top panels, 5 μm; bottom panels, 50 μm.
To evaluate which cysteines within the C terminus of myomaker undergo palmitoylation, single cysteines were re-introduced to the 217,218,220S (STLSSTSV) mutant to generate STLCSTSV, STLSCTSV, and STLSSTCV. Acyl-RAC detection of each mutant indicated that individual C-terminal cysteines are able to be palmitoylated (Fig. 9C). Localization analysis of the constructs revealed that the addition of single cysteines resulted in vesicular myomaker and no Golgi staining, suggesting that at least two palmitoylated cysteines are required for Golgi retention (Fig. 9D, top panel). Functional analysis revealed a significant improvement in their fusion ability compared with cysteine to alanine (STLAATAV) or cysteine to serine (STLSSTSV) mutants (Fig. 9, D, lower panel, and E). However, this improvement did not reach the levels achieved with WT myomaker.
C-terminal palmitoylation and a hydrophobic leucine maximize myomaker-mediated fusion
Various soluble and membrane proteins that require association with the plasma membrane for their function contain regions for lipid modification that facilitate membrane anchoring. However, in some cases lipid modification is not sufficient and other hydrophobic amino acids adjacent to these lipid-modified domains facilitate proper membrane interactions. For example, SNAP-25 not only requires cysteine palmitoylation but also surrounding hydrophobic amino acids to optimally associate with the membrane to participate in vesicle fusion at synapses (25). To evaluate the contribution of the C-terminal hydrophobic amino acids for myomaker function, we added two conserved amino acids Leu216 and Thr215 back onto the mutant that contains only the C-terminal cysteines (SAACCACA) to generate SALCCACA and STACCACA constructs. The acyl-RAC analysis was positive with each of these proteins indicating that Leu216 or Thr215 do not impact palmitoylation (Fig. 10A). Moreover, SALCCACA and STACCACA localized to Golgi and intracellular vesicles similar to WT myomaker (Fig. 10B, top panel). Surprisingly, the construct that contains both cysteines and the hydrophobic leucine (SALCCACA) was able to rescue myomaker KO myoblasts to a level achieved with WT myomaker (Fig. 10, B, lower panel, and C). In contrast, addition of Thr215 with the cysteines (STACCACA) did not enhance activity above SAACCACA (Fig. 10, B, lower panel, and C). These results indicate that cysteine palmitoylation and the side chain hydrophobicity of the C-terminal leucine cooperate for maximal myomaker function.
Figure 10.

A C-terminal leucine influences palmitoylation-dependent function of myomaker. A, two additional myomaker constructs were generated that contain either Leu216 or Thr215 within the myomaker mutant that contains the cysteines but not the other amino acids within the C-terminal region (SAACCACA). Acyl-RAC analysis indicates each mutant was palmitoylated. B, localization of each of these mutants through co-immunostaining with myomaker and GM130 antibodies revealed that re-addition of Leu216 or Thr215 did not significantly alter the relative amounts of myomaker in the Golgi or intracellular vesicles, compared with the SAACCACA mutant (top panels). In contrast, re-addition of Leu216 to the SAACCACA mutant significantly enhanced fusion compared with Thr215 (bottom panels). C, quantification of fusion revealed no significant differences between WT myomaker and SALCCACA. Data are presented as mean ± S.D. *, p < 0.05. N.S., not significant. Scale bars, B, top panels, 5 μm; bottom panels, 50 μm.
Discussion
Accumulating evidence indicates that myomaker is a central effector of myoblast fusion, however, multiple questions exist regarding its regulation and mechanism of action. Here, we show that myomaker is enriched in the Golgi and intracellular vesicles, and present on the plasma membrane suggesting an unidentified regulatory axis that governs myomaker localization and trafficking. We also demonstrate that the presence of myomaker at the plasma membrane is required for fusion. We uncovered that palmitoylation of myomaker C-terminal cysteines localize myomaker to the Golgi and this lipidation is required in concert with additional hydrophobic amino acids for function. Table 1 summarizes the mutants generated here and their associated fusogenic activity. Overall, our results indicate that myoblast fusion is at least partially controlled through proper localization of myomaker.
Table 1.
Summary of myomaker C-terminal mutants generated and their associated fusogenic activity and localization
The fusion index is based on the ability of the mutant to rescue myomaker KO myoblasts and is expressed as a percentage of the number of nuclei within a myosin + myotube (with three or more nuclei) to the total number of myosin + nuclei. n.t., not tested.

One intriguing observation emanating from this study is that myomaker is localized to the Golgi. The precise relevance of this localization is not known, however, multiple possibilities exist including a potential function for myomaker within the Golgi, which this study does not completely exclude. Another possibility is that myomaker is contained in the Golgi to provide a concentrated reservoir of myomaker vesicles when fusion pathways are activated. Despite no definitive role for myomaker in the Golgi, we do show that myomaker is required at the plasma membrane through use of a blocking antibody during the synchronized fusion assay. It should be noted that antibody binding to proteins present at the cell surface at the time and place of fusion can, in principle, inhibit fusion by steric hindrance even if the proteins recognized by the antibodies are not involved in fusion. Nonetheless, these are the first set of data defining a role for myomaker at the membrane, however, further investigations will be needed to delineate the mechanisms by which myomaker promotes fusion.
Although we show that the G12 antibody specifically recognizes myomaker in immunofluorescent experiments, the mechanism of this binding is not clear. Based on the linear topology model, there are only three amino acids in the extracellular region, however, it is possible that the tertiary structure of the protein allows more of the epitope to be accessible. Moreover, it is well established, especially in the field of structural biology of viruses, that the protein structure is dynamic and some virus-neutralizing antibodies bind epitopes that, based on protein structures, are not expected to be accessible. Proteins constantly sample related conformations at equilibrium. Antibodies bind and trap some conformations not predicted by the existing structural models as these transient conformations become accessible on the surface (26).
In this study we also show that myomaker localizes to intracellular vesicles. The precise identity of these vesicles and their contents requires further investigation. Although we did not observe colocalization with Rab8 or Rab10 vesicles, it is possible that these vesicles are representative of anterograde myomaker trafficking. We also did not detect dramatic localization of myomaker with the endosomal pathway suggesting that if these vesicles represent retrograde transport of myomaker, they do not utilize the canonical endosomal pathway. The observation of vesicular myomaker is similar to recent work on the fusion of epithelial cells in Caenorhabditis elegans and potentially suggests a conserved mechanism to control the localization of central fusion proteins. Indeed, epithelial fusion failure (Eff-1), a potent C. elegans fusogen is contained in endosomes and regulated by canonical endocytic proteins, Rab5 and dynamin (27). Also, a role for exocytic vesicles in myoblast fusion has been previously proposed where electron dense vesicles were present at contact sites between two fusing cells (28). The biochemical composition of these vesicles, whether myomaker is contained in these vesicles, and their precise function is not understood, but could include trafficking of fusogenic proteins or lipids to the cell surface to promote membrane merger.
Our palmitoylation studies provide further insights into the regulation of myomaker trafficking. Palmitoylation of at least two of the three C-terminal cysteines is required for Golgi localization and is also important for function. Our results indicate that each cysteine has the capacity to undergo palmitoylation, however, our study did not investigate whether they are dynamically regulated or if a particular cysteine is palmitoylated only at a certain cellular location. For instance, because palmitoylation is a reversible modification it is possible that two cysteines are palmitoylated at the Golgi, and one becomes de-palmitoylated to allow release to vesicles, then the final cysteine undergoes palmitoylation, which aids in function at the plasma membrane. Additionally, given the exact motifs for palmitoylation are not completely known, myomaker may undergo complex acylation where one of the cysteines is prenylated and another palmitoylated similar to what occurs with Cdc42 (29). Although myomaker also contains a consensus myristoylation site, we did not directly assay for the presence of myristic acid on the myomaker protein but did demonstrate that mutation of the myristoylation site did not have a biologically relevant impact on fusogenic activity. One caveat for this interpretation is that our assay involves overexpression, thus myristoylation may be necessary for the function of myomaker in a more physiological system.
Finally, new insights were obtained regarding the role of the C terminus for myomaker activity, however, its function remains to be completely elucidated. Our results suggest a model whereby the C-terminal region may associate with the membrane for proper activity. Palmitoylation is essential but alone not sufficient for function, and overall hydrophobicity of the region potentially impacts membrane association. The hydrophobic amino acid leucine 216 cooperates with palmitoylated cysteines to fully rescue fusion, which could signify an enhancement of the membrane association of this region. This membrane association may promote interaction of myomaker with a necessary protein partner or place myomaker in a proper lipid domain within the membrane such as lipid rafts (30, 31). Another possibility is that sequence alteration at the C terminus impacts recognition by DHHC palmitoyltransferases, which would reduce the extent of palmitoylation and alter myomaker localization and function.
It is also plausible that palmitoylation of myomaker causes localization within the Golgi at specific subdomains that facilitates proper sorting to proper microdomains of the plasma membrane. Consistent with this possibility, the C-terminal mutant (SAAAAAAA) does not localize to the Golgi and is able to transit to the plasma membrane but does not possess fusogenic activity. This reveals that simply the presence of myomaker on the plasma membrane does not render cells fusogenic.
It was recently demonstrated that mutations of myomaker cause a congenital myopathy in humans (32). However, the mechanism by which these mutations reduce myomaker activity is not known. Given that our results indicate that myomaker trafficking and localization must be precisely controlled, it would be interesting in the future to understand if the human myomaker mutations are pathogenic due to defective localization.
Experimental procedures
Cell culture
C2C12 cells were purchased from American Type Culture Collection and was propagated in growth media, DMEM (HyClone) containing 10% heat inactivated bovine growth serum (BGS) (HyClone) supplemented with antibiotics. Myomaker knock-out (KO) C2C12 cells were generated through CRISPR/Cas9 genome editing as described previously (10). Primary myoblasts were isolated from the forelimbs and hindlimbs of three to four 5-day-old pups of wild-type C57BL/6 mice (NCI, Frederick, MD) and characterized, as described in Ref. 19. The cells were cultured on collagen-treated dishes (BD Biosciences) in F10 medium (Invitrogen) supplemented with basic fibroblast growth factor (10 ng/ml of bFGF; PeproTech) and 10% fetal calf serum (HyClone). To induce differentiation, C2C12 cells and primary myoblasts at 75% confluence were placed into DMEM containing antibiotics and 2% heat-inactivated horse serum (Invitrogen).
Mutagenesis, retroviral infection, and cell fusion assay
Site-directed mutagenesis was achieved through introduction of mutations into the pBabeX-myomaker plasmid using standard PCR-based strategies. All recombinant plasmids were confirmed by sequencing prior to use. The Tmem8b template used to engineer the C-terminal region of myomaker was described previously (10). For retroviral generation, Platinum E cells (Cell Biolabs) were plated on 100-mm culture dishes at a density of 5 × 106 cells per dish 24 h before transfection. Ten micrograms of the retroviral plasmid DNA was transfected into Platinum E cells using FuGENE 6 (Promega). After 48 h of transfection the viral media was collected, filtered through 0.45-μm filter, and mixed with Polybrene (Sigma) at a final concentration of 6 μg/ml. The C2C12 cells were plated at a density of 1.2 × 106 on 100-mm dish and infected with 9 ml of the viral media. After 24 h of infection the viral media was removed, washed with PBS, and cells were trypsinized and propagated for further analysis. For evaluation of transduction efficiency immunocytochemistry for myomaker was performed as described below. Specifically, 2 × 104 cells were plated per well of an 8-well μ-slide (Ibidi) and the percentage of infection was calculated. If the percentage of infection was less than 80%, all cell types for that particular experiment were re-infected. For the cell fusion assay, 4 × 105 cells were plated on a 35-mm culture dish and the cultures were differentiated the next day and allowed to undergo myogenesis for 4 days.
Immunocytochemistry
The myomaker custom antibody was generated through YenZym Antibodies LLC. Rabbits were immunized with amino acids 137–152 of mouse myomaker (MKEKKGLYPDKSIYTQ) after conjugation to keyhole limpet hemocyanin and then bled and antibodies were affinity purified.
Co-immunostaining experiments with α-GM130, α-EEA1, α-Lamp1, and α-PDI were done using custom generated antibody using the following protocol. Cells were fixed with 4% paraformaldehyde/PBS, blocked with 0.01% Triton X-100, 0.2% gelatin, 10% FBS in PBS. The following primary antibodies were used at the dilutions indicated and incubated for at least 1 h at room temperature in the antibody solution (0.01% Triton X-100, 0.2% gelatin, 1% FBS in PBS): α-GM130 (mouse; BD Biosciences; 1:100), α-EEA1 (mouse; BD Bioscience; 1:100), α-Lamp1 (rat; Abcam; 1:100), α-PDI (mouse; BD Bioscience; 1:100), α-myomaker (rabbit; custom generated; 1:100).
The co-immunostaining experiments with α-Golgin 97 and all Rab markers were done with the TMEM8C antibody G12 (goat; Santa Cruz, 1:20, catalog number SC-244459) using the following protocol. Cells were fixed with 4% paraformaldehyde/PBS, and blocked with 0.01% Triton X-100, 0.2% gelatin, 10% FBS in PBS. The cells were first incubated with the G12 antibody for 10 min at 37 °C. After three 1× PBS washes, the following primary antibodies were used at the dilutions indicated and incubated for at least 1 h at room temperature in the antibody solution: α-Golgin 97 (rabbit; Cell Signaling Technology; 1:100), α-Rab5 (rabbit; Cell Signaling Technology; 1:100), α-Rab7 (rabbit; Cell Signaling Technology; 1:100), α-Rab8 (rabbit; Cell Signaling Technology; 1:100), α-Rab9 (rabbit; Cell Signaling Technology; 1:100), α-Rab10 (mouse; EMD Millipore, 1:100), α-Rab11 (rabbit; Cell Signaling Technology; 1:100).
For analysis of fusion, cells were immunostained with α-Myosin (MY32; Sigma; 1:200). Alexa Fluor (Invitrogen; 1:400) were used as the secondary antibodies and were incubated for 30 min at room temperature. Nuclei were stained with Hoechst (Invitrogen). Samples were imaged with a with a Nikon Eclipse Ti inverted microscope with A1R confocal running NIS Elements and images were analyzed with Fiji (33).
For super-resolution microscopy, images were taken using a confocal microscope (Nikon A1 LUN-A inverted) with a ×100 objective NA 1.45. Image planes were oversampled in XY using a 0.03-μm pixel size (∼4 times Nyquist). Confocal stacks were acquired with a 0.4 AU pinhole yielding a 0.35-μm optical section at 0.1-μm intervals and 2× integration avoiding pixel saturation. Images were deconvolved with NIS elements using 15 iterations of the Landweber method. Images shown are a single focal plane.
For live staining of C2C12 cells, the myomaker G12 antibody from Santa Cruz was used. For the secondary antibodies in immunofluorescence experiments on live cells, we used the Alexa Fluor 546 rabbit anti-goat antibody (number A21085, ThermoFisher Scientific). TMEM8C antibody (G12) were added to C2C12 cells or to myomaker KO C2C12 cells for 10 min at 37 °C at a concentration of 20 μg/ml in 200 μl of differentiation medium. The cells were washed 5 times with PBS and then fixed with 4% formalin/PBS. The cells were washed, blocked with 10% FBS for 10 min at room temperature, and the secondary antibodies were added for 1 h at 1:400 dilution. The cells were washed 6 times and imaged.
Synchronized fusion assay
We uncoupled the cell-cell fusion stage of myogenic differentiation from the pre-fusion stages using a fusion-synchronization approach described in Ref. 19. In brief, we placed fusion-committed cells into complete medium supplemented with 150 μm lauroyl LPC (number 855475P, Avanti Polar Lipids). Fusion ensued when 16 h later LPC was removed in three washes with LPC-free complete medium. C2C12 cells and primary myoblasts grown to 75% confluence in a 10-cm dish were labeled with either fluorescent lipid the Vybrant DiI or membrane-permeant Green 5-chloromethylfluorescein diacetate cell tracker (number V22885 and C7025, ThermoFisher Scientific) as recommended by the manufacturer and described in Ref. 19. Differently labeled cells were co-plated in a 1:1 ratio in complete medium before application of LPC. We labeled C2C12 cells 48 h after placement in the differentiation medium. Differently labeled cells were allowed to attach for 2–3 h after co-plating before application of LPC. LPC was removed 16 h later, i.e. ∼67 h after triggering of myogenic differentiation. We scored fusion 30 min after LPC removal and thus ∼68 h after triggering myogenic differentiation. In the experiments on primary myoblasts, we labeled proliferating cells in the growth medium, lifted them with versene, co-plated differently labeled cells, and grew them for 16 more hours before placing the cells into the differentiation medium. We applied LPC 7 h later, kept the cells in LPC-supplemented differentiation medium for 16 h, and then applied LPC-free medium to score fusion ∼24 h after triggering myogenic differentiation. Antibodies to myomaker and antibodies to human syncytin 1 (Santa Cruz number SC-30640) were applied by addition to the LPC-free medium in which the cells were kept after LPC withdrawal. Myoblast fusion was quantified as described previously (19). For each condition, ≥8 randomly chosen fields of view compiled from 3 independent experiments were analyzed. Formation of multinucleated myotubes was quantified as the percentage of cell nuclei present in myotubes normalized to the total number of cell nuclei. In the membrane merger assay, we detected early fusion stages as the redistribution of membrane probe (DiI) and cytosolic probes (cell tracker) between differently labeled cells and quantified membrane merger as the percentage of nuclei in co-labeled cells (including both mono- and multinucleated cells) compared with the total number of cell nuclei. The percentage of nuclei in multinucleated cells (syncytium formation assay) and the percentage of co-labeled cells (membrane merger assay) were normalized to those in the parallel control experiments.
Western blot analysis
Cells were scraped in 1 ml of cold PBS, pelleted at 1,200 rpm for 5 min, and resuspended in 100 μl of lysis buffer (50 mm Tris-HCl, pH 6.8, 1 mm EDTA, 2% SDS). Lysates were then sonicated, insoluble material was pelleted by centrifugation at 15,000 rpm for 15 min at room temperature, and the supernatant was used to determine the total protein concentration through BCA assay kit (Pierce). Equal amounts (100 μg) were prepared with 5× Laemmli loading dye (300 mm Tris-HCl, pH 6.8, 10% SDS, 50% glycerol, 0.25% bromphenol blue) and each sample was boiled at 95 °C for 5 min prior to separation on a 12% SDS-PAGE. The gel was transferred to a pre-equilibrated PVDF membrane (Bio-Rad) and the membrane was blocked with 5% milk in TBS-Tween 20 (TBS-T) for 30 min at room temperature. After three washes with TBS-T, the membrane was incubated with the custom myomaker antibody (1:250), overnight at 4 °C. GAPDH (Millipore; 1:10,000) was used as a loading control. IRDye® 800CW secondary antibodies (1:10,000; LI-COR Biosciences) were used for detection on the Odyssey® infrared detection system (LI-COR Biosciences).
Subcellular fractionation
C2C12 cells were harvested on day 2 of differentiation in ice-cold hypotonic buffer (10 mm Tris-HCl, pH 8, 2 mm EDTA) and lysed using a Dounce homogenizer. Lysates were then centrifuged at 800 × g for 5 min at 4 °C to separate nuclei and cell debris. Differential centrifugation of supernatants was performed at 5,000 × g for 10 min, 17,000 × g for 10 min, and 100,000 × g for 20 min. The final supernatant was collected as the cytosolic fraction and all pellets were resuspended in lysis buffer (50 mm Tris-HCl, pH 6.8, 1 mm EDTA, 2% SDS) at volumes equal to the supernatant. Fractions were separated by SDS-PAGE and analyzed for the presence of myomaker, caveolin-3, and tubulin. Caveolin-3 antibody (BD Transduction Laboratories number 610421) was used at 1:6700 and tubulin (Santa Cruz number SC-8035) at 1:50.
Acyl-RAC detection of myomaker
To detect whether myomaker is S-acylated, cells were collected and washed with cold PBS at 48 h after differentiation followed by the acyl-RAC protocol adapted from a method previously described (23). Cells were lysed in RIPA buffer (50 mm Tris, pH 7.4, 1% Triton X-100, 1% sodium deoxycholate, 1 mm EDTA, 0.1% SDS) and sonicated five times on ice. Cell lysates were centrifuged at 5000 rpm for 5 min at 4 °C to clear the supernatant. Total protein was quantified as described above and each extract was diluted to 3 mg/ml with blocking buffer that contains methyl methanethiosulfonate (MMTS), which blocks free thiols in the protein mixture (100 mm HEPES, pH 7.4, 1 mm EDTA, 2.5% SDS, and 0.3% MMTS). The samples were incubated at 40 °C for 20 min in a rotating hybridization oven to obtain a homogenous mixture. Three volumes of ice-cold acetone were added to remove unreacted MMTS, and proteins were allowed to precipitate at −20 °C for 20 min. The pellet was collected through centrifugation at 10,000 × g for 10 min, followed by three washes with cold 70% acetone. The dried pellet was resuspended in 1 ml of binding buffer (100 mm HEPES, 1 mm EDTA, 1% SDS) and incubated at 37 °C for 20 min. Total protein concentration was measured and 1 mg of total protein was added to 25 μl of prewashed thiolpropyl-Sepharose beads (Sigma). To this mixture, 300 μl of either freshly prepared 1 m hydroxylamine, pH 7.4, or 50 mm Tris, pH 7.4, were added to facilitate the cleavage of the thioester linkage or as a negative control. To reduce nonspecific binding, an additional 200 μl of high salt buffer (100 mm HEPES, pH 7.4, 1 mm EDTA, 500 mm NaCl) was added. The binding reaction was carried out on a rotator at room temperature for 2.5 h. The resin was washed at least eight times with the washing buffer (100 mm HEPES, pH 7.4, 1 mm EDTA, 0.3% SDS). For immunoblot analysis, 20 μl of binding buffer was added along with 5 μl of 5× Laemmli loading buffer to the resin and heated to 95 °C for 5 min. Bands were separated using 12% SDS-PAGE and analyzed for myomaker as described above. 100 μg of each supernatant was used as the total input, mixed with Laemmli loading buffer, and heated for 95 °C for 5 min.
Quantification of fusion index, myomaker localization, and statistical analysis
The fusion index was calculated as a percentage of the number of nuclei within myosin+ myotubes (with three or more nuclei) to the total number of myosin+ nuclei. Myosin+ nuclei were counted manually. Myomaker localization in the Golgi and vesicles was quantified by analyzing mean fluorescence intensity in each region using NIS elements. All data are presented as mean ± S.D. and were compiled from at least two independent experiments performed in duplicate. p values were obtained using an unpaired t test with Prism 6 software and values were considered statistically significant when p < 0.05.
Author contributions
D. P. M., L. V. C., D. G. G., and E. L. conceived the study and designed experiments. D. G. G., E. L., M. E. Q., and A. R. performed experiments and analyzed the data. D. P. M., D. G. G., L. V. C., and E. L. wrote the manuscript, whereas all authors offered constructive comments.
Acknowledgments
We thank the Cincinnati Children's Hospital Medical Center (CCHMC) Confocal Imaging Core for microscopy assistance and members of the Molkentin lab at CCHMC for technical assistance.
This work was supported by grants from the Cincinnati Children's Hospital Research Foundation, NIAMD, National Institutes of Health Grant R01AR068286, the Muscular Dystrophy Association, and Pew Charitable Trusts (to D. P. M.) and the Intramural Research Program of the Eunice Kennedy Shriver NICHD, National Institutes of Health, and United States-Israel Binational Science Foundation (BSF) Grant 2013151 (to L. V. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- LPC
- lysophosphatidylcholine
- acyl-RAC
- acyl-resin associated capture
- α-PDI
- α-protein-disulfide isomerase
- MMTS
- methyl methanethiosulfonate.
References
- 1. Kim J. H., Jin P., Duan R., and Chen E. H. (2015) Mechanisms of myoblast fusion during muscle development. Curr. Opin. Genet. Dev. 32, 162–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Millay D. P., O'Rourke J. R., Sutherland L. B., Bezprozvannaya S., Shelton J. M., Bassel-Duby R., and Olson E. N. (2013) Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature 499, 301–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Millay D. P., Sutherland L. B., Bassel-Duby R., and Olson E. N. (2014) Myomaker is essential for muscle regeneration. Genes Dev. 28, 1641–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Landemaine A., Rescan P. Y., and Gabillard J. C. (2014) Myomaker mediates fusion of fast myocytes in zebrafish embryos. Biochem. Biophys. Res. Commun. 451, 480–484 [DOI] [PubMed] [Google Scholar]
- 5. Luo W., Li E., Nie Q., and Zhang X. (2015) Myomaker, regulated by MYOD, MYOG and miR-140–3p, promotes chicken myoblast fusion. Int. J. Mol. Sci. 16, 26186–26201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang W., and Roy S. (2017) Myomaker is required for the fusion of fast-twitch myocytes in the zebrafish embryo. Dev. Biol. 423, 24–33 [DOI] [PubMed] [Google Scholar]
- 7. Quinn M. E., Goh Q., Kurosaka M., Gamage D. G., Petrany M. J., Prasad V., and Millay D. P. (2017) Myomerger induces fusion of non-fusogenic cells and is required for skeletal muscle development. Nat. Commun. 8, 15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Q., Vashisht A. A., O'Rourke J., Corbel S. Y., Moran R., Romero A., Miraglia L., Zhang J., Durrant E., Schmedt C., Sampath S. C., and Sampath S. C. (2017) The microprotein Minion controls cell fusion and muscle formation. Nat. Commun 8, 15664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bi P., Ramirez-Martinez A., Li H., Cannavino J., McAnally J. R., Shelton J. M., Sánchez-Ortiz E., Bassel-Duby R., and Olson E. N. (2017) Control of muscle formation by the fusogenic micropeptide myomixer. Science 356, 323–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Millay D. P., Gamage D. G., Quinn M. E., Min Y. L., Mitani Y., Bassel-Duby R., and Olson E. N. (2016) Structure-function analysis of myomaker domains required for myoblast fusion. Proc. Natl. Acad. Sci. U.S.A. 113, 2116–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Resh M. D. (2016) Fatty acylation of proteins: the long and the short of it. Prog Lipid Res 63, 120–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Blaskovic S., Blanc M., and van der Goot F. G. (2013) What does S-palmitoylation do to membrane proteins? FEBS J. 280, 2766–2774 [DOI] [PubMed] [Google Scholar]
- 13. Bologna G., Yvon C., Duvaud S., and Veuthey A. L. (2004) N-Terminal myristoylation predictions by ensembles of neural networks. Proteomics 4, 1626–1632 [DOI] [PubMed] [Google Scholar]
- 14. Schejter E. D. (2016) Myoblast fusion: experimental systems and cellular mechanisms. Semin. Cell Dev. Biol. 60, 112–120 [DOI] [PubMed] [Google Scholar]
- 15. Zhen Y., and Stenmark H. (2015) Cellular functions of Rab GTPases at a glance. J. Cell Sci. 128, 3171–3176 [DOI] [PubMed] [Google Scholar]
- 16. Hutagalung A. H., and Novick P. J. (2011) Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 91, 119–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Huber L. A., Pimplikar S., Parton R. G., Virta H., Zerial M., and Simons K. (1993) Rab8, a small GTPase involved in vesicular traffic between the TGN and the basolateral plasma membrane. J. Cell Biol. 123, 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sano H., Eguez L., Teruel M. N., Fukuda M., Chuang T. D., Chavez J. A., Lienhard G. E., and McGraw T. E. (2007) Rab10, a target of the AS160 Rab GAP, is required for insulin-stimulated translocation of GLUT4 to the adipocyte plasma membrane. Cell Metab. 5, 293–303 [DOI] [PubMed] [Google Scholar]
- 19. Leikina E., Melikov K., Sanyal S., Verma S. K., Eun B., Gebert C., Pfeifer K., Lizunov V. A., Kozlov M. M., and Chernomordik L. V. (2013) Extracellular annexins and dynamin are important for sequential steps in myoblast fusion. J. Cell Biol. 200, 109–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Coleman J., Inukai M., and Inouye M. (1985) Dual functions of the signal peptide in protein transfer across the membrane. Cell 43, 351–360 [DOI] [PubMed] [Google Scholar]
- 21. Käll L., Krogh A., and Sonnhammer E. L. (2004) A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 338, 1027–1036 [DOI] [PubMed] [Google Scholar]
- 22. Kobilka B. K. (1995) Amino and carboxyl terminal modifications to facilitate the production and purification of a G protein-coupled receptor. Anal. Biochem. 231, 269–271 [DOI] [PubMed] [Google Scholar]
- 23. Forrester M. T., Hess D. T., Thompson J. W., Hultman R., Moseley M. A., Stamler J. S., and Casey P. J. (2011) Site-specific analysis of protein S-acylation by resin-assisted capture. J. Lipid Res. 52, 393–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vivian J. P., Hastings A. F., Duggin I. G., Wake R. G., Wilce M. C., and Wilce J. A. (2003) The impact of single cysteine residue mutations on the replication terminator protein. Biochem. Biophys. Res. Commun. 310, 1096–1103 [DOI] [PubMed] [Google Scholar]
- 25. Greaves J., Prescott G. R., Fukata Y., Fukata M., Salaun C., and Chamberlain L. H. (2009) The hydrophobic cysteine-rich domain of SNAP25 couples with downstream residues to mediate membrane interactions and recognition by DHHC palmitoyl transferases. Mol. Biol. Cell 20, 1845–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pierson T. C., and Kuhn R. J. (2012) Capturing a virus while it catches its breath. Structure 20, 200–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Smurova K., and Podbilewicz B. (2016) RAB-5- and DYNAMIN-1-mediated endocytosis of EFF-1 fusogen controls cell-cell fusion. Cell Rep. 14, 1517–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim S., Shilagardi K., Zhang S., Hong S. N., Sens K. L., Bo J., Gonzalez G. A., and Chen E. H. (2007) A critical function for the actin cytoskeleton in targeted exocytosis of prefusion vesicles during myoblast fusion. Dev. Cell 12, 571–586 [DOI] [PubMed] [Google Scholar]
- 29. Nishimura A., and Linder M. E. (2013) Identification of a novel prenyl and palmitoyl modification at the CaaX motif of Cdc42 that regulates RhoGDI binding. Mol. Cell. Biol. 33, 1417–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lorent J. H., and Levental I. (2015) Structural determinants of protein partitioning into ordered membrane domains and lipid rafts. Chem. Phys. Lipids 192, 23–32 [DOI] [PubMed] [Google Scholar]
- 31. Yang X., Kovalenko O. V., Tang W., Claas C., Stipp C. S., and Hemler M. E. (2004) Palmitoylation supports assembly and function of integrin-tetraspanin complexes. J. Cell Biol. 167, 1231–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Di Gioia S. A., Connors S., Matsunami N., Cannavino J., Rose M. F., Gilette N. M., Artoni P., de Macena Sobreira N. L., Chan W. M., Webb B. D., Robson C. D., Cheng L., Van Ryzin C., Ramirez-Martinez A., Mohassel P., et al. (2017) A defect in myoblast fusion underlies Carey-Fineman-Ziter syndrome. Nat. Commun 8, 16077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]