

Abstract
Clostridium difficile is a major nosocomial pathogen that produces two exotoxins, TcdA and TcdB, with TcdB thought to be the primary determinant in human disease. TcdA and TcdB are large, multidomain proteins, each harboring a cytotoxic glucosyltransferase domain that is delivered into the cytosol from endosomes via a translocation domain after receptor-mediated endocytosis of toxins from the cell surface. Although there are currently no known host cell receptors for TcdA, three cell-surface receptors for TcdB have been identified: CSPG4, NECTIN3, and FZD1/2/7. The sites on TcdB that mediate binding to each receptor are not defined. Furthermore, it is not known whether the combined repetitive oligopeptide (CROP) domain is involved in or required for receptor binding. Here, in a screen designed to identify sites in TcdB that are essential for target cell intoxication, we identified a region at the junction of the translocation and the CROP domains that is implicated in CSPG4 binding. Using a series of C-terminal truncations, we show that the CSPG4-binding site on TcdB extends into the CROP domain, requiring three short repeats for binding and for full toxicity on CSPG4-expressing cells. Consistent with the location of the CSPG4-binding site on TcdB, we show that the anti-TcdB antibody bezlotoxumab, which binds partially within the first three short repeats, prevents CSPG4 binding to TcdB. In addition to establishing the binding region for CSPG4, this work ascribes for the first time a role in TcdB CROPs in receptor binding and further clarifies the relative roles of host receptors in TcdB pathogenesis.
Keywords: antibody, cell surface, receptor, toxin, trafficking
Introduction
TcdA (308 kDa; 2710 residues) and TcdB (270 kDa; 2366 residues) are the primary virulence factors of Clostridium difficile, the leading cause of healthcare-associated diarrhea (1). Upon colonization in the colon, C. difficile produces TcdA and TcdB, which cause disruption of the gut epithelial barrier, leading to pseudomembranous colitis and in extreme cases death (2). Either TcdA or TcdB causes disease in rodents, whereas TcdB may be the primary disease-causing toxin in pig and humans (3–5). TcdA and TcdB share 48% sequence identity and are structurally organized into 4 functionally distinct domains: an N-terminal glucosyltransferase domain (GTD), an autoprocessing domain (APD), a translocation/pore-forming domain, and a C-terminal combined repetitive oligopeptide repeat (CROP)3 domain. The CROP domains of the two toxins are composed of multiple short repeats (32 in TcdA and 20 in TcdB) interspersed with a smaller number of long repeats (7 in TcdA and 4 in TcdB) (supplemental Fig. S1). Upon secretion, the toxins enter colonic epithelial cells via receptor-mediated endocytosis (6) and glucosylate Rac and Rho GTPases (7–9). Rac/Rho glucosylation triggers actin depolymerization, cell rounding, and eventually cell death, also referred to as the cytopathic effect (10). At high concentrations, TcdB can trigger necrosis, causing colonic tissue damage independent of the glucosylation activity of the toxin (11, 12).
Research over the past decade has provided great insight into the structure and function of the C. difficile toxins, in particular for the individual toxin domains and the key processes that they carry out once inside host cells. Our understanding of how each toxin recognizes and binds target cells, however, is incomplete. Historically, the CROP domain was assumed to be the sole receptor-binding domain in both TcdA and TcdB (13, 14), although the discovery of TpeL from Clostridium perfringens, a homologue of TcdA/TcdB that naturally lacks the CROP domain (15), and the observation that TcdA/TcdB truncations with the CROP domains deleted are capable of intoxicating cells (16, 17) have called the role of the CROPs into question. Recent efforts have begun to focus outside the CROP domain to find receptor-binding determinants (17–19). From these studies, a multiple receptor model for host cell entry has been proposed (17, 20). According to this model, it is suggested that toxin docks onto the cell surface by binding to a low affinity receptor/oligosaccharide via its CROP domain followed by binding to high affinity CROP independent receptor(s), a model suggested for both TcdA and TcdB (13, 14).
Recently, three distinct cell-surface receptors for TcdB were identified: poliovirus receptor like 3 (PVRL3, or NECTIN3), chondroitin sulfate proteoglycan 4 (CSPG4), and members of the Frizzled protein family (FZD1, FZD2, and FZD7) (21–23). Remarkably, none of these receptors appears to bind TcdA despite the substantial sequence identity shared by the toxins. Although NECTIN3 has been shown to be important for the necrosis phenotype induced by higher concentrations of TcdB, CSPG4 and FZD have been shown to be important for the cytopathic effects of the toxin that are induced at lower doses of TcdB. That NECTIN3 and FZD proteins were shown to directly interact with TcdB1–1830 indicates that toxin entry via these receptors does not require the presence of the CROP domain (22, 23). For CSPG4, the binding determinants on TcdB are not as clear. Based on the ability of CSPG4 to bind TcdB1500–2366 but not TcdB1852–2366, Yuan et al. (21) proposed that CSPG4 is a CROPs-independent receptor that binds in a region spanning amino acid residues 1500–1852. Tao et al. (23), on the other hand, proposed that CSPG4 is a CROPs-dependent receptor due to lack of binding of CSPG4 to TcdB1–1830. Direct binding of CSPG4 to TcdB1830–2366, however, was not tested.
In this study we set out initially with the goal of identifying regions in the TcdB delivery domain (amino acids 800–1850), outside the previously characterized hydrophobic region (amino acids 956–1128), that were required for pore formation/translocation. Through this analysis, we identified two residues, Tyr-1824 and Asn-1839, at the junction of the C-terminal end of the translocation domain and the CROP region that were essential for functional intoxication by TcdB. Rather than being involved in pore formation or translocation, however, we discovered that residues in this region were implicated in binding to the TcdB receptor CSPG4. Unexpectedly, CSPG4-binding–defective mutants, although still able to bind NECTIN3 and FZD7, showed reduced binding to the surface of cells expressing all three receptors, suggesting that the other TcdB receptors are unable to fully compensate for reductions in CSPG4 binding. Using C-terminal truncations of TcdB and binding of a CROPs-targeted antibody, we established that CSPG4 binding (and full cellular toxicity by TcdB) requires at most the first three short oligopeptide repeats from the CROPs. In addition to identifying the CSPG4 receptor-binding determinants, these findings help reconcile previous seemingly contradictory findings about the CROPs dependence for CSPG4 binding. Furthermore, this work provides evidence that the majority of the CROPs region beyond residue 1900 is dispensable for full cellular intoxication by TcdB.
Results
Mutations at the boundary of the translocation and the CROPs domain affected TcdB function
As part of our ongoing efforts to elucidate the mechanism by which TcdB intoxicates host cells, we recently developed a platform that enables rapid generation and screening of site-specific perturbations in TcdB (24). We used this platform here to interrogate a poorly defined region of TcdB encompassing the junction of the translocation domain and the CROP domain (Fig. 1A). Despite a wealth of structures available for various TcdA and TcdB fragments containing either the translocation domain (25) or the CROPs (26, 27), none provided any structural information for residues between 1803 and 1834. Based on the prevalence of hydrophobic residues in this region, between 1823 and 1845 (FYINNGFMMVSGLIYINDSLYYF), we initially posited that this region might insert into the membrane during the events in the endosome that involve acid-induced unfolding and formation of the translocation pore (24). Cysteine and lysine substitutions were introduced at several conserved residues (across clostridia toxins) and screened, initially from Escherichia coli lysates, for their ability to intoxicate Vero cells, which are widely used for assessing TcdB activity due to their high sensitivity to this toxin. Through this analysis, we identified two sets of mutations, Y1824C/Y1824K and N1839C/N1839K, which significantly decreased the ability of these toxins to intoxicate CHO-K1 cells (Fig. 1B). Purified Y1824K and N1839K toxins were 120-fold and 360-fold less toxic than WT TcdB, respectively (Fig. 1C). Surprisingly, other intervening residues had no impact on toxin activity, arguing against the notion that this entire region was inserting into the membrane.
Figure 1.

Identification of functional defects at the boundary of the CROPs and translocation domain. A, schematic drawing of TcdB organized into four domains: N-terminal glucosyltransferase domain (red), autoprotease domain (blue), translocation/pore formation domain (yellow), with the hydrophobic region (residues 956–1128) shown in orange, and the C-terminal CROPs repeats domain (green). The box represents the hydrophobic region at the junction under investigation (N1820-F1856). B, effects of Cys and Lys substitutions on cellular toxicity. Viability of each Cys and Lys mutant was tested by exposing titrated mutant soluble lysates onto CHO-K1 cells (3-fold titration, 3 days) and quantified using the PrestoBlue fluorescence viability assay. Fold shift of mutant to wild type was calculated by dividing the half-maximum effective concentration (EC50) of mutants by the EC50 of wild type. n = 3. ***, p < 0.001, two-way analysis of variance. C, toxicity of mutant and wild type toxins on Vero cells was quantified by titrating purified proteins onto Vero cells (4-fold titration) in 96-well plates and incubated for 24 h at 37 °C (n = 3). Forty-eight hours later, the cell viability of treated cells was quantitated by SRB staining.
Defective mutants showed decreased host cell-surface binding
To establish the mechanistic basis for the observed defects in activity for mutants, we employed a series of assays analyzing Rac-1 glucosylation (glucosyltransferase activity), autoproteolytic cleavage, pore formation, pH-dependent unfolding, and cell-surface–binding activities. Both defective mutants were equally active as WT toxin in Rac-glucosylation and autoproteolytic cleavage assays, and each showed a similar pattern to WT TcdB in the pH-dependent unfolding assay (supplemental Fig. S2, A–D), demonstrating the toxins were functional and not misfolded. We next measured the pore-formation functionality of the Y1824K and N1839K mutants using the standard 86Rb release assay (Fig. 2A) (24). Consistent with the atypical pattern of sensitivity to mutants highlighted above, Y1824K and N1839K were minimally defective in pore formation. By comparison, defective point mutations in the hydrophobic region of the translocation domain that decreased intoxication efficiency by >10-fold, such as L1106K, gave major defects in pore formation in this assay (i.e. >90%) (24).
Figure 2.
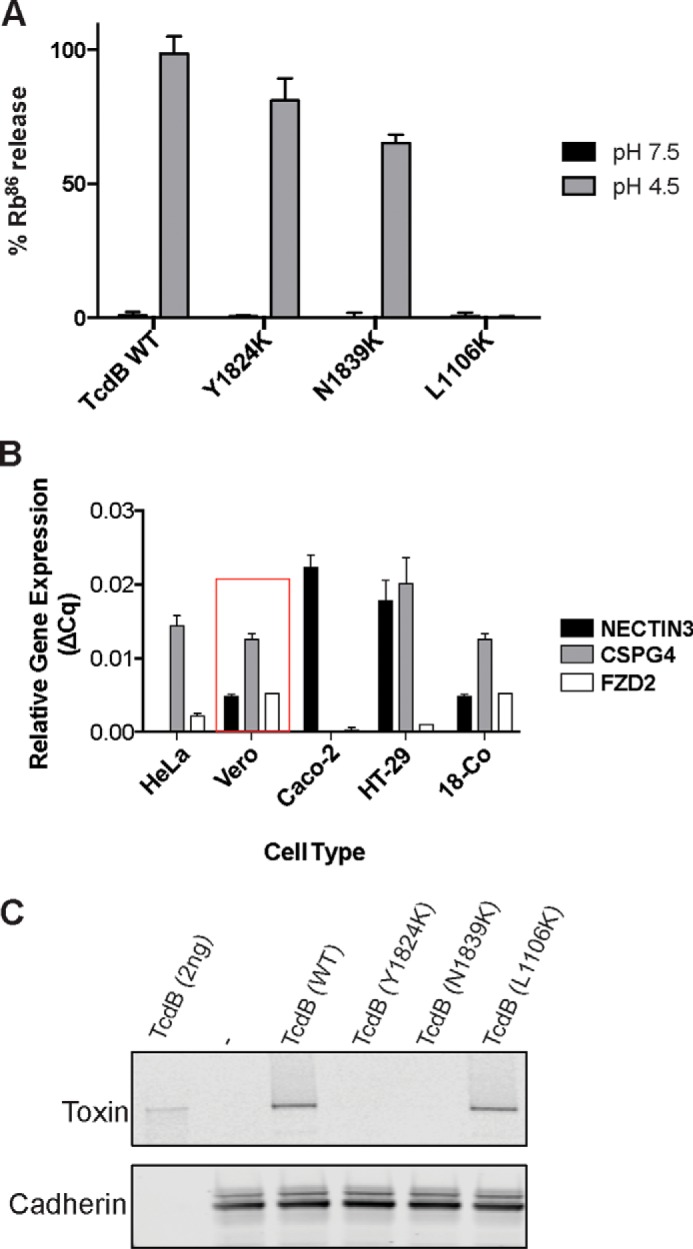
Characterization of defective purified TcdB mutants Y1824K and N1839K. A, pore formation of purified mutant toxins was tested on CHO cells preloaded with 86Rb+. Pore formation was induced by acidification of the external medium (black bars, control, pH 7.5; gray bars, pH 4.5) n = 4. B, relative gene expression of TcdB receptors. The relative gene expression of identified TcdB receptors NECTIN3, CSPG4, and FZDs on different mammalian cell lines was assessed using a ΔΔCq method determined from quantitative PCR data (31) using β-actin, ACTB, as the reference gene (n = 2). Vero cells that were used in the cell viability assay are highlighted in the red box. C, cell-surface binding of defective mutant toxins to target cells. Shown is an immunoblot analysis of TcdB wild type and mutants bound to the Vero cell surface at 4 °C. The cells were exposed to 200 ng/ml TcdB for 30 min before being lysed for immunoblot analysis. Membrane-bound proteins were detected by anti-TcdB antibody and rabbit anti-cadherin antibody as loading control.
Finally, we measured the ability of defective mutant toxins to bind target cells. To examine this, we investigated their binding to Vero cells, which express all three TcdB protein receptors, NECTIN3 (22), CSPG4 (21), and FZD2 (23), but predominantly, CSPG4 (Fig. 2B). Cells were incubated in the cold in the presence of toxin to allow binding. After extensive washing to remove unbound toxin, membrane fractions were then isolated and probed for toxin by Western blotting. As shown in Fig. 2C, Y1824K and N1839K were impaired in their ability to bind to Vero cells compared with WT TcdB and the pore-formation–defective mutant L1106K (24). Rather than being involved in formation of the translocation pore, these findings suggest that Tyr-1824 and Asn-1839 are involved in binding to target cells. We next undertook to decipher which receptor(s) was involved in binding to this region.
Defective TcdB mutants decreased binding to CSPG4 but not NECTIN3 or FZD7
To elucidate the molecular cause of the defective cell-surface binding observed for the Y1824K and N1839K mutants, we tested the binding of the mutant toxins to the ectodomains of NECTIN3 and FZD7, a member of the FZD family shown to act as a receptor for TcdB (23) as well as full-length CSPG4. By co-immunoprecipitation analysis, WT TcdB, Y1824K, and N1839K toxins were able to interact equally with the extracellular domain of NECTIN3 (Fig. 3A). Similarly, mutant toxins formed stable complexes with FZD7 as measured by gel filtration, demonstrating effective binding to the receptor (Fig. 3B). These findings are consistent with previous work indicating that NECTIN3 and FZD proteins bind upstream of TcdB residue 1830 (17, 23).
Figure 3.

Characterization of receptor binding of defective mutants. A, association of ectodomain of NECTIN3 with TcdB wild type and mutants, assayed by co-immunoprecipitation (CO-IP) analysis. His-tagged TcdB (3 μg) was incubated with 5 μg of the His-tagged NECTIN3 ectodomain at 4 °C for 16 h. TcdB and NECTIN3 interaction was immunoprecipitated and detected using anti-His antibody. B, interaction of Fzd7 with TcdB by gel filtration assay. Purified Fzd7-CRD mVenus was mixed with TcdB at 1:5 molar ratios of TcdB:Fzd7 and incubated at room temperature for 30 min and run on a Superose 6 Increase 10/300 GL column. Elution peaks of only TcdB (black) and the complex with both TcdB and Fzd7 (red) were monitored by absorbance at 280 nm, and the complex was visualized on SDS-PAGE. Note that all samples were run on the same gel; however, to simplify the figure, the relevant portion of the gel was shown with the corresponding column elution profile. Accordingly, the molecular weight marker was reused in all panels. C, binding of CSPG4 to TcdB wild type and mutants, assayed by co-immunoprecipitation analysis. TcdB (3 μg) was mixed incubated with 150 μg of cell lysate from HEK293 cells overexpressing FLAG-tagged CSPG4 at 4 °C for 2 h. The complex was immunoprecipitated using anti-FLAG magnetic beads and visualized by anti-FLAG (CSPG4) and anti-His (TcdB) antibodies. D, immunoblot analysis of the wild type (CSPG4+/+) and CSPG4 knock-out (CSPG4−/−) Vero cell lysates for expression of CSPG4 and GAPDH (control). E, sensitivity of wild type (CSPG4+/+) and CSPG4−/− Vero cells toward TcdB wild type and mutants. Indicated Vero cells were exposed to TcdB (wild type, Y1824K, and N1839K) and subjected to cell survival using the SRB assay. F, toxicity of TcdA wild type and mutants (Y1822K and N1837K) on CHO cells, measured by Prestoblue fluorescence assay as described.
Next, we tested whether the defective mutant toxins were able to bind the CSPG4 receptor. Co-immunoprecipitation studies between mutant toxins and CSPG4 showed that Y1824K and N1839K were defective in binding to CSPG4 compared with WT and L1106K mutant toxins (Fig. 3C). To confirm this finding, we next generated CSPG4 knock-out Vero cells (Vero/CSPG4−/−) using CRISPR-Cas9 technology (Fig. 3D). Consistent with these residues playing a role in binding to CSPG4, WT TcdB was defective in intoxicating Vero/CSPG4−/− relative to Vero/CSPG4+/+, whereas Lys-1824 and Lys-1839 toxins were both equally active on Vero/CSPG4−/− as on Vero/CSPG4+/+ cells (Fig. 3E). The relevance of the reduced activity observed for N1839K relative to WT TcdB on Vero/CSPG4−/− is not known but may indicate the presence of an additional host cell receptor or factor that binds in this region. Overall, these data indicate that CSPG4 binding at least in part requires determinants that are in both the translocation domain and in the CROP domain and as such help reconcile previous studies regarding whether CSPG4 was a CROPs-dependent (23) or CROPs-independent receptor (21).
Finally, because CSPG4 is a TcdB-specific receptor, we generated the equivalent mutations to Tyr-1824 and Asn-1839 in the homologous toxin TcdA, which was shown previously not to bind CSPG4 (21). TcdA-Lys-1822 and TcdA-Lys-1837 showed no defects compared with WT TcdA in intoxicating mammalian cells (Fig. 3F), further demonstrating that these residues are involved in CSPG4 binding and not any other more general aspect of intoxication.
TcdB required three oligopeptide repeats for CSPG4-binding and full cellular activity
With part of the CSPG4-binding determinants coming from the N-terminal boundary of the CROPs domain of TcdB, we set out to determine how much of the CROPs was required for binding to CSPG4. To this end we engineered, expressed, and purified a series of C-terminally truncated toxins of increasing length (TcdB1–1834 (B1834), TcdB1–1900 (B1900), TcdB1–2034 (B2034), TcdB1–2366 (WT TcdB)) and a CROPs only construct (TcdB1834–2366 (CROP)) (Fig. 4A). To test for an interaction between truncated TcdB constructs and CSPG4, His-tagged TcdB, B1834, B1900, B2034, and the TcdB-CROP were incubated with lysate from cells overexpressing FLAG-tagged CSPG4. CSPG4 was then immunoprecipitated with an anti-FLAG antibody, and co-immunoprecipitation of the toxin constructs was analyzed by Western blotting with an anti-His antibody. As expected, neither B1834 nor the CROPs-alone construct interacted with CSPG4 (Fig. 4B). By contrast, WT TcdB, B2034, and B1900 were all pulled down with CSPG4, indicating that just three short repeats were sufficient for binding to CSPG4 (Fig. 4B). Importantly, the observed binding of truncated toxin to CSPG4 directly correlated with the ability of these toxin truncations to intoxicate CSPG4-expressing Vero cells (Fig. 4C). Conversely, in Caco-2 cells, which do not express appreciable levels of CSPG4 (Fig. 2B), B1834 was as potent as the longer TcdB constructs, confirming that both CSGP4 was responsible for the observed defects and that the truncated toxins were otherwise functional (Fig. 4C).
Figure 4.
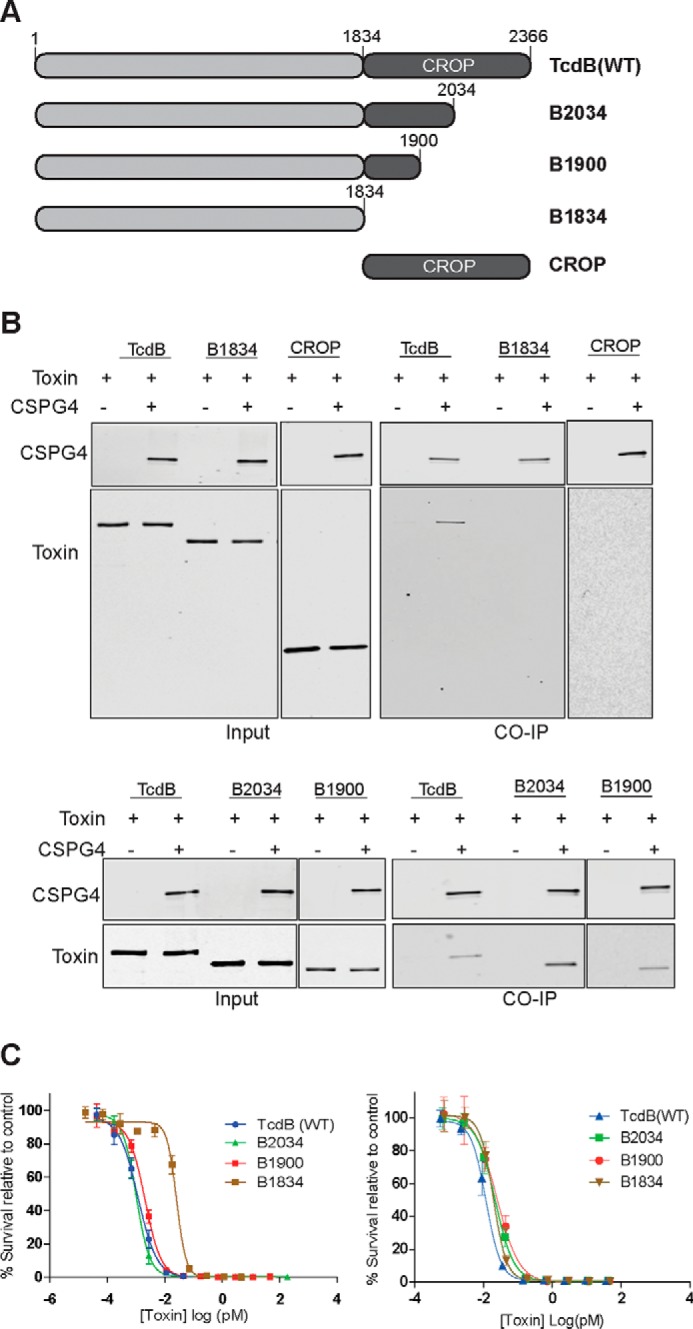
Characterization of TcdB C-terminal truncations. A, schematic drawings of TcdB C-terminal truncated constructs, B2034 (TcdB1–2034), B1900 (TcdB1–1900), B1834 (TcdB1–1834), and CROP domain (TcdB1834–2366). B, interaction between truncated TcdB constructs and CSPG4. His-tagged TcdB constructs were incubated with lysate from cells overexpressing FLAG-tagged CSPG4 and subjected to immunoprecipitation (CO-IP) by anti-FLAG antibody and visualized by immunoblot as described. C, sensitivity of Vero and Caco-2 cells toward TcdB C-terminal truncations. Vero cells (left) and Caco-2 cells (right) were exposed to TcdB-truncated toxins, and cell survival was assessed by the SRB assay as previously described.
The CROP domain from TcdB was not sufficient for cell-surface binding
Our binding data and functional studies with these constructs indicate that the region spanning the junction of the TM and CROP domains is important for binding to CSPG4 but not to the other TcdB receptors NECTIN3 and FZD, which bind upstream of the CROPs (22, 23). In addition, because B1834 is equally active as WT TcdB on Caco-2 cells, which do not express CSPG4 (Fig. 4C), CSPG4 appears to be the only receptor that requires at least part of the CROP domain for binding. Because of this we questioned whether the TcdB CROP domain by itself has any cell-surface–binding activity. To probe this, we analyzed cell-surface–binding activity of the CROP domains from both TcdA and TcdB. Although the TcdA-CROP bound to Vero cells at molar concentrations of 5- and 10-fold greater (15–30 nm) than TcdA (3 nm), no cell binding was detected for TcdB-CROP at up to 100-fold greater concentration (80 nm) than TcdB (0.8 nm) (Fig. 5A and B). To rule out the possibility that a lack of detectable binding was not due to low sensitivity of our binding assay, we carried out a competition assay with the toxins and CROP domains. A fixed amount of either TcdA or TcdB was added to Vero cells in the presence of an increasing concentration of the respective CROP domain. Consistent with the cell-surface–binding data, the TcdA CROP domain inhibited TcdA activity in a dose-dependent manner. However, TcdB activity was unaffected by up to a 20,000-fold molar excess of the TcdB CROP domain (Fig. 5, C and D). These data strongly suggest that the TcdB CROP domain by itself cannot bind to cells and that CSPG4 is the only CROP specific receptor for TcdB.
Figure 5.

Cell-surface–binding activity of the CROP domains from both TcdA and TcdB. Vero cells were incubated in A with TcdA (3 nm) and TcdA CROP (15 nm, 30 nm) and in B with TcdB (0.8 nm) and TcdB CROP (16 nm, 80 nm), and surface-bound proteins were subjected to immunoblot analysis by an anti-TcdA and anti-TcdB antibody, respectively. C, inhibition of TcdA cytotoxicity at a fixed concentration by the A-CROP domain in molar excess was measured by SRB assay. D, inhibition of TcdB cytotoxicity at a fixed concentration by the B-CROP domain in molar excess was measured by SRB assay.
Antibody binding to TcdB CROPs occluded CSPG4 binding
The anti-TcdB antibody bezlotoxumab, which neutralizes TcdB activity both in vitro and in vivo (3, 4, 27), was shown recently using hydrogen-deuterium mass spectrometry and X-ray crystallography to have two distinct binding sites within the CROP domain of TcdB: E1 and E2, spanning residues 1878–1961 and 2018–2093, respectively (27). A homology model of the segment of TcdB for which there is no structural information (i.e. 1800–1833) was built using sequence and structural alignments via Phyre 2.0 (28). Interestingly, the region that directly precedes the CROPs, 1814–1833, and that contains the defective mutant 1824 models as another short oligopeptide repeat (Fig. 6A). An alignment of this region and the downstream CROPs supports this model (supplemental Fig. S3). Mapping the defective mutants identified in this study onto this model and structure obtained by Orth et al. (27) shows that defective mutants cluster to the same face of the CROPs and in close proximity to the E1-binding site of bezlotoxumab. Given that the CSPG4-binding region established here partially overlaps a portion of E1, we reasoned that bezlotoxumab should prevent binding of CSPG4 to TcdB. To examine this and determine whether bezlotoxumab and CSPG4 binding is mutually exclusive or not, we measured complex formation between full-length TcdB and CSPG4 in the absence and presence of bezlotoxumab and actoxumab (an anti-TcdA antibody used here as a control). Indeed, bezlotoxumab, but not actoxumab, prevented CSPG4 from binding to TcdB (Fig. 6).
Figure 6.
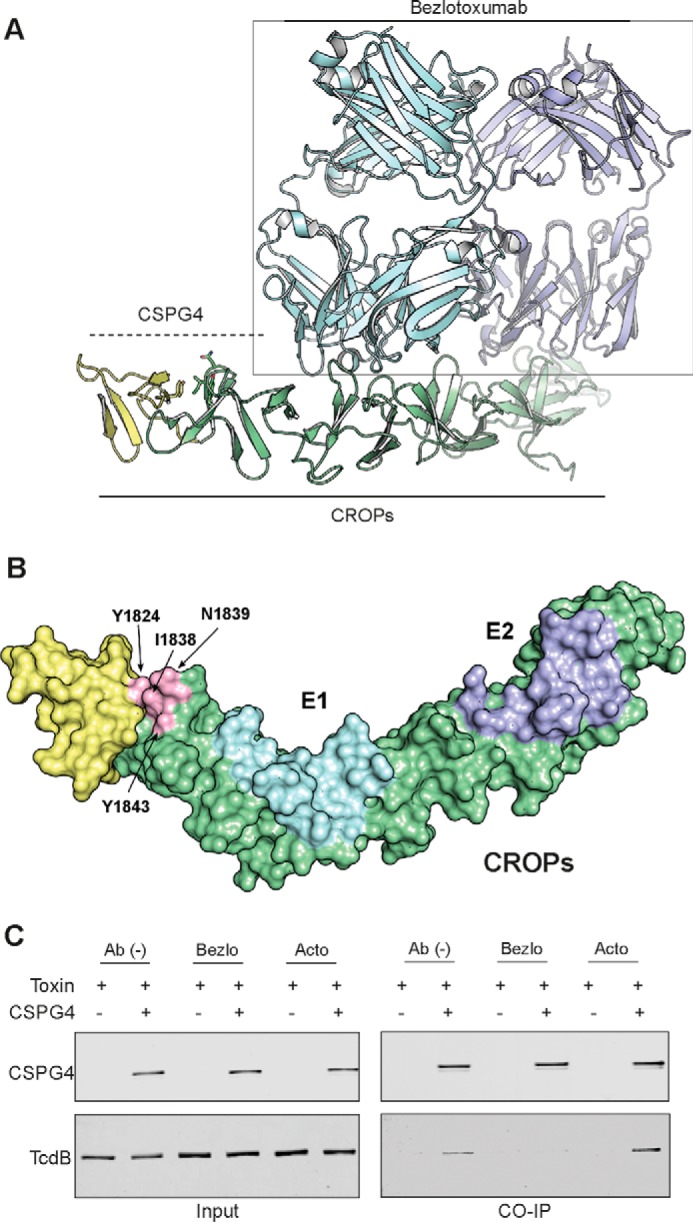
Binding model of CSPG4 and bezlotoxumab to TcdB. A, a homology model of the TcdB segment 1800–1833 with downstream CROP domain was built based on sequence and structural alignment via Phyre 2.0 (yellow) and the crystal structure of CROPs (green) with two Fab domains of bezlotoxumab from Orth et al. (27). B, model of structure of TcdB segment 1800–1833 and CROP domain with bezlotoxumab-binding sites, E1 and E2, and identified key residues mapped to the structure. C, inhibition of CSPG4 binding to TcdB by bezlotoxumab and actoxumab (control) by immunoprecipitation (CO-IP) assay as described previously.
Discussion
In this study we identified a region at the boundary of the translocation domain and the CROP domain that was critical for TcdB intoxication of mammalian cells. Two residues, Tyr-1824 and Asn-1839, flanking a stretch of ∼20 largely hydrophobic residues, were highly sensitive to mutation, reducing the ability of TcdB to intoxicate Vero and CHO cells by >2 orders of magnitude (Fig. 1). Contrary to our initial hypothesis that these residues were involved in pore formation or translocation, we traced the source of the defects on intoxication to an inability of mutant toxins to bind Vero cells (Fig. 2). We showed using either the ectodomain or full-length version of the three known TcdB receptors that mutant toxins were specifically defective in their ability to bind CSPG4 receptor but not to NECTIN3 or to FZD7 (Fig. 3). Consistent with this, we found that cells lacking CSPG4, either naturally or through targeted removal via CRISPR/Cas9-mediated deletion, were less sensitive to both WT TcdB and TcdB mutants to an equal extent. Along with confirming that mutant toxins are folded and otherwise functional, these data help to further establish that Tyr-1824 and Asn-1839 are involved in binding to CSPG4. Another important observation from these studies is that when present on the surface of a given cell, CSPG4 is the primary receptor for TcdB, as neither FZD7 nor NECTIN3 could fully compensate for its loss.
The location of these two critical CSPG4-binding mutations, at the historically defined boundary between the translocation domain and the CROPs, prompted us to explore whether more of the CROPs were involved in receptor binding. Using a series of C-terminal truncations, we found that keeping just three short repeats (i.e. B1900) was sufficient for TcdB binding to CSPG4 (Fig. 4). B1900, missing the majority of the CROPs, is as potent as WT TcdB on Vero cells, indicating that this represents a fully functional form of TcdB on cells expressing CSPG4. These findings suggested that the CROPs play a subtle role, if any at all, in the context of cellular intoxication. This was reinforced by our experiments testing the ability of the TcdA and TcdB CROPs to bind to cells (Fig. 5). Whereas the TcdA CROPs were able to bind to the surface of cells and completely inhibit binding and intoxication by TcdA, the TcdB CROPs showed no evidence of binding to cells and was unable to competitively inhibit TcdB intoxication even up to a molar excess of 30,000-fold. These data argue against the notion that TcdB utilizes the CROP domain to dock to the host cell surface by interacting with oligosaccharides followed by binding with specific cellular receptors (17, 20). Based on these data alone, however, we cannot exclude the possibility that the CROPs in TcdB beyond 1900 (i.e. 1901–2366) play a different role in the context of an in vivo infection. What is clear from our data here and has been reported previously (18) is that TcdA utilizes its CROP domain to interact with cell-surface receptors and that this interaction is, at the very least, more important for TcdA intoxication than it is for TcdB intoxication. These findings further highlight the functional differences between TcdA and TcdB.
The data presented here suggest that the majority of the CROP domain in TcdB does not play a role in receptor binding; however, it is well established that neutralizing antibodies, which bind the CROP domain, block TcdB activity both in vitro and in vivo (4, 27, 29). Our finding that CSPG4 binding required three short repeats of the CROPs that overlap the E1-binding site of bezlotoxumab helps reconcile this apparent discrepancy (Fig. 6). Consistent with the proposed binding site, in the presence of bezlotoxumab, CSPG4 can no longer bind full-length TcdB, suggesting binding of the antibody interferes with the potential binding pocket of CSPG4. It also supports our finding that the CSPG4-binding site is at the junction between residues 1810–1850, where two key residues are located, highlighting the importance of this junction. Unfortunately, there is no structural information on this junction from existing crystal structural data from both TcdA (TcdA 1–1810) and TcdB CROP domain (1834–2366). Future studies should consider keeping this junction intact for binding and structural analysis.
The notion of defining a “receptor-binding domain” for TcdB, let alone receptor-binding domains for each of the individual receptors of TcdB, has remained elusive for TcdB. This arises in part from issues associated with studying individual domains that have been truncated at previously defined domain boundaries, which presume a lack of functionality for these boundaries. Indeed, truncating TcdB at either of the historical boundary sites (i.e. 1852 and 1834) yields two toxin fragments, neither of which can bind CSPG4. Our finding that CSPG4 requires determinants from the translocation domain and the CROPs is satisfying as it helps reconcile the somewhat contradictory conclusions regarding CSPG4's CROPs dependence (21, 23). We opted to not explore the N-terminal boundary of CSPG4 binding in detail beyond what had been done previously (21) because we were most interested in describing the functional determinants of binding, which required upstream factors from TcdB. Nevertheless, from previous work showing that TcdB1500–2366 co-precipitates CSPG4, we can minimally define the CSPG4-binding region to within these boundaries. A more detailed description of the binding site likely awaits a high-resolution structure of the complex of the CSPG4 with TcdB or fragments thereof, which this work will help guide.
Lastly, our findings here also highlight the importance of carefully considering cell lines for studying toxin binding and cytotoxicity analysis. Our gene expression analysis shows very different receptor expression profiles for the different mammalian cell lines that are widely used (Fig. 2B); for example, HeLa and Vero express high level of CSPG4 and low level of NECTIN3, whereas Caco-2 highly expresses NECTIN3 but no CSPG4, and HT-29 expresses both similarly. As shown in our cytotoxicity data, cellular expression of specific receptors could have significant impact on cellular sensitivity to the toxins. Previous findings show that both FZDs and NECTIN3 are found predominantly on colonic epithelial cells, whereas CSPG4 is expressed mainly in subepithelial myofibroblast cells like Co-18 cells (22, 23, 30). In combination, they could serve as targets for different stages of toxin entry. The physiological roles and clinical relevance of all three receptors remains to be elucidated.
Experimental procedures
Cell lines and reagents
Vero, Caco-2, HT29, 18Co, HEK293, and CHO-K1 cell lines were purchased from the American Type Culture Collection (ATCC). All cell lines were grown at 37 °C in 5% CO2. Vero and 18Co cells were maintained in Eagle's minimal essential medium supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 units/ml streptomycin. Caco-2 cells were maintained in Eagle's minimal essential medium supplemented with 10% FBS, 100 units/ml penicillin, 100 units/ml streptomycin, nonessential amino acids, and 0.75% sodium bicarbonate. HT29 cells were cultured in McCoy's 5A Modified Medium supplemented with 10% fetal bovine serum, 2 mm glutamine, 0.75% sodium bicarbonate, 100 units/ml penicillin, and 100 units/ml streptomycin. HEK 293 cells were cultured in Dulbecco's minimal essential medium (DMEM) (ATCC) containing 10% FBS, penicillin (100 units/ml), and streptomycin (100 units/ml). Chinese hamster ovary cells CHO-K1 cells were cultured in Ham's F-12 medium (Wisent) with 10% FBS and 100 units/ml penicillin and 100 units/ml streptomycin. The C. difficile VPI 10463 strain (ribotype 087) was purchased from the ATCC. Toxin fragments were expressed in Bacillus megaterium. All other chemicals and reagents were purchased from ThermoFisher Scientific unless otherwise stated.
Cloning and mutagenesis
Plasmids for recombinant expression of TcdB, TcdB 1–2034, and TcdB 1–1900 were generated by Gibson cloning methodology. DNA fragments encoding full-length TcdB and the C-terminal truncations were amplified by PCR using genomic DNA from C. difficile 10463 strain as the template. The amplified fragments were cloned in-frame with the His6 tag in the plasmid pHis1522 (MoBiTec, catalog no. BMEG10) using the Gibson cloning kit (New England BioLabs, catalog no. E2611). Cloning of plasmids expressing TcdB1–1834 (LaFrance et al.; Ref. 22), TcdB1834–2366 (Orth et al.; Ref. 27), and TcdA1832–2710 (Hernandez et al.; Ref. 29) was previously described. Single point mutations were made in the TcdB codon-optimized sequence using QuikChange lightning multimutagenesis kit (Agilent Technologies). Plasmids with correct mutations were transformed and expressed using the same conditions as wild type.
Expression and purification of recombinant TcdB from B. megaterium
Proteins were expressed using the B. megaterium expression system as previously described (Yang et al.; Ref. 32) and were purified by sequential nickel affinity chromatography using a HisTrap FF column (GE Healthcare) and ion exchange chromatography using a Q FF column (GE Healthcare). Fractions containing TcdB were verified and pooled with a 100,000 molecular weight cutoff ultrafiltration device. Ten percent of glycerol was added; protein concentration was calculated by densitometry (Image Lab 3.0).
Small-scale expression of TcdB mutants
Overnight cultures were prepared in 24-well blocks (BD Biosciences) in 5 ml of Luria broth. A total of 250 μl of overnight culture was inoculated into 5 ml of LB with kanamycin and induced at A600 of 0.6 with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at 37 °C for 4 h. Cells were harvested by centrifugation and resuspended in buffer (20 mm Tris, 500 mm NaCl, pH 8.0, and protease inhibitor; Sigma) and lysed by lysozyme (Bioshop) according to the manufacturer's instructions followed by centrifugation at 4000 × g for 20 min. Supernatants were collected. The concentration of each full-length mutant protein in the lysates was determined by densitometry (Image Lab 3.0).
Cell viability assay
CHO-K1 cells were seeded at a concentration of 8000 cells per well in 96-well CellBind plates (Corning). The next day medium was exchanged with serum-free medium, and cells were intoxicated by adding TcdB toxins at a serial dilution of 1/3 starting at 1 nm. After intoxication, cells were incubated at 37 °C, 5% CO2 for 48 h. Serum (FBS) was added back to cells 24 h after intoxication to a final concentration of 10% (v/v). The cell viability after 48 h was assessed by PrestoBlue Cell Viability Reagent (Life Technologies). Fluorescence was read on a Spectramax M5 plate reader (Molecular Devices).
Toxin cell death assay
Effect of toxin constructs on cell survival was assessed as previously described using the sulforhodamine B (SRB) assay (Orth et al.; Ref. 27); Hernandez et al.; Ref. 29)). Briefly, serial dilutions of toxins in cell culture medium were added to cells previously grown overnight in 96-well plates. After a 24-h incubation at 37 °C, cells were washed with phosphate-buffered saline and allowed to grow for further 48 h. Cells were fixed by adding 10% cold TCA followed by incubation for 60 min at 4 °C. TCA was removed, wells were washed 4 times with distilled water, and 100 μl/well of 2 mg/ml sulforhodamine B (Sigma) in 1% acetic acid was added. Plates were incubated for 20 min at room temperature and then washed 4 times with 1% acetic acid and air-dried. 150 μl/well of 10 mm Tris was added, and plates were incubated with shaking at room temperature for an additional 10 min. Plates were read in a SpectraMax plate reader (Molecular Biosystems) at an absorbance wavelength of 570 nm.
To test for competition between the CROP domain and the toxin, purified toxin was diluted in the culture medium to a final concentration that resulted in a ∼90% decrease in cell viability in the absence and presence of various concentrations of the CROP domain and then added to cells. After 2 h of incubation at 37 °C, cells were washed with PBS, allowed to grow for further 48 h, and processed as described above.
Rubidium release assay
86Rb+ release assay was performed as previously reported (Genisyuerek et al., Ref. 19) with slight modifications. Briefly, CHO-K1 cells were seeded in 96-well plates in the medium (Ham's F-12 with 10% FBS) and supplemented with 1 μCi/ml 86Rb+ (PerkinElmer Life Sciences) at a density of 1 × 104 cells per well. Cells were incubated at 37 °C, 5% CO2 overnight. Medium was exchanged with fresh growth medium with 100 nm bafilomycin A1 (Sigma) and continued to incubate for another 20 min. Then cells were chilled on ice, and ice-cold medium containing TcdB mutants (10 nm) was added. Cells were kept on ice for toxin binding for 1 h at 4 °C before they were washed with ice-cold PBS twice to remove unbound toxins. pH-dependent insertion into the plasma membrane was induced by warm, acidified growth medium (37 °C, pH 4.5 or pH 7.5) for 5 min at 37 °C. After 1 h of further incubation on ice, medium containing released 86Rb+ was removed from the cell plate, and the amount of 86Rb+ released was determined by liquid scintillation counting with TopCount NXT (PerkinElmer Life Sciences).
Cysteine protease assay
TcdB self-cleavage by its intrinsic cysteine protease activity was measured by incubating TcdB WT and mutants with 100 μm inositol hexakisphosphate (InsP6) and 5 mm DTT at 37 °C for 3 h. Cleavage was visualized by electrophoresis of the samples on SDS-polyacrylamide gels and staining with Coomassie Blue R250.
2-p-Toluidinylnaphthylene-6-sulfonate fluorescence assay
pH-induced conformational changes of TcdB were assessed as described previously (Lanis et al.; Ref. 33). A total of 2 μg of TcdB prepared in buffer with a pH ranging from 4 to 7.2 ((p-toluidiny)-naphthalene-6-sulfonic acid, sodium salt (2,6–2-p-toluidinylnaphthylene-6-sulfonate (TNS); Invitrogen) was added at a final concentration of 150 μm. The final volume was 250 μl and was mixed in a 96-well black plate (Corning). Mixtures were incubated at 37 °C for 20 min. The plate was analyzed in SpectramaxM5 plate reader (Molecular Devices) with excitation of 366 nm and an emission scan of 380–500 nm.
Differential scanning fluorometry
Differential scanning fluorometry was performed in a similar manner as described previously (Tam et al.; Ref. 34). TcdB protein was diluted in phosphate buffer (100 mm K3PO4, 150 mm NaCl, pH 7) containing 5× SYPRO Orange (Invitrogen). A Bio-Rad CFX96 quantitative real-time-PCR thermocycler was used to establish a temperature gradient from 15 °C to 95 °C in 30-s increments while simultaneously recording the increase in SYPRO Orange fluorescence as a consequence of binding to hydrophobic regions exposed on unfolded proteins. The Bio-Rad CFX Manager 3.1 software was used to integrate the fluorescence curves to calculate the melting point.
Glucosyltransferase Western blot assays
For each reaction, 0.8 μm GSTRac1 was mixed with 25 μm UDP-glucose and followed by the addition of 20 nm TcdB. The reaction was stopped after a 60-min reaction time with an equal volume of Laemmli loading buffer plus β-mercaptoethanol (Bio-Rad) heated to 90 °C before immediately loading on an SDS-polyacrylamide gel. After electrophoresis, samples were transferred to nitrocellulose using an iBlot device (Invitrogen), blocked with 5% milk/Tris-buffered saline (TBS), and probed with a 1/1000 dilution of either Mab102 or anti-GST antibodies. After an overnight incubation with the primary antibody, the blot was washed with TBS, 0.1% Tween 20 and incubated with a 1:5000 dilution of anti-mouse horseradish peroxidase for 60 min. After the final washes in Tris-buffered saline with Tween 20, chemiluminescent detection was carried out using SuperSignal substrate (Thermo Pierce) and exposing to BioMax MR film (Eastman Kodak Co.).
Generation of CSPG4-expressing HEK 293 cell line
pCMV6-CSPG4 vector expressing full-length Myc-DDK-tagged CSPG4 was purchased from OriGene Technologies (catalog no. RC218462). HEK-293 cells were transfected with pCMV6-CSPG4 plasmid using Lipofectamine 2000 according to the manufacturer's protocol (ThermoFisher Scientific). 2 days after transfection, the medium was replaced with DMEM supplemented with 10% FBS, penicillin (100 units/ml), streptomycin (100 units/ml), and G418 (1 μg/ml). G418-resistant cells were pooled, expanded, and then analyzed for expression of CSPG4 expression by Western blotting with anti-DDK antibody (OriGene Technologies, catalog no.TA5011-100).
CRISPR mutagenesis
Vero CSPG4 knock-out cells were generated via CRISPR-Cas9 technology using Geneart CRISPR Nuclease (CD4 Enrichment) Vector kit from ThermoFisher Scientific (catalog no. A21175). Briefly, two complementary oligonucleotides (5′-AACGCCTCCTCGCAGTCCCGTTTT-3′, 5′-GGGACTGCAGAGGAGGCGTTCGGTG-3′) encoding a guide RNA fragment were used (Sigma). The protospacer sequence (5′-AACGCCTCCTCTGCAGTCCC-3′) was complementary to a sequence in exon 3 of the CSPG4 gene. The two oligonucleotides were annealed and cloned into CRISPR nuclease CD4 vector supplied with the kit according to manufacturer's instructions, and the resulting plasmid was transfected into Vero cells using Lipofectamine 2000. Transfected cells were first enriched using Dynabeads CD4 magnetic beads (ThermoFisher Scientific, catalog no. 11331D) and then treated with TcdB for the selection of toxin-resistant Vero CSPG4 knock-out cells. A single CSPG4 knock-out clone was isolated by limited dilution. Loss of CSPG4 expression was confirmed by Western blot analysis, and a base pair insertion in the exon 3 of the CSPG4 gene was confirmed by Sanger sequencing of the region targeted by the guide RNA.
TcdB cell-surface–binding assay
Binding of TcdB to the Vero cell surface was assessed as described previously (Orth et al.; Ref. 27). Briefly, 10-cm dishes of confluent Vero cells were pre-chilled on ice. 200 ng/ml TcdB or the mutants in Vero cell culture medium were added to the cells. Plates were incubated on ice to allow toxin binding. After 30 min, plates were washed 3 times with cold PBS, and cells were harvested by scraping. Membrane proteins were isolated in the cold using the Mem-PER Plus membrane protein extraction kit (ThermoFisher Scientific, catalog no. 89842) according to the manufacturer's instructions. In the final step membrane proteins were solubilized in a total volume of 100 μl of solubilization buffer and analyzed by immunoblotting using bezlotoxumab to detect TcdB and rabbit anti-cadherin polyclonal antibody (Cell Signaling, catalog no. 4068) to ensure equal amounts of protein were loaded in each lane.
Co-immunoprecipitation assays
Cell lysates for immunoblotting and co-immunoprecipitation studies were prepared by lysing 5 million cells in 300 μl of Nonidet P-40 lysis buffer (ThermoFisher Scientific, catalog no. FNN0021) by incubating on ice for 30 min. For studying the interaction of TcdB with CSPG4, 3 μg of TcdB or TcdB fragments were incubated with 150 μg of total cells lysate from HEK 293 cells overexpressing myc-DDK-tagged CSPG4 or containing empty vector in 20 mm Tris, pH 7.4, 150 mm NaCl at 4 °C for 2 h. For TcdB, the CSPG4 complex was immunoprecipitated using anti-FLAG M2 magnetic beads using the manufacturer's protocol (Sigma, catalog no. M8823). Briefly, proteins mixtures were incubated with 50 μl of beads at 4 °C for 1 h. Beads were washed 3 times with 50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.15% Triton X-100. FLAG-tagged proteins were eluted by incubating the beads with 100 μl of 150 ng/μl 3× FLAG peptide (Sigma, catalog no. F4799) at 15 °C for 30 min. For studying TcdB NECTIN3 interaction, 3 μg of His-tagged TcdB (WT and the mutants) were incubated with 5 μg of His-tagged NECTIN3 ectodomain, amino acids 1–400 (ThermoFisher Scientific, catalog no. 10852-H08H-25) in 20 mm Tris, pH 7.4, 150 mm NaCl at 4 °C for 16 h. For TcdB, the NECTIN3 complex was immunoprecipitated using 1G10 antibody, targeting TcdB CROP domain (Babcock et al; Ref. 35) coupled to protein A magnetic beads (ThermoFisher Scientific, catalog no. 10001D). 10 μg of antibody was incubated with 50 μl of Protein A beads in 200 μl of PBS with 0.02% Tween 20 for 10 min at room temperature. Beads were washed to remove excess unbound antibody and incubated with TcdB-NECTIN3 protein complex at 4 °C for 1 h. After 3 washes with PBS + 0.02% Tween 20, proteins were eluted by resuspending the beads in 100 μl of Laemmli sample buffer. Eluted proteins were subjected to immunoblotting analysis.
Immunoblotting
Proteins were resuspended in Laemmli sample buffer, boiled, and separated by SDS-gel electrophoresis on 4–12% NuPage Tris-glycine gels. Proteins were transferred to a nitrocellulose membrane using the iBLOT blotting system from ThermoFisher Scientific. The nitrocellulose membrane containing transferred protein was blocked in Odyssey blocking buffer (LI-COR Biosciences, catalog no. 927-40000) for 30 min followed by incubation with appropriate primary antibody for 1 h at room temperature. TcdB and NECTIN3 proteins were detected using anti-His antibody (Cell Signaling, catalog no. 2365) and CSPG4 with anti-FLAG (OriGene Technologies, catalog no. TA5011-100). After washing, the blot was incubated with a secondary IgG antibody coupled to IRDye 800CW or 680RD (LI-COR Biosciences) for 30 min at room temperature. After additional washing, bands were visualized using the Odyssey Imaging System (LI-COR Biosciences).
Fzd7 purification
The gene for the CRD construct of human Fzd7 (residues 42–179) with a C-terminal monoVenus tag followed by a tandem His12 tag was codon-optimized for expression in human cells (Life Technologies) and cloned into the pHLsec vector. Human Fzd7-CRD was expressed in HEK 293F cells and purified using nickel-nitrilotriacetic acid affinity chromatography. The protein was eluted with an increasing gradient of imidazole with a maximum concentration of 500 mm followed by gel-filtration chromatography (Superdex 200 Increase, GE Healthcare) in 20 mm Tris, pH 8.0, and 150 mm NaCl buffer.
Gel filtration assay
Purified hFzd7-CRD mVenus was mixed with wild-type and TcdB mutants at 1:5 molar ratios of TcdB:Fzd7 and incubated at room temperature for 30 min. The mixture was run on a Superose 6 Increase 10/300 GL column (GE Healthcare) equilibrated in 20 mm Tris, pH 8.0, and 150 mm NaCl. Eluting peaks were monitored by absorbance at 280 nm.
Author contributions
P. G. and Z. Z. performed and analyzed the experiments and helped write the paper. S. N. S.-M., J. T., S. R., N. M., and K. B. provided technical assistance and contributed to the preparation of the figures. J. J.-P. coordinated and analyzed the experiments shown in Fig. 3. H. K. K. and D. B. L. designed and helped construct the vectors and proteins used in this study. A. G. T., L. D. H., and R. A. M. conceived and coordinated the study and wrote the paper.
Supplementary Material
This work was supported by Canadian Institutes of Health Research and by Merck, Inc. P. G., N. M., K. B., A. G. T., and L. D. H. are employees of Merck, Inc.
This article contains supplemental Figs. S1–S3.
- CROP
- combined repetitive oligopeptide
- SRB
- sulforhodamine B
- CRD
- cysteine-rich domain.
References
- 1. Kelly C. P., and LaMont J. T. (2008) Clostridium difficile: more difficult than ever. N. Engl. J. Med. 359, 1932–1940 [DOI] [PubMed] [Google Scholar]
- 2. Voth D. E., and Ballard J. D. (2005) Clostridium difficile toxins: mechanism of action and role in disease. Clin. Microbiol. Rev. 18, 247–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wilcox M., Gerding D. N., Poxton I., Kelly C., Nathan R., Cornely O., Rahav G., Lee C., Eves K., Pedley A., Tipping R., Guris D., Kartsonis N., and Dorr M. B. (2015) Bezlotoxumab (BEZ) alone and with actoxumab (ACT) for prevention of recurrent C. difficile infection (rCDI) in patients on standard of care (SoC) antibiotics: integrated results of 2 phase 3 studies (MODIFY I and MODIFY II). In IDWeek 2015, San Diego, CA [Google Scholar]
- 4. Yang Z., Ramsey J., Hamza T., Zhang Y., Li S., Yfantis H. G., Lee D., Hernandez L. D., Seghezzi W., Furneisen J. M., Davis N. M., Therien A. G., and Feng H. (2015) Mechanisms of protection against Clostridium difficile infection by the monoclonal antitoxin antibodies actoxumab and bezlotoxumab. Infect. Immun. 83, 822–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Steele J., Mukherjee J., Parry N., and Tzipori S. (2013) Antibody against TcdB, but not TcdA, prevents development of gastrointestinal and systemic Clostridium difficile disease. J. Infect. Dis. 207, 323–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Papatheodorou P., Zamboglou C., Genisyuerek S., Guttenberg G., and Aktories K. (2010) Clostridial glucosylating toxins enter cells via clathrin-mediated endocytosis. PLoS ONE 5, e10673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Reineke J., Tenzer S., Rupnik M., Koschinski A., Hasselmayer O., Schrattenholz A., Schild H., and von Eichel-Streiber C. (2007) Autocatalytic cleavage of Clostridium difficile toxin B. Nature 446, 415–419 [DOI] [PubMed] [Google Scholar]
- 8. Just I., Selzer J., Wilm M., von Eichel-Streiber C., Mann M., and Aktories K. (1995) Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375, 500–503 [DOI] [PubMed] [Google Scholar]
- 9. Just I., Wilm M., Selzer J., Rex G., von Eichel-Streiber C., Mann M., and Aktories K. (1995) The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J. Biol. Chem. 270, 13932–13936 [DOI] [PubMed] [Google Scholar]
- 10. Qa'Dan M., Ramsey M., Daniel J., Spyres L. M., Safiejko-Mroczka B., Ortiz-Leduc W., and Ballard J. D. (2002) Clostridium difficile toxin B activates dual caspase-dependent and caspase-independent apoptosis in intoxicated cells. Cell Microbiol. 4, 425–434 [DOI] [PubMed] [Google Scholar]
- 11. Chumbler N. M., Farrow M. A., Lapierre L. A., Franklin J. L., Haslam D. B., Haslam D., Goldenring J. R., and Lacy D. B. (2012) Clostridium difficile toxin B causes epithelial cell necrosis through an autoprocessing-independent mechanism. PLoS Pathog. 8, e1003072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Farrow M. A., Chumbler N. M., Lapierre L. A., Franklin J. L., Rutherford S. A., Goldenring J. R., and Lacy D. B. (2013) Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc. Natl. Acad. Sci. U.S.A. 110, 18674–18679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krivan H. C., Clark G. F., Smith D. F., and Wilkins T. D. (1986) Cell surface binding site for Clostridium difficile enterotoxin: evidence for a glycoconjugate containing the sequence Gal α1–3Gal β1–4GlcNAc. Infect. Immun. 53, 573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tucker K. D., and Wilkins T. D. (1991) Toxin A of Clostridium difficile binds to the human carbohydrate antigens I, X, and Y. Infect. Immun. 59, 73–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guttenberg G., Hornei S., Jank T., Schwan C., Lü W., Einsle O., Papatheodorou P., and Aktories K. (2012) Molecular characteristics of Clostridium perfringens TpeL toxin and consequences of mono-O-GlcNAcylation of Ras in living cells. J. Biol. Chem. 287, 24929–24940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gerhard R., Frenzel E., Goy S., and Olling A. (2013) Cellular uptake of Clostridium difficile TcdA and truncated TcdA lacking the receptor binding domain. J. Med. Microbiol. 62, 1414–1422 [DOI] [PubMed] [Google Scholar]
- 17. Manse J. S., and Baldwin M. R. (2015) Binding and entry of Clostridium difficile toxin B is mediated by multiple domains. FEBS Lett. 589, 3945–3951 [DOI] [PubMed] [Google Scholar]
- 18. Olling A., Goy S., Hoffmann F., Tatge H., Just I., and Gerhard R. (2011) The repetitive oligopeptide sequences modulate cytopathic potency but are not crucial for cellular uptake of Clostridium difficile toxin A. PLoS ONE 6, e17623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Genisyuerek S., Papatheodorou P., Guttenberg G., Schubert R., Benz R., and Aktories K. (2011) Structural determinants for membrane insertion, pore formation, and translocation of Clostridium difficile toxin B. Mol. Microbiol. 79, 1643–1654 [DOI] [PubMed] [Google Scholar]
- 20. Schorch B., Song S., van Diemen F. R., Bock H. H., May P., Herz J., Brummelkamp T. R., Papatheodorou P., and Aktories K. (2014) LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. Proc. Natl. Acad. Sci. U.S.A. 111, 6431–6436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuan P., Zhang H., Cai C., Zhu S., Zhou Y., Yang X., He R., Li C., Guo S., Li S., Huang T., Perez-Cordon G., Feng H., and Wei W. (2015) Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res. 25, 157–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. LaFrance M. E., Farrow M. A., Chandrasekaran R., Sheng J., Rubin D. H., and Lacy D. B. (2015) Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proc. Natl. Acad. Sci. U.S.A. 112, 7073–7078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tao L., Zhang J., Meraner P., Tovaglieri A., Wu X., Gerhard R., Zhang X., Stallcup W. B., Miao J., He X., Hurdle J. G., Breault D. T., Brass A. L., and Dong M. (2016) Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 538, 350–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Z., Park M., Tam J., Auger A., Beilhartz G. L., Lacy D. B., and Melnyk R. A. (2014) Translocation domain mutations affecting cellular toxicity identify the Clostridium difficile toxin B pore. Proc. Natl. Acad. Sci. U.S.A. 111, 3721–3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chumbler N. M., Rutherford S. A., Zhang Z., Farrow M. A., Lisher J. P., Farquhar E., Giedroc D. P., Spiller B. W., Melnyk R. A., and Lacy D. B. (2016) Crystal structure of Clostridium difficile toxin A. Nat. Microbiol. 1, 15002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ho J. G., Greco A., Rupnik M., and Ng K. K. (2005) Crystal structure of receptor-binding C-terminal repeats from Clostridium difficile toxin A. Proc. Natl. Acad. Sci. U.S.A. 102, 18373–18378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Orth P., Xiao L., Hernandez L. D., Reichert P., Sheth P. R., Beaumont M., Yang X., Murgolo N., Ermakov G., DiNunzio E., Racine F., Karczewski J., Secore S., Ingram R. N., Mayhood T., Strickland C., and Therien A. G. (2014) Mechanism of action and epitopes of Clostridium difficile toxin B-neutralizing antibody bezlotoxumab revealed by X-ray crystallography. J. Biol. Chem. 289, 18008–18021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kelley L. A., and Sternberg M. J. (2009) Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 29. Hernandez L. D., Kroh H. D. Hsieh E., Yang X., Beaumont M., Sheth P. R., DiNunzio E., Rutherford S. A., Ohi M. D., Ermakov G., Xiao L., Secore S., Karczewski J., Racine F., Mayhood T., Fischer P., Sher X., Gupta P., Lacy D. B., and Therien A. G. (2017) Epitopes and mechanism of action of the Clostridium difficile toxin A-neutralizing antibody actoxumab. J. Mol. Biol. 429, 1030–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Terada N., Ohno N., Murata S., Katoh R., Stallcup W. B., and Ohno S. (2006) Immunohistochemical study of NG2 chondroitin sulfate proteoglycan expression in the small and large intestines. Histochem. Cell Biol. 126, 483–490 [DOI] [PubMed] [Google Scholar]
- 31. Haimes J., and Kelley M. C. (2010) Demonstration of a ΔΔCq calculation method to compute relative gene expression from qPCR data. GE Healthcare. dharmacon.gelifesciences.com/uploadedfiles/resources/delta-cq-solaris-technote.pdf
- 32. Yang G., Zhou B., Wang J., He X., Sun X., Nie W., Tzipori S., and Feng H. (2008) Expression of recombinant Clostridium difficile toxin A and B in Bacillus megaterium. BMC Microbiol. 8, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lanis J. M., Barua S., and Ballard J. D. (2010) Variations in TcdB activity and the hypervirulence of emerging strains of Clostridium difficile. PLoS Pathog. 6, e1001061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tam J., Beilhartz G. L., Auger A., Gupta P., Therien A. G., and Melnyk R. A. (2015) Small molecule inhibitors of Clostridium difficile toxin B-induced cellular damage. Chem. Biol. 22, 175–185 [DOI] [PubMed] [Google Scholar]
- 35. Babcock G. J., Broering T. J., Hernandez H. J., Mandell R. B., Donahue K., Boatright N., Stack A. M., Lowy I., Graziano R., Molrine D., Ambrosino D. M., and Thomas W. D. Jr. (2006) Human monoclonal antibodies directed against toxins A and B prevent Clostridium difficile-induced mortality in hamsters. Infect. Immun. 74, 6339–6347 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.