

Abstract
Neuronal excitation can induce new mRNA transcription, a phenomenon called excitation–transcription (E-T) coupling. Among several pathways implicated in E-T coupling, activation of voltage-gated L-type Ca2+ channels (LTCCs) in the plasma membrane can initiate a signaling pathway that ultimately increases nuclear CREB phosphorylation and, in most cases, expression of immediate early genes. Initiation of this long-range pathway has been shown to require recruitment of Ca2+-sensitive enzymes to a nanodomain in the immediate vicinity of the LTCC by an unknown mechanism. Here, we show that activated Ca2+/calmodulin-dependent protein kinase II (CaMKII) strongly interacts with a novel binding motif in the N-terminal domain of CaV1 LTCC α1 subunits that is not conserved in CaV2 or CaV3 voltage-gated Ca2+ channel subunits. Mutations in the CaV1.3 α1 subunit N-terminal domain or in the CaMKII catalytic domain that largely prevent the in vitro interaction also disrupt CaMKII association with intact LTCC complexes isolated by immunoprecipitation. Furthermore, these same mutations interfere with E-T coupling in cultured hippocampal neurons. Taken together, our findings define a novel molecular interaction with the neuronal LTCC that is required for the initiation of a long-range signal to the nucleus that is critical for learning and memory.
Keywords: Ca2+/calmodulin-dependent protein kinase II (CaMKII), calcium channel, cAMP-response element-binding protein (CREB), neuron, protein-protein interaction
Introduction
Neuronal excitation can lead to activity-dependent gene expression, a process known as excitation–transcription (E-T)4 coupling (1, 2). E-T coupling is thought to be essential for learning and memory consolidation (3). In particular, phosphorylation of CREB transcription factor at Ser133 is a key regulatory step in the transcription of several immediate early genes encoding proteins, such as c-Fos, BDNF, Arc, and Homer1a (4), and is important for synaptic plasticity and long-term memory (5, 6). Multiple signaling mechanisms have been implicated in CREB phosphorylation and E-T coupling, and their relative roles appear to depend on the cell type, the specific stimulating signal, and the strength and duration of stimulation (7–9).
One major pathway to trigger CREB phosphorylation at Ser133 is the activation of L-type Ca2+ channels (LTCCs) (10–12). LTCC activation can induce global increases of neuronal Ca2+ concentrations. However, at least under some conditions, the initiation of LTCC signaling to trigger nuclear CREB phosphorylation appears to require increased Ca2+ concentrations only within a nanodomain in the immediate vicinity of the channel. In this paradigm, Ca2+ binds to the ubiquitous Ca2+ sensor, calmodulin, within the LTCC nanodomain, and Ca2+/calmodulin then translocates to the nucleus to induce CREB phosphorylation (13, 14).
Molecular and pharmacological studies have provided robust evidence that Ca2+/calmodulin-dependent protein kinase II (CaMKII) is required for LTCC-dependent E-T coupling. CaMKII is specifically recruited to neuronal LTCCs during E-T coupling (11, 15). In fact, recent studies indicate that E-T coupling requires precisely coordinated recruitment and activation of two CaMKII holoenzymes within the LTCC nanodomain (10). CaMKII has been reported to directly interact with multiple proteins within LTCC complexes, including the pore-forming α1 (CaV1.2 or CaV1.3) subunits, auxiliary β1 or β2 subunits, and associated scaffolding proteins, such as densin (16–20). CaMKII interactions with β2 and densin play a role in modulating Ca2+-dependent facilitation of CaV1.2 and CaV1.3 LTCCs, respectively (16, 19). However, the roles, if any, of these interactions in E-T coupling are unclear.
Here we identify a novel direct interaction between CaMKII and the N-terminal domain (NTD) of LTCC α1 subunits. Residues in the CaV1.3 NTD and in CaMKII that are critical for the interaction were identified by site-directed mutagenesis. Finally, these mutated proteins were used to show that CaMKII–NTD interaction is important for the association of CaMKII with CaV1.3 and for LTCC-dependent E-T coupling in neurons.
Results
CaV1.3 NTD directly binds activated CaMKII
CaMKII has been suggested to interact with the pore-forming α1 subunits of CaV1.2 and CaV1.3 LTCCs (17–20), although there are conflicting data about the specific domains involved. To address this question, we expressed and purified a family of GST fusion proteins containing each intracellular domain of the CaV1.3 α1 subunit (Fig. 1A). Despite extensive efforts to further optimize the experimental conditions, some of these proteins were partially degraded; however, full-length proteins were readily detected (asterisks in Fig. 1, B and C). Because direct interactions of CaMKII with several neuronal proteins are differentially modulated by CaMKII activation (21–23), we tested for direct binding of these GST fusion proteins to purified mouse CaMKIIα following preincubation to induce different conformations. There was no consistently detectable binding of inactive CaMKII to any of the intracellular domains above the level of GST control, but the CaV1.3 NTD directly and specifically interacts with activated CaMKII conformations induced by pre-autophosphorylation at Thr-286 (Fig. 1B) or by the binding of Ca2+/calmodulin and ADP (Fig. 1C). The fact that binding of Ca2+/calmodulin and ADP to CaMKII is sufficient to induce interaction with the NTD shows that Thr-286 phosphorylation is not necessary for binding; rather, an open, activated conformation of CaMKII is required. Thus, these data show that activated CaMKII directly interacts with the NTD of CaV1.3 with very high selectivity.
Figure 1.
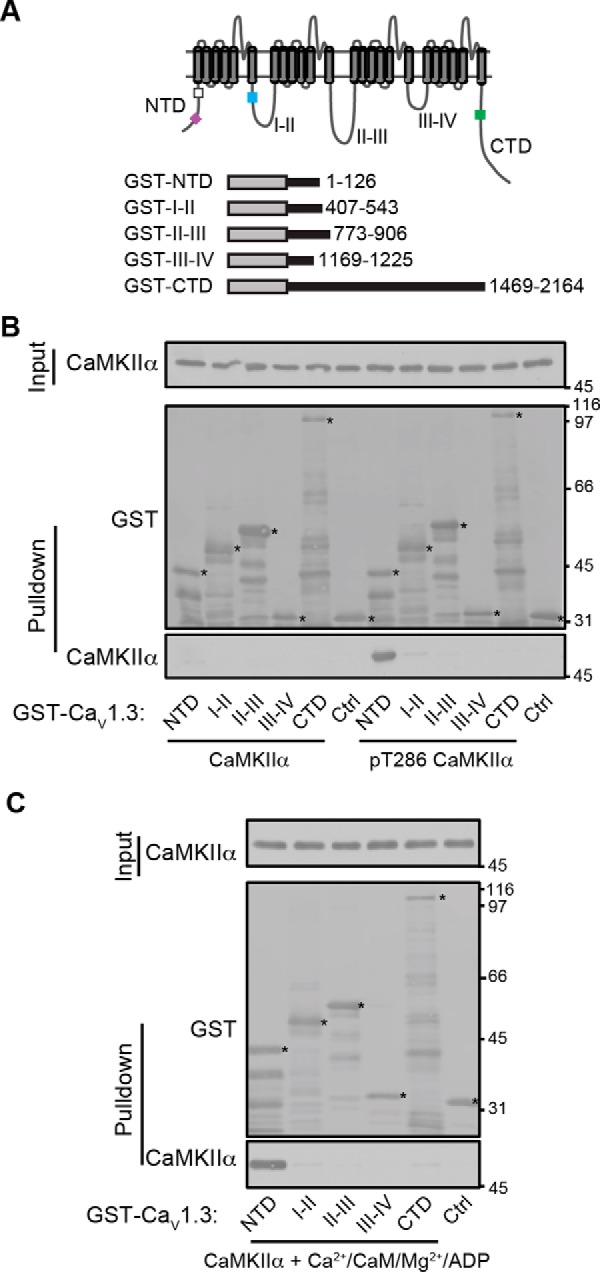
Activated CaMKII specifically binds to the LTCC NTD. A, domain structure of LTCCs and GST fusion proteins used here. Rectangular boxes in the intracellular domains indicate approximate positions of previously reported calmodulin (NSCaTE (24)) (purple box)-binding and CaMKII (20) (white box)-binding domains in the NTD, the α subunit interaction domain (AID, for β subunit interaction) in the I/II linker (50) (blue box), and overlapping calmodulin- and CaMKII-binding sites in the CTD (17) (green box). B, glutathione-agarose co-sedimentation assays show that there is no reliably detectable interaction of inactive (non-autophosphorylated) conformations of CaMKIIα with any of the CaV1.3 intracellular domains but that activated (pre-autophosphorylated) CaMKIIα specifically binds to the NTD. C, activation of CaMKIIα by binding of Ca2+/calmodulin and Mg-ADP is sufficient for interaction with the CaV1.3 NTD. The immunoblots shown are representative of three independent experiments.
CaMKII specifically binds to the LTCC NTDs
To investigate the specificity of this novel CaMKII interaction, we compared the amino acid sequences of NTDs from all 10 human VGCC α1 subunits. The NTDs are quite divergent in the initial membrane-distal sections but become more conserved in the membrane-proximal region (Fig. 2A). A similar conservation pattern holds true for mouse and rat VGCCs. To test binding specificity, we expressed and purified GST-tagged NTDs from CaV1.2, CaV2.2, and CaV3.2. As noted above, preactivated purified CaMKII robustly interacts with the CaV1.3 NTD, and there was a slightly weaker interaction with the CaV1.2 NTD. However, interaction of preactivated CaMKII with the CaV2.2 and CaV3.2 NTDs was barely detected above the GST negative control (Fig. 2, B and C). These data show that activated CaMKII selectively interacts with NTDs of the LTCCs.
Figure 2.

CaMKII specifically binds to LTCC NTDs. A, alignment of membrane-proximal regions of the NTDs from all human VGCCs. Ca2+ channel NTDs are more conserved in membrane-proximal regions but become more divergent in the distal regions. B, representative glutathione-agarose co-sedimentation assay comparing the binding of activated CaMKII to GST-NTDs from CaV1, CaV2, and CaV3 Ca2+ channels. C, quantitation of 2–3 independent experiments similar to those shown in B. The CaV1.3 NTD shows the strongest binding to CaMKIIα, followed by CaV1.2, whereas interactions with CaV2.2 and CaV3.2 are barely detected. All values were normalized to CaV1.3 NTD pulldown. Error bars, S.E.
Molecular determinants for CaV1.3 NTD interaction with CaMKII
Previous studies indicate that the CaV1.2 and CaV1.3 NTDs contain conserved binding sites for calmodulin (residues Ser52–Lys64 in CaV1.3), termed NSCaTE (24, 25), and for CaMKII (Lys110–Trp123 in CaV1.2) (20) (Fig. 3A). To investigate the potential roles of these domains in the CaMKII binding detected here, we mapped the site of direct CaMKII interaction in the CaV1.3 NTD. There was no detectable interaction between preactivated CaMKII and the membrane-distal fragment (NT-A; amino acids 1–68) containing the NSCaTE domain (Fig. 3B), but the membrane-proximal fragment (NT-B; amino acids 69–126) robustly interacts with preactivated CaMKII. Further dissection of NT-B revealed that preactivated CaMKII interacts with a GST-tagged fragment containing residues 69–93 (NT-B1), but not with two fragments containing more membrane-proximal residues 94–110 or 111–126 (NT-B2 and NT-B3; Fig. 3C).
Figure 3.
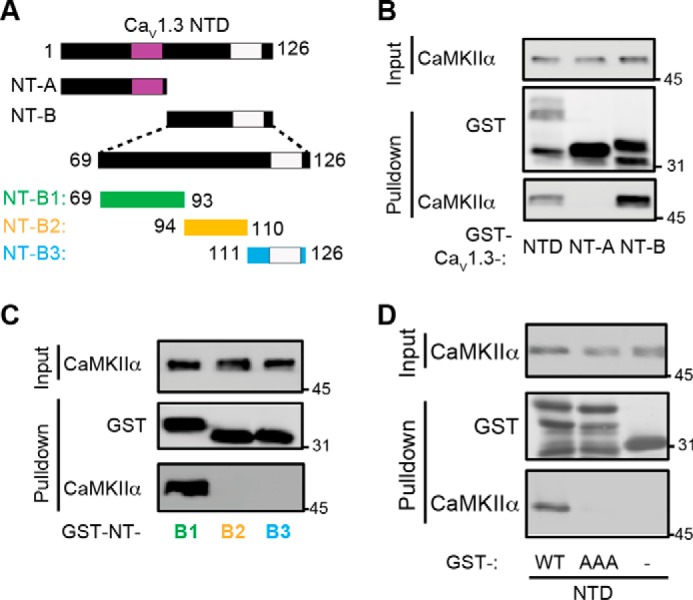
Characterization of the CaV1.3 NTD CaMKII-binding domain. A, truncations used to map the CaMKII interaction site in the CaV1.3 NTD. Purple and white rectangles indicate approximate positions of previously defined NSCaTE calmodulin-binding and CaMKII-binding domains, respectively (see legend to Fig. 1 and “Results”). B, glutathione-agarose co-sedimentation assay comparing binding of activated CaMKIIα to the full-length CaV1.3 NTD, the membrane-distal part (NT-A), and the membrane-proximal part (NT-B). C, analysis of further NTD truncations reveals that the NT-B1 region (residues 69–93) is sufficient for binding of activated CaMKIIα. D, mutation of amino acids 83RKR85 to AAA within the full-length CaV1.3 NTD blocks CaV1.3–CaMKIIα interaction. These immunoblots are representative of at least three independent replicates.
The amino acid sequence of CaV1.3 residues 69–93 shares little identifiable similarity with known CaMKII-binding domains in other proteins (16, 21, 26). However, we identified three basic amino acids in CaV1.3 (Arg83-Lys84-Arg85) that are largely conserved in NTDs of CaV1.2 and other LTCC α1 subunits, but not in the NTDs of CaV2.2 or CaV3.2 (which do not bind CaMKII) or of other α1 subunits (Fig. 2A). Replacement of this RKR motif with three alanines in the CaV1.3 NTD almost completely abrogated binding of preactivated CaMKII (Fig. 3D). These data identify three amino acids in the CaV1.3 NTD that are required for strong and direct in vitro interactions with preactivated CaMKII.
Preferential interactions of preactivated CaMKII with several other CaMKII-associated proteins (CaMKAPs) are mediated by the catalytic domain. Therefore, to identify CaMKIIα residues critical for binding to the CaV1.3 NTD, we screened previously characterized as well as novel CaMKII mutations in the catalytic domain (Fig. 4A) using a fluorescence-based 96-well plate binding assay (see “Experimental procedures”). An I205K mutation, previously shown to disrupt binding to GluN2B and the densin-IN domain (26, 27), also reduced binding to the CaV1.3 NTD by ∼80% (Fig. 4B). We identified two additional CaMKII mutations (V102E and E109K) that also significantly interfere with binding to the CaV1.3 NTD (Fig. 4B), whereas another mutation (Y210E) had no significant impact. Strikingly, the CaMKIIα-V102E mutation had no significant effect on binding to the β2a subunit of VGCCs, to the densin-IN or -CTD domains (Fig. 4C), or to GluN2B (not shown). In combination, these data suggest that the mechanism underlying binding of activated conformations of CaMKII to the CaV1.3 NTD is partially distinct from the mechanisms for binding to other known CaMKAPs.
Figure 4.
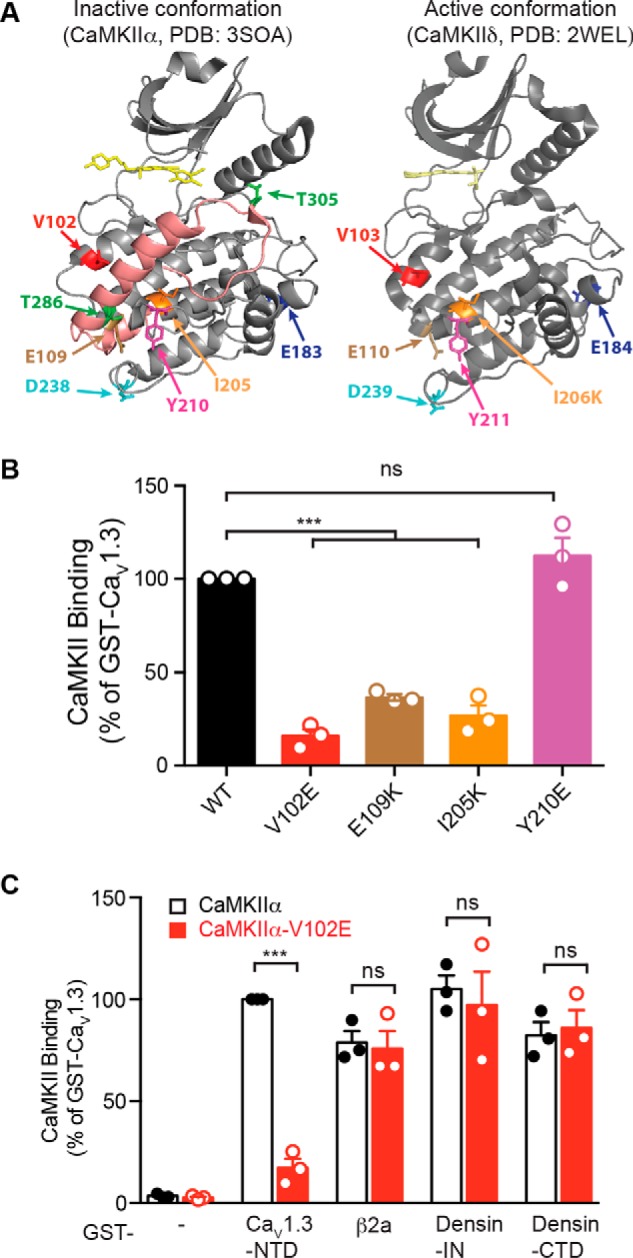
Identification of a CaMKII mutation that specifically disrupts binding to the CaV1.3 NTD. A, CaMKII structures. Left, a single CaMKIIα subunit in an inactive (autoinhibited) conformation with an inhibitor (Bosutinib, yellow) bound in the nucleotide binding site (PDB entry 3SOA (51)). Right, a single CaMKIIδ subunit in an activated conformation (displaced regulatory domain) with a bound inhibitor (SU6656, yellow) (PDB entry 2WEL (52)). The catalytic and regulatory domains are shown in gray and pink, respectively. For clarity of presentation, C-terminal holoenzyme association domains are not shown, and the displaced regulatory domain with bound Ca2+/calmodulin is not shown in PDB entry 2WEL. Thr286 and Thr305 (green) are two regulatory autophosphorylation sites. Mutation of Ile205/206 (orange) to Lys disrupts CaMKII interaction with GluN2B and densin-IN (22, 26), whereas mutation of Asp238/239 (cyan) to Arg disrupts GluN2B binding but spares interactions with densin-IN (22). A naturally occurring de novo Glu183 (purple) to Val mutation in CaMKIIα is linked to autism spectrum disorder and disrupts CaMKII interaction with multiple CaMKAPs (48, 53). B, a 96-well glutathione plate assay to screen activated mApple-tagged CaMKIIα mutants for interactions with GST-tagged CaV1.3 NTD. C, binding of activated mApple-tagged WT and V102E-CaMKIIα to multiple GST-CaMKAP proteins in the 96-well plate assay. A V102E mutation selectively disrupts CaMKIIα binding to the CaV1.3 NTD; Val102/103 is highlighted in red in A. Data from three independent experiments were analyzed by one-way ANOVA (for B) and two-way ANOVA followed by Sidak's multiple-comparison test (for C), respectively. ***, p < 0.001; ns, not significant (p > 0.05). Error bars, S.E.
The CaV1.3 NTD is important for CaMKII association with LTCC complexes
To begin to address the importance of the CaV1.3 NTD in CaMKII targeting to LTCC complexes, we first directly compared CaMKII binding to GST fusion proteins containing the CaV1.3 NT-B1 fragment or previously defined minimal CaMKII-binding domains in the VGCC β2 subunit (residues 485–505) (16) and a CaV1.2 CTD fragment (residues 1639–1660) that is fully conserved in CaV1.3 (17). Although we did not detect CaMKII binding to the full-length CaV1.3 CTD (Fig. 1, B and C), we rationalized that the full-length CTD may adopt a conformation that prevents CaMKII interaction with this previously defined domain, perhaps due to binding of a C-terminal modulatory domain to the calmodulin-binding IQ domain (28). Similar levels of preactivated CaMKII bound to the CaV1.3 NT-B1 and β2 (residues 485–505) fragments, but we could not detect an interaction with the CaV1.3 CTD (residues 1639–1660) fragment under these conditions (Fig. 5A). Nevertheless, these data indicate that CaMKII can directly interact with multiple components of native LTCC complexes.
Figure 5.

The NTD is important for CaMKII association with LTCC complexes. A, preactivated CaMKIIα robustly interacts with the minimal CaMKII-binding sites from the CaV1.3 NTD and the β2 auxiliary subunit, but not with a previously reported minimal CaMKII-binding site in the CaV1.2 CTD that is identical in CaV1.3. B, a schematic diagram showing the structure of chimeric CaVx.x NTD-CaV1.3 channels in which the CaV1.3 NTD was substituted by NTDs from CaV2.2 or CaV3.2. C, equal aliquots of lysates from cells expressing CaMKIIα with WT or NTD chimeric HA-tagged CaV1.3s were immunoprecipitated using anti-HA antibodies without (EDTA) or with the addition of excess Ca2+/calmodulin/Mg2+-ATP. C2 plots levels of immunoprecipitated HA-CaV1.3 proteins (black) and CaMKII (purple) in the presence of Ca2+/calmodulin/Mg2+/ATP normalized to levels isolated in the presence of EDTA in each experiment. C3 compares levels of immunoprecipitated CaMKIIα normalized to immunoprecipitated HA proteins in the presence of EDTA and Ca2+/calmodulin/Mg2+-ATP. D, similar analysis of the co-immunoprecipitation of WT or V102E-CaMKIIα with WT, Δ69–93, or RKR-AAA HA-CaV1.3 in the presence of EDTA or Ca2+/calmodulin/Mg2+/ATP. Levels of immunoprecipitated HA-CaV1.3 and CaMKIIα are compared in D2, and normalized CaMKIIα/HA-CaV1.3 ratios are shown in D3. Data are from 3–4 independent experiments and analyzed by two-way ANOVA followed by Sidak's multiple-comparison test. *, p < 0.05; ***, p < 0.001; ns, not significant (p > 0.05). Error bars, S.E.
The most abundant VGCC auxiliary β subunit in the brain appears to be β3 (29). Therefore, we investigated whether the NTD is important for CaMKII targeting to CaV1.3 LTCC complexes containing the β3 subunit. CaMKIIα, β3, and the α2δ subunit were co-expressed in HEK293T cells with the HA-tagged WT CaV1.3 α1 subunit or with chimeric α1 subunits in which the CaV1.3 NTD was replaced with NTDs from either CaV2.2 or CaV3.2 NTD (Fig. 5B). Antibodies to the HA tag were then used to immunoprecipitate α1 subunits from aliquots of the same cell lysate in the presence of EDTA or following the addition of excess Ca2+/CaM and Mg2+-ATP to activate CaMKIIα (see “Experimental procedures”). Immunoblotting revealed that the HA immune complexes isolated in the presence of Ca2+/calmodulin/Mg2+-ATP contained significantly more HA-CaV1.3 α1 subunit and CaMKIIα, relative to immune complexes isolated in parallel in the presence of excess EDTA. Similar data were obtained from two independent sets of experiments (Fig. 5, C and D). Combining the quantitative analysis of these two data sets revealed that Ca2+/CaM/Mg2+-ATP increased the levels of HA-CaV1.3 and CaMKIIα by 2.5 ± 0.3- and 12.8 ± 2.6-fold, respectively (mean ± S.E., n = 7; p < 0.01 for both, one-sample t test compared with a theoretical value of 1.00 indicating no change). Therefore, the addition of Ca2+/calmodulin/Mg2+/ATP significantly increases the ratio of CaMKII to HA-CaV1.3 in the immune complexes by 6.7 ± 2.3-fold (n = 7; p < 0.001).
To explore the mechanism underlying these changes, we first found that the addition of Ca2+/calmodulin/Mg2+/ATP failed to increase the levels of immunoprecipitated HA-CaV1.3 in the absence of co-expressed CaMKIIα (data not shown). Moreover, replacement of the entire CaV1.3 NTD with corresponding NTDs from CaV2.2 or CaV3.2 (Fig. 5B) abrogated Ca2+/calmodulin/Mg2+/ATP-induced increases in the levels of both HA-tagged channels and CaMKIIα in the HA immune complexes, as well as in the CaMKIIα/HA-CaV1.3-chimera ratio (Fig. 5C). Similarly, significant Ca2+/calmodulin/Mg2+/ATP-induced increases in levels of CaMKII and HA-CaV1.3 and in the CaMKII/HA-CaV1.3 ratio in HA immune complexes were prevented by the deletion of residues 69–93 or by mutation of 83RKR85 to AAA within the CaV1.3 NTD (Fig. 5D). Finally, the CaMKIIα-V102E mutation also prevented Ca2+/calmodulin/Mg2+/ATP-induced increases in levels of HA-CaV1.3 and CaMKIIα and in the CaMKIIα/HA-CaV1.3 ratio in HA immune complexes (Fig. 5D). Because all of the molecular changes tested here disrupt the CaMKII–NTD interaction in vitro, these data collectively indicate that CaMKIIα interaction with the NTD is required for activity-dependent association of CaMKIIα with intact HA-CaV1.3 complexes as well as for a more modest increase in the levels of immunoprecipitated HA-CaV1.3.
The CaV1.3 NTD Δ69–93 deletion does not affect Ca2+ influx
To assess the impact of the NTD CaMKII-binding site on LTCC activity, we compared Ca2+ influx via WT and Δ69–93 CaV1.3 LTCCs containing β3 and α2δ subunits in HEK293T cells. Whole-cell currents were elicited by step depolarizations from the holding voltage of −70 mV to a series of test voltages, which were held for 50 ms (Fig. 6A). Analysis of the current–voltage relationships indicated that deletion of amino acids 69–93 had no significant effect on voltage-dependent activation (WT: V½ = −11.4 ± 3.2 mV, k = 5.3 ± 2.8 mV, n = 13; Δ69–93: V½ = −9.2 ± 5.2 mV, k = 8.8 ± 4.7 mV, n = 15; p > 0.05 for both, two-tailed unpaired Student's t test) or on the maximal current density (10.9 ± 2.6 pA/picofarads for WT versus 8.6 ± 2.5 pA/picofarads for Δ69–93, p = 0.49, two-tailed unpaired Student's t test; Fig. 6, A–C). Furthermore, there was no difference in residual current measured 30 ms after depolarization to 0 mV, indicating that fast inactivation kinetics also were unaffected (Fig. 6D). These data suggest that the deletion of NTD residues 69–93 has no significant effect on the properties of CaV1.3/β3/α2δ LTCCs.
Figure 6.
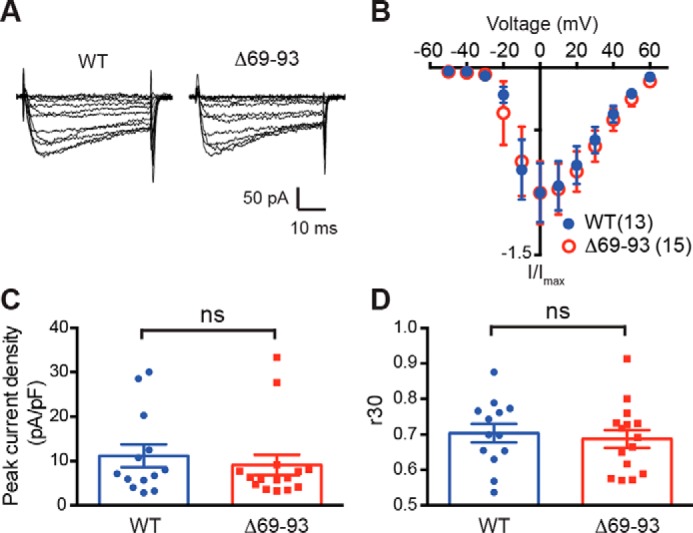
Deletion of residues 69–93 from the CaV1.3 NTD does not affect Ca2+ influx via CaV1.3 LTCCs. A, representative Ca2+ currents elicited by step depolarizations (50 ms) to various voltages for CaV1.3-WT (left) and CaV1.3-Δ69–93 LTCCs. Scale bars, 10 ms (horizontal) and 50 pA (vertical), respectively. B, no significant difference in current-voltage (I-V) relationships for CaV1.3-WT and CaV1.3-Δ69–93 LTCCs (p > 0.05, two-way ANOVA followed by Sidak's multiple-comparison test). C, no significant difference in peak current densities of CaV1.3-WT and CaV1.3-Δ69–93 LTCCs. D, no significant difference in fast inactivation of CaV1.3-WT and CaV1.3-Δ69–93 LTCCs, based on the fraction of residual current measured 30 ms after depolarization from −70 to 0 mV. Data were collected from five independent transfections, n = 13 for WT and n = 15 for Δ69–93. Data were analyzed by two-tailed unpaired Student's t test; ns, not significant (p > 0.05). Error bars, S.E.
CaV1.3 NTD is required for LTCC- and CaMKII-mediated nuclear signaling
We then tested whether the NTD–CaMKII interaction is important for LTCC-mediated downstream signaling to increase Ser133 phosphorylation of the CREB transcription factor in primary cultures of hippocampal neurons. We first established a stimulation paradigm to induce LTCC-dependent increases of Ca2+ concentrations in neurons based on Fura-2–based Ca2+ imaging. Neurons were preincubated in 5 mm K+ Tyrode's solution containing APV and CNQX to block the activation of NMDA- and AMPA-type glutamate receptors and with tetrodotoxin (TTX) to inhibit voltage-dependent sodium channels. Neuronal depolarization by replacing the solution with 40 mm K+ Tyrode's solution in the presence of APV, CNQX, and TTX induced a significant increase in intracellular (somatic) Ca2+, which is reduced by ∼80% in the presence of 10 μm nimodipine, a highly selective LTCC antagonist (Fig. 7A). In parallel, we showed that depolarization with 40 mm KCl in the presence of APV, CNQX, and TTX for 90 s induces a robust increase of nuclear staining using a phospho-Ser133–specific CREB antibody (pCREB staining) that can be completely blocked by 10 μm nimodipine (Fig. 7, B and C), consistent with previous findings (15, 30, 31).
Figure 7.
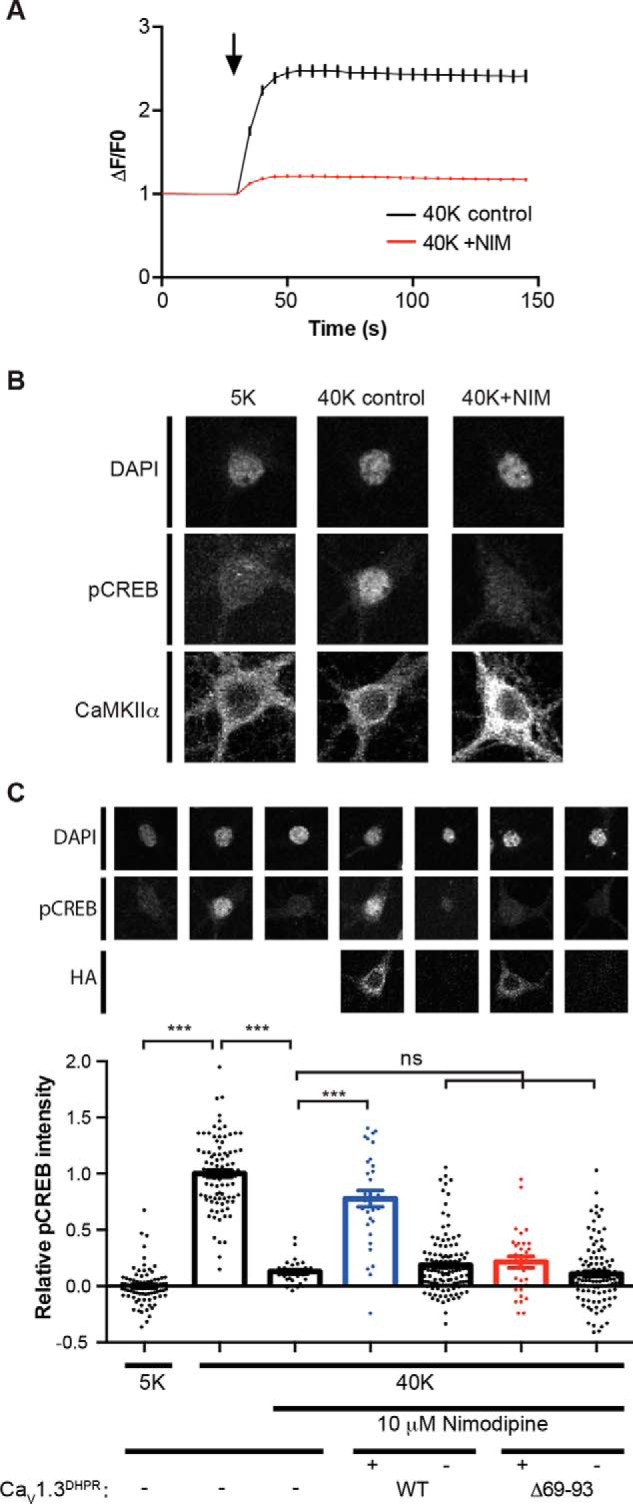
Role of the LTCC NTD in high K+-induced CREB Ser133 phosphorylation. A, Ca2+ imaging showing that nimodipine largely prevents the high K+-induced increase of somatic Ca2+. Neurons were incubated with Tyrode's solution containing 5 mm KCl (5K) for 30 s and switched to Tyrode's solution containing 40 mm KCl for 2 min in the absence (40K control, n = 193) or presence (40K + NIM, n = 215) of 10 μm nimodipine. The black arrow indicates the buffer switch, and data were plotted as mean ± S.E. A total of five dishes from two independent cultures were analyzed per group. B, depolarization of cultured hippocampal neurons induced LTCC-dependent Ser133 phosphorylation of CREB. In the columns from left to right, neurons were incubated with 5K Tyrode's solution and then switched to 5K, 40K control, or 40K + NIM Tyrode's solution, respectively, for 90 s. Neurons were then fixed and stained for DAPI, CREB Ser133 phosphorylation (pCREB), and CaMKIIα. Incubation with 40 mm KCl induced an increase of pCREB that was blocked by the LTCC antagonist nimodipine. C, deletion of the CaMKII-binding domain in the CaV1.3 NTD disrupts nuclear signaling. Expression of a nimodipine-resistant CaV1.3-T1033Y mutant rescues the nimodipine blockade of pCREB induction by 40 mm KCl. However, deletion of the CaMKII-binding domain (Δ69–93) prevents the rescue of pCREB signaling by CaV1.3-T1033Y. Each data point represents analysis of a single cell collected from 3–4 independent neuronal cultures/transfections. Pooled data were analyzed by one-way ANOVA followed by Tukey's multiple-comparison test. ***, p < 0.001; ns, not significant (p > 0.05). All confocal images show a 40 × 40-μm area. Error bars, S.E.
We then used a pharmacological knock-in approach (32) to compare the E-T coupling efficiency of wild-type CaV1.3 with CaV1.3-Δ69–93, which compromises CaMKII binding. We expressed an HA-tagged nimodipine-resistant CaV1.3 mutant (T1033Y, designated as CaV1.3DHPR) to allow for activation of exogenous CaV1.3 channels while using nimodipine to block all of the endogenous CaV1.2 and CaV1.3 LTCCs. The expression of CaV1.3DHPR almost completely rescued the increase of pCREB staining in the presence of nimodipine (transfected neurons were identified by staining for the HA epitope), but this rescue of pCREB signaling was disrupted by the deletion of NTD residues 69–93 from CaV1.3DHPR (Fig. 7C), which prevents CaMKII binding.
To complement these findings, we examined the impact of the CaMKIIα-V102E mutation using an shRNA knockdown and rescue strategy that has been used previously to demonstrate a key role for CaMKIIα/β (15). We first verified that shRNA knockdown of CaMKIIα and CaMKIIβ expression had no significant effect on LTCC-dependent somatic Ca2+ responses to stimulation with 40 mm K+ Tyrode's solution. Moreover, re-expression of shRNA-resistant wild-type CaMKIIα (CaMKIIαR-WT) or CaMKIIαR-V102E also did not alter the amplitude or kinetics of Ca2+ responses (Fig. 8A). However, CaMKIIα/β knockdown significantly attenuated the increase of pCREB staining induced by 40 mm KCl, and the effect of this knockdown was largely rescued by re-expression of CaMKIIαR-WT but not by CaMKIIαR-K42R (a kinase-dead mutant). Notably, CaMKIIαR-V102E, which cannot bind to the CaV1.3 NTD but retains full kinase activity (data not shown), was also unable to rescue the pCREB staining (Fig. 8B). In contrast, pilot experiments using two independent neuronal cultures found that increases of nuclear pCREB staining elicited using a longer (5 min) and more robust (identical except lacking CNQX) stimulation paradigm were partially independent of LTCCs (5 mm KCl control: 0.00 ± 0.02, n = 56 cells; 40 mm KCl: 1.00 ± 0.05, n = 68; 40 mm KCl plus 10 μm nimodipine: 0.51 ± 0.05, n = 71; mean ± S.E., p < 0.01, one-way ANOVA followed by Tukey's multiple-comparison test) and were independent of CaMKIIα/β (control/untransfected cells: 1.00 ± 0.10, n = 50; CaMKIIα/β shRNA: 0.74 ± 0.27, n = 15; mean ± S.E., p = 0.25, unpaired Student's t test). Taken together, data obtained by expressing nimodipine-resistant channels and using CaMKII knockdown/rescue approaches suggest that the NTD-CaMKII interaction is essential for LTCC-mediated E-T coupling.
Figure 8.
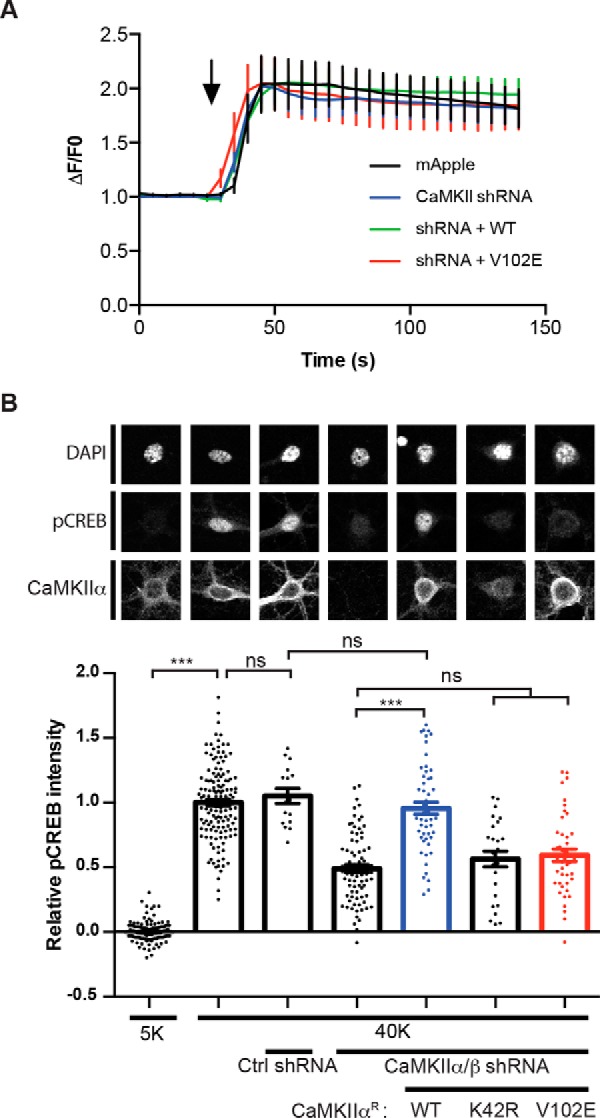
CaMKII-binding to the CaV1.3 NTD is required for high K+-induced CREB Ser133 phosphorylation. A, CaMKIIα/β knockdown or re-expression/rescue has no effect on high K+-induced increases of somatic Ca2+. Cultured hippocampal neurons were transfected with mApple only (n = 17), mApple with CaMKIIα/β shRNA (n = 23), mApple/CaMKIIα/β shRNA with shRNA-resistant CaMKIIαR-WT (n = 26), or CaMKIIαR-V102E cDNA (n = 17), respectively. Transfected neurons were then monitored for Ca2+ influx in response to 40 mm K+-induced depolarization (black arrow; see Fig. 7A). B, the CaMKIIα-V102E mutant does not support nuclear signaling. Expression of CaMKIIα/CaMKIIβ shRNAs significantly reduces nuclear CREB phosphorylation following 40 mm KCl treatment. This reduction in CREB phosphorylation is rescued by co-expression of shRNA-resistant CaMKIIαR-WT, but not CaMKIIαR-K42R (kinase-dead) or CaMKIIαR-V102E (deficient in CaV1.3 NTD binding). Each data point represents analysis of a single cell collected from 3–4 independent neuronal cultures/transfections. Pooled data were analyzed by one-way ANOVA followed by Tukey's multiple-comparison test. ***, p < 0.001; ns, not significant (p > 0.05). All confocal images show a 40 × 40-μm area. Error bars, S.E.
Discussion
Ca2+ influx into neurons via ligand- and voltage-gated Ca2+ channels plays a key role in a variety of processes, including synaptic plasticity and long-term memory formation. Elucidation of the molecular mechanisms that underlie the specificity and efficiency of signaling downstream of the different channels is critical to understanding their biological roles. Our data identify a novel CaMKII binding site in the NTD of LTCCs that is important for the coupling of LTCCs to a nuclear response. The specificity of this interaction for CaV1 LTCCs over CaV2 and CaV3 VGCCs presumably contributes to their preferential role in E-T coupling (30).
We systematically compared CaMKII binding to all intracellular domains of the CaV1.3 α1 subunit, expressed as GST fusion proteins. Activated CaMKII bound with high specificity to the CaV1.3 NTD. This interaction was conserved with CaV1.2 NTDs, but not with NTDs from CaV2.2 or CaV3.2 VGCCs. The fact that we failed to detect CaMKII binding to the full-length CaV1.3 CTD or to the previously defined minimal CTD CaMKII-binding domain that is 100% conserved between CaV1.2 and CaV1.3 was somewhat surprising, because it was previously reported that CaMKII binding to the CTD is important for Ca2+-dependent facilitation (17). Another prior study detected a putative CaMKII-binding site in the membrane-proximal region of the CaV1.2 NTD (residues 110–123) that is conserved in CaV1.3 based on co-immunoprecipitations from cell lysates and found that mutation of this motif disrupted membrane trafficking yet enhanced Ca2+ influx (20). However, this study provided no evidence for a direct interaction of CaMKII with this domain (e.g. with purified proteins). Moreover, neither of these prior studies established that the domains identified are important for CaMKII association with intact LTCC complexes. Reasons for the discrepancies between the present studies and these prior studies are unclear, but it is possible that the conditions used here favor detection of more specific, high-affinity, direct interactions.
Comparison of NTD with previously identified CaMKII-binding domains
Like several other CaMKAPs, the LTCC NTD preferentially interacts with activated CaMKII. However, the molecular bases for these interactions appear to be distinct. CaMKII-binding domains in the NMDA receptor GluN2B subunit and β1/β2 subunits of VGCCs share sequence similarity with the CaMKII autoregulatory domain, including the presence of a (auto)phosphorylation site (16). However, these domains share no sequence similarity with the internal CaMKII-binding domain in densin, which resembles CaMKIIN, a naturally occurring CaMKII inhibitor protein (22). Moreover, neither of these classes of CaMKII-binding domain shares noticeable sequence similarity with CaMKII-binding domains in the CaV1.3 and CaV1.2 NTDs identified here. Furthermore, we identified a V102E mutation in the CaMKIIα catalytic domain that substantially reduced interactions with the CaV1.3 NTD, without significantly affecting interactions with the β1/β2 subunits, densin or GluN2B. In contrast, an I205K mutation in the CaMKIIα catalytic domain (I206K in CaMKIIβ, -γ, and -δ) previously shown to interfere with binding to GluN2B (26) and densin (22), also disrupts CaMKII binding to β1/β2 subunits and the CaV1.3 NTD (Fig. 4B). Taken together, despite some overlap in the CaMKII residues required for binding, our data suggest that the newly identified CaV1.3 NTD CaMKII-binding domain may represent a new class of CaMKAP.
Roles of the NTD and other CaMKAPs in LTCC complexes
It is well-established that CaMKII associates with LTCCs in cardiomyocytes and neurons, as revealed by co-immunoprecipitation and/or by co-localization (17, 18, 30), but the molecular basis for this interaction is unclear. In one series of studies, CaMKII was shown to bind directly to β1 and β2 LTCC auxiliary subunits, but not to β3 or β4 (16), and co-immunoprecipitated from brain extracts with β1 subunits, but not β4 subunits (18). Moreover, mutation of the CaMKII-binding domain in the β2 subunit reduces CaMKII co-immunoprecipitation with CaV1.2 channels in heterologous cells (18) as well as Ca2+-dependent facilitation of CaV1.2 (33). In contrast, another CaMKAP, the synaptic scaffolding protein densin, forms ternary complexes with CaMKII and CaV1.3 LTCCs in the brain and is necessary for CaMKII- and Ca2+-dependent facilitation of CaV1.3 (19). Here, we found that the α1 subunit NTD is important for CaMKII association with intact CaV1.3 LTCC complexes by co-immunoprecipitation of activated CaMKII with HA-tagged CaV1.3 LTCCs. Co-immunoprecipitation of CaMKII was substantially reduced by replacement of the CaV1.3 NTD with NTDs from CaV2.2 or CaV3.2, which do not significantly bind CaMKII in vitro. Similarly, co-immunoprecipitation was substantially reduced by either deletion of residues 69–93 or mutation of 83RKR85 to AAA in the CaV1.3 NTD or by the CaMKIIα-V102E mutation. Thus, the present findings demonstrate the importance of a direct CaMKII interaction with a novel CaMKII-binding domain in the CaV1.3 α1 subunit NTD, significantly extending our understanding of biochemical mechanisms involved in LTCC signaling.
It is important to note that our heterologous cell studies were conducted using HA-tagged CaV1.3 and the β3 auxiliary subunit, which does not directly interact with CaMKII (16). β3 is thought to be the most abundant in brain (29), but the other three β subunits also are expressed in neurons. It seems likely that the association of β subunit variants with the I-II linker domains of VGCC α1 subunits is determined in part by their relative expression levels. However, VGCCs may exhibit selectivity for the β subunits in cells, consistent with data showing that overexpressed β subunit variants are differentially localized in cultured hippocampal neurons (34). Nevertheless, we posit that neurons contain multiple subpopulations of CaV1.3 LTCC complexes associated with different β subunit variants. CaV1.3 LTCCs containing β3 or β4 may rely only on the NTD for CaMKII association, whereas those containing β1 or β2 have a second interaction site. Indeed, although this novel CaMKII-binding domain is highly conserved, CaV1.2 NTD binding to CaMKII is somewhat weaker than that of CaV1.3 NTD (Fig. 2C), and we previously reported that CaMKII association with CaV1.2 channel complexes depends in part on interaction with β2 subunits (18).
Neuronal Ca2+ channels are often part of larger complexes containing other proteins. For example, a canonical PDZ domain–binding motif at the C terminus of the CaV1.3 α1 subunit interacts with PDZ domains in synaptic scaffolding proteins like densin or Shank3 (19, 35). CaMKII interactions with such scaffolding proteins may represent an additional mechanism for targeting CaMKII to certain subpopulations of neuronal CaV1.3 LTCCs (in addition to α1 subunit NTDs and β1/2 subunits). Indeed, the key role for densin in targeting CaMKII to promote Ca2+-dependent facilitation of CaV1.3 LTCCs was noted above (19). Taken together, these observations indicate that several “flavors” of neuronal CaV1.3 LTCC complexes with distinct protein compositions may associate with CaMKII in different ways, perhaps conferring distinct roles for CaMKII in regulating LTCCs and/or downstream signaling.
Role of CaMKII binding to the CaV1.3 NTD in excitation–transcription coupling
As noted above, the precise regulation of CREB phosphorylation at Ser133 is critical for the regulation of gene expression during normal learning and memory consolidation. Although Ser133 phosphorylation may not be sufficient for gene expression under all conditions (36), it is frequently used as a readout for E-T coupling to CREB, as in the current studies. It is well-established that the selective activation of several different receptors and ion channels can engage diverse signal transduction pathways (e.g. cyclic AMP, Ca2+, and MAPKs) to increase neuronal CREB phosphorylation at Ser133. Moreover, distinct Ca2+-dependent pathways can be engaged to increase Ser133 phosphorylation, depending on the specific channel that generates the Ca2+ signal. Among the known mechanisms, selective LTCC activation is sufficient for immediate early gene expression in vivo (37) and for CREB Ser133 phosphorylation in cultured neurons (30, 38). The stimulation of CREB phosphorylation can be driven by increased nuclear Ca2+ concentrations, which can be induced using some stimulation paradigms (e.g. NMDA receptor activation) (39). However, the initiation of E-T coupling to CREB by moderate LTCC activation seems to be independent of increases in nuclear Ca2+ and only requires increased Ca2+ concentrations in the immediate vicinity of the channel itself (12, 30). Stronger, more prolonged, stimulation paradigms may overcome this requirement for Ca2+ signaling within the LTCC nanodomain by recruiting additional mechanisms. This may be evident in superior cervical ganglion neurons (30), where the global increase of Ca2+ in response to modest stimulation (40 mm KCl) involves similar contributions from CaV1 and CaV2 channels, but the resulting increase of CREB phosphorylation at Ser133 is preferentially coupled to CaV1 LTCCs, correlating with the co-localization of CaMKII with CaV1, but not CaV2, channels under these conditions. CaV2 channel-dependent increases of global Ca2+ in superior cervical ganglion neurons are shaped by mitochondria and the endoplasmic reticulum, but more robust stimulation induces CaV2-dependent increases of CREB phosphorylation by a mechanism that is only partially dependent on CaMKII (30). Sufficiently robust CaV2-dependent increases in global Ca2+ may also increase nuclear Ca2+, potentially contributing to the stimulation of CREB phosphorylation (39).
In this study, we focused on an E-T coupling paradigm that has been shown to depend on signaling within the LTCC nanodomain (12, 15). We found that mutations in the CaV1.3 NTD that disrupt CaMKII binding in vitro interfere with E-T coupling in cultured neurons. Similarly, mutation of the CaMKII catalytic domain to selectively disrupt binding to the NTD interferes with E-T coupling. Notably, although CaMKIIα/β knockdown significantly interfered with LTCC-dependent E-T coupling, there was no significant effect on the Ca2+ signal detected in the soma. Thus, the data presented herein show that a novel and unique CaMKII binding site in the N-terminal domain of CaV1.3 LTCCs plays an important role in E-T coupling, apparently by initiating a local mechanism within the LTCC nanodomain rather than modulating the global Ca2+ signal.
Our current data do not preclude additional roles for CaMKII binding to other components of LTCC complexes (see above) in E-T coupling. This may be particularly germane in light of a recent study indicating that Ca2+ influx drives activation and Thr286/287 autophosphorylation of CaMKIIα/β holoenzymes within the LTCC nanodomain, which in turn trans-autophosphorylate a CaMKIIγ holoenzyme at Thr287, trapping bound calmodulin to be shuttled to the nucleus (10). Given that trans-holoenzyme autophosphorylation is very inefficient in solution (40), it is tempting to speculate that this process is facilitated by simultaneous targeting of CaMKIIα/β and CaMKIIγ holoenzymes within the LTCC nanodomain. Our data indicate that CaMKIIα/β holoenzymes can dock to the NTD, and it is possible that a CaMKIIγ holoenzyme interacts with other components of the larger LTCC complex (see above). Interestingly, CREB phosphorylation was reported to be preferentially coupled to CaV1.3 over CaV1.2 LTCCs (31). Notably, Shank3 selectively associates with CaV1.3 LTCCs and has been shown to play an important role in E-T coupling (35). Moreover, Shank3 was identified as an abundant component of synaptic CaMKII complexes in a recent proteomics study (41). Multiple CaMKII docking sites in the LTCC complex also may be linked to voltage-dependent conformational changes that appear to be required to initiate E-T coupling (11). Such changes may be required to facilitate docking of CaMKII holenzyme(s) and appropriately position CaMKII relative to other components of the nanodomain. Additional studies are clearly required to explore these ideas and provide further insight into the molecular mechanisms underlying the initiation of E-T coupling.
Experimental procedures
Animals
Timed pregnant Sprague-Dawley rats were purchased from Charles River Laboratories. Embryonic day 18.5 pregnant rats were euthanized in a CO2 chamber before embryos were removed from the uterus. All animal experiments were approved by the Vanderbilt University institutional animal care and use committee and were carried out following the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
DNA constructs
Rat CaV1.3 complete coding sequence (GenBankTM accession number AF370010) was a gift from Dr. Diane Lipscombe (Brown University, Providence, RI). The intracellular domains contain NTD (Met1–Lys126), I-II (Gly407–Val543), II-III (Asp773–His906), III-IV (Gly1169–Ser1225), and CTD (Met1469–Leu2164). Rabbit CaV1.2 cDNA (a gift from Dr. William Thiel, GenBankTM accession number X15539) was used to amplify a fragment encoding the CaV1.2 CTD (Asp1507–Leu2171). DNAs encoding all other CaV1.2 intracellular domains were amplified from a rat CaV1.2 cDNA construct (a gift from Dr. Gerald W. Zamponi (University of Calgary), GenBankTM accession number NM_012517): NTD (Met1–Lys124), I-II (Ser405–Asn524), II-III (Gln754–Ile901), III-IV (Val1167–Tyr1220). DNAs encoding the CaV2.2 NTD (Met1–Pro95) and CaV3.2 NTD (Met1–Asp140) were amplified from a bovine cDNA construct (a gift from Dr. Aaron Fox (University of Chicago) and a human cDNA construct (a gift from Dr. Edward Perez-Reyes (University of Virginia, Charlottesville, VA), Addgene plasmid ID 45809), respectively. DNAs encoding rat CaV1.3 NTD fragments Met69–Leu93, Gln94–Ser110, and Leu111–Lys126 were also amplified. DNAs encoding previously defined minimal CaMKII-binding domains in the CaV1.2 CTD (Gly1639–Lys1660 (17)) and the rat Ca2+ channel β2a subunit (His485–Glu505) (16) were also generated. All cDNAs were inserted into pGEX-4T1 using traditional ligation or sequence- and ligation-independent cloning (42).
A plasmid encoding CaV1.3 with an N-terminal HA-tag (pCGNH-CaV1.3, for co-immunoprecipitation) was made by inserting rat CaV1.3 cDNA into pCGN vector (a gift from Dr. Winship Herr, Université de Lausanne, Switzerland, Addgene plasmid ID 53308). CaV1.3 chimeric constructs were made in the following way: pCGNH-CaV1.3 was used as a template to delete the N-terminal domain of CaV1.3, leaving the XbaI site intact (pCGNH-CaV1.3-ΔNTD); cDNAs encoding the CaV2.2 and CaV3.2 NTDs were then inserted into the XbaI site using sequence- and ligation-independent cloning. Plasmid encoding CaV1.3 with an extracellular HA tag was generated by first removing the sequence encoding the N-terminal HA in pCGNH-CaV1.3 and then inserting the sequence encoding an HA tag flanked by flexible linkers on both ends between Gln693 and Lys694 (pCGN0-CaV1.3-sHA). The inserted amino acid sequence is TRHYPYDVPDYAVTFDEMQ, where the HA sequence is in boldface type (43). The nimodipine-resistant CaV1.3 was then generated by mutagenesis of pCGN0-CaV1.3-sHA to generate a T1033Y mutant (32, 44). The region encoding Met69–Leu93 was deleted from pCGN0-CaV1.3-sHA plasmid to remove the CaMKII-binding domain. Site-directed mutagenesis, epitope insertions, and all deletions were done following the one-step mutagenesis protocol described by Liu and Naismith (45).
CaMKII shRNA constructs for pCREB staining were expressed with GFP using a pLL3.7 plasmid (a gift from the Luk Van Parijs laboratory, Massachusetts Institute of Technology, Cambridge, MA) that was modified to replace the CMV promoter with a 0.4-kb fragment of the mouse CaMKIIα promoter (designated as pLLCK) that is primarily active only in excitatory neurons (46). The shRNA sequences were designed following Ma et al. (10). The shRNA-targeted sequence in the mouse CaMKIIα cDNA contains two mismatches from the corresponding rat sequence, rendering it resistant to the shRNA. Knockdown and shRNA resistance were confirmed by Western blotting and immunostaining. All constructs were confirmed by DNA sequencing.
Recombinant mouse CaMKIIα and GST-tagged protein purification
Expression and purification of recombinant mouse CaMKIIαhas been described previously (47). pGEX-4T1 plasmids were transformed into BL21(DE3) bacteria cells to express GST-tagged proteins. Cells were grown in LB medium at 37 °C to reach an OD of ∼0.6. Isopropyl 1-thio-β-d-galactopyranoside (0.2 mm) was then added to induce the protein expression at room temperature for 2 h. We found that the CaV1.2 and CaV1.3 full-length C-terminal domain fragments do nottk;1 express well in BL21(DE3) cells. We identified several rare codons in the cDNAs encoding both CTDs and found that their expression was substantially improved in Rosetta 2(DE3) BL21 cells engineered to contain rare tRNAs (EMD Millipore, catalog no. 71400). Expressed proteins were purified using Pierce glutathione-agarose beads (catalog no. 16101) following the manufacturer's instructions. Eluted proteins were then dialyzed in 10 mm HEPES, pH 7.5, 25 μm PMSF, 62.5 μm benzamidine, 62.5 μm EDTA, 0.1% Triton X-100 overnight with one buffer change.
CaMKII autophosphorylation and GST pulldown
Purified mouse CaMKIIα was incubated with 50 mm HEPES, pH 7.5, 10 mm Mg(CH3COO)2, 0.5 mm CaCl2, 2.5 μm calmodulin, 40 μm ATP on ice for 90 s before the addition of EDTA (20 mm final) to terminate phosphorylation by chelation of Mg2+ and Ca2+. The reaction was then diluted 10-fold using 1× GST pulldown buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% (v/v) Triton X-100). A final protein concentration of 125 nm was used for both CaMKIIα and GST-tagged proteins. An aliquot (5%) of each incubation was saved as input followed by the addition of 5 μl of prewashed glutathione magnetic beads (Pierce, catalog no. 88821, 25% (v/v)). After incubating at 4 °C for 1 h, beads were separated magnetically and washed three times with GST pulldown buffer. GST protein complexes were eluted by incubation with 40 μl of 20 mm glutathione (pH 8.0) in GST pulldown buffer at 4 °C for 10 min.
Cell culture, transfection, and co-immunoprecipitation
Mouse CaMKIIα pcDNA was co-transfected with pcDNA empty vector (control) or pCGNH-CaV1.3, β3, and α2-δ subunits. A total of 10 μg of DNA were transfected into one 6-cm dish of HEK293T cells. Amounts of DNA transfected were as follows: CaMKIIα, 2 μg; pCGNH-CaV1.3 (WT, mutant or chimeras), 4 μg; pcDNA-β3, 2 μg; pcDNA-α2δ, 2 μg. After 48 h of transfection, cells were lysed in 50 mm Tris-HCl, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1 mm DTT, 1% Nonidet P-40 (v/v), 1 mm Microcystin-LR, and protease inhibitor mixtures. Cell lysates were cleared by low-speed centrifugation (500 × g), and supernatant was used for subsequent co-immunoprecipitation. Where indicated, lysates were supplemented with 2 mm CaCl2, 2 mm MgCl2, 1 mm ATP, and 1 μm calmodulin (final concentrations) to activate CaMKII before immunoprecipitation. Cell lysates were incubated at 4 °C for 1 h with rabbit anti-HA (Santa Cruz Biotechnology, Inc., catalog no. sc805; 1:500) and 10 μl of prewashed Dynabeads Protein A (Thermo Fisher Scientific, catalog no. 10001D; 25% (v/v)). The beads were isolated magnetically and washed three times using lysis buffer before eluting proteins using 1× Laemmli sample buffer.
Fluorescent plate-binding assay
The fluorescent plate-binding assay has been described previously (48). Briefly, GST fusion proteins (200 pmol in 0.1 ml of plate binding buffer: 50 mm Tris-HCl, pH 7.5, 200 mm NaCl, 0.1 mm EDTA, 5 mm 2-mercaptoethanol, 0.1% (v/v) Tween 20, 5 mg/ml bovine serum albumin) were added to the wells of glutathione-coated 96-well plates (Thermo Fisher Scientific, catalog no. 15340). After incubation overnight at 4 °C, the wells were washed with plate-binding buffer before adding lysates of transfected HEK293FT cells (100 μl) containing ∼150 nm mApple-CaMKIIα WT or mutant proteins and supplemented with 2.5 mm CaCl2, 1 μm calmodulin, 10 mm MgCl2, and 400 μm ADP. After an additional 2-h incubation at 4 °C, wells were washed in wash buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.5% (v/v) Triton X-100, and 2.4 mm CaCl2) two times, and bound mApple-CaMKIIα was detected using a fluorescent plate reader at 592 nm.
Electrophysiology
HEK293T cells in 35-mm dishes were transfected with 2 μg of CaV1.3 WT or Δ69–93 pcDNAs together with 1 μg of β3, 1 μg of α2δ, and 0.05 μg of enhanced green fluorescent protein pcDNAs. Cells were split into new dishes 36 h after transfection, and whole-cell Ca2+ currents were recorded at room temperature 48 h after transfection. Data were collected through an Axopatch 200B amplifier and pCLAMP10 software (Molecular Devices). Pipette resistance was 4–6 megaohms when loaded with the intracellular solution and immersed in the extracellular solution. Series resistance and membrane capacitance were compensated up to 80%. The intracellular solution contained 132 mm CsCl, 10 mm tetraethylammonium chloride, 10 mm EGTA, 1 mm MgCl2, 3 mm Mg-ATP, 5 mm HEPES, pH 7.3 adjusted by CsOH. The external solution contained 112 mm NaCl, 20 mm tetraethylammonium chloride, 10 mm CaCl2, 5 mm CsCl, 1 mm MgCl2, 10 mm HEPES, 5 mm glucose, pH 7.3 adjusted by NaOH. The osmolarity is 300 mosm for the intracellular solution and 305 mosm for the extracellular solution. For current-voltage protocols, the membrane voltage was depolarized in 50-ms steps from −70 mV to various voltages in 10-s intervals. A P/4 protocol was used for leak subtraction.
Primary hippocampal neuron cultures and pCREB assay
Dissociated rat embryonic day 18 hippocampal neurons were prepared as described previously (49) and transfected after 6–8 days in vitro (DIV). A total of 1 μg of DNA was transfected into each well of a 12-well plate using Lipofectamine 2000 with a DNA/lipid ratio of 1:1. For the pharmacological knock-in experiment, 0.6 μg of pCGN0-CaV1.3-sHA T1033Y, 0.2 μg of pcDNA-β3, and 0.2 μg of pcDNA-α2δ were transfected. For shRNA experiments, 0.4 μg of pLLCK-ratCaMKIIα shRNA, 0.4 μg of pLLCK-ratCaMKIIβ shRNA, and 0.2 μg of pcDNA msCaMKIIα were transfected. Transfection complexes were incubated with neurons for 3 h before switching back to conditioned medium. After an additional 72 h, neurons were preincubated with 5K Tyrode's solution (150 mm NaCl, 5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 10 mm glucose, and 10 mm HEPES, pH 7.5 (∼313 mosm)) with 1 μm TTX, 10 μm APV, and 50 μm NBQX to suppress intrinsic neuronal activity by blocking sodium channels, NMDA receptors, and AMPA receptors, respectively. Neurons were then treated with either 5K Tyrode's or 40K Tyrode's solution (adjusted to 40 mm KCl and 115 mm NaCl) containing TTX, APV, and NBQX for 90 s. For nimodipine treatment, neurons were incubated in 5K Tyrode's solution containing 10 μm nimodipine (plus TTX, APV, and NBQX) for about 2 min before switching to 40K Tyrode's solution plus 10 μm nimodipine, TTX, APV, and NBQX. Neurons were fixed using ice-cold 4% paraformaldehyde, 4% sucrose in 0.1 m phosphate buffer, pH 7.4, for 8 min; washed three times with PBS; permeabilized with PBS + 0.2% Triton X-100; and then incubated with block solution for 1 h (1× PBS, 0.1% Triton X-100 (v/v), 2.5% BSA (w/v), 5% normal donkey serum (w/v), 1% glycerol (v/v)). Neurons were then incubated overnight with primary antibodies: rabbit anti-pCREB (1:1000; Cell Signaling, catalog no. 9198), and either mouse anti-HA (1:1000, Biolegend catalog no. 901502) for the pharmacological knock-in experiment or mouse anti-CaMKIIα (1:2000; Thermo Fisher Scientific, catalog no. MA1-048) for shRNA experiments. The next morning, neurons were washed three times in PBS + 0.2% Triton X-100 and incubated with secondary antibody for 1 h. Secondary antibodies (from Thermo Fisher Scientific) were diluted 1:1000 in block solution: donkey anti-rabbit Alexa Fluor 647 (catalog no. A-31573) and donkey anti-mouse Alexa Fluor 546 (catalog no. A-10036). After washing three times in PBS, neurons were mounted on slides using Prolong Gold Antifade Mountant with DAPI (Thermo Fisher Scientific, catalog no. P36931).
Neuronal pCREB imaging and quantification
Images were collected using an Olympus FV-1000 inverted confocal microscope with a 40×/1.30 numeric aperture Plan-Neofluar oil lens. The binocular lens was used to identify transfected neurons based on the Alexa 546 signal from the HA staining or enhanced green fluorescent protein from the shRNA construct. The DAPI channel was then used to focus on the z plane that yielded the highest DAPI signal (one that presumably runs across the nuclei) for image acquisition. Images were then collected in all of the channels, and MetaMorph Microscope Automation and Image Analysis Software (Molecular Devices) was used to quantify the pCREB signal. Briefly, nuclei were identified by thresholding the DAPI channel to create and select the nuclear regions of interest. The regions of interest were then transferred to other channels to measure the average pCREB intensity.
The relative pCREB intensity was computed as (pCREBx-pCREB5K)/(pCREB40K-pCREB5K), where pCREBx is the pCREB signal being calculated, and pCREB5K and pCREB40K are the average signals of the 5K and 40 K conditions in that batch of cultured neurons, respectively. Data shown were collected from images of the indicated total number of neurons from 3–4 independent cultures.
Neuronal Ca2+ imaging
Dissociated rat hippocampal neurons cultured in coated 29-mm glass bottom dishes (Cellvis, catalog no. D29-10-1.5-N) were transfected with a total of 2 μg of DNA/dish after 8 DIV. All neurons (nontransfected for Fig. 7A, transfected with shRNA vectors for Fig. 8A) were imaged on DIV 13–14. Because GFP interferes with Fura-2 imaging, CaMKII shRNA constructs lacking the CaMKII promoter and GFP were co-expressed with mApple to label transfected cells. Cells were incubated at 37 °C for 20 min in culture medium (neural basal medium with 2% B27, 0.25% glutamax, and 1% penicillin-streptomycin) supplemented with 2 μm Fura-2 acetoxymethyl ester (Thermo Fisher Scientific, catalog no. F1221), washed twice with 5K Tyrode's solution, and then incubated at 37 °C for 15 min in 5K Tyrode's solution with TTX, APV, and NBQX (as above). For nimodipine-treated groups, this medium was replaced with 5K Tyrode's solution containing 10 μm nimodipine (in addition to TTX, APV, and NBQX) ∼5 min before imaging. Fura-2 fluorescence images were collected using a Nikon Eclipse TE2000-U microscope equipped with an epifluorescence illuminator (Sutter Instrument Co.) and an HQ2 CCD camera (PhotoMetrics Inc.). Baseline Ca2+ was recorded for 30 s in 5K Tyrode's solution before replacing by 40K Tyrode's solution. Cell somas were selected as regions of interest using Nikon Elements software; transfected neurons were selected based on mApple fluorescence. The ratios of emitted fluorescence (505 nm) intensities at excitation wavelengths of 340 and 380 nm (F340/F380) were measured every 5 s. Responses of individual cells at each time point were quantified as the change in fluorescence ratio above baseline (ΔF = (340/380 value)/(baseline 340/380 value)). The peak change in fluorescence ratio was used to compare responses between cells within each group (ΔF = (maximum 340/380 value)/(baseline 340/380 value)), and outlier cells were excluded based on a ROUT outliers test (Q = 1%). The numbers of excluded outliers were as follow: Fig. 7A, 31 of 246 cells from the 40K + NIM group; Fig. 8A, 1 of 18 cells from the control group (mApple only) and 1 of 24 cells from the CaMKIIα/β shRNA group.
Author contributions
X. W. helped conceive of the studies, created most of the reagents, designed experiments, conducted most of the experimental work, and wrote drafts of the manuscript. T. L. P. and C. R. M. designed/created reagents and conducted experiments related to Fig. 4. C. R. M. performed the Fura-2 calcium imaging studies and analyzed these data (Figs. 7A and 8A). T. N., A. L., and D. A. J. provided reagents and assisted with experimental design and data analysis/interpretation. R. J. C. conceived and coordinated the studies, designed experiments, analyzed/interpreted the data, and helped write the manuscript. All authors reviewed the results, helped edit the manuscript, and approved the final version.
Acknowledgments
We thank Kevin P. M. Currie for helpful discussions, Nicholas C. Vierra for help with whole-cell patch clamp, Keeley Spiess for assistance with Ca2+ imaging, and Elena I. Zaika for assistance with rat hippocampal neuron cultures. We also thank Diane Lipscombe, William Thiel, Gerald W. Zamponi, Aaron Fox, Edward Perez-Reyes, Winship Herr, and Luk Van Parijs for providing various plasmids, as detailed throughout. Confocal imaging and analysis were performed in part through the use of the Vanderbilt Cell Imaging Shared Resource (supported by National Institutes of Health Grants CA68485, DK20593, DK58404, DK59637, and EY08126).
This work was supported in part by National Institutes of Health Grants R01-MH063232 and R01-NS078291 (to R. J. C.), R01-DK097392 (to D. A. J.), R01-DC009433 and R01-NS084190 (to A. L.), R01-HD061543 (to T. N.), and F31-MH109196 (to C. R. M.) and American Heart Association predoctoral fellowships 14PRE18420020 (to X. W.) and 15PRE25110020 (to C. R. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- E-T
- excitation–transcription
- LTCC
- L-type Ca2+ channel
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- VGCC
- voltage-gated Ca2+ channel
- APV
- dl-2-amino-5-phosphonopentanoic acid
- CNQX
- 6-cyano-7-nitroquinoxaline-2,3-dione
- TTX
- tetrodotoxin
- DIV
- days in vitro
- PDB
- Protein Data Bank.
References
- 1. Hagenston A. M., and Bading H. (2011) Calcium signaling in synapse-to-nucleus communication. Cold Spring Harb. Perspect. Biol. 3, a004564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Deisseroth K., Mermelstein P. G., Xia H., and Tsien R. W. (2003) Signaling from synapse to nucleus: the logic behind the mechanisms. Curr. Opin. Neurobiol. 13, 354–365 [DOI] [PubMed] [Google Scholar]
- 3. Bading H. (2013) Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 14, 593–608 [DOI] [PubMed] [Google Scholar]
- 4. Flavell S. W., and Greenberg M. E. (2008) Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu. Rev. Neurosci. 31, 563–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alberini C. M. (2009) Transcription factors in long-term memory and synaptic plasticity. Physiol. Rev. 89, 121–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Athos J., Impey S., Pineda V. V., Chen X., and Storm D. R. (2002) Hippocampal CRE-mediated gene expression is required for contextual memory formation. Nat. Neurosci. 5, 1119–1120 [DOI] [PubMed] [Google Scholar]
- 7. West A. E., and Greenberg M. E. (2011) Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb. Perspect. Biol. 10.1101/cshperspect.a005744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cohen S. M., Ma H., Kuchibhotla K. V., Watson B. O., Buzsáki G., Froemke R. C., and Tsien R. W. (2016) Excitation-transcription coupling in parvalbumin-positive interneurons employs a novel CaM kinase-dependent pathway distinct from excitatory neurons. Neuron 90, 292–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cohen S. M., Li B., Tsien R. W., and Ma H. (2015) Evolutionary and functional perspectives on signaling from neuronal surface to nucleus. Biochem. Biophys. Res. Commun. 460, 88–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma H., Groth R. D., Cohen S. M., Emery J. F., Li B., Hoedt E., Zhang G., Neubert T. A., and Tsien R. W. (2014) γCaMKII shuttles Ca2+/CaM to the nucleus to trigger CREB phosphorylation and gene expression. Cell 159, 281–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li B., Tadross M. R., and Tsien R. W. (2016) Sequential ionic and conformational signaling by calcium channels drives neuronal gene expression. Science 351, 863–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deisseroth K., Bito H., and Tsien R. W. (1996) Signaling from synapse to nucleus: postsynaptic CREB phosphorylation during multiple forms of hippocampal synaptic plasticity. Neuron 16, 89–101 [DOI] [PubMed] [Google Scholar]
- 13. Deisseroth K., Heist E. K., and Tsien R. W. (1998) Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature 392, 198–202 [DOI] [PubMed] [Google Scholar]
- 14. Ma H., Cohen S., Li B., and Tsien R. W. (2012) Exploring the dominant role of Cav1 channels in signalling to the nucleus. Biosci. Rep. 33, 97–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wheeler D. G., Barrett C. F., Groth R. D., Safa P., and Tsien R. W. (2008) CaMKII locally encodes L-type channel activity to signal to nuclear CREB in excitation-transcription coupling. J. Cell Biol. 183, 849–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Grueter C. E., Abiria S. A., Wu Y., Anderson M. E., and Colbran R. J. (2008) Differential regulated interactions of calcium/calmodulin-dependent protein kinase II with isoforms of voltage-gated calcium channel β subunits. Biochemistry 47, 1760–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hudmon A., Schulman H., Kim J., Maltez J. M., Tsien R. W., and Pitt G. S. (2005) CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J. Cell Biol. 171, 537–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Abiria S. A., and Colbran R. J. (2010) CaMKII associates with CaV1.2 L-type calcium channels via selected β subunits to enhance regulatory phosphorylation. J. Neurochem. 112, 150–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jenkins M. A., Christel C. J., Jiao Y., Abiria S., Kim K. Y., Usachev Y. M., Obermair G. J., Colbran R. J., and Lee A. (2010) Ca2+-dependent facilitation of Cav1.3 Ca2+ channels by densin and Ca2+/calmodulin-dependent protein kinase II. J. Neurosci. 30, 5125–5135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Simms B. A., Souza I. A., Rehak R., and Zamponi G. W. (2015) The Cav1.2 N terminus contains a CaM kinase site that modulates channel trafficking and function. Pflugers Arch. 467, 677–686 [DOI] [PubMed] [Google Scholar]
- 21. Strack S., McNeill R. B., and Colbran R. J. (2000) Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-d-aspartate receptor. J. Biol. Chem. 275, 23798–23806 [DOI] [PubMed] [Google Scholar]
- 22. Jiao Y., Jalan-Sakrikar N., Robison A. J., Baucum A. J. 2nd, Bass M. A., and Colbran R. J. (2011) Characterization of a central Ca2+/calmodulin-dependent protein kinase IIα/β binding domain in densin that selectively modulates glutamate receptor subunit phosphorylation. J. Biol. Chem. 286, 24806–24818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jin D. Z., Guo M. L., Xue B., Fibuch E. E., Choe E. S., Mao L. M., and Wang J. Q. (2013) Phosphorylation and feedback regulation of metabotropic glutamate receptor 1 by calcium/calmodulin-dependent protein kinase II. J. Neurosci. 33, 3402–3412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dick I. E., Tadross M. R., Liang H., Tay L. H., Yang W., and Yue D. T. (2008) A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 451, 830–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tadross M. R., Dick I. E., and Yue D. T. (2008) Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell 133, 1228–1240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bayer K. U., De Koninck P., Leonard A. S., Hell J. W., and Schulman H. (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805 [DOI] [PubMed] [Google Scholar]
- 27. Bayer K. U., LeBel E., McDonald G. L., O'Leary H., Schulman H., and De Koninck P. (2006) Transition from reversible to persistent binding of CaMKII to postsynaptic sites and NR2B. J. Neurosci. 26, 1164–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Singh A., Gebhart M., Fritsch R., Sinnegger-Brauns M. J., Poggiani C., Hoda J. C., Engel J., Romanin C., Striessnig J., and Koschak A. (2008) Modulation of voltage- and Ca2+-dependent gating of CaV1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J. Biol. Chem. 283, 20733–20744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ludwig A., Flockerzi V., and Hofmann F. (1997) Regional expression and cellular localization of the α1 and β subunit of high voltage-activated calcium channels in rat brain. J. Neurosci. 17, 1339–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wheeler D. G., Groth R. D., Ma H., Barrett C. F., Owen S. F., Safa P., and Tsien R. W. (2012) CaV1 and CaV2 channels engage distinct modes of Ca2+ signaling to control CREB-dependent gene expression. Cell 149, 1112–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang H., Fu Y., Altier C., Platzer J., Surmeier D. J., and Bezprozvanny I. (2006) Ca1.2 and CaV1.3 neuronal L-type calcium channels: differential targeting and signaling to pCREB. Eur. J. Neurosci. 23, 2297–2310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dolmetsch R. E., Pajvani U., Fife K., Spotts J. M., and Greenberg M. E. (2001) Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294, 333–339 [DOI] [PubMed] [Google Scholar]
- 33. Koval O. M., Guan X., Wu Y., Joiner M. L., Gao Z., Chen B., Grumbach I. M., Luczak E. D., Colbran R. J., Song L. S., Hund T. J., Mohler P. J., and Anderson M. E. (2010) CaV1.2 β-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc. Natl. Acad. Sci. U.S.A. 107, 4996–5000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Obermair G. J., Schlick B., Di Biase V., Subramanyam P., Gebhart M., Baumgartner S., and Flucher B. E. (2010) Reciprocal interactions regulate targeting of calcium channel β subunits and membrane expression of α1 subunits in cultured hippocampal neurons. J. Biol. Chem. 285, 5776–5791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang H., Maximov A., Fu Y., Xu F., Tang T. S., Tkatch T., Surmeier D. J., and Bezprozvanny I. (2005) Association of CaV1.3 L-type calcium channels with Shank. J. Neurosci. 25, 1037–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hardingham G. E., Chawla S., Cruzalegui F. H., and Bading H. (1999) Control of recruitment and transcription-activating function of CBP determines gene regulation by NMDA receptors and L-type calcium channels. Neuron 22, 789–798 [DOI] [PubMed] [Google Scholar]
- 37. Hetzenauer A., Sinnegger-Brauns M. J., Striessnig J., and Singewald N. (2006) Brain activation pattern induced by stimulation of L-type Ca2+-channels: contribution of CaV1.3 and CaV1.2 isoforms. Neuroscience 139, 1005–1015 [DOI] [PubMed] [Google Scholar]
- 38. Bading H., Ginty D. D., and Greenberg M. E. (1993) Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260, 181–186 [DOI] [PubMed] [Google Scholar]
- 39. Hardingham G. E., Arnold F. J., and Bading H. (2001) Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat. Neurosci. 4, 261–267 [DOI] [PubMed] [Google Scholar]
- 40. Hanson P. I., Meyer T., Stryer L., and Schulman H. (1994) Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron 12, 943–956 [DOI] [PubMed] [Google Scholar]
- 41. Baucum A. J. 2nd, Shonesy B. C., Rose K. L., and Colbran R. J. (2015) Quantitative proteomics analysis of CaMKII phosphorylation and the CaMKII interactome in the mouse forebrain. ACS Chem. Neurosci. 6, 615–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li M. Z., and Elledge S. J. (2007) Harnessing homologous recombination in vitro to generate recombinant DNA via SLIC. Nat. Methods 4, 251–256 [DOI] [PubMed] [Google Scholar]
- 43. Altier C., Dubel S. J., Barrère C., Jarvis S. E., Stotz S. C., Spaetgens R. L., Scott J. D., Cornet V., De Waard M., Zamponi G. W., Nargeot J., and Bourinet E. (2002) Trafficking of L-type calcium channels mediated by the postsynaptic scaffolding protein AKAP79. J. Biol. Chem. 277, 33598–33603 [DOI] [PubMed] [Google Scholar]
- 44. He M., Bodi I., Mikala G., and Schwartz A. (1997) Motif III S5 of L-type calcium channels is involved in the dihydropyridine binding site: a combined radioligand binding and electrophysiological study. J. Biol. Chem. 272, 2629–2633 [DOI] [PubMed] [Google Scholar]
- 45. Liu H., and Naismith J. H. (2008) An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol. 8, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dittgen T., Nimmerjahn A., Komai S., Licznerski P., Waters J., Margrie T. W., Helmchen F., Denk W., Brecht M., and Osten P. (2004) Lentivirus-based genetic manipulations of cortical neurons and their optical and electrophysiological monitoring in vivo. Proc. Natl. Acad. Sci. U.S.A. 101, 18206–18211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. McNeill R. B., and Colbran R. J. (1995) Interaction of autophosphorylated Ca2+/calmodulin-dependent protein kinase II with neuronal cytoskeletal proteins: characterization of binding to a 190-kDa postsynaptic density protein. J. Biol. Chem. 270, 10043–10049 [DOI] [PubMed] [Google Scholar]
- 48. Stephenson J. R., Wang X., Perfitt T. L., Parrish W. P., Shonesy B. C., Marks C. R., Mortlock D. P., Nakagawa T., Sutcliffe J. S., and Colbran R. J. (2017) A novel human CAMK2A mutation disrupts dendritic morphology and synaptic transmission, and causes ASD-related behaviors. J. Neurosci. 37, 2216–2233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sala C., Futai K., Yamamoto K., Worley P. F., Hayashi Y., and Sheng M. (2003) Inhibition of dendritic spine morphogenesis and synaptic transmission by activity-inducible protein Homer1a. J. Neurosci. 23, 6327–6337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pragnell M., De Waard M., Mori Y., Tanabe T., Snutch T. P., and Campbell K. P. (1994) Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature 368, 67–70 [DOI] [PubMed] [Google Scholar]
- 51. Chao L. H., Stratton M. M., Lee I. H., Rosenberg O. S., Levitz J., Mandell D. J., Kortemme T., Groves J. T., Schulman H., and Kuriyan J. (2011) A mechanism for tunable autoinhibition in the structure of a human Ca2+/calmodulin- dependent kinase II holoenzyme. Cell 146, 732–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rellos P., Pike A. C., Niesen F. H., Salah E., Lee W. H., von Delft F., and Knapp S. (2010) Structure of the CaMKIIδ/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS Biol. 8, e1000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. De Rubeis S., He X., Goldberg A. P., Poultney C. S., Samocha K., Cicek A. E., Kou Y., Liu L., Fromer M., Walker S., Singh T., Klei L., Kosmicki J., Shih-Chen F., Aleksic B., et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 [DOI] [PMC free article] [PubMed] [Google Scholar]