

Abstract
Voltage-gated Na+ (NaV) channels are key regulators of myocardial excitability, and Ca2+/calmodulin-dependent protein kinase II (CaMKII)-dependent alterations in NaV1.5 channel inactivation are emerging as a critical determinant of arrhythmias in heart failure. However, the global native phosphorylation pattern of NaV1.5 subunits associated with these arrhythmogenic disorders and the associated channel regulatory defects remain unknown. Here, we undertook phosphoproteomic analyses to identify and quantify in situ the phosphorylation sites in the NaV1.5 proteins purified from adult WT and failing CaMKIIδc-overexpressing (CaMKIIδc-Tg) mouse ventricles. Of 19 native NaV1.5 phosphorylation sites identified, two C-terminal phosphoserines at positions 1938 and 1989 showed increased phosphorylation in the CaMKIIδc-Tg compared with the WT ventricles. We then tested the hypothesis that phosphorylation at these two sites impairs fibroblast growth factor 13 (FGF13)-dependent regulation of NaV1.5 channel inactivation. Whole-cell voltage-clamp analyses in HEK293 cells demonstrated that FGF13 increases NaV1.5 channel availability and decreases late Na+ current, two effects that were abrogated with NaV1.5 mutants mimicking phosphorylation at both sites. Additional co-immunoprecipitation experiments revealed that FGF13 potentiates the binding of calmodulin to NaV1.5 and that phosphomimetic mutations at both sites decrease the interaction of FGF13 and, consequently, of calmodulin with NaV1.5. Together, we have identified two novel native phosphorylation sites in the C terminus of NaV1.5 that impair FGF13-dependent regulation of channel inactivation and may contribute to CaMKIIδc-dependent arrhythmogenic disorders in failing hearts.
Keywords: Ca2+/calmodulin-dependent protein kinase II (CaMKII), calmodulin (CaM), heart, phosphoproteomics, phosphorylation, sodium channel, FGF13, Nav1.5, channel inactivation
Introduction
Voltage-gated Na+ (NaV) channels are critical determinants of myocardial excitability, and defects in NaV channel functioning or regulation in the context of inherited or acquired cardiac disease increase the risk of life-threatening arrhythmias (1). Under physiological conditions, the primary ventricular NaV channel subunits, the NaV1.5 channels, activate and inactivate rapidly to generate the transient peak Na+ current, INa, responsible for the depolarization phase and the propagation of action potentials. However, a small proportion of NaV channels inactivates slowly to generate a late or persistent Na+ current, referred to as INaL, contributing markedly to determining action potential waveform, duration, and refractoriness. Among the various determinants recognized to cause NaV channel dysfunctions and consequent increased risk of acquired arrhythmias is the activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII)3 (2–7). A prime example of this dysregulation is heart failure in which an increased INaL has been linked to the activation of CaMKII (2, 4–7) and associated alteration in NaV1.5 phosphorylation (7). This increased INaL in failing hearts is sometimes accompanied by a decreased NaV channel availability caused by a hyperpolarizing shift of the voltage dependence of steady-state INa inactivation (2, 7). Three phosphorylation sites in the first linker loop of NaV1.5, at positions 516 (8, 9), 571 (10–12), and 594 (8), have been suggested to play causative roles in these deleterious CaMKII-dependent mechanisms. Nonetheless, the global native phosphorylation pattern of NaV1.5 channels associated with CaMKII activation in failing hearts as well as the molecular mechanisms associated with altered phosphorylation that underlie the defects in channel inactivation remain unknown.
The understanding of NaV1.5 channel inactivation has received much attention over the past decade and has recently been buttressed by the generation of several crystal structures of the cytoplasmic C-terminal domain (CTD) of NaV1.5 (as well as of other NaV subunits) in complex with Ca2+-free or Ca2+-bound calmodulin (CaM) and/or a member of the intracellular fibroblast growth factor (iFGF) family of proteins (13–16). The Ca2+-binding protein CaM acts as a sensor for Ca2+ and regulates inactivation of NaV channels, while underlying mechanisms still elude consensus. Depending on whether CaM is loaded with Ca2+, it binds different sites within and around the IQ motif in the CTD and/or the third linker loop of NaV channels, thereby inducing conformational changes and affecting channel inactivation and the late Na+ current (13–19). iFGFs, which include FGF11–14, are also constitutive NaV channel-associated proteins, binding the CTD of NaV channels, just upstream of the IQ motif (14–16, 20–22). The most consistent effect of iFGFs is to increase the availability of NaV channels by shifting the voltage dependence of steady-state inactivation toward depolarized potentials (15, 20–26). The central role of the NaV1.5 CTD as well as of iFGFs and CaM in regulating the inactivation properties of NaV1.5 channels is highlighted by the effects of several mutations observed in the arrhythmic long QT3 (LQT3) and Brugada syndromes that map to their binding interfaces (14, 27–29).
This study uses a mass spectrometry (MS)-based phosphoproteomic analysis to identify and quantify in situ the native phosphorylation sites in the NaV1.5 proteins purified from failing CaMKIIδc-overexpressing (CaMKIIδc-Tg) versus non-failing wild-type (WT) mouse ventricles. The rationale for using this transgenic and failing mouse model is to identify the NaV1.5 phosphorylation sites that participate in the channel inactivation defects associated with the activation of CaMKIIδc in heart failure. Further biochemical and electrophysiological approaches in human embryonic kidney 293 (HEK293) cells were then used to investigate the impact of phosphorylation at two C-terminal serine residues on the interaction of FGF13 and CaM with NaV1.5 and on the inactivation properties of NaV1.5 channels.
Results
Purification and characterization of NaV channel complexes from WT and CaMKIIδc-Tg mouse ventricles
Total lysates from four adult WT and four CaMKIIδc-Tg (30) mouse (m) ventricles were prepared, pooled, and used in eight distinct IPs (four WT IPs and four CaMKIIδc-Tg IPs) using the mαNaVPAN-specific antibody. As illustrated in Fig. 1A and consistent with previous findings (7), Western blot analyses of total lysates showed greater NaV1.5 protein expression in the CaMKIIδc-Tg compared with the WT ventricles. This difference in total protein expression resulted in significantly (p < 0.05) higher NaV1.5 protein abundance (3.8-fold) in the mαNaVPAN-IPs from the CaMKIIδc-Tg than from the WT ventricles (Fig. 1, A and B). Accordingly, analyses of mαNaVPAN-IPs on SYPRO Ruby-stained gels revealed the presence of a band corresponding to the molecular weight of NaV α subunits (31), the intensities of which are higher in the CaMKIIδc-Tg IPs than in the WT IPs (Fig. 1C).
Figure 1.

Immunoprecipitation of NaV channel complexes from adult WT and CaMKIIδc-Tg mouse ventricles. A, representative NaV α Western blots of total lysates and immunoprecipitated proteins from adult WT and CaMKIIδc-Tg mouse ventricles with the anti-NaVPAN monoclonal antibody (mαNaVPAN-IPs) probed with the anti-NaV1.5 rabbit polyclonal (RbαNaV1.5) and the mαNaVPAN antibodies, respectively. B, mean ± S.E. relative NaV α protein abundance in WT (n = 4) and CaMKIIδc-Tg (n = 4) IPs. *, p < 0.05, Mann-Whitney test. C, SYPRO Ruby-stained gel of mαNaVPAN-IPs from WT and CaMKIIδc-Tg mouse ventricles. Relative abundance of proteins running at the molecular weight of NaV α subunits is higher in CaMKIIδc-Tg IPs than in WT IPs.
The protein components of isolated NaV channel complexes from WT and CaMKIIδc-Tg ventricles were identified by MS using three distinct mass spectrometers: an LTQ-Orbitrap XL, an LTQ-Orbitrap Elite, and a TripleTOF 5600 Plus. The NaV1.5 protein was the most abundant protein in the mαNaVPAN-IPs from both WT and CaMKIIδc-Tg ventricles with average numbers of total exclusive MS/MS spectra acquired increasing from 95 using the Orbitrap XL to 177 using the Orbitrap Elite and 524 using the TripleTOF (Fig. 2A). These greater sensitivities of the Orbitrap Elite and TripleTOF mass spectrometers improved the NaV1.5 amino acid sequence coverage from 27 to 28 and 32%, respectively, and from 36 to 38 and 43% with the transmembrane domains removed. NaV1.4 was the second most abundant NaV α subunit with much fewer spectra acquired: 5, 7, and 30 using the Orbitrap XL, Orbitrap Elite, and TripleTOF, respectively. Most interestingly, the greater sensitivity of the TripleTOF mass spectrometer allowed the identification of three additional NaV α subunits, NaV1.7 (four spectra), NaV1.3 (three spectra), and NaV1.8 (two spectra). These experiments also led to the reliable identification of several previously identified NaV1.5 channel-associated/regulatory proteins (32), including the δ, β, and γ subunits of CaMKII, CaM, FGF13, and ankyrin-G (Fig. 2C). The supplemental Tables 1–3 provide the complete lists of identified peptides and proteins using the Orbitrap XL, Elite, and the TripleTOF mass spectrometers, respectively.
Figure 2.

MS protein identification in immunoprecipitated NaV channel complexes from WT and CaMKIIδc-Tg mouse ventricles. A, NaV α subunits identified using the LTQ-Orbitrap XL, LTQ-Orbitrap Elite, and TripleTOF 5600 Plus mass spectrometers. The average numbers of exclusive unique peptides and total spectra for each NaV α subunit and the percent amino acid sequence coverages obtained for NaV1.5, including or excluding transmembrane domains (TD), are presented. In addition to NaV1.5, which is the most abundant protein in the mαNaVPAN-IPs, NaV1.4 is also detected, and the greater sensitivity of the Orbitrap Elite and TripleTOF mass spectrometers allowed the identification of NaV1.7, NaV1.8, and NaV1.3. B, amino acid sequence coverage obtained for the (mouse) NaV1.5 protein (NP_001240789). Detected peptides are highlighted in yellow; identified phosphorylation sites are highlighted in blue (sites already identified in our previous MS analyses) and red (newly identified sites in the present study); transmembrane segments (S1–S6) in each domain (I–IV) are in bold and underlined in black; loops I, II, and III correspond to interdomains I and II, II and III, and III and IV, respectively; and binding sites for iFGF and calmodulin (IQ-motif) are boxed in green and orange, respectively. C, relative abundances of NaV α subunits and previously characterized NaV1.5 channel-associated/regulatory proteins in the CaMKIIδc-Tg IPs (n = 4) versus the WT IPs (n = 4) were calculated from the entire (Orbitrap XL) MS1 peptide data set using the DAnTE statistical software (**, p < 0.01; ***, p < 0.001).
The relative abundances of identified proteins in the mαNaVPAN-IPs from the CaMKIIδc-Tg versus the WT ventricles were calculated from the MS1 peptide data (Orbitrap XL) using the DAnTE statistical analysis tool (33–35). Consistent with the biochemistry data (Fig. 1), the NaV1.5 protein is 2.9-fold (p < 0.001) more represented in the CaMKIIδc-Tg IPs than in the WT IPs (Fig. 2C). Interestingly, this quantitative MS analysis also revealed that of the 10 NaV1.5-associated/regulatory proteins identified, the abundance ratios of CaMKIIδ, β2-syntrophin, and dystrophin in the CaMKIIδc-Tg versus the WT IPs are substantially greater than the 2.9-fold abundance ratio obtained for NaV1.5. On the contrary, the CaMKIIβ and γ subunits as well as CaM and FGF13 are relatively less represented compared with NaV1.5. The abundance ratios obtained for ankyrin-G, plakophilin-2, and α1-syntrophin are close to the NaV1.5 abundance ratio. Taken together, these observations demonstrate that NaV1.5 is more expressed in the CaMKIIδc-Tg than in the WT mouse ventricles, which, as a consequence, led to greater NaV1.5 IP yields from the CaMKIIδc-Tg than from the WT ventricles. These analyses also suggest different relative compositions of associated/regulatory proteins in NaV1.5 channel complexes in the CaMKIIδc-Tg compared with the WT ventricles.
Identification of native NaV1.5 phosphorylation sites from WT and CaMKIIδc-Tg mouse ventricles
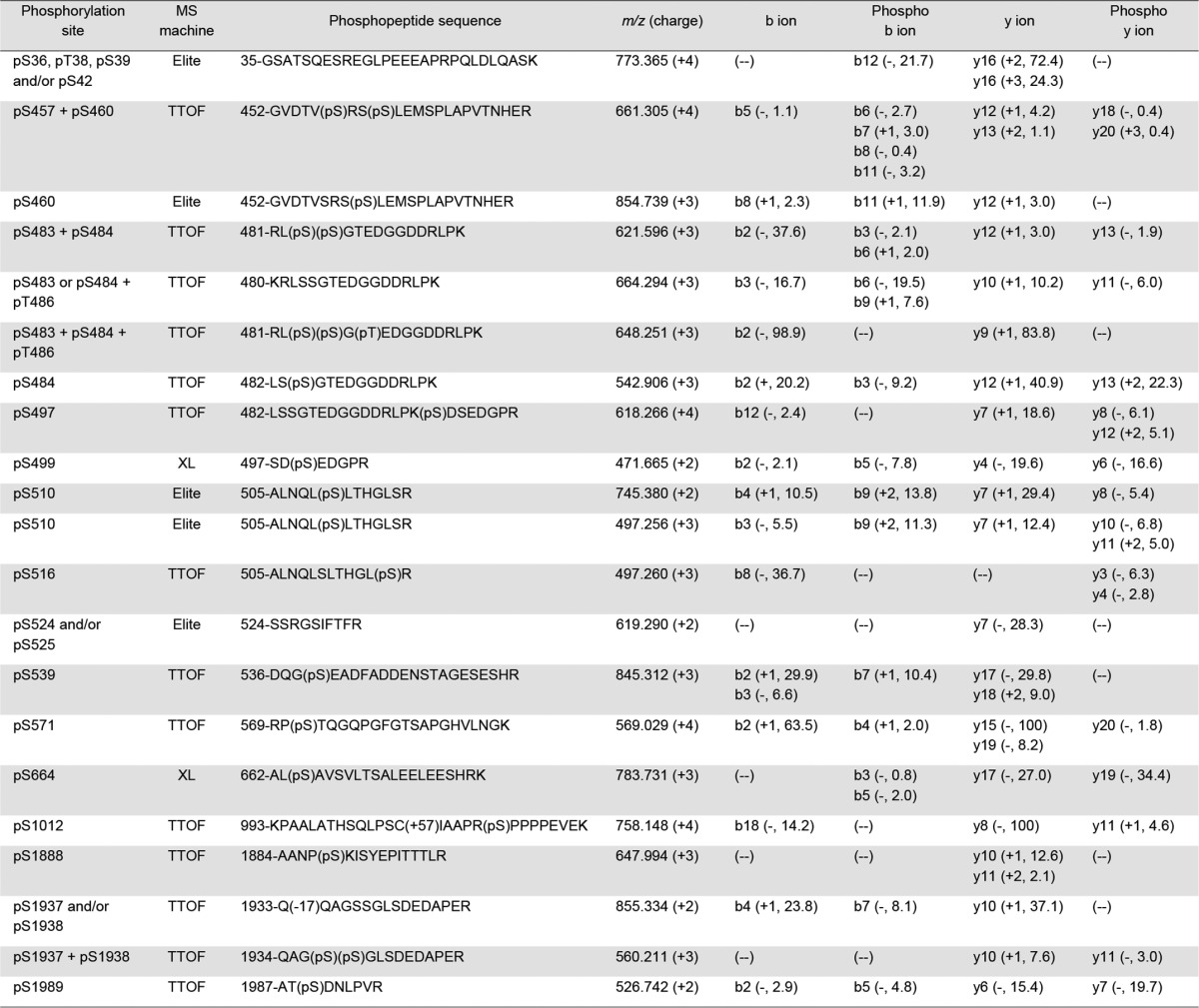
To determine the global native phosphorylation pattern of NaV1.5 channels associated with the overexpression of CaMKIIδc, a quantitative phosphoproteomic analysis of the NaV1.5 proteins purified from the CaMKIIδc-Tg and the WT mouse ventricles was performed. This phosphoproteomic analysis unambiguously allowed the identification of 19 native phosphorylation sites in the mouse ventricular NaV1.5 protein (Figs. 2B and 3). Table 1 lists the phosphopeptides enabling the best phosphorylation site assignment(s), with the mass spectrometer used for the identification and the percentages of maximum intensities of site-discriminating ions, for each phosphorylation site. Representative MS/MS (and MS1) phosphopeptide spectra are presented in supplemental Fig. 1. Descriptions of all detected site-discriminating and supporting ions (calculated mass errors, confirmations of charge states) are given in supplemental Tables 4. Among the 19 NaV1.5 phosphoserines (pSer) or phosphothreonines (pThr) identified, 10 (in blue) had already been identified in our previous phosphoproteomic analyses (31) and nine (in red) are novel (Figs. 2B and 3). These nine novel phosphorylation sites are located in the first (pThr-486, pSer-499, pSer-516, and pSer-539) and second (pSer-1012) intracellular linker loops as well as in the CTD (pSer-1888, pSer-1937, pSer-1938, and pSer-1989) of NaV1.5. Interestingly, the three C-terminal pSer-1888, pSer-1937, and pSer-1938 are in close proximity to the binding sites for the iFGFs (14–16, 20–22) and CaM (IQ-motif) (13–19).
Figure 3.

Localization of MS-identified in situ phosphorylation sites on the mouse ventricular NaV1.5 α subunit protein. Among the 19 phosphorylation sites identified, 10 (in blue) had already been identified in our previous MS analyses and 9 (in red) are novel. Four and two phosphorylation site locations are possible at amino acids 36–42 and 524–525, respectively. The three newly identified C-terminal phosphoserines at positions 1888, 1937, and 1938 (pSer-1888, pSer-1937, and pSer-1938) are in close proximity to the binding sites for iFGF and calmodulin (IQ-motif).
Table 1.
Phosphorylation sites, phosphopeptides, and site-discriminating ions identified in immunoprecipitated NaV1.5 α subunits from adult WT and/or CaMKIIδc-Tg mouse ventricles using MS
The site-discriminating ions observed in the high-resolution (LTQ-Orbitrap Elite or TripleTOF 5600 Plus) or low-resolution (LTQ-Orbitrap XL) MS/MS spectra of each annotated NaV1.5 phosphopeptide support the assignment of the indicated phosphorylation site(s). The charge state as well as the percentage of maximum intensity of each observed unphosphorylated and phosphorylated site-discriminating (b and y) ion are reported in parentheses; the minus symbol indicates that the charge state could not be determined.
To determine whether one or several of these MS-identified NaV1.5 phosphorylation sites are associated with the overexpression of CaMKIIδc, the relative abundance of each NaV1.5 phosphopeptide in the CaMKIIδc-Tg versus the WT mαNaVPAN-IPs was determined by label-free quantification of MS1 data (36). As illustrated in Fig. 4A, and consistent with the quantification of the biochemical (Fig. 1) and the MS protein (Fig. 2C) data, the unphosphorylated NaV1.5 peptides are 3.6-fold more represented in the CaMKIIδc-Tg versus the WT IPs. Of the 86 unphosphorylated and 32 phosphorylated NaV1.5 peptides (118 peptides total), only the three phosphopeptides AT(pS)DNLPVR, RL(pS)(pS)GTEDGGDDR, and AL(pS)AVSVLTSALEELEESHRK (marked with a † in Fig. 4A), exhibiting phosphorylation(s) on serines 1989 (pSer-1989), 483 and 484 (pSer-483 and pSer-484), and 664 (pSer-664), respectively, present fold change ratios (8.96-, 7.13-, and 0.58-fold) significantly different from the median ratio (Tukey whisker analysis, Fig. 4B). Nonetheless, the other phosphopeptides assigning pSer-483 and/or pSer-484 do not show any significant abundance changes compared with the median ratio (Fig. 4, A and B); and the abundance change observed for the phosphopeptide identifying pSer-664 is not significantly different between the mαNaVPAN-IPs from the CaMKIIδc-Tg and the WT ventricles (Fig. 4A). In addition, the phosphopeptide Q(−17.03)QAGSSGLSDEDAPER, assigning pSer-1937 and/or pSer-1938 (Table 1), is present in the CaMKIIδc-Tg IPs (n = 3/4) and absent in the WT IPs (n = 0/4) (Fig. 4C). Relative abundances of all the other NaV1.5 phosphopeptides are comparable with the median (or mean) relative abundance found for the unphosphorylated NaV1.5 peptides (Fig. 4, A and B).
Figure 4.

Quantification analysis of NaV1.5 phosphorylation sites in the CaMKIIδc-Tg versus the WT mαNaVPAN-IPs. A, relative abundances of 32 NaV1.5 phosphopeptides allowing assignments of the listed phosphorylation sites, in the CaMKIIδc-Tg (n = 4) versus the WT (n = 4) IPs, were calculated using label-free quantification of the (Orbitrap XL) MS1 data. The mean ± S.E. relative abundance of unphosphorylated NaV1.5 peptides in the CaMKIIδc-Tg versus the WT IPs was calculated from 86 unphosphorylated NaV1.5 peptides (minimum Scaffold peptide probability scores of 95%). Consistent with the biochemistry data (Fig. 1) and the DAnTE protein statistical analysis (Fig. 2C), the unphosphorylated NaV1.5 peptides are 3.6-fold more represented in the CaMKIIδc-Tg IPs than in the WT IPs (red dashed line). The relative abundances of individual NaV1.5 phosphopeptides are significantly (*, p < 0.05, Mann-Whitney test) different in the CaMKIIδc-Tg compared with the WT IPs. B, Tukey whisker analysis of NaV1.5 peptide relative abundance in CaMKIIδc-Tg versus WT IPs. Of the 118 (86 unphosphorylated and 32 phosphorylated) NaV1.5 peptides, only the phosphopeptides AT(pS)DNLPVR, RL(pS)(pS)GTEDGGDDR, and AL(pS)AVSVLTSALEELEESHRK (marked with † in A), exhibiting phosphorylation(s) on serines 1989 (pSer-1989), 483 and 484 (pSer-483 and pSer-484), and 664 (pSer-664), respectively, present fold change ratios (8.96-, 7.13-, and 0.58-fold) significantly different from the median ratio. C, mean ± S.E. intensities of phosphopeptides Q(−17.03)QAGSSGLSDEDAPER, assigning pSer-1937 and/or pSer-1938 (absence in WT IPs and presence in CaMKIIδc-Tg IPs), and AT(pS)DNLPVR, assigning pSer-1989 (8.96-fold change ratio), in WT (n = 4) and CaMKIIδc-Tg (n = 4) IPs. *, p < 0.05, Mann-Whitney test. D, conservation of the two C-terminal NaV1.5 serines 1938 and 1989 across orthologs.
Amino acid sequence alignment of the flanking regions of pSer-1937/38 and pSer-1989 showed a good conservation of Ser-1938 and Ser-1989 across orthologous sequences (Ser-1937 is not well-conserved) and, interestingly, revealed that Ser-1989 conforms to the consensus CaMKII phosphorylation site (RXX(S/T) Fig. 4D (37)). Taken together, these phosphoproteomic analyses identified nine novel native NaV1.5 phosphorylation sites, of which two conserved serines in the CTD, at positions 1938 and 1989, show increased phosphorylation in the CaMKIIδc-Tg compared with the WT ventricles.
Phosphorylation at serines 1933 and 1984 impairs FGF13-dependent regulation of NaV1.5 channel inactivation
To determine the impact of phosphorylation at these two C-terminal (mouse) serines 1938 and 1989 on the gating properties of NaV1.5 channels, the orthologous human serine to glutamate (NaV1.5-S1933E/S1984E, NaV1.5-EE) or serine to alanine (NaV1.5-S1933A/S1984A, NaV1.5-AA) double NaV1.5 phosphomutants were generated and analyzed by whole-cell voltage-clamp analyses in transiently transfected HEK293 cells. Because pSer-1938 and pSer-1989 are located in close proximity to the binding site for the iFGFs (14–16, 20–22), we tested the hypothesis that phosphorylation at these two sites disrupts the interaction of the iFGFs with NaV1.5 and the associated iFGFs-dependent regulation of NaV1.5 channel function. To this purpose, initial experiments were aimed at exploring the effects of FGF13 on the current density and biophysical properties of heterologously expressed NaV1.5 channels. The isoform 2 of FGF13 (FGF13–2) was chosen as the iFGF in these experiments as it is the isoform detected in the mαNaVPAN-IPs. As illustrated in Fig. 5, these whole-cell voltage-clamp analyses demonstrated that FGF13 significantly decreases the peak Na+ current (INa) density (p < 0.05, Fig. 5, A and B) and shifts the voltage dependence of steady-state current inactivation toward depolarized potentials (p < 0.01, Fig. 5D, see detailed densities, properties, and statistics in Table 2). In contrast, no significant differences in the voltage dependence of activation (Fig. 5C) or the kinetics of activation, inactivation, and recovery from inactivation were observed upon FGF13 co-expression (Table 2). To determine the effect of NaV1.5 phosphorylation at serines 1933 and 1984 on these FGF13-dependent regulations, these analyses were repeated from cells co-expressing NaV1.5-EE or NaV1.5-AA and FGF13. These recordings revealed that the voltage dependence of steady-state inactivation in cells co-expressing NaV1.5-EE and FGF13 is significantly (p < 0.01) shifted toward hyperpolarized potentials compared with cells co-expressing NaV1.5-WT and FGF13 and similar to cells expressing NaV1.5-WT alone (Fig. 5D and Table 2). In contrast, the NaV1.5-AA phosphomutant co-expressed with FGF13 showed voltage dependence of inactivation properties similar to those recorded from cells co-expressing NaV1.5-WT and FGF13. No changes in peak INa density, voltage dependence of activation, or kinetics of activation, inactivation, and recovery from inactivation were observed with either the NaV1.5-EE or NaV1.5-AA phosphomutants compared with the NaV1.5-WT co-expressed with FGF13 (Fig. 5, A–C, and Table 2). In addition, no changes in the voltage dependences of inactivation were observed between NaV1.5-WT (V½ = −84.5 ± 0.6 mV, n = 24), NaV1.5-EE (V½ = −83.9 ± 0.5 mV, n = 18), and NaV1.5-AA (V½ = −84.1 ± 0.7 mV, n = 13) in the absence of FGF13 (Fig. 5E), suggesting that FGF13 plays a pivotal role in mediating this phosphorylation-dependent effect. Finally, mimicking or abolishing phosphorylation at only one of the two phosphorylation sites (in cells co-expressing NaV1.5-S1933E, NaV1.5-S1933A, NaV1.5-S1984E, or NaV1.5-S1984A with FGF13) did not show any significant changes on peak INa density or channel biophysical properties compared with cells co-expressing NaV1.5-WT and FGF13 (Table 2).
Figure 5.

Phosphorylation at serines 1933 and 1984 disrupts the FGF13-dependent increase in steady-state NaV1.5 channel availability. A, representative whole-cell voltage-gated Na+ currents recorded from transiently transfected HEK293 cells. Currents were obtained 48 h following transfection of HEK293 cells with NaV1.5-WT (black), NaV1.5-WT + FGF13 (green), NaV1.5-S1933E/S1984E + FGF13 (NaV1.5-EE + FGF13, red), NaV1.5-S1933A/S1984A + FGF13 (NaV1.5-AA + FGF13, blue), NaV1.5-S1933E/S1984E (NaV1.5-EE, purple), and NaV1.5-S1933A/S1984A (NaV1.5-AA, pink) using the protocols illustrated in each panel. B, mean ± S.E. peak Na+ current (INa) densities are plotted as a function of test potential. C, voltage dependence of current activation. Mean ± S.E. normalized conductances (GNa) are plotted as a function of test potential and fitted using a Boltzmann equation. D, voltage dependence of steady-state current inactivation. Mean ± S.E. normalized current amplitudes are plotted as a function of prepulse potential and fitted using a Boltzmann equation. FGF13 significantly (p < 0.01 versus NaV1.5-WT, one-way ANOVA followed by the Dunnett's post hoc test) shifts the voltage dependence of NaV1.5 channel inactivation toward depolarized potentials, an effect reversed with the NaV1.5-EE phosphomutant (p < 0.01 versus NaV1.5-WT + FGF13, one-way ANOVA followed by the Dunnett's post hoc test). E, no significant changes in voltage dependence of steady-state current inactivation were observed between NaV1.5-WT, NaV1.5-EE, and NaV1.5-AA in the absence of FGF13. Detailed densities, properties, and statistics are provided in Table 2.
Table 2.
Voltage-gated Na+ current densities and properties in transiently transfected HEK293 cells
The peak Na+ current density (INa), time to peak, and time course of inactivation properties presented were determined from analyses of records obtained on depolarizations to −20 mV (HP = −120 mV). All values are means ± S.E. The number of cells analyzed is provided in parentheses. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus NaV1.5-WT; ##, p < 0.01 versus NaV1.5-WT + FGF13; one-way ANOVA followed by the Dunnett's post-hoc test.

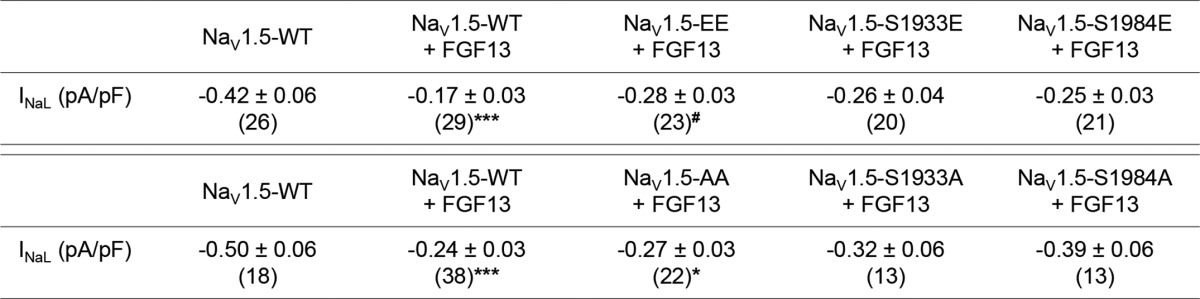
Additional voltage-clamp experiments were designed to examine the effects of FGF13 and of NaV1.5 phosphomutants on the late Na+ current. These analyses showed that the TTX-sensitive late Na+ current (INaL) density is significantly (p < 0.001) smaller in cells co-expressing NaV1.5 and FGF13 than in cells expressing NaV1.5 alone (Fig. 6, A and B, and Table 3). Interestingly, this effect was significantly (p < 0.05) abrogated in cells co-expressing NaV1.5-EE and FGF13 (Fig. 6, A and B, and Table 3). However, no such abrogation of the FGF13 effect was observed with the NaV1.5-AA phosphomutant (Fig. 6, C and D, and Table 3). In addition, similar to the biophysical properties, no significant effects on INaL could be detected in the absence of FGF13 (INaL = −0.32 ± 0.04 pA/pF, n = 27 for NaV1.5-WT; INaL = −0.46 ± 0.07 pA/pF, n = 28 for NaV1.5-EE; and INaL = −0.25 ± 0.05 pA/pF, n = 15 for NaV1.5-AA, see Fig. 6E) or with the simple phosphomutants (Table 3). Taken together, these results suggest that simultaneous phosphorylation at serines 1933 and 1984 on NaV1.5 impairs the effects of FGF13 on channel inactivation properties, which results in decreased channel availability and increased INaL.
Figure 6.

Phosphorylation at serines 1933 and 1984 disrupts the FGF13-dependent decrease in INaL. TTX-sensitive late Na+ current (INaL) recordings were obtained 48 h after transfection of HEK293 cells in three different data sets. The first data set (A and B) was obtained from cells expressing NaV1.5-WT (black), NaV1.5-WT + FGF13 (green), and NaV1.5-S1933E/1984E + FGF13 (NaV1.5-EE + FGF13, red); the second data set (C and D) from cells expressing NaV1.5-WT (black), NaV1.5-WT + FGF13 (green), and NaV1.5-S1933A/S1984A + FGF13 (NaV1.5-AA + FGF13, blue); and the third data set (E) from cells expressing NaV1.5-WT (black), NaV1.5-S1933E/S1984E (NaV1.5-EE, purple), and NaV1.5-S1933A/S1984A (NaV1.5-AA, pink). A and C, representative TTX-sensitive INaL recordings evoked by prolonged depolarizations (350 ms at −20 mV) from a holding potential of −120 mV. The scale bars indicate a current amplitude of 10 pA, which corresponds to a current density of 0.8 pA/pF and time (50 ms). B, D, and E, distributions and mean ± S.E. TTX-sensitive late Na+ current (INaL) densities. *, p < 0.05; ***, p < 0.001 versus NaV1.5-WT; #, p < 0.05 versus NaV1.5-WT + FGF13; ns, non-significant; Kruskal-Wallis one-way ANOVA followed by the Dunn's post hoc test. Detailed densities and statistics are provided in Table 3.
Table 3.
Late Na+ current densities in transiently transfected HEK293 cells
The TTX-sensitive late Na+ current (INaL) densities were measured at −20 mV (HP = −120 mV). All values are means ± S.E. The number of cells analyzed is provided in parentheses. *, p < 0.05; ***, p < 0.001 versus NaV1.5-WT; #, p < 0.05 versus NaV1.5-WT + FGF13; Kruskal-Wallis one-way ANOVA followed by the Dunn's post-hoc test.
Phosphorylation at serines 1933 and 1984 decreases the interaction of FGF13 and CaM with NaV1.5
To explore the hypothesis that phosphorylation at serines 1933 and 1984 in NaV1.5 impairs the regulation of channel inactivation mediated by FGF13 by altering the binding of FGF13 and/or CaM to the channel, co-immunoprecipitation experiments were completed using the same experimental paradigm as above. As illustrated in Fig. 7A, FGF13 and (endogenous) CaM co-immunoprecipitate with NaV1.5, whether in the WT, NaV1.5-AA, or NaV1.5-EE forms, in HEK293 cells. Interestingly, however, the relative abundances of FGF13 (Fig. 7B) and CaM (Fig. 7C) are significantly (p < 0.01 and p < 0.001, respectively) lower in the NaV1.5 immunoprecipitates from cells expressing the NaV1.5-EE phosphomutant compared with cells expressing the WT channels. No changes were observed with the NaV1.5-AA phosphomutant. Parallel negative controls of co-immunoprecipitations obtained from cells transiently transfected with FGF13 or NaV1.5-WT alone confirmed the specificity of detected signals. To determine whether phosphorylation at only one site was sufficient to decrease the interaction of FGF13 or CaM with the channel, co-immunoprecipitations were performed with the single alanine or glutamate phosphomutant channels. Consistent with the electrophysiological findings, no significant changes in binding affinity were observed with any of the single phosphomutant channels (Fig. 7, B and C).
Figure 7.

Phosphorylation at serines 1933 and 1984 decreases the interaction of FGF13 and consequently of CaM with NaV1.5. Forty eight hours following transfection of HEK293 cells with NaV1.5-WT, NaV1.5-S1933A/S1984A (NaV1.5-AA), NaV1.5-S1933E/S1984E (NaV1.5-EE), NaV1.5-S1933A (1933A), NaV1.5-S1933E (1933E), NaV1.5-S1984A (1984A), NaV1.5-S1984E (1984E), FGF13, and/or CaM, cell lysates were prepared and used for IPs with the mαNaVPAN antibody. A, D, and F, representative Western blots of the lysates (left panel) and the immunoprecipitates (right panel) with the monoclonal anti-NaVPAN, anti-FGF13, and/or anti-CaM antibodies. Relative mean ± S.E. FGF13 (B) and CaM (C) abundances in mαNaVPAN-IPs from cells expressing NaV1.5-WT (WT, n = 8), NaV1.5-AA (AA, n = 8), and NaV1.5-EE (EE, n = 8); NaV1.5-WT (WT, n = 5 and 4, respectively), 1933A (n = 6 and 4, respectively), and 1933E (n = 6 and 4, respectively); and NaV1.5-WT (WT, n = 6), 1984A (n = 6), and 1984E (n = 6). E, relative mean ± S.E. CaM abundances in mαNaVPAN-IPs from cells expressing NaV1.5-WT (n = 8) and NaV1.5-WT + FGF13 (n = 12), and FGF13 abundances in mαNaVPAN-IPs from cells expressing NaV1.5-WT + FGF13 (n = 4) and NaV1.5-WT + FGF13 + CaM (n = 4). **, p < 0.01; ***, p < 0.001 versus NaV1.5-WT, Kruskal-Wallis one-way ANOVA followed by the Dunn's post hoc test (B and C), or Mann-Whitney test (E). FGF13 and CaM abundances in each IP were first normalized to immunoprecipitated NaV1.5 and then expressed relative to FGF13 or CaM abundances in IPs from control conditions.
Additional co-immunoprecipitation experiments were then designed to determine whether FGF13 and CaM influence each other for the binding to the channel. Interestingly, these experiments revealed that co-expression of FGF13 significantly (p < 0.01) increases the binding of CaM to NaV1.5 (Fig. 7, D and E). Conversely, overexpression of CaM did not influence the binding of FGF13 to the channel (Fig. 7, D and E). Additionally, no changes in the binding of CaM to NaV1.5-EE or NaV1.5-AA, compared with NaV1.5-WT, were observed in the absence of FGF13 (Fig. 7F). Together, these biochemical analyses suggest that FGF13 potentiates the binding of CaM to NaV1.5 and that phosphorylation at both serines 1933 and 1984 decreases the interaction of FGF13 and, consequently, of CaM with the channel.
Discussion
The results presented here provide two novel phosphorylation maps of native mouse NaV1.5 channel subunits purified from control WT and failing CaMKIIδc-overexpressing ventricles, and they delineate two novel C-terminal phosphoserines, at positions 1938 and 1989, that are up-regulated in the failing CaMKIIδc-overexpressing ventricles. Mechanistic analyses in HEK293 cells revealed that mimicking phosphorylation at both sites (in the human sequence) impairs the regulation of NaV1.5 channels by FGF13, resulting in decreased channel availability and increased late Na+ current. Co-immunoprecipitation experiments demonstrated that FGF13 potentiates the binding of CaM to NaV1.5 and that FGF13 and, consequently, CaM bindings are decreased when phosphorylation at both sites is mimicked. Overall, these results provide evidence for a novel phosphorylation-dependent mechanism that acts at the level of the NaV1.5 channel macromolecular complex through regulation of specific protein/protein interactions.
CaMKIIδc-dependent phosphorylation map of native mouse NaV1.5 channels
The present phosphoproteomic analysis confidently identified a total of 19 native phosphorylation sites in the NaV1.5 channel proteins purified from mouse ventricles, of which nine are novel. Consistent with our previous MS analysis (31), and with another phosphoproteomic analysis of human NaV1.5 channels purified from HEK293 cells (9), the great majority (13 of 19) of identified phosphorylation sites are located in the first intracellular linker loop of the channel, suggesting critical roles for this region in mediating phosphorylation-dependent regulation of cardiac NaV1.5 channels. However, none of these 13 phosphorylation sites identified in the first linker loop, including phosphoserine 571 which was reported to be CaMKIIδc-dependent (11, 12), appeared to be regulated in the CaMKIIδc-Tg ventricles compared with the WT ventricles. Note that no relative quantification could be obtained for the low abundance phosphopeptides assigning phosphoserines 516 and 1888, and that the region surrounding threonine 594 was not covered (Fig. 2B), precluding possible detection and quantification of phosphothreonine 594. The only two/three phosphorylation sites showing significant and consistent abundance changes in the CaMKIIδc-Tg compared with the WT ventricles are located in the CTD of NaV1.5, at position(s) 1937 and/or 1938 and at position 1989. Although localization(s) of phosphorylation could not be discriminated between serines 1937 and 1938 in the singly phosphorylated peptide allowing quantification (presence in the CaMKIIδc-Tg IPs, and absence in the WT IPs), we focused our attention on serine 1938 (human serine 1933) because serine 1937 is not well-conserved across species and is notably absent in human. In addition, the human NaV1.5 phosphoserine 1933 has previously been shown to be CaMKIIδc-dependent in in vitro phosphoproteomic analyses (9), underscoring the potential involvement of CaMKIIδc in phosphorylating this site. Finally, it is also important to note here that the relative quantification of the doubly phosphorylated peptide supporting phosphorylation at both serines 1937 and 1938 (Table 1) could not be obtained because of low abundance, yet this peptide was only detected in the CaMKIIδc-Tg IPs. The number of phosphorylation sites identified here in association with the overexpression of CaMKIIδc in mouse ventricles (two phosphoserines) could seem little compared with the results of the previous phosphoproteomic study of human NaV1.5 channels in which 23 phosphorylation sites, of the 34 identified, were found to be phosphorylated in vitro by CaMKIIδc. These seemingly distinct findings might reflect the fact that different experimental approaches were employed. Indeed, although phosphorylation sites were identified from native, in situ phosphorylated NaV1.5 channels in this study, previous MS analysis was performed from heterologously-expressed NaV1.5 channels that were immunoaffinity-purified and subsequently subjected to in vitro phosphorylation by recombinant CaMKIIδc. It is also stressed here that although the present MS analysis was performed from ventricles in which CaMKIIδc is overexpressed, we cannot exclude the involvement of other kinases/phosphatases whose activity may also be changed. Consistent with the direct implication of CaMKIIδc, however, the in vitro phosphoproteomic analyses of human NaV1.5 demonstrated that human phosphoserine 1933 is CaMKIIδc-dependent (9). Likewise, it is interesting to note that mouse phosphoserine 1989 (and human phosphoserine 1984) is located in a well-conserved consensus CaMKII phosphorylation site (37). Together, these MS analyses from mouse ventricles therefore suggest that the two NaV1.5 phosphoserines at positions 1938 and 1989 are associated with the overexpression of CaMKIIδc.
FGF13-dependent regulation of NaV1.5 channel inactivation
Because our MS findings converged on the possible involvement of NaV1.5 C-terminal phosphorylation sites in mediating CaMKIIδc-dependent channel regulation and that the CTD of NaV channels is well-recognized for its role in regulating channel inactivation through the binding of iFGFs (15, 20–26) and CaM (13–19), we first characterized the roles of FGF13 in regulating NaV1.5 channels in HEK293 cells. Our findings are in accordance with previous data demonstrating a key role for FGF13 in increasing steady-state NaV1.5 channel availability, although the amplitude of the presently observed effect (∼2 mV) is smaller than previously reported effects of FGF13 or other iFGFs (∼5–10 mV) (15, 20–26). Interestingly, our data also provide a novel role for FGF13 in decreasing the late Na+ current in HEK293 cells. The role of FGF13, and in particular of FGF13-2, in regulating the late Na+ current had only been investigated once in tsA201 cells, and it was shown to have no effect (26). These different findings perhaps are due to differences in the heterologous expression systems used or are due to the fact that no NaVβ subunits were included in the previous study compared with this study in which NaV1.5 channels were co-expressed with FGF13–2 and NaVβ1. Consistent with this negative effect of FGF13 on the late Na+ current, the conditional knock-out of FGF13 in murine hearts revealed longer action potential durations (25), which could well be caused by an increased late Na+ current. The answer to this question must await further analysis of the late Na+ current from this or other FGF13 animal models. The observed decrease in peak Na+ current density upon FGF13 co-expression was somewhat surprising in light of previous findings in native cardiomyocytes demonstrating a decreased NaV1.5 channel cell-surface expression upon FGF13 knockdown (21) or a reduced peak Na+ current density in ventricular myocytes isolated from FGF13 conditional knock-out mice (25). This lack of consistency is nonetheless reminiscent of previous investigations of iFGF-dependent regulation of NaV channels from Pitt and co-workers, who reported the same disparate effects on peak Na+ current density in native (increase) (21, 25) and heterologous (decrease) (26) cells, and it may reflect differences in channel cellular and molecular environment. In addition, note that no differences in peak Na+ current densities were observed in ventricular myocytes isolated from FGF13 knock-out and WT mice (24). In summary, these results suggest that FGF13 primarily regulates the inactivation properties of NaV1.5 channels from both the closed state (increased availability) and the open state (decreased late Na+ current).
Impairment of FGF13-dependent NaV1.5 channel inactivation by phosphorylation
Determination of the impact of phosphorylation at serines 1933 and 1984 in the regulation of NaV1.5 channels first revealed that the binding of FGF13 and CaM to NaV1.5 is reduced with the double-glutamate phosphomutant compared with the WT channels, suggesting that phosphorylation at these serines disables the interaction of FGF13 and CaM with the channel. Most importantly, this reduced interaction, as assessed by co-immunoprecipitation, correlates with an impairment of functional modulation. Indeed, all the effects of FGF13 on NaV1.5-generated currents, but those on the peak density were nearly completely abolished with the double-glutamate phosphomutant, therefore resulting in decreased channel availability and increased late Na+ current. Together, these analyses show that mimicking phosphorylation at serines 1933 and 1984, which according to these and previous findings (9) are not phosphorylated at baseline in HEK293 cells, impairs FGF13-dependent regulation of NaV1.5 channel inactivation. Of note, these functional effects associated with the decreased FGF13 and CaM bindings are similar to those previously observed with several NaV1.5 mutants in which the binding of CaM to the NaV1.5 CTD is disrupted (17, 19, 29). Mimicking phosphorylation at only one site, however, did not lead to any consistent alterations in the interaction of FGF13 or CaM with the channel nor in the functional regulation by FGF13, suggesting that phosphorylation at both sites is necessary to disable FGF13/CaM bindings and associated dysregulation of channel inactivation. Consistent with these findings in HEK293 cells, our MS analyses also revealed different relative compositions of associated/regulatory proteins in NaV1.5 channel complexes in the CaMKIIδc-Tg ventricles compared with the WT ventricles. Of particular interest, indeed, the FGF13 and CaM relative abundances in the CaMKIIδc-Tg IPs compared with the WT IP, are lower (2-fold abundance ratios) than the 3-fold abundance ratios obtained for NaV1.5 or ankyrin-G. Together, therefore, these analyses suggest that phosphorylation at serines 1933 and 1984 impairs the binding of FGF13 and CaM to NaV1.5 and abrogates FGF13-dependent regulation of channel inactivation.
FGF13 potentiates the binding of CaM to NaV1.5, and phosphorylation-dependent disruption of FGF13 binding to NaV1.5 results in reduced CaM binding
To further delineate the relationship between FGF13 and CaM for the binding to NaV1.5 and subsequent phosphorylation-dependent regulation, the same electrophysiological and biochemical analyses were performed in the absence of FGF13. Interestingly, neither the channel availability nor the late Na+ current or the CaM/NaV1.5 interaction were altered in the absence of FGF13, highlighting the primary role of FGF13 in this phosphorylation-dependent regulation. These findings also suggest that the decreased CaM binding to the double-glutamate phosphomutant channel is subsequent to the decreased FGF13 binding. In this respect, further co-immunoprecipitation experiments were performed to examine the relationship between FGF13 and CaM for the binding to NaV1.5. Remarkably, these analyses revealed that FGF13 substantially increases the binding of CaM to the channel, demonstrating for the first time that the binding of CaM to NaV1.5 is potentiated by the binding of FGF13. Inversely, no changes in FGF13 binding were observed upon CaM overexpression, suggesting that CaM does not influence the binding of FGF13. Whether phosphorylation or FGF13 influence directly the Ca2+/CaM dependence of NaV1.5 channel inactivation remains to be investigated. Interestingly, these novel findings are evocative of recent observations from the Pitt group (29) demonstrating that the increased late Na+ current associated with several LQT3 syndrome mutations within the NaV1.5 CTD correlates with a decreased binding of apoCaM to the NaV1.5 CTD. In line with these findings, the recently reported LQT3-causing mutation at p.H1849R in the NaV1.5 CTD is also associated with an impairment of iFGF binding and iFGF-mediated regulation of NaV1.5 channels (28). Together with these two previous studies, our results herein support the overall hypothesis that the binding of FGF13 and/or CaM to the NaV1.5 CTD is pivotal in the regulation of channel inactivation, as unveiled by C-terminal phosphorylation and disease mutations within the NaV1.5 CTD, and that impairment of iFGF and/or CaM modulation may constitute a common mechanism to inherited (CTD-mutated LQT3 patients) and acquired (CaMKIIδc-associated) arrhythmias.
Potential broader implication of phosphorylation-dependent regulation of NaV1.5 channels in heart failure
Evidence suggests that some of the arrhythmogenic abnormalities associated with heart failure are triggered by the activation of CaMKII (2, 30) and a subsequent CaMKII-dependent increase in INaL (4–7). By identifying two novel NaV1.5 phosphorylation sites up-regulated in CaMKIIδc-Tg ventricles, which are failing (30), and by demonstrating roles for these sites in altering NaV1.5 channel inactivation, our results provide evidence for a model in which increased CaMKIIδc-dependent NaV1.5 phosphorylation at serines 1933 and 1984 may provide an arrhythmic substrate in heart failure. Nonetheless, it is unclear whether the functional consequences of impaired FGF13 and CaM binding to NaV1.5 by phosphorylation, as observed here in HEK293 cells, fully match up with the NaV channel defects associated with heart failure. Indeed, although the increased late Na+ current is a consistent finding in failing cardiomyocytes from both human and animal models (2, 4–7, 38), evidence for a decreased NaV channel availability has only been observed, to our knowledge, in the CaMKIIδc-Tg mice (7) and in a failing dog model induced by left-bundle branch ablation and right atrial pacing (2). Besides, the activation of CaMKII has consistently been associated with an increased INaL and a decreased NaV1.5 channel availability (3, 7, 8, 11, 12). Overall, the molecular mechanisms elucidated here may be relevant in conditions of increased CaMKIIδc expression and/or activity and may partly contribute to the arrhythmias associated with heart failure.
Experimental procedures
Animals were handled in accordance with the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes. Experimental protocols were approved by the local animal care and use committee (Comité d'Ethique pour l'Expérimentation Animale des Pays de la Loire, authorization CEAA.2010.9). Generation and characterization of the CaMKIIδc-Tg mouse line have been described previously (30). Cardiac tissues for in vitro experiments were harvested after euthanasia of the mice by cervical dislocation.
Immunoprecipitations of NaV channel complexes
Flash-frozen ventricles from four 13-month-old CaMKIIδc-Tg and four age- and sex-matched WT mice were homogenized as described previously (31) in ice-cold lysis buffer containing 20 mm HEPES (pH 7.4), 150 mm NaCl, 0.5% amidosulfobetaine (Sigma), 1× complete protease inhibitor mixture tablet (Roche Applied Science), 1 mm phenylmethylsulfonyl fluoride (PMSF, Interchim), 0.7 μg/ml pepstatin A (Thermo Fisher Scientific), and 1× Halt phosphatase inhibitor mixture (Thermo Fisher Scientific). After a 15-min rotation at 4 °C, 8 mg of the pooled WT or CaMKIIδc-Tg ventricular soluble protein fractions were pre-cleared with 200 μl of protein G-magnetic beads (Pierce) for 1 h and subsequently used for IPs with 48 μg of an anti-NaVPAN mouse monoclonal antibody (mαNaVPAN, Sigma, S8809), raised against the SP19 epitope (39) located in the third intracellular linker loop and common to all NaV α subunits. Prior to the IP, antibodies were cross-linked to 200 μl of protein G-magnetic beads using 20 mm dimethyl pimelimidate (Thermo Fisher Scientific) (40). Protein samples and antibody-coupled beads were mixed for 2 h at 4 °C. Magnetic beads were then collected and washed rapidly four times with ice-cold lysis buffer, and isolated protein complexes were eluted from the beads in 2% Rapigest (41) (Waters), 8 m urea (Sigma), 100 mm Tris (pH 8.5) at 37 °C for 30 min.
For co-immunoprecipitations of heterologously expressed proteins, HEK293 cells were washed twice with PBS and lysed in lysis buffer (as above) 48 h after transfection. Soluble protein fractions were collected and incubated with 12.5 μl of mαNaVPAN-coupled magnetic beads (as above). After a 2-h incubation at 4 °C, beads were washed four times in lysis buffer, and protein complexes were eluted with 1× SDS sample buffer at 60 °C for 5 min.
Gel electrophoreses and Western blot analyses
Ten percent of the immunoprecipitated mouse ventricular NaV channel protein complexes were fractionated on one-dimensional polyacrylamide gels and analyzed using either SYPRO Ruby (Life Technologies, Inc.) staining or Western blotting using the mαNaVPAN antibody (1:2000, Sigma, S8809) as described previously (31). The total ventricular lysates were blotted with a rabbit polyclonal anti-NaV1.5 antibody (1:2000, RbαNaV1.5, Alomone, ASC-005). Western blot analyses of protein eluates from co-immunoprecipitations of heterologously expressed proteins were performed using the following primary antibodies: mouse monoclonal anti-NaVPAN (1:2000, mαNaVPAN, Sigma, S8809); mouse monoclonal anti-FGF13 (1:300, clone N91/27, NeuroMab Facility, University of California Davis, NINDS/NIMH, National Institutes of Health); and rabbit monoclonal anti-CaM (1:300, Abcam, EP799Y). Bound antibodies were detected using horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse secondary antibodies (Santa Cruz Biotechnology), and protein signals were visualized using the SuperSignal West Dura Extended Duration Substrate (Pierce). The intensities of co-immunoprecipitated FGF13 or CaM bands were normalized to the intensities of immunoprecipitated NaV1.5 bands from the same IP sample, and FGF13 or CaM abundances in immunoprecipitations from experimental conditions are expressed relative to abundances in immunoprecipitations from control conditions.
In-solution endoprotease digestions
Samples for mass spectrometry were prepared as described previously (31). Briefly, eluted proteins were precipitated using the 2D protein clean-up kit (GE Healthcare). The resulting pellets were dissolved in 8 m urea, 100 mm Tris (pH 8.5), reduced with 5 mm tris(2-carboxyethyl)phosphine (pH 8.0) for 30 min at room temperature, and alkylated with 10 mm iodoacetamide (Bio-Rad) for 30 min at room temperature. Samples were then digested with 1 μg of endoproteinase Lys-C (Roche Applied Science) overnight at 37 °C and subsequently with 4 μg of trypsin (Sigma) overnight at 37 °C. Peptides were acidified with formic acid to a final concentration of 1%, extracted with NuTip porous graphite carbon wedge tips (Glygen), and eluted with aqueous acetonitrile (ACN, 60%) containing formic acid (FA, 0.1%). The extracted peptides were dried, dissolved in aqueous ACN/FA (1%/1%), stored at −80 °C, and subsequently analyzed using one-dimensional liquid chromatography-tandem mass spectrometric experiments (LC-MS/MS).
Mass spectrometric analyses
Peptide mixtures were analyzed using nano-LC-MS on three high-resolution hybrid mass spectrometers, a linear quadrupole ion trap Orbitrap XL (LTQ-Orbitrap XL) (31), an LTQ-Orbitrap Elite (42) (both from Thermo Fisher Scientific), and a TripleTOF® 5600 Plus (SCIEX) (43). The chromatograph was a 2D Plus (Eksigent) LC with a Nanoflex module and AS2 autosampler in dual cHiPLC columns (ChromXP C18 200 μm × 15 cm; particle size 3 μm, 120 Å) configuration. The mobile phases were 1% FA in water (A) and 1% FA in ACN (B). The liquid chromatographs were interfaced to the mass spectrometers through a nanospray source (PicoView PV550, New Objective). The samples were loaded in a volume of 5–10 μl at a flow rate of 1.5 μl/min followed by organic gradient elution of peptides (800 nl/min). The LC conditions for analyses on the LTQ-Orbitrap XL and LTQ-Orbitrap Elite were performed after equilibrating the columns in 98% solvent A and 2% solvent B followed by 2% B, 0–5 min; 2- 25% B, 5–110 min; 25–80% B, 110–170 min; 80–2% B, 170–175 min; and isocratic elution at 2% B, 175–190 min. The survey scans (m/z 350–2000) (MS1) were acquired at high resolution (60,000 at m/z 400) in the Orbitrap XL, and the MS2 spectra were acquired in the linear ion trap at low resolution, both in profile mode. The maximum injection times for the MS1 scans in the Orbitrap and the LTQ were 500 and 200 ms, respectively. The automatic gain control targets for the Orbitrap and the LTQ were 5 × 105 and 3 × 104, respectively, with maximum injection times of 100 ms for the MS2 scans. The MS1 scans were followed by three MS2 events in the linear ion trap with collision activation in the ion trap (parent threshold 1000, isolation width 2.0 Da, normalized collision energy 30%, activation Q 0.250, and activation time 30 ms). Dynamic exclusion was enabled (−0.20/+1.0 Da) for 90 s after MS2 acquisitions. A repeat count of 1, a repeat duration of 45 s, and a maximal exclusion list size of 500 were used. The following ion source parameters were used: capillary temperature 200 °C, source voltage 3.0 kV, source current 100 μA, capillary voltage 33 V, and tube lens 120 V. Data were acquired using XCalibur, version 2.2 SP1 (Thermo Fisher Scientific). For data acquisition on the LTQ-Orbitrap Elite, the following modifications were employed. The survey scans (m/z = 300–1650) were acquired at a resolution of 120,000 with a target value of 1e6 ions. The HCD MS2 acquisitions were performed for the top 15 most intense ions at a resolution of 15,000, a target value of 40,000 and a low mass setting of 120 m/z. For collision-induced dissociation, a collision energy of 40% was used with an activation time of 100 ms. Dynamic exclusion was enabled (30 s) without MS2 acquisition of +1 charged parent ions. The data were acquired using Xcalibur, version 2.0.7 (Thermo Fisher Scientific).
The LC-MS analysis on the TripleTOF® 5600 Plus mass spectrometer was obtained using the following gradient elution program: 0 time, 98% A, 2% B; 5 min, 2% A, 98% B; 415 min, 65% A, 35% B; and 440 min, 20% A, 80% B. Initial chromatographic conditions were restored in 5 min and maintained for 20 min. The ion spray voltage was set at 2.9 kV, the curtain gas at 10 p.s.i., the nebulizer gas at 14 p.s.i., and the interface heater temperature set at 175 °C. The MS1 data were acquired at a resolution of 25,000 with a scan range of 400–1200 m/z in 250 ms. The top 50 product ions were selected for MS2 acquisition with a dwell time of 100 ms. Four time bins were summed for each scan at a frequency of 15.4 kHz (through monitoring of the 40 GHz multichannel TDC detector with four-anode/channel detection). A rolling collision energy (CE) was applied to all precursor ions for collision-induced dissociation using the following equation: CE = slope·m/z + intercept, where the slope for all charge states above +2 is 0.0625 and the intercept is −3, −5, and −6 for +2, +3, and +4, respectively.
MS data processing and analyses
The LC-MS raw files from the LTQ-Orbitrap XL and Elite were processed using MASCOT Distiller (version 2.3.02, Matrix Science) with settings previously described (31). The TripleTOF data were processed using the SCIEX MS Data Converter (version 1.3, SCIEX), converting the raw data files (*.wiff) to mgf files. The resulting MS2 centroid files were used for database searching with MASCOT (version 2.3.02) against the UNIPROT mouse protein database (downloaded on May 2, 2011, with 72,510 entries for the XL data, and on December 20, 2011, with 77,200 entries for the Elite and the TripleTOF data) using the following parameters: trypsin as the enzyme, MS tolerances of 50 (XL), 10 (Elite), and 25 (TripleTOF) ppm; MS/MS tolerances of 0.8 (XL), 0.05 (Elite), and 0.1 (TripleTOF) Da, with a fixed carbamidomethylation of Cys residues and variable modifications being oxidation (Met), pyro-glutamination (Gln), and phosphorylation (Ser, Thr, and/or Tyr), a maximal number of missed cleavages of 4, and +1, +2, +3, and +4 charge states. Scaffold (versions 3.1.4.1 (XL), 4.4.3 (Elite) and 3.6.4 (TripleTOF), Proteome software) was used to validate MS2-based peptide and protein identifications with the Peptide (44) and Protein (45) Prophet algorithms using thresholds of 50 and 95%, respectively. The supplemental Tables 1–3 provide the complete lists of identified peptides and proteins using the LTQ-Orbitrap XL, Elite, and the TripleTOF mass spectrometers, respectively.
Phosphopeptide spectra were manually interpreted by comparing the observed mass values from the spectrum in XCalibur (LTQ-Orbitrap XL and Elite data) or PeakView (TripleTOF data) with the theoretical parent and fragment NaV1.5 ion mass values from MS-Product. Annotations of MS2 spectra were first automated using java-based software, which matches observed m/z values with theoretical fragment masses from MS-Product, and definitive annotations were subsequently obtained by manual verification and interpretation. Mass accuracy tolerances of 20 ppm or 0.5 Da were used as guidelines for high- and low-resolution spectral annotations, respectively, and only those ions with mass errors within these ranges were included to determine residue coverage and location of phosphorylation site(s). Additionally, for spectral annotation of high-resolution MS2 data, charge states of observed parent and fragment ions were determined, and only precursor, b- and y-ions with confirmed charge states (i.e. with at least the presence of the 13C isotope peak) were used. The phosphorylation site assignments were based on the presence or absence of the unphosphorylated and phosphorylated b- and y-ions flanking the site(s) of phosphorylation, ions that we call site-discriminating ions throughout this study. When site-discriminating ions were not all detected, assignments of phosphorylation sites were narrowed down to one, two (for pSer-524 and/or pSer-525), or four (for pSer-36, pThr-38, pSer-39, and/or pSer-42) possibility(ies) by elimination. In addition to mass accuracy and charge state, the relative percentage of maximum intensity of site-discriminating ions or of any supporting ions in each analyzed spectrum was also considered for spectral annotation. Representative MS1 and MS2 spectra used for each of the phosphorylation site assignments, mass errors of parent ions (in ppm), and Mascot Ion scores for each phosphopeptide are presented in supplemental Fig. 1. The definition of all observed site-discriminating ions, as well as the calculated mass errors and charge state confirmations for all supporting b- and y-ions (as well as for, when detected, the loss of phosphoric acid peaks) are summarized in Table 1 and supplemental Tables 4.
The label-free quantitative MS analysis of protein abundance in the IPs was performed from the entire (LTQ-Orbitrap XL) MS1 peptide data set using the DAnTE (Data Analysis Tool Extension) software (33–35). To perform label-free quantitative analyses of mass spectra of precursor ion (MS1 peptides) data, the LTQ-Orbitrap XL MS1 and MS2 data from quadruplicate analyses of mαNaVPAN-IPs from WT and CaMKIIδc-Tg mouse ventricles were imported into Rosetta ElucidatorTM (version 3.3, Rosetta Biosoftware) for retention time and m/z alignment of the peptide ion chromatograms using previously described parameters (36). Peak intensities of MS1 peptide features were quantified, and normalization of intensities across samples was performed using the average signal intensities obtained in each sample. Ion chromatograms and isotopic distributions of aligned NaV1.5 peptides were all visually inspected.
Plasmids
The simple and double NaV1.5 phosphomutant constructs were generated by mutating the serines 1933 and/or 1984 to alanine(s) or glutamate(s) by site-directed mutagenesis of the pCI-NaV1.5 plasmid containing the human NaV1.5 hH1C cDNA (46) (NCBI Reference Sequence NM_000335) using the QuikChange II XL site-directed mutagenesis kit (Agilent Technologies) with the following primers (with mutations underlined): NaV1.5-S1933A-F, 5′-cagcaggcgggcgccggcctctccga, and NaV1.5-S1933A-R, 5′-tcggagaggccggcgcccgcctgctg; NaV1.5-S1933E-F, 5′-cgtcagcaggcgggcgaaggcctctccgaagag, and NaV1.5-S1933E-R 5′-ctcttcggagaggccttcgcccgcctgctgacg; NaV1.5-S1984A-F,5′-tgtcactagagccaccgccgataacctccaggtg, and NaV1.5-S1984A-R 5′-cacctggaggttatcggcggtggctctagtgac; NaV1.5-S1984E-F 5′-agtgtcactagagccaccgaagataacctccaggtgcgg, and NaV1.5-S1984E-R5′-ccgcacctggaggttatcttcggtggctctagtgacact. The mutated constructs were then digested with restriction endonucleases to excise the mutated fragments, which were then subcloned into the original pCI-NaV1.5 plasmid. The human NaVβ1 (NM_001037, a gift from A. L. George) and rat CaM (NM_017326, a gift from I. Deschenes) cDNAs were subcloned into pRc/CMV and pcDNA3, respectively. The human transcript variant 2 of FGF13 (FGF13–2, NM_001139500), subcloned into pCMV6-XL5, was purchased from Amsbio. All constructs were sequenced to ensure that no unintentional mutations were introduced.
Culture and transient transfections
HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Life Technologies, Inc.) supplemented with 10% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin, in 37 °C, 5% CO2, 95% air incubator. Cells were transiently transfected at 70–80% confluence in 35-mm dishes with 0.9 μg of the WT or phosphomutant NaV1.5 plasmid, 0.45 μg of the NaVβ1 plasmid, with or without 0.45 μg of the FGF13 plasmid using 2 μl of Lipofectamine 2000 (Life Technologies, Inc.) following the manufacturer's instructions. For patch-clamp recordings, transfections also contained 0.2 μg of the pEGFP plasmid (enhanced green fluorescent protein plasmid, Clontech), so that the enhanced GFP expression serves as a marker of transfection. The absolute amounts of the various constructs were calculated, and the empty pcDNA3.1 plasmid (Life Technologies, Inc.) was used as a filler plasmid to keep the total DNA constant at 2 μg in each transfection.
Electrophysiological recordings
Whole-cell NaV currents were recorded at room temperature from transiently transfected HEK293 cells using an Axopatch 200A amplifier (Axon Instruments) 48 h after transfection. Voltage-clamp protocols were applied using the pClamp 10.2 software package (Axon Instruments) interfaced to the electrophysiological equipment using a Digidata 1440A digitizer (Axon Instruments). Current signals were filtered at 10 kHz prior to digitization at 50 kHz and storage. Patch-clamp pipettes were pulled from borosilicate glass (outer diameter is 1.5 mm and inner diameter is 0.86 mm, Sutter Instrument) using a P-97 micropipette puller (Sutter Instrument), coated with wax, and fire-polished to a resistance between 1.5 and 2.5 megohms when filled with internal solution. The internal solution contained (in mm): NaCl 5, CsCl 105, HEPES 10, glucose 5, EGTA 10, CaCl2 8.7 (1 μm free [Ca2+], calculated with MaxChelator) and Mg-ATP 5 (1 mm free [Mg2+], MaxChelator) (pH 7.2 with CsOH, ∼300 mosm). The external (low Na+) solution contained (in mm): NaCl 25, CsCl 94, tetraethylammonium chloride 25, HEPES 10, glucose 5, CaCl2 1, MgCl2 2 (pH 7.4 with CsOH, ∼300 mosm). All the chemicals were purchased from Sigma. After establishing the whole-cell configuration, 3 min were allowed to ensure stabilization of the voltage dependence of inactivation properties, at which time 25-ms voltage steps to ±10 mV from a holding potential (HP) of −70 mV were applied to allow measurement of whole-cell membrane capacitance, input, and series resistances. Only cells with access resistance <7 megohms were used, and input resistances were typically >5 giga-ohms. After compensation of series resistance (80%), the membrane was held at a HP of −120 mV, and the voltage-clamp protocols were carried out as indicated below. Leak currents were always <200 pA at the HP (−120 mV) and corrected off line. Cells exhibiting peak current amplitudes <500 or >5000 pA were excluded from analyses.
Data were compiled and analyzed using ClampFit 10.2 (Axon Instruments), Microsoft Excel, and Prism (GraphPad Software). Whole-cell membrane capacitance (Cm) was determined by analyzing the decays of capacitive transients elicited by the 25-ms voltage steps to ± 10 mV from the HP (−70 mV) prior to compensation. Cm was calculated by dividing the integrated capacitive transients by the voltage. Input resistance was calculated from the steady-state currents elicited by the same ±10-mV steps (from the HP). Series resistance was calculated by dividing the decay time constants of the capacitive transients (fitted with a single exponential) by the Cm. To determine the current-voltage relationships, currents were elicited by 50-ms depolarizing pulses from −80 to +40 mV in 5-mV increments from a HP of −120 mV (5-s inter-pulse duration). Peak current amplitudes at each voltage step were defined as the maximal current amplitudes. Current amplitudes were corrected by leak currents (calculated at each voltage step from the leak current measured at the HP) and normalized to the Cm. To analyze the voltage dependence of activation properties, current amplitudes at each voltage step were transformed to conductances (G), and conductance-voltage relationships were fitted with the Boltzmann equation G = Gmax/(1 + exp (− (Vm − V½)/k)), in which V½ is the membrane potential for half-activation, and k is the slope factor. The time course of inactivation of macroscopic current was fitted with the double exponential function I(t) = Aslow × exp(−t/τslow) + Afast × exp(−t/τfast) + A0, where Aslow and Afast are the amplitudes of the slow and fast inactivating current components, respectively, and τslow and τfast are the decay time constants of Aslow and Afast, respectively. A standard two-pulse protocol was used to generate the voltage dependence of steady-state inactivation curves; from a HP of −120 mV, cells were stepped to 1-s conditioning potentials varying from −120 to −50 mV (prepulse) in 5-mV increments, followed by 20-ms test pulses to −20 mV (5-s interpulse duration). Current amplitudes measured at each test pulse were normalized to the maximal current amplitude (Imax), and the steady-state inactivation curves were fitted with the Boltzmann equation I = Imax/(1 + exp ((Vm − V½)/k)), in which V½ is the membrane potential for half-inactivation, and k is the slope factor. Recovery from inactivation was assessed using a three-pulse protocol (5-s interpulse duration) with a test pulse (at −20 mV) at variable times (from 1 to 200 ms at −120 mV) after a 1-s conditioning pulse to −20 mV (from a HP of −120 mV). The time course of recovery from inactivation was analyzed by fitting the current amplitudes measured at each test pulse normalized to the current amplitudes measured during each conditioning pulse with the single exponential function I(t) = A × (1 − exp(−t/τrec)).
In experiments aimed at recording the TTX-sensitive late Na+ current, cells were bathed in external (full Na+) solution containing (in mm) the following: NaCl 140, CsCl 5, HEPES 10, glucose 5, CaCl2 1, MgCl2 2 (pH 7.4 with CsOH, ∼300 mosm). Repetitive 350-ms test pulses to −20 mV from a HP of −120 mV (5-s interpulse duration) were applied to cells superfused locally with external (full Na+) solutions supplemented with 20 mm mannitol, in the absence and in the presence of 30 μm TTX (Tocris Bioscience). Only cells exhibiting peak current amplitudes of >4000 pA were used (those with peak current of <4000 pA did not show measurable late Na+ current), and cells with difference in leak current amplitudes before and after TTX application of >2 pA at −20 mV (calculated from leak currents at −120 mV) were excluded from analyses. TTX-sensitive currents from individual cells were determined by off-line digital subtraction of average leak-subtracted currents obtained from five recordings in the absence and in the presence of TTX after achieving steady state. The amplitude of TTX-sensitive late Na+ current was defined as the steady-state current amplitude (A0) obtained by fitting the inactivation decay of macroscopic TTX-sensitive current with the double exponential function I(t) = Aslow × exp(−t/τslow) + Afast × exp(−t/τfast) + A0. For each cell, the TTX-sensitive late Na+ current amplitude was normalized to the Cm, and expressed as TTX-sensitive late Na+ current density (in pA/pF).
Statistical analyses
Results are expressed as means ± S.E. Data were first tested for normality using the D'Agostino and Pearson normality test. Depending on the results of normality tests, statistical analyses were then performed using the Mann-Whitney nonparametric test, the one-way ANOVA followed by the Dunnett's post hoc test, or the Kruskal-Wallis one-way ANOVA followed by the Dunn's post hoc test as indicated in the figures and tables. All of these analyses, the Tukey whisker analysis and the plots, were performed using Prism (GraphPad software).
Author contributions
C. M. designed the study and wrote the paper. C. M., M. R. M., C. F. L., J. M. N., R. R. T., and L. S. M. designed, performed, and/or analyzed the mass spectrometry experiments shown in Figs. 1–4 and Table 1. S. B., F. C. C., M. L., G. L., F. C., and C. M. designed, performed, and/or analyzed the experiments shown in Figs. 5–7 and Tables 2 and 3. J. H. B. and L. S. M. provided the CaMKIIδc-Tg mice. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
The expert technical assistance of Aurore Girardeau, Petra Erdmann-Gilmore, Alan E. Davis, and James P. Malone is gratefully acknowledged.
This work was supported in part by Agence Nationale de la Recherche Grants ANR-15-CE14-0006-01 (to C. M.) and ANR-12-BSV1-0013-01 (to F. C.), Marie Curie 7th Framework Program of the European Commission Grant PIRG06-GA-2009-256397 (to C. M.), the Fondation d'Entreprise Genavie (to C. M.), National Institutes of Health Grants UL1 TR000448 and P41 GM103422-35 (to R. R. T.) and R01 HL034161 (to J. M. N.), Groupe de Réflexion sur la Recherche Cardiovasculaire-Société Française de Cardiologie Predoctoral Fellowship eOTP 1518DASS (to S. B.), and the Deutsche Forschungsgemeinschaft Ma 1982/5-1 (to L. S. M.). The authors declare that they have no conflicts of interest with the contents of this article.The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Tables S1–S4 and Fig. S1.
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- CaMKIIδc-Tg
- transgenic mouse overexpressing the cytosolic isoform of the δ subunit of Ca2+/calmodulin-dependent protein kinase II
- CTD
- C-terminal domain of NaV channels
- iFGF
- intracellular fibroblast growth factor
- IP
- immunoprecipitation
- MS1
- mass spectrum of precursor ions
- mαNaVPAN
- anti-NaV α subunit monoclonal antibody
- NaV α subunit
- voltage-gated Na+ (NaV) channel pore-forming (α) subunit
- TTX
- tetrodotoxin
- FA
- formic acid
- ACN
- acetonitrile
- ANOVA
- analysis of variance
- pF
- picofarad
- HP
- holding potential
- CaM
- calmodulin
- m
- mouse.
References
- 1. Remme C. A., and Bezzina C. R. (2010) Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc. Ther. 28, 287–294 [DOI] [PubMed] [Google Scholar]
- 2. Aiba T., Barth A. S., Hesketh G. G., Hashambhoy Y. L., Chakir K., Tunin R. S., Greenstein J. L., Winslow R. L., Kass D. A., and Tomaselli G. F. (2013) Cardiac resynchronization therapy improves altered Na channel gating in canine model of dyssynchronous heart failure. Circ. Arrhythm. Electrophysiol. 6, 546–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dybkova N., Wagner S., Backs J., Hund T. J., Mohler P. J., Sowa T., Nikolaev V. O., and Maier L. S. (2014) Tubulin polymerization disrupts cardiac β-adrenergic regulation of late INa. Cardiovasc. Res. 103, 168–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maltsev V. A., Reznikov V., Undrovinas N. A., Sabbah H. N., and Undrovinas A. (2008) Modulation of late sodium current by Ca2+, calmodulin, and CaMKII in normal and failing dog cardiomyocytes: similarities and differences. Am. J. Physiol. Heart Circ. Physiol. 294, H1597–H1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Maltsev V. A., and Undrovinas A. (2008) Late sodium current in failing heart: friend or foe? Prog. Biophys. Mol. Biol. 96, 421–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Toischer K., Hartmann N., Wagner S., Fischer T. H., Herting J., Danner B. C., Sag C. M., Hund T. J., Mohler P. J., Belardinelli L., Hasenfuss G., Maier L. S., and Sossalla S. (2013) Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J. Mol. Cell. Cardiol. 61, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wagner S., Dybkova N., Rasenack E. C., Jacobshagen C., Fabritz L., Kirchhof P., Maier S. K., Zhang T., Hasenfuss G., Brown J. H., Bers D. M., and Maier L. S. (2006) Ca2+/calmodulin-dependent protein kinase II regulates cardiac Na+ channels. J. Clin. Invest. 116, 3127–3138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ashpole N. M., Herren A. W., Ginsburg K. S., Brogan J. D., Johnson D. E., Cummins T. R., Bers D. M., and Hudmon A. (2012) Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1.5 gating by multiple phosphorylation sites. J. Biol. Chem. 287, 19856–19869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Herren A. W., Weber D. M., Rigor R. R., Margulies K. B., Phinney B. S., and Bers D. M. (2015) CaMKII phosphorylation of Na(V)1.5: novel in vitro sites identified by mass spectrometry and reduced S516 phosphorylation in human heart failure. J. Proteome Res. 14, 2298–2311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Glynn P., Musa H., Wu X., Unudurthi S. D., Little S., Qian L., Wright P. J., Radwanski P. B., Gyorke S., Mohler P. J., and Hund T. J. (2015) Voltage-gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation 132, 567–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hund T. J., Koval O. M., Li J., Wright P. J., Qian L., Snyder J. S., Gudmundsson H., Kline C. F., Davidson N. P., Cardona N., Rasband M. N., Anderson M. E., and Mohler P. J. (2010) A β(IV)-spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J. Clin. Invest. 120, 3508–3519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koval O. M., Snyder J. S., Wolf R. M., Pavlovicz R. E., Glynn P., Curran J., Leymaster N. D., Dun W., Wright P. J., Cardona N., Qian L., Mitchell C. C., Boyden P. A., Binkley P. F., Li C., et al. (2012) Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation 126, 2084–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chagot B., and Chazin W. J. (2011) Solution NMR structure of apo-calmodulin in complex with the IQ motif of human cardiac sodium channel NaV1.5. J. Mol. Biol. 406, 106–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gabelli S. B., Boto A., Kuhns V. H., Bianchet M. A., Farinelli F., Aripirala S., Yoder J., Jakoncic J., Tomaselli G. F., and Amzel L. M. (2014) Regulation of the NaV1.5 cytoplasmic domain by calmodulin. Nat. Commun. 5, 5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang C., Chung B. C., Yan H., Lee S. Y., and Pitt G. S. (2012) Crystal structure of the ternary complex of a NaV C-terminal domain, a fibroblast growth factor homologous factor, and calmodulin. Structure 20, 1167–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang C., Chung B. C., Yan H., Wang H. G., Lee S. Y., and Pitt G. S. (2014) Structural analyses of Ca2+/CaM interaction with NaV channel C-termini reveal mechanisms of calcium-dependent regulation. Nat. Commun. 5, 4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim J., Ghosh S., Liu H., Tateyama M., Kass R. S., and Pitt G. S. (2004) Calmodulin mediates Ca2+ sensitivity of sodium channels. J. Biol. Chem. 279, 45004–45012 [DOI] [PubMed] [Google Scholar]
- 18. Sarhan M. F., Tung C. C., Van Petegem F., and Ahern C. A. (2012) Crystallographic basis for calcium regulation of sodium channels. Proc. Natl. Acad. Sci. U.S.A. 109, 3558–3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shah V. N., Wingo T. L., Weiss K. L., Williams C. K., Balser J. R., and Chazin W. J. (2006) Calcium-dependent regulation of the voltage-gated sodium channel hH1: intrinsic and extrinsic sensors use a common molecular switch. Proc. Natl. Acad. Sci. U.S.A. 103, 3592–3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goetz R., Dover K., Laezza F., Shtraizent N., Huang X., Tchetchik D., Eliseenkova A. V., Xu C. F., Neubert T. A., Ornitz D. M., Goldfarb M., and Mohammadi M. (2009) Crystal structure of a fibroblast growth factor homologous factor (FHF) defines a conserved surface on FHFs for binding and modulation of voltage-gated sodium channels. J. Biol. Chem. 284, 17883–17896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang C., Hennessey J. A., Kirkton R. D., Wang C., Graham V., Puranam R. S., Rosenberg P. B., Bursac N., and Pitt G. S. (2011) Fibroblast growth factor homologous factor 13 regulates Na+ channels and conduction velocity in murine hearts. Circ. Res. 109, 775–782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang C., Wang C., Hoch E. G., and Pitt G. S. (2011) Identification of novel interaction sites that determine specificity between fibroblast growth factor homologous factors and voltage-gated sodium channels. J. Biol. Chem. 286, 24253–24263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bosch M. K., Carrasquillo Y., Ransdell J. L., Kanakamedala A., Ornitz D. M., and Nerbonne J. M. (2015) Intracellular FGF14 (iFGF14) is required for spontaneous and evoked firing in cerebellar Purkinje neurons and for motor coordination and balance. J. Neurosci. 35, 6752–6769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Park D. S., Shekhar A., Marra C., Lin X., Vasquez C., Solinas S., Kelley K., Morley G., Goldfarb M., and Fishman G. I. (2016) Fhf2 gene deletion causes temperature-sensitive cardiac conduction failure. Nat. Commun. 7, 12966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang X., Tang H., Wei E. Q., Wang Z., Yang J., Yang R., Wang S., Zhang Y., Pitt G. S., Zhang H., and Wang C. (2017) Conditional knockout of Fgf13 in murine hearts increases arrhythmia susceptibility and reveals novel ion channel modulatory roles. J. Mol. Cell. Cardiol. 104, 63–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang J., Wang Z., Sinden D. S., Wang X., Shan B., Yu X., Zhang H., Pitt G. S., and Wang C. (2016) FGF13 modulates the gating properties of the cardiac sodium channel Nav1.5 in an isoform-specific manner. Channels 10, 410–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gabelli S. B., Yoder J. B., Tomaselli G. F., and Amzel L. M. (2016) Calmodulin and Ca2+ control of voltage gated Na+ channels. Channels 10, 45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Musa H., Kline C. F., Sturm A. C., Murphy N., Adelman S., Wang C., Yan H., Johnson B. L., Csepe T. A., Kilic A., Higgins R. S., Janssen P. M., Fedorov V. V., Weiss R., Salazar C., et al. (2015) SCN5A variant that blocks fibroblast growth factor homologous factor regulation causes human arrhythmia. Proc. Natl. Acad. Sci. U.S.A. 112, 12528–12533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yan H., Wang C., Marx S. O., and Pitt G. S. (2017) Calmodulin limits pathogenic Na+ channel persistent current. J. Gen. Physiol. 149, 277–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang T., Maier L. S., Dalton N. D., Miyamoto S., Ross J. Jr., Bers D. M., and Brown J. H. (2003) The δC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ. Res. 92, 912–919 [DOI] [PubMed] [Google Scholar]
- 31. Marionneau C., Lichti C. F., Lindenbaum P., Charpentier F., Nerbonne J. M., Townsend R. R., and Mérot J. (2012) Mass spectrometry-based identification of native cardiac Nav1.5 channel α subunit phosphorylation sites. J. Proteome Res. 11, 5994–6007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Abriel H., Rougier J. S., and Jalife J. (2015) Ion channel macromolecular complexes in cardiomyocytes: roles in sudden cardiac death. Circ. Res. 116, 1971–1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Karpievitch Y., Stanley J., Taverner T., Huang J., Adkins J. N., Ansong C., Heffron F., Metz T. O., Qian W. J., Yoon H., Smith R. D., and Dabney A. R. (2009) A statistical framework for protein quantitation in bottom-up MS-based proteomics. Bioinformatics 25, 2028–2034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Polpitiya A. D., Qian W. J., Jaitly N., Petyuk V. A., Adkins J. N., Camp D. G. 2nd, Anderson G. A., and Smith R. D. (2008) DAnTE: a statistical tool for quantitative analysis of -omics data. Bioinformatics 24, 1556–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Taverner T., Karpievitch Y. V., Polpitiya A. D., Brown J. N., Dabney A. R., Anderson G. A., and Smith R. D. (2012) DanteR: an extensible R-based tool for quantitative analysis of -omics data. Bioinformatics 28, 2404–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Neubert H., Bonnert T. P., Rumpel K., Hunt B. T., Henle E. S., and James I. T. (2008) Label-free detection of differential protein expression by LC/MALDI mass spectrometry. J. Proteome Res. 7, 2270–2279 [DOI] [PubMed] [Google Scholar]
- 37. Songyang Z., Lu K. P., Kwon Y. T., Tsai L. H., Filhol O., Cochet C., Brickey D. A., Soderling T. R., Bartleson C., Graves D. J., DeMaggio A. J., Hoekstra M. F., Blenis J., Hunter T., and Cantley L. C. (1996) A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol. Cell. Biol. 16, 6486–6493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Valdivia C. R., Chu W. W., Pu J., Foell J. D., Haworth R. A., Wolff M. R., Kamp T. J., and Makielski J. C. (2005) Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell. Cardiol. 38, 475–483 [DOI] [PubMed] [Google Scholar]
- 39. Vassilev P. M., Scheuer T., and Catterall W. A. (1988) Identification of an intracellular peptide segment involved in sodium channel inactivation. Science 241, 1658–1661 [DOI] [PubMed] [Google Scholar]
- 40. Schneider C., Newman R. A., Sutherland D. R., Asser U., and Greaves M. F. (1982) A one-step purification of membrane proteins using a high efficiency immunomatrix. J. Biol. Chem. 257, 10766–10769 [PubMed] [Google Scholar]
- 41. Yu Y. Q., Gilar M., and Gebler J. C. (2004) A complete peptide mapping of membrane proteins: a novel surfactant aiding the enzymatic digestion of bacteriorhodopsin. Rapid Commun. Mass Spectrom. 18, 711–715 [DOI] [PubMed] [Google Scholar]
- 42. Michalski A., Damoc E., Lange O., Denisov E., Nolting D., Muller M., Viner R., Schwartz J., Remes P., Belford M., Dunyach J. J., Cox J., Horning S., Mann M., and Makarov A. (2012) Ultra high-resolution linear ion trap Orbitrap mass spectrometer (Orbitrap Elite) facilitates top down LC MS/MS and versatile peptide fragmentation modes. Mol. Cell. Proteomics 11, O111–013698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Andrews G. L., Simons B. L., Young J. B., Hawkridge A. M., and Muddiman D. C. (2011) Performance characteristics of a new hybrid quadrupole time-of-flight tandem mass spectrometer (TripleTOF 5600). Anal. Chem. 83, 5442–5446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Keller A., Nesvizhskii A. I., Kolker E., and Aebersold R. (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 45. Nesvizhskii A. I., Keller A., Kolker E., and Aebersold R. (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 46. Makielski J. C., Ye B., Valdivia C. R., Pagel M. D., Pu J., Tester D. J., and Ackerman M. J. (2003) A ubiquitous splice variant and a common polymorphism affect heterologous expression of recombinant human SCN5A heart sodium channels. Circ. Res. 93, 821–828 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.