

Abstract
The neonatal Fc receptor FcRn plays a critical role in the trafficking of IgGs across tissue barriers and in retaining high circulating concentrations of both IgG and albumin. Although generally beneficial from an immunological perspective in maintaining IgG populations, FcRn can contribute to the pathogenesis of autoimmune disorders when an abnormal immune response targets normal biological components. We previously described a monoclonal antibody (DX-2507) that binds to FcRn with high affinity at both neutral and acidic pH, prevents the simultaneous binding of IgG, and reduces circulating IgG levels in preclinical animal models. Here, we report a 2.5 Å resolution X-ray crystal structure of an FcRn–DX-2507 Fab complex, revealing a nearly complete overlap of the IgG–Fc binding site in FcRn by complementarity-determining regions in DX-2507. This overlap explains how DX-2507 blocks IgG binding to FcRn and thereby shortens IgG half-life by preventing IgGs from recycling back into circulation. Moreover, the complex structure explains how the DX-2507 interaction is pH-insensitive unlike normal Fc interactions and how serum albumin levels are unaffected by DX-2507 binding. These structural studies could inform antibody-based therapeutic approaches for limiting the effects of IgG-mediated autoimmune disease.
Keywords: antibody, autoimmune disease, Fc receptor, immunoglobulin G (IgG), inhibitor, receptor recycling
Introduction
Antibody-mediated autoimmune diseases are associated with the presence of circulating IgGs that erroneously bind to “self” targets rather than those that are foreign. Such interactions damage the tissues containing these antigen targets and are the foundation of multiple disease pathologies that include rheumatoid arthritis, systemic lupus erythematosis, pemphigus vulgaris, and myasthenia gravis, to name a few (1). These tend to be serious diseases with no known cures and can be insufficiently treated with general immune system-modifying drugs. A recent focus for treating diseases mediated by pathologic IgGs has been to target the neonatal Fc2 receptor (FcRn). FcRn is a cellular receptor that, despite the name, is responsible throughout mammalian life for maintaining elevated plasma concentrations of IgG and serum albumin (HSA), as well as for the directional transport of these molecules across endothelial barriers (2). Attenuated FcRn function has been shown to decrease overall circulating IgGs without altering the ability to mount a normal immune defense when needed (3). Thus, FcRn antagonists may accelerate the clearance of pathologic IgGs in autoimmune diseases without globally suppressing immune function.
FcRn is a heterodimer composed of the β2-microglobulin (β2M) light chain and a membrane-anchored heavy chain that is related to the α-chain of the class I major histocompatibility complex (4). Biochemical and structural studies of rat and human FcRn reveal a pH-dependent binding of the CH2–CH3 interface in the Fc region of IgG to the α2 region of the FcRn heavy chain, with a single IgG molecule able to simultaneously bind two FcRn molecules at acidic pH but negligible binding at neutral pH (5, 6). Such studies helped to elucidate the recycling and transport mechanism of FcRn. Specifically, IgG passively enters cells from the neutral extracellular fluid by pinocytosis and binds membrane-embedded FcRn only at the lower endosomal pH (pH ∼5–6). In contrast to unbound proteins that are then diverted to the lysosomal degradation pathway, FcRn-bound IgGs are either transported to the distal cell surface (transcytosis) or recycled and released back to circulation once the complex is exposed to neutral extracellular pH. This latter mechanism, which also applies to a distinct binding interaction with HSA, considerably increases the half-life of recycled molecules. This is beneficial from the perspective of normal IgG accumulation, as it helps to maintain steady IgG levels in the blood that promote healthy immune system function. However, this same mechanism can turn pathogenic when the body aberrantly produces self-targeting autoantibodies, which, if left in circulation, increase the amount of cellular damage that they may cause.
Current treatments to combat antibody-mediated autoimmune disease include the use of corticosteroids, immunosuppressants such as purine synthesis inhibitors (e.g. azathioprine), and monoclonal antibody therapeutics that deplete B cells (7, 8) or inhibit complement (9, 10), all of which carry the risk of generalized immune suppression. In addition, immunomodulatory therapies are used to deplete serum antibodies (immunoabsorption and plasmapheresis procedures) or effectively dilute serum levels by treatment with a large quantity of IgG supplied intravenously (IvIg). Such procedures are considered cumbersome and invasive, can result in severe treatment-related complications (11), and, in the case of IvIg, may rely on a limited supply of plasma from healthy blood donors and carry the risks associated with human-derived products (12, 13).
For more directed therapies, significant efforts are now aimed at targeting the FcRn-mediated IgG-recycling mechanism. Experiments in β2M knock-out mice, which lack active FcRn and are specifically depleted of normal IgG levels, highlight the role of FcRn in propagating autoimmune disease due to the fact that these mice were protected from pathogenic IgGs (14, 15). Consequently, numerous groups have been investigating ways to specifically block IgG binding to FcRn with competing peptides (16), engineered high-affinity IgG–Fc variants (17), and experimental monoclonal antibodies (18) directed at FcRn. Although a number of these FcRn-targeting strategies have proven efficacious in preclinical animal models of autoimmune disease, clinical studies are in the early phases.
We previously used the CDR sequence diversity of an antibody phage display library (19) to discover fully human monoclonal IgGs (DX-2504 and a sequence-related variant, DX-2507) that bind FcRn potently (KD ∼2 nm) at both acidic and neutral pH (3). Competitive flow cytometry experiments demonstrate that these antibodies can block IgG binding to HEK cells overexpressing human FcRn. Consistent with these findings, administration of DX-2504 to both human transgenic mice (TG32B) and cynomolgus monkeys increased the rate of total IgG catabolism, whereas plasma concentrations of HSA, IgM, and IgA were unchanged. Importantly, dosed primates were still able to mount primary and secondary immune responses despite the decrease in overall IgG plasma levels.
To better understand how DX-2507 interacts with FcRn and thereby blocks IgG binding, X-ray crystallography was used to structurally define the interface between an isolated Fab fragment of the DX-2507 IgG with a soluble form of the FcRn–β2M heterodimer. The Fab fragment, shown here to biochemically mimic the potent and pH-insensitive FcRn binding by the IgG from which it derives, binds and completely covers the canonical IgG–Fc interface of FcRn using all six CDRs from both the heavy and light variable domains and would thus directly compete out IgG binding and consequent FcRn-mediated recycling. In addition, the complex structure (i) explains the lack of pH sensitivity of the interactions between FcRn and DX-2507 compared with the canonical IgG–Fc interface; (ii) provides a rationale for the binding preference for FcRn homologues from different preclinical species; and (iii) confirms that there is no overlap between DX-2507 and HSA epitopes. Taken together, the complex structure can be used to mechanistically rationalize our in vitro and in vivo findings that DX-2507 inhibits IgG binding to FcRn, provides a template for future protein engineering efforts, and supports the use of DX-2507 for the therapeutic treatment of disease driven by pathologic IgGs.
Results
Purification and characterization of DX-2507 Fab domain
We previously isolated and developed a fully human antibody (DX-2507) that binds to the FcRn receptor with high affinity and in a pH-independent manner (3). To characterize this interaction between DX-2507 and FcRn with atomic resolution, a DX-2507 Fab′ preparation was generated by limited proteolysis of whole DX-2507 IgG to isolate a more compact inhibitor domain more amenable to crystallography. Treatment of DX-2507 IgG with endoproteinase Lys-C protease liberated individual Fab domains that were subsequently purified to homogeneity and shown to interact in a 1:1 stoichiometric manner with soluble human FcRn (a recombinant FcRn–β2M construct that is truncated to exclude the transmembrane domain and intracellular C-terminal tail, hereby referred to as “shFcRn”). Specifically, analytical size-exclusion chromatography (SEC) experiments demonstrated that molar equivalents of DX-2507 and shFcRn lead to the nearly complete loss of their individual elution peaks, with the concurrent appearance of a single peak consistent with a larger complex species (data not shown).
To more finely probe the interaction of DX-2507 Fab with shFcRn, we employed surface plasmon resonance (SPR) experiments that monitor the binding of shFcRn to biosensor-immobilized DX-2507 IgG and Fab. At neutral pH, shFcRn bound to the DX-2507 IgG (KDapp = 1.95 ± 0.03 nm; Fig. 1A) and Fab′ (KDapp = 1.81 ± 0.04 nm, Fig. 1B) with similar affinities. Consistent with previously reported values for the DX-2507 IgG (3), these affinities far exceed the negligible binding of native IgG–Fc interactions at neutral pH, as well as the ∼100 nm reported binding affinity of IgG1 at pH 6 (20, 21). Such interactions were also observed when similar experiments were performed in lower-pH solutions, with DX-2507 IgG and Fab binding shFcRn with fitted KDapp values of 1.28 ± 0.03 and 1.33 ± 0.04 nm, respectively, at pH 5.5 (Table 1). Although there was a minimal but consistent increase in affinity at more acidic pH for both IgG and Fab′, it is important to note that this must be related to changes in antibody CDR interactions and not an increased contribution of standard pH-dependent antibody Fc interactions, given that the DX-2507 Fab construct does not have an Fc domain.
Figure 1.

Equivalent targeting of human FcRn by the DX-2507 IgG and Fab constructs. A, a representative SPR sensorgram of human sFcRn injected at multiple concentrations over surface-immobilized DX-2507 IgG. Color lines show the measured RU value during the association and dissociation phases, whereas the solid black line is the best fit to a 1:1 Langmuir model. Average fits are presented in Table 1. B, SPR sensorgram as in A, but the biosensor surface was coated with DX-2507 Fab. C, flow cytometry experiments show that increasing concentrations of DX-2507 IgG (blue circles) and Fab (red circles) can compete with fluorescent IgG-AF488 binding to HEK293T cells expressing human FcRn (expressed as a shift in the mean FITC peak), in contrast to a control IgG (DX-2300; green triangles) that only targets FcRn through the canonical Fc interaction. In the absence of expressed FcRn, there is a low IgG-AF488 signal that is not competed by DX-2507. Error bars, S.D. of replicates (n ≥ 2); solid lines, fit to an IC50 equation.
Table 1.
Comparison of DX-2507 Fab and IgG binding affinities to FcRn
| DX-2507 IgG |
DX-2507 Fab |
|||||
|---|---|---|---|---|---|---|
| ka | kd | KDapp | ka | kd | KDapp | |
| m−1 s−1 | s−1 | nm | m−1 s−1 | s−1 | nm | |
| pH 7.5b | ||||||
| Averagec | 1.42E + 05 | 2.78E-04 | 1.95 | 1.71E + 05 | 3.08E-04 | 1.81 |
| S.D.c | 2.00E + 03 | 5.00E-07 | 0.03 | 2.50E + 03 | 1.50E-06 | 0.04 |
| pH 6.0 | ||||||
| Average | 2.10E + 05 | 2.92E-04 | 1.39 | 2.41E + 05 | 3.30E-04 | 1.37 |
| S.D. | 2.00E + 03 | 1.00E-06 | 0.01 | 4.00E + 03 | 1.50E-06 | 0.01 |
| pH 5.5 | ||||||
| Average | 2.64E + 05 | 3.36E-04 | 1.28 | 2.93E + 05 | 3.87E-04 | 1.33 |
| S.D. | 6.00E + 03 | 2.00E-06 | 0.03 | 8.00E + 03 | 5.00E-07 | 0.04 |
a Kinetic values KDapp, ka, and kd were determined by fits to SPR sensorgrams like those shown in Fig. 1 (A and B).
b pH was established by varying running and dilution buffers as follows: pH 7.5 = 20 mm HEPES, pH 7.5, 150 mm NaCl, and 0.005% surfactant P20 (HBS-P buffer); pH 6.0 = 20 mm HEPES, pH 7.5, 150 mm NaCl, and 0.005% surfactant P20; pH 5.5 = 20 mm acetate, pH 5.5, 150 mm NaCl, and 0.005% surfactant P20.
c Average and S.D. values were calculated for at least two replicate data sets.
In addition, the isolated DX-2507 Fab effectively binds full-length FcRn that is expressed at the surface of HEK293 cells. Flow cytometry experiments monitored the binding of a fluorescently labeled Alexa Fluor® 488 IgG conjugate (IgG-AF488) to FcRn-expressing cells in the presence of various competitors. As demonstrated in Fig. 1C, IgG-AF488 exhibits only background binding to control HEK293 cells but elevated binding to HEK293 cells that overexpress FcRn (HEK293c11 cells). FcRn-mediated binding of IgG-AF488 is potently competed by increasing concentrations of DX-2507 IgG (IC50 = 0.36 ± 0.1 nm) or Fab (IC50 = 1.1 ± 0.1 nm). The competitive advantage of the IgG format over the Fab fragment of DX-2507 is probably due to differences in the stoichiometry of inhibitory domains per mole of inhibitor (a single IgG molecule contains two inhibitory Fab domains and thus twice the molar potency of a Fab-only construct). In contrast to the DX-2507 constructs, a control IgG with an identical Fc region but variant Fv sequence (the IgG “DX-2300”) does not compete within the concentration range shown (up to 14 nm) even at the experimental pH of 6 (Fig. 1C). Instead, the standard Fc-mediated interaction of DX-2300 with FcRn competes with an IC50 >100-fold greater than DX-2507 (data not shown).
Taken together, the isolated Fab portion of DX-2507 retains its ability to bind both recombinant and cell surface–expressed FcRn with a high affinity that is independent of solution pH. This interaction of DX-2507 with FcRn completely inhibits IgG–Fc interactions.
X-ray crystal structure of DX-2507–FcRn complex
To better understand this high-affinity and pH-independent interaction, the DX-2507 Fab was crystallized both alone and in complex with shFcRn, and X-ray diffraction data were collected to 2.15 and 2.5 Å, respectively (Table 2). Overall, complexed and apo-Fab both adopt a conventional immunoglobulin fold, varying little from one another (RMSD 0.733 Å) except for regions involved in the paratope–epitope interface (Fig. 2A). Temperature factor analysis reveals that the Fab–FcRn interface exhibits low B-factors and is generally more well-ordered (lower local B-factors) relative to the paratope residues in the apo-Fab structure (Fig. 2B). Complexed FcRn is also folded much like previously reported FcRn structures, including apo-FcRn heavy chain (PDB entry 1EXU (22), RMSD 0.677 Å) and FcRn–β2M complexed simultaneously with IgG and HSA (PDB code 4N0U (23), RMSD 0.544 Å). In addition to protein, the final complex model contains two CHES molecules and two ethylene glycol molecules from the solvent as well as 254 water molecules. Interestingly, one of the two CHES buffer molecules binds along the MHC-like binding groove of the FcRn heavy chain (data not shown).
Table 2.
X-ray crystallographic collection and refinement statistics
| PDB code | 5WHJ | 5WHK |
| Crystallized species | DX-2507 Fab | DX-2507 Fab + shFcRn |
| Beam line | APS LS-CAT 21 ID-G | APS LS-CAT 21 ID-F |
| Space group | P4322 | P212121 |
| Unit cell | ||
| a, b, c (Å) | 116.2, 116.2, 79.2 | 59.6, 70.5, 256.6 |
| α, β, γ(degrees) | 90, 90, 90 | 90, 90, 90 |
| Solvent content (%) | 58 | 58 |
| Vm (Å3/Da) | 2.96 | 2.95 |
| Resolution (Å) | 50–2.15 (2.21–2.15)a | 50–2.50 (2.56–2.50) |
| I/σ | 23.8 (3.8) | 16.3 (2.4) |
| Completeness (%) | 99.3 (100) | 99.8 (100) |
| Rmerge | 0.057 (0.589) | 0.072 (0.629) |
| Multiplicity | 7.3 (7.4) | 4.9 (5.0) |
| Reflections | 28,879 (2197) | 38,383 (2772) |
| Mosaicity | 0.2 | 0.2 |
| Refinement | ||
| R | 0.194 (0.229) | 0.172 (0.229) |
| Rfree | 0.233 (0.235) | 0.225 (0.308) |
| Validationb (%) | ||
| Ramachandran favored | 96.7 | 97.3 |
| Ramachandran outliers | 0.0 | 0.0 |
| Rotamer outliers | 2.7 | 2.3 |
| Clash score | 1.33 (100th percentile) | 3.5 (99th percentile) |
| MolProbity score | 1.38 (98th percentile) | 1.55 (98th percentile) |
a Statistics for highest resolution shell data are shown in parentheses throughout the table.
b Validation statistics were obtained from MolProbity (55).
Figure 2.
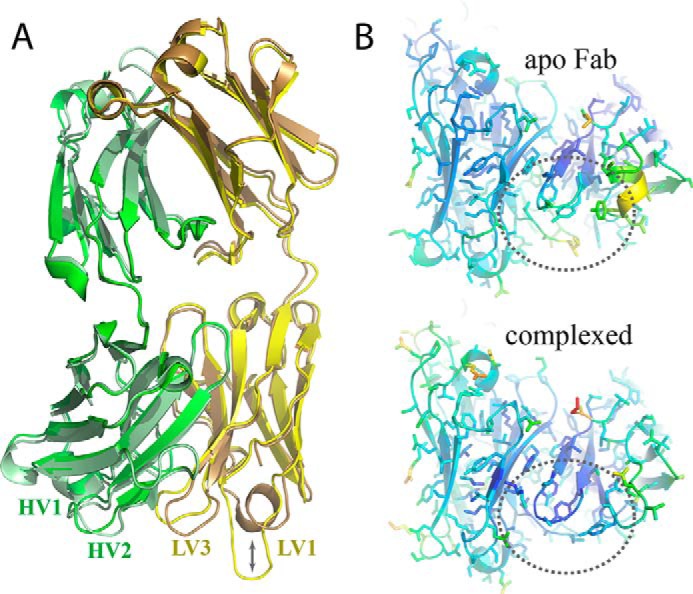
Crystal structures of apo and complexed DX-2507 Fab. A, overlay of the apo-DX-2507 Fab structure (PDB code 5WHJ) with the Fab domain that is bound to FcRn (PDB code 5WHK, RMSD = 0.733 Å). The Fab light chain is shown in brown and yellow ribbon for apo and complexed Fab, respectively, and the heavy chain is shown in pale green and green ribbon for apo and complexed Fab, respectively. Certain CDR loops (e.g. HV2) highlight the region of the DX-2507 Fab that interacts with FcRn. Extension of LV1 from a helical conformation to an extended loop is the most apparent difference, although some minor changes in LV3 and HV2 are evident. B, temperature factor analysis reveals that the FcRn interface residues are generally more ordered in the complex than in the Fab crystallized alone. The dashed circle region highlights the central region of the Fab that interacts with FcRn.
The DX-2507–FcRn complex interface is located adjacent to the MHC-like groove and has an extensive total contact area (buried paratope and epitope surface) of 10,354 Å2 made with both the FcRn heavy chain (9374 Å2) and β2M (980 Å2) components (Fig. 3, A and B). Quite surprisingly, all DX-2507 CDRs in both the Fab heavy chain variable (HV) and light chain variable (LV) are involved in the complex interface to some extent (Fig. 3B), with LV interactions (8249-Å2 total contact area) dominating the interface over HV interactions (1872 Å2). As shown in Fig. 3C, the DX-2507 epitope covers most of the antibody Fc–binding region of FcRn, definitively confirming that the Fab region at a minimum competitively inhibits IgG–Fc–mediated interactions.
Figure 3.

X-ray crystal structure of the DX-2507 Fab bound to human FcRn. A, ribbon model of the DX-2507–FcRn complex (PDB code 5WHK). The FcRn heavy chain is colored in gray, and the β2M subunit is shown in cyan. The DX-2507 heavy chain is colored green, and the light chain is yellow. B, close-up view of the DX-2507 CDRs, showing all of them in close proximity to FcRn. C, FcRn is shown as a surface schematic with coloring as in A. Left, the canonical IgG–Fc interface residues are highlighted in blue, and (right) superimposed with residues interacting with the DX-2507 heavy (green) and light (yellow) chains. The DX-2507 epitope covers most of the IgG–Fc binding region of FcRn.
The DX-2507 Fab contacts several linear stretches of FcRn primary sequence, composed of a total of 20 FcRn residues. One stretch spans FcRn residues Gly129–Gln139 with major interactions at Asp130, Trp131, Pro132, and Glu133 (Fig. 4A). In particular, Trp131 is sandwiched by both the heavy chain variable CDR2 (HV CDR2) and light chain variable CDR3 (LV CDR3) chains of DX-2507, forming a major point of contact. In addition to van der Waals and water-mediated contacts, direct hydrogen bonds are observed between Asp130 of FcRn and Ser54 of DX-2507 HV CDR2 as well as between Glu133 of FcRn and Tyr93 and Gly95 from DX-2507 LV CDR3. Other major sites of interaction include (i) the stretch of FcRn residues Ala81–Tyr88, which interacts with LV CDR1 and CDR2 and with LV framework 3 residue Lys68 (Fig. 4B); (ii) FcRn Leu112–Asn113, which interacts with LV1; and (iii) FcRn Glu115, which makes several water-mediated interactions with LV CDR1 (Ser31) and CDR3 (Ser96), and HV CDR2 (Lys59). In the center of these interactions also lies a minor hydrophobic groove formed by FcRn residues Ala136, Leu82, Leu112, and Tyr88, with an adjacent hydrophobic ridge composed of Trp131, Pro132, and Leu135. DX-2507 counters this patch with a series of hydrophobics from LV1 (Tyr32 and Leu34), LV3 (Tyr93), HV1 (Tyr32 and Ala33), HV2 (aliphatic portion of Gln57 and Lys59), and HV3 (Leu99, Ala100, and Ile101). Interestingly, in all reported structures of human FcRn (22, 24), including those docked with Fc (23) or inhibitory peptides (25), Trp131 is tucked against the hydrophobic groove. In the DX-2507 complex reported here, Trp131 is instead flipped away from the hydrophobic groove to contact the above noted HV2 Gln57 and Lys59 as well as HV Ala33 (Fig. 4, A and C). The most significant conformational change from the apo to complexed state arises from the DX-2507 LV CDR1 as it uncoils from a condensed α-helical secondary structure to an extended loop that interacts with both the FcRn heavy chain and the β2M subunits (Fig. 4D). In particular, Val29 extends ∼11.5 Å to form hydrophobic interactions with both Pro32 and Val85 of the β2M chain. These LV1 interactions may not be a major source for affinity, however, because LV1 exhibited the highest B-factors and therefore the most thermal motion of all of the CDRs (Fig. 2).
Figure 4.
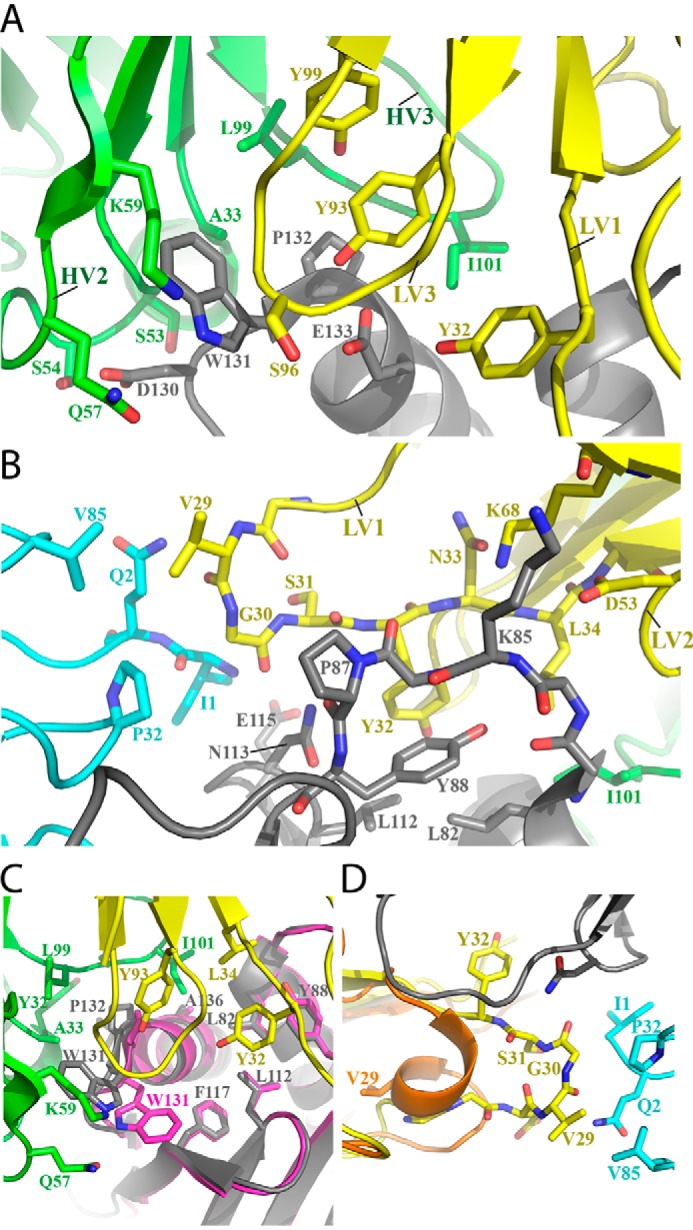
A detailed look at the DX-2507–FcRn complex paratope–epitope interface. A, the central interaction between DX-2507 HV2, HV3, LV1, and LV3 and the FcRn stretch of residues 130DWPE133 drives complex formation. The color scheme is as in Fig. 3. B, DX-2507 interacts with a second major linear stretch of FcRn residues Ala81–Tyr88. C, a view of the extensive hydrophobic interactions that define the center of the complex interface. In addition, it is notable that Trp131 is flipped toward the DX-2507 paratope relative to all other reported human FcRn structures, including the Fc-complexed FcRn structure aligned here (magenta; PDB code 4N0U). D, DX-2507 LV1 unfurls from an α-helical conformation in the apo-Fab structure (orange) to an extended loop, as the complexed Fab (yellow) now contacts residues in the β2M subunit of FcRn (cyan).
DX-2507–FcRn complex structure explains DX-2507 pH insensitivity
As noted, the DX-2507 epitope almost completely occludes the region of FcRn that is involved in Fc interactions (Fig. 3C). However, the residues and types of interactions used to drive DX-2507 binding are surprisingly distinct from those used in Fc interactions. The prototypical Fc interaction uses numerous salt bridges in addition to interactions mediated by several Fc-derived histidine residues (23). One of these histidine residues (His310) contributes to the well-characterized pH dependence of the standard IgG–Fc–FcRn interaction (23). In contrast, the DX-2507 interface does not utilize any histidine residues or salt bridge interactions to bind FcRn. Instead, the interaction is largely driven by hydrophobic, van der Waals, water-mediated bonding, and hydrogen-bonding interactions. For instance, the binding of IgG–Fc to FcRn strongly depends on the interaction of the FcRn heavy chain residue Glu115 with a protonated Fc residue, His310. As shown above, FcRn Glu115 makes water-mediated contacts with several DX-2507 CDRs. The only interaction that could be considered a shared property of DX-2507 and Fc targeting of FcRn is the placement of hydrophobic residues in contact with the FcRn hydrophobic groove described above. However, the IgG–Fc counters this groove with hydrophobic residues Leu251, Tyr252, Ile253, and Leu314, a more subtle contact in comparison with the extensive hydrophobic interactions contributed by DX-2507 (Fig. 4C). In total, the DX-2507–FcRn interface is composed of bonding events that would not be expected to change significantly within the range of typical physiological pH values encountered by FcRn.
Species-specific targeting by DX-2507
The DX-2507–FcRn complex structure explains why DX-2507 exhibits an affinity bias against rodent FcRn but not cyno FcRn. To determine the cross-reactivity of DX-2507 with these FcRn homologues, the binding of cyno and rat equivalents of the shFcRn construct to surface-immobilized DX-2507 was explored. DX-2507 bound to cyno FcRn as potently as human FcRn at pH 7.5 (KDapp = 1.71 ± 0.02 nm; Fig. 5A) but had a markedly reduced affinity to rat FcRn, such that the normal binding sensorgrams could not be used to accurately determine the affinity (data not shown). For a more quantitative comparison of rat FcRn binding, a competition SPR experiment was performed in which 5 nm DX-2507 Fab was preincubated with a dilution series of soluble human, cyno, and rat FcRn and then flowed over biosensor surface-immobilized shFcRn (Fig. 5, B–D). Cyno FcRn competed for DX-2507 Fab binding (IC50pH 6 = 4.9 ± 0.2 nm; IC50pH 7.5 = 5.5 ± 0.2 nm) similarly as human FcRn (IC50pH 6 = 4.1 ± 0.3 nm; IC50pH 7.5 = 4.2 ± 0.2 nm), whereas rat FcRn competition was reduced ∼100-fold (IC50pH 6 = 535 ± 60 nm; IC50pH 7.5 = 397 ± 33 nm). Of note, an equivalent recombinant mouse FcRn construct exhibited similarly low affinity to DX-2507, but data are excluded here due to poor experimental reproducibility. The lack of DX-2507 binding to rat FcRn was not due to improper folding of the recombinant construct but reflects a true DX-2507 bias. Specifically, in SPR experiments like those reported above, soluble rat FcRn bound nonspecific rat and human IgG at pH 6 and thus through low-pH Fc interactions (data not shown). Moreover, the rat FcRn construct used in these experiments interacted with similar affinity (KD = 3.0 nm; data not shown) to an antibody (1G3) previously demonstrated to compete for IgG binding on rat FcRn (reported KDpH 7.4 = 5.8 nm (18)).
Figure 5.

DX-2507 potently binds cyno FcRn but not rat FcRn. A, an example SPR sensorgram of soluble cyno FcRn interacting with surface-immobilized DX-2507 Fab. Color lines show the experimental RU value for the association and dissociation phases, with the solid black line representing the best fit to a 1:1 Langmuir model. B, a competition SPR experiment probes the ability of added FcRn to compete away binding of DX-2507 Fab analyte to surface-immobilized human FcRn. Error bars, S.D. of replicates (n ≥ 2); solid lines, fit to an IC50 equation = 4.1 ± 0.3 nm (pH 6.0) and 4.2 ± 0.2 nm (pH 7.5). C, competition SPR as in B but with increasing concentrations of cyno FcRn as the competitor. Fitted IC50 = 4.9 ± 0.2 nm (pH 6.0) and 5.5 ± 0.2 nm (pH 7.5). D, rat FcRn competition SPR demonstrates a significant decrease in affinity for this species, with resulting IC50 fits = 535 ± 60 nm (pH 6.0) and 397 ± 33 nm (pH 7.5). E, alignment of the DX-2507–shFcRn complex structure (colored as in Fig. 3) with rat FcRn (colored magenta; PDB code 3FRU) shows generally good agreement between the structures (RMSD = 1.07 Å). F, close-up of the alignment of rat and human FcRn at the DX-2507 interface reveals a small but significant extension of α-helix 1 in the rat FcRn that would protrude into DX-2507 LV1 and LV2, probably explaining why this interaction has reduced affinity relative to human FcRn.
With respect to the DX-2507 epitope on FcRn, it is not surprising that the binding affinity is similar for cyno and human FcRn, given the complete sequence identity in this region. Overall, there is good agreement between rat and human primary and structural sequence at the DX-2507 epitope (Fig. 5E), but there are critical differences. The most central FcRn stretch, 130DWPE133, which includes the buried Trp131 discussed above, is nearly conserved in rat (sequence EWPE). In addition, there is no change in the FcRn Leu112–Asn113 residues that contact LV1 nor in any of the β2M residues that interact with this extended LV1 loop. In general, the hydrophobic patch is also present in rats, with a few residue differences making the groove slightly more hydrophobic in rats (human to rat Y88F and A136I). However, Leu135 in human FcRn is instead an aspartate in rat FcRn, which not only removes a minor hydrophobic interaction but also probably sets up a steric clash between the rat aspartate and the polar/acidic Glu31 and Tyr32 residues of DX-2507 HV1. This clash could in turn influence interactions mediated by the adjacent 130(D/E)WPE133 FcRn residues. Finally, the stretch of residues from human FcRn 82LGGKGPY88 is significantly different in rat FcRn (LENQINGTF), but with the terminal Leu and Tyr/Phe residues anchored in similar positions when comparing this region between human and rat FcRn crystal structures (22). An interaction of the human FcRn Lys85 with the backbone of light chain framework 3 would be absent in rats. More significantly though, this sequence difference causes the rat FcRn helix 1 to be extended by a half-turn, which would consequently also extend the loop that comprises much of human FcRn Ala81–Tyr88 into DX-2507 LV CDR1 and CDR2 (Fig. 5F), probably straining the interaction. Because of the significant differences in affinity to rodent FcRn, preclinical experiments on DX-2507 will require an alternative approach, such as the use of a recently reported human FcRn transgenic mouse model (26).
Distinct epitope for DX-2507 targeting in relation to HSA-binding site
An ideal competitor for Fc binding to FcRn would have no impact on the physiological interaction between FcRn and HSA. It is also important from a pharmacologic standpoint that the normally prevalent binding of HSA to FcRn does not influence targeting of DX-2507. Toward this end, the DX-2507 Fab–shFcRn complex crystal structure shows that DX-2507– and HSA-binding sites (23, 24, 27, 28) are distal from one another, with the non-overlapping epitopes separated by >17 Å (Fig. 6A). Moreover, the geometry of the bound Fab is angled such that the remainder of the DX-2507 IgG (Fc and second Fab domains) would still probably not impact normal HSA-binding interactions. To confirm this finding in solution, a competitive ELISA experiment was performed in the presence and absence of 50 mg/ml (∼800 μm) HSA, a concentration at the higher end of the normal range in human blood (29) and at least 160-fold greater than reported KD values for the HSA–FcRn interaction (24, 28). Specifically, the ELISA probes the ability of titrated antibody to competitively inhibit IgG-HRP from binding microplate-immobilized FcRn. The DX-2300 control IgG that is identical in Fc composition as DX-2507, but with different CDR residues, very poorly competes for IgG-HRP binding relative to DX-2507, as expected for a low-affinity Fc–FcRn interaction even at the low experimental pH 6.0. In accord with the distinct DX-2507 and HSA epitopes observed in the complex structure, the potency of DX-2507 was observed to be nearly identical in the absence (IC50 = 1.27 ± 0.08 nm) and presence (IC50 = 1.14 ± 0.08 nm) of saturating HSA (Fig. 6B). Consistent with this result, it was previously reported that albumin levels were unchanged in cyno monkeys that were dosed intravenously with 20 mg/kg of the equivalent DX-2507 precursor, DX-2504 (3). In contrast, the same DX-2507 dose reduced serum IgG levels by >60%, following the competitive inhibition of IgG binding to FcRn and consequent reduction in IgG recycling. Thus, the targeting of human FcRn by DX-2507 is not expected to prevent or reduce HSA binding; nor should the presence of the normally high circulating concentration of albumin influence the interaction of DX-2507 with FcRn.
Figure 6.

Binding of DX-2507 to FcRn is independent of HSA binding. A, structural model of the simultaneous binding of DX-2507 and HSA (orange) based on the structural alignment of FcRn from the complex structure in this study (PDB code 5WHK) and FcRn in complex with HSA (PDB code 4K71). HSA and DX-2507 Fab target distinct regions of FcRn. B, the addition of 50 mg/ml HSA (at the high end of the normal physiological concentration in blood) does not influence the ability of DX-2507 to compete away IgG-HRP binding to microplate immobilized FcRn, as exhibited in the competition ELISA experiment shown here. At pH 6.0, a control IgG (DX-2300) competes only at high IgG (Fc) concentrations relative to DX-2507. Error bars, S.D. of replicates (n ≥ 2); solid lines, fit to an IC50 equation.
Discussion
Given the role FcRn plays in maintaining elevated IgG levels, there is increasing interest in FcRn as a target for therapeutic intervention in diseases propagated by pathogenic IgG autoantibodies. We and others have sought to directly target the FcRn-mediated IgG-recycling mechanism to accelerate the clearance of such antibodies. Toward this end, we developed an IgG (DX-2507) that potently binds FcRn through pH-insensitive variable domain interactions, which blocks the binding of other IgGs and thereby prevents their recycling back into circulation (3). In this report, the pH-insensitive binding of the isolated Fab′ portion of DX-2507 to FcRn is characterized, and through the X-ray crystallographic structure of the DX-2507 Fab–shFcRn complex, it is shown how DX-2507 directly competes with the canonical IgG–Fc binding site on FcRn.
This approach is comparable with previous reports of engineered Fc domains with improved affinities for FcRn (30). In a notable study, phage display of targeted IgG–Fc residues resulted in the selection of a variant IgG (M252Y/S254T/T256E, often referred to as the “YTE” mutation) with a ∼10-fold improvement in binding affinity toward FcRn at pH 6 but less substantial improvement of pH 7.5 binding affinity (31). When dosed into non-human primates, a YTE variant ultimately increased serum half-life by 4-fold (32). Ward and colleagues (33) further investigated H433L and N434F mutations added to this YTE background (resulting in a combination they refer to as an “MST-HN” variant, or “Abdegs”), which binds to mouse FcRn potently at both pH 6.0 (KD = 1.2 nm) and pH 7.2 (KD = 7.4 nm) but with a somewhat lower affinity to human FcRn at pH 6 (KD = 15.5 nm) and pH 7 (poor but improved binding relative to wild type). They further demonstrate that mice dosed with the MST-HN enhanced Fc variant showed a significant reduction in both exogenously supplied 125I-labeled IgG1 and total endogenous IgG in comparison with animals dosed with wild-type human IgG1. Interestingly, a majority of the MST-HN residue changes provide additional hydrophobic contacts with (M253Y/S245T) or adjacent to (H433L/N434H) the FcRn hydrophobic patch centered around the C-terminal side of α-helix 1 and the N-terminal side of α-helix 2 (Fig. 4C). The Y256E mutation stands alone in that it adds an additional salt bridge interaction with Gln2 of the β2M subunit of FcRn. In fact, a number of other reported IgG–Fc mutations that enhance its affinity to FcRn also employ the addition of hydrophobic contacts, directly or indirectly, including the P257I and T307A mutations, which allow more favorable interaction with β2M subunit residue Ile1 (30, 34, 35).
Mezo et al. (16) alternatively used phage display to identify a class of peptides (SYN1327 and dimeric derivatives) that bind and compete for IgG–Fc binding to FcRn. The peptides bind potently to FcRn in a pH-independent manner (reaching subnanomolar affinity) and result in a significant decrease in endogenous IgG levels (up to 80%) when dosed into cynomolgus monkeys. Interestingly, X-ray structures of the SYN peptides bound to FcRn (25) reveal critical interactions of three hydrophobic residues in the inhibitor peptides (His6, Phe7, and Tyr11) with the hydrophobic patch that is central to the DX-2507 interaction (Fig. 4). Peptide residue Phe7, in particular, is wedged between Tyr88, Leu112, and Trp131 of FcRn. In DX-2507, LV Tyr32 is also wedged against Tyr88 and Leu112 (albeit at a different angle), but with Trp131 flipped out of the pocket to make interactions with other LV and HV residues (Fig. 4, A and C).
As highlighted throughout this study, hydrophobic interactions are an important driver of FcRn targeting by DX-2507. In fact, the electrostatic interactions that help mediate IgG–Fc binding to FcRn are absent in the DX-2507 interface, despite the significant overlap in epitopes (Fig. 3C). Given that hydrophobic interactions are insensitive to pH changes in the neutral to slightly acidic range, it is perhaps not too surprising then that varying competitors selected to bind FcRn in a pH-independent manner (Abdegs, peptides, and DX-2507) all rely on hydrophobic interaction energy.
Now that several distinct competitive tools are available to probe the efficacy of FcRn blockade in preventing endogenous IgG recycling, it will be important to compare this efficacy with their biochemical properties. For instance, is there a benefit to having specific antagonist potencies, and if so, are they equally important at both neutral and endosomal pH? Do different competitor-binding modalities alter trafficking of FcRn in a negative or beneficial way? Most importantly, can blockade of IgG binding alone prove efficacious toward human autoimmune disease?
Competitive inhibition of IgG binding to FcRn has been considered a potential mode of action for the IvIg augmentation therapy, whereby the dosing of individuals with excess exogenous IgG floods FcRn receptors, preventing endogenous IgGs (including neutral, beneficial, and pathologic ones) from binding FcRn (36, 37). The unbound IgGs are consequently excluded from the FcRn-mediated salvage pathway and are now trafficked toward lysosomal degradation. IvIg augmentation therapy has been used to treat a variety of immunological disorders, including idiopathic thrombocytopenia purpura, Guillain-Barrè syndrome, Kawasaki disease, chronic inflammatory demyelinating polyneuropathy, myesthenia gravis, autoimmune neutropenia, and rheumatoid arthritis, among others (38). Although the delivery of exogenous IgG could in theory ameliorate the effects of pathologic autoantibodies through several mechanisms, including the modulation of Fc receptor expression and effector function and the supply of protective anti-idiotype antibodies (39, 40), recent studies in preclinical species have highlighted the promise of FcRn blockade alone as an effective mode of action for certain diseases. For instance, administration of a monoclonal antibody that targets rat FcRn and consequently blocks IgG binding (antibody 1G3) was shown to inhibit a model of experimental autoimmune myasthenia gravis in rats (18). A number of notable studies with enhanced Fc variants have also proven efficacious in animal models of autoimmune disease, including (i) a reduction in arthritis induced in mice by passive transfer with injected plasma from a human patient with active rheumatoid arthritis (41) as well as arthritis induced in normal mice with the K/BxN serum transfer method (17) and (ii) amelioration of disease severity in a murine model of experimental autoimmune encephalomyelitis (42).
Despite this mounting evidence, it appears that not all autoimmune diseases may be corrected by directly targeting FcRn-mediated IgG recycling, even in those thought to be driven specifically by the presence of pathogenic IgGs. For instance, idiopathic thrombocytopenia purpura (ITP) is induced by the pathologic activity of anti-platelet antibodies. Individuals with ITP have long been treated by IvIg (43, 44), and saturation of pathogenic IgG binding to FcRn has been theorized to play a role in the efficacy of this treatment (1, 45). However, experiments in FcRn-deficient mice demonstrate that ITP is still induced following injection with an anti-platelet antibody, indicating that FcRn function is not necessary to propagate disease in this animal model (46). In addition, it has been shown in a mouse arthritis model that reduction of disease by administration of IvIg requires glycosylation of the IgG population (47), a finding that points toward a functional role of FcγR receptors rather than FcRn, given that, unlike the FcγRs, binding of IgG to FcRn is independent of glycosylation status (35, 48). These discrepancies highlight the need not only to better understand the etiology behind specific autoimmune diseases but also to begin studying disease in higher organisms where the pathologies and treatments are more likely to replicate human disease. Such studies will consequently require inhibitor tools that effectively target non-human primate and human FcRn.
We believe that DX-2507 or a derivative thereof is poised to address this need. It exhibits potent binding of FcRn at neutral and endosomal pH (Fig. 1) and directly competes for IgG binding (Figs. 3 and 4) without influencing the binding of HSA (or vice versa) (Fig. 6, A and B). Experiments in non-human primates reveal that this targeting can reduce endogenous IgG levels down to <40% of normal and does not appear to be immunosuppressive (3). Future studies, however, will need to address the safety and efficacy of this approach in human disease.
Experimental procedures
Proteins
Recombinant cyno, rat, and mouse FcRn were obtained from ACRO Biosystems, as was the soluble human FcRn used in ELISA experiments. Human serum albumin was purchased from Sigma-Aldrich (catalog no. A3782). Whole-molecule human IgG both with and without conjugated HRP was purchased from Jackson Laboratory. For flow cytometry experiments, human IgG1 was conjugated with a 10-fold molar excess of Alexa Fluor® 488 NHS ester following the manufacturer's procedures (Thermo Fisher Scientific), resulting in a 5:1 ratio of fluorophore/IgG conjugate (IgG-AF488). Anti-rat FcRn monoclonal antibody 1G3 was purchased from BioXCell.
Soluble human FcRn in complex with β2M (shFcRn) used for crystallography and Biacore SPR experiments was expressed and purified by Beryllium (Bainbridge Island, WA). Specifically, a soluble human FcRn receptor heterodimer composed of a truncated version of the FcRn heavy chain component (residues 1–297) and full β2M sequence was expressed in baculovirus-infected Trichoplusia ni insect cells as a secreted complex using a multiplicity of infection of 5 and harvested after 48 h. Both sequences used the native leader sequence. After medium exchange into 50 mm sodium phosphate, pH 5.8, 30 mm NaCl and concentration using a tangential flow filtration system, the FcRn–β2M complex was purified by IgG-Sepharose followed by ion exchange on a HiTrap Q FF column. The final protein was purified by SEC and concentrated to 10 mg/ml in 50 mm Hepes, pH 7.0, 75 mm NaCl and stored at −80 °C until used in binding analysis or crystallography.
The DX-2507 IgG and the DX-2300 IgG used as a negative control in flow cytometry and competitive ELISA experiments were expressed and purified as described (3, 49). The purified Fab fragment of DX-2507 was generated by limited proteolysis with endoproteinase Lys-C. Specifically, one vial of lyophilized endoproteinase Lys-C from Lysobacter enzymogenes (Sigma) was resuspended with 1.0 ml of 20 mg/ml DX-2507 IgG (that was previously adjusted to pH 8.0) and incubated at 37 °C with mixing (250 rpm) for 4 h. The solution was then flowed over a MabSelect SuRe Protein A (GE Healthcare) column equilibrated with 10 column volumes of 1× phosphate-buffered saline, pH 7.2 (PBS) at a residence time of 3 min. Flow-through containing removed Fab domains was collected and subsequently loaded onto a 1 ml MabSelect Protein A (GE Healthcare) column equilibrated with 10 column volumes of 1× PBS at a residence time of 3 min. The resin was washed with 10 column volumes of 1× phosphate-buffered saline, 0.4 m sodium chloride, pH 7.2, followed by a 10-column volume wash with PBS. Bound Fab was then eluted with 0.1 m citrate, pH 3.2, and buffer-exchanged into 0.1 m citrate-phosphate, 50 mm sodium chloride, pH 6.0.
Surface plasmon resonance
To determine the affinity of DX-2507 IgG and Fab preparations for FcRn, SPR measurements were performed at 25 °C using a Biacore 3000 (GE Healthcare). DX-2507 IgG and Fab were directly conjugated by NHS-EDC chemistry to distinct flow cells on a CM5 biosensor chip to densities of 450 and 1100 RU, respectively, with unreacted NHS quenched by the addition of 1 m ethanolamine. A reference flow cell was activated and blocked in a mock amine-coupling reaction. Varying concentrations of shFcRn was flowed over the DX-2507 surface and regenerated as described previously (3), with replicate experiments using distinct sample and running buffers, including HBS-P (20 mm HEPES, pH 7.5, 150 mm NaCl, and 0.005% surfactant P20), HBS-P, pH 6.0, and ABS-P (20 mm acetate, pH 5.5, 150 mm NaCl, and 0.005% surfactant P20). Experiments with soluble cyno, mouse, and rat FcRn directly followed these procedures for shFcRn in HBS-P, with the exception that significantly higher mouse and rat FcRn was required to observe DX-2507 binding.
Competition SPR experiments employed the reverse configuration for binding detection, with ∼800 RU of shFcRn directly immobilized on a CM5 chip by NHS-EDC coupling. 5 nm DX-2507 solutions (pH 7.5 or 6 HBS-P buffers) were preincubated for at least 30 min with and without competing human, cyno, or rat soluble FcRn before injection over the shFcRn surface at 20 μl/min. RU values were recorded after 12 min of the association phase (near binding saturation and defined as RUeq), and surfaces were regenerated with a 20-μl pulse of 7.5 mm NaOH at 100 μl/min. Repeat controls without competing FcRn were included throughout experiments to ensure surface stability, and the average RUeq values of these runs (RUeqNoComp) were used to establish the relative level of competition for DX-2507 binding, where the percentage of starting RUeq = RUeqcomp/RUeqNoComp × 100, with RUeqcomp equivalent to the RUeq of the experimental sample containing competitor. Fits of percentage of starting RUeq versus DX-2507 concentration were fit to a four-parameter logistic curve, Y = RUeqmin + (RUeqmax − RUeqmin)/(1 + ([competitor]/IC50)−B) in SigmaPlot, where RUeqmax and RUeqmin are the maximum and minimum RUeq of DX-2507 binding in the presence of competing FcRn, [competitor] is the concentration of FcRn competitor, and B is the Hill slope.
Flow cytometry
Detection of IgG1 binding to control HEK293T and FcRn-expressing HEK293c11 (16) cells utilized an Intellicyte iQue Screener flow cytometer. Specifically, 1 × 105 cells were aliquoted to a 96-well polypropylene microplate, pelleted by spinning for 5 min at 200 × g, resuspended in Buffer A (20 mm HEPES, pH 6.0, 150 NaCl, 0.1% Tween 20, 2% BSA (additional 5 m HCl added to final buffer to achieve pH 6.0)), pelleted once more as above, and resuspended in 90 μl of Buffer A solutions containing a 1:2 serial dilution of DX-2507 IgG or Fab or a control IgG (DX-2300 (49)) that does not specifically target FcRn through interactions involving CDRs (but can target FcRn through typical Fc interactions). The top undiluted concentration of IgG or Fab competitor was 2 and ∼0.67 μg/ml, respectively (equivalent to 14 μm) unless otherwise noted. Cells were incubated at 4 °C for 45 min, after which 10 μl of a 10 μg/ml solution of IgG-AF488 (diluted in Buffer A) was added to a final concentration of 1 μg/ml. Reference cells contained only buffer and no IgG-AF488 or antibody competitor. Cells were incubated for an additional 45 min at 4 °C in the dark, pelleted with a 5-min spin at 200 × g, and resuspended in 100 μl of Buffer A. Each well of the 96-well plate was sampled in the flow cytometer with a 6-s sip time and 0.5-s up time, with ≥2000 cells analyzed with a 530/30-nm filter. ForeCyt screening software (IntelliCyt Corp.) was used for data analysis; the population cluster of HEK cells was visually identified from the scatter profiles (plot of forward versus side scatter), gates were manually drawn, and histograms for each fluorescence parameter were constructed for the identified cells, whereby geometric mean fluorescence values were calculated. Data were fit to a generalized IC50 equation.
Competitive ELISA
Competitive ELISA experiments were initiated by coating a Nunc Immobilizer streptavidin F96 clear microplate with recombinant shFcRn (ACRO Biosciences) that was conjugated with NHS-PEG4-biotin (Thermo Fisher Scientific) to a degree-of-labeling of 3 biotin/FcRn. 100 μl of 500 ng/ml FcRn-biotin diluted in Buffer B (100 mm MES, pH 6.0, 150 mm NaCl, 0.05% Tween 20) was added to each well of the microplate and incubated overnight at 4 °C. Plates were washed in Buffer C (20 mm MES, pH 6.0, 150 mm NaCl, 0.02% Tween 20), and 90 μl of 500 ng/ml IgG-HRP diluted in Buffer B (with or without supplemented 50 mg/ml HSA) was added to sample wells. 10 μl of a concentrated competitor dilution series (DX-2507 or DX-2300 in Buffer B with or without 50 mg/ml HSA) at a top concentration of 40 μg/ml was immediately added to sample wells (thus 4 μg/ml final) and incubated for 60 min at room temperature. Plates were subsequently washed in Buffer C, and bound IgG-HRP was detected by the addition of 100 μl of Ultra-TMB (Thermo Scientific), the reaction was quenched with 100 μl of 1 m phosphoric acid, and absorbance values were read at 450 nm (A450) in a Molecular Devices Spectramax Plus plate reader. A450 values were normalized to the percentage value of the absorbance measured in the absence of competitor (%A450) by the calculation (A450Competitor)/(A450Initial) × 100, where A450Initial and A450Competitor are the 450-nm absorbance values in the absence and presence of competitor, respectively. Such normalization was applied, given that the presence of very high HSA concentrations led to slight differences in the overall ELISA signal (whereby excessive HSA probably acts as a “blocking” protein). Plots of %A450 versus competitor concentration were fit to a four-parameter logistic curve, Y = %A450min + (%A450max − %A450min)/(1 + ([competitor]/IC50)−B) in SigmaPlot, where %A450max and %A450min are the maximum and minimum %A450, respectively; [competitor] is the concentration of IgG competitor (DX-2507 or DX-2300); and B is the Hill slope.
X-ray crystallography
Crystals of DX-2507 Fab′ were obtained from sitting-drop vapor diffusion at 16 °C in Morpheus screen condition G1, which contains 10% (w/v) PEG 10,000, 20% (w/v) PEG 550 MME, 0.02 m carboxylic acids (sodium formate, ammonium acetate, trisodium citrate, sodium potassium l-tartrate, sodium oxamate), 0.1 m MES/imidazole, pH 6.5. Diffraction data were collected from resulting crystals directly (no cryoprotectant) at the Advanced Photon Source LS-CAT beamline 21 ID-G (collection Table 2), and the final structural model was obtained after molecular replacement in Phaser with a generic humanized Fab with the CDRs removed (PDB code 3IDX (50)), followed by iterative rounds of manual model building in Coot and refinement in Refmac (51–53).
Before co-crystallization of the DX-2507 Fab–shFcRn complex, analytical SEC was used to determine the optimal ratio to generate homogeneous sample. The Fab was titrated into a sample of shFcRn, and the optimal ratio was determined to be 1.0:1.0 in PBS buffer with 0.5 mm tris(2-carboxyethyl)phosphine run on an Agilent 1100 HPLC with a Superdex 200HR10.300 GL column. shFcRn had a retention time of ∼32 min, DX-2507 Fab had a retention time of ∼31 min, and the complex had a retention time of ∼28 min. Based on this result, co-crystallization trials for the DX-2507 Fab′–shFcRn complex were set up at a 1:1 molar ratio at 16 °C. Initial crystals were formed by the vapor diffusion method in the JCSG+ screen condition A7 (20% PEG 8,000, 0.1 m CHES, pH 9.5) and diffracted anisotropically to ∼4 Å resolution. A gradient-grid optimization screen, in which the PEG (2% (w/v)/step) was varied against the CHES pH with microseeding from the original hit, resulted in a 2.85 Å resolution data set. A second round of gradient grid screening with smaller incremental changes in PEG concentration (1% (w/v)/step) was done with seeding from the first round of optimization. Optimal crystals (18% PEG final) were cryoprotected with 20% (v/v) ethylene glycol, and diffraction data were collected at the Advanced Photon Source LS-CAT beamline 21 ID-F. The resulting 2.5 Å resolution data set was solved by molecular replacement in Phaser using the structures of DX-2507 Fab′ alone (above) and FcRn–β2M (PDB code 3M17 (25)). The structure contains one complex in the asymmetric unit, and the final model was obtained after multiple iterative rounds of manual model building in Coot and refinement in Refmac and Phenix (54). Collection and refinement statistics for apo-Fab and complex are presented in Table 2.
Author contributions
J. A. K. and T. E. E. drafted the manuscript. J. A. K., B. M. T., G. P. C., J. C., K. J. K., A. P. L., S. R. C., K. A., and J. B. designed, performed, and analyzed experiments. J. A. K. prepared all of the figures. C. T., B. A. A., D. J. S., T. E. E., and A. E. N. participated in experimental design and data analysis/interpretation. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Heather S. Duffy for manuscript revisions.
This investigation was supported by Dyax Corp., now a wholly owned subsidiary of Shire Pharmaceuticals. J. A. K., G. P. C., J. C., K. J. K., A. P. L., S. R. C., C. T., B. A. A., D. J. S., and A. E. N. were employees of Dyax at the time of the study. J. A. K. and D. J. S. are currently Shire employees.
The atomic coordinates and structure factors (codes 5WHJ and 5WHK) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Fc
- fragment crystallizable region of an antibody
- FcRn
- neonatal Fc receptor
- β2M
- β2-microglobulin
- CDR
- complementarity-determining region
- Fab
- fragment antigen-binding domain
- HSA
- human serum albumin
- HV
- antibody heavy chain variable domain
- IvIg
- intravenous IgG
- LV
- antibody light chain variable domain
- SEC
- size-exclusion chromatography
- shFcRn
- soluble human FcRn
- SPR
- surface plasmon resonance
- EDC
- 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- RMSD
- root mean square deviation
- PDB
- Protein Data Bank
- cyno
- cynomolgus
- ITP
- idiopathic thrombocytopenia purpura
- RU
- resonance units.
References
- 1. Sesarman A., Vidarsson G., and Sitaru C. (2010) The neonatal Fc receptor as therapeutic target in IgG-mediated autoimmune diseases. Cell Mol. Life Sci. 67, 2533–2550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pyzik M., Rath T., Lencer W. I., Baker K., and Blumberg R. S. (2015) FcRn: the architect behind the immune and nonimmune functions of IgG and albumin. J. Immunol. 194, 4595–4603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nixon A. E., Chen J., Sexton D. J., Muruganandam A., Bitonti A. J., Dumont J., Viswanathan M., Martik D., Wassaf D., Mezo A., Wood C. R., Biedenkapp J. C., and TenHoor C. (2015) Fully human monoclonal antibody inhibitors of the neonatal fc receptor reduce circulating IgG in non-human primates. Front. Immunol. 6, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Simister N. E., and Mostov K. E. (1989) An Fc receptor structurally related to MHC class I antigens. Nature 337, 184–187 [DOI] [PubMed] [Google Scholar]
- 5. Martin W. L., West A. P. Jr, Gan L., and Bjorkman P. J. (2001) Crystal structure at 2.8 Å of an FcRn/heterodimeric Fc complex: mechanism of pH-dependent binding. Mol. Cell 7, 867–877 [DOI] [PubMed] [Google Scholar]
- 6. Abdiche Y. N., Yeung Y. A., Chaparro-Riggers J., Barman I., Strop P., Chin S. M., Pham A., Bolton G., McDonough D., Lindquist K., Pons J., and Rajpal A. (2015) The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity. MAbs 7, 331–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Castillo J., Milani C., and Mendez-Allwood D. (2009) Ofatumumab, a second-generation anti-CD20 monoclonal antibody, for the treatment of lymphoproliferative and autoimmune disorders. Expert Opin. Investig. Drugs 18, 491–500 [DOI] [PubMed] [Google Scholar]
- 8. Joly P., Mouquet H., Roujeau J. C., D'Incan M., Gilbert D., Jacquot S., Gougeon M. L., Bedane C., Muller R., Dreno B., Doutre M. S., Delaporte E., Pauwels C., Franck N., Caux F., et al. (2007) A single cycle of rituximab for the treatment of severe pemphigus. N. Engl. J. Med. 357, 545–552 [DOI] [PubMed] [Google Scholar]
- 9. Erkan D., Aguiar C. L., Andrade D., Cohen H., Cuadrado M. J., Danowski A., Levy R. A., Ortel T. L., Rahman A., Salmon J. E., Tektonidou M. G., Willis R., and Lockshin M. D. (2014) 14th International Congress on Antiphospholipid Antibodies: task force report on antiphospholipid syndrome treatment trends. Autoimmun. Rev. 13, 685–696 [DOI] [PubMed] [Google Scholar]
- 10. Huda R., Tüzün E., and Christadoss P. (2014) Targeting complement system to treat myasthenia gravis. Rev. Neurosci. 25, 575–583 [DOI] [PubMed] [Google Scholar]
- 11. Szczeklik W., Wawrzycka K., Włudarczyk A., Sega A., Nowak I., Seczyńska B., Fajfer I., Zaja̧c K., Królikowski W., and Kózka M. (2013) Complications in patients treated with plasmapheresis in the intensive care unit. Anaesthesiol. Intensive Ther. 45, 7–13 [DOI] [PubMed] [Google Scholar]
- 12. Bayry J., Kazatchkine M. D., and Kaveri S. V. (2007) Shortage of human intravenous immunoglobulin: reasons and possible solutions. Nat. Clin. Pract. Neurol. 3, 120–121 [DOI] [PubMed] [Google Scholar]
- 13. Kessary-Shoham H., Levy Y., Shoenfeld Y., Lorber M., and Gershon H. (1999) In vivo administration of intravenous immunoglobulin (IVIg) can lead to enhanced erythrocyte sequestration. J. Autoimmun. 13, 129–135 [DOI] [PubMed] [Google Scholar]
- 14. Liu Z., Roopenian D. C., Zhou X., Christianson G. J., Diaz L. A., Sedmak D. D., and Anderson C. L. (1997) β2-Microglobulin-deficient mice are resistant to bullous pemphigoid. J. Exp. Med. 186, 777–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Akilesh S., Petkova S., Sproule T. J., Shaffer D. J., Christianson G. J., and Roopenian D. (2004) The MHC class I-like Fc receptor promotes humorally mediated autoimmune disease. J. Clin. Invest. 113, 1328–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mezo A. R., McDonnell K. A., Hehir C. A., Low S. C., Palombella V. J., Stattel J. M., Kamphaus G. D., Fraley C., Zhang Y., Dumont J. A., and Bitonti A. J. (2008) Reduction of IgG in nonhuman primates by a peptide antagonist of the neonatal Fc receptor FcRn. Proc. Natl. Acad. Sci. U.S.A. 105, 2337–2342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patel D. A., Puig-Canto A., Challa D. K., Perez Montoyo H., Ober R. J., and Ward E. S. (2011) Neonatal Fc receptor blockade by Fc engineering ameliorates arthritis in a murine model. J. Immunol. 187, 1015–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu L., Garcia A. M., Santoro H., Zhang Y., McDonnell K., Dumont J., and Bitonti A. (2007) Amelioration of experimental autoimmune myasthenia gravis in rats by neonatal FcR blockade. J. Immunol. 178, 5390–5398 [DOI] [PubMed] [Google Scholar]
- 19. Hoet R. M., Cohen E. H., Kent R. B., Rookey K., Schoonbroodt S., Hogan S., Rem L., Frans N., Daukandt M., Pieters H., van Hegelsom R., Neer N. C., Nastri H. G., Rondon I. J., Leeds J. A., Hufton S. E., Huang L., Kashin I., Devlin M., Kuang G., Steukers M., Viswanathan M., Nixon A. E., Sexton D. J., Hoogenboom H. R., and Ladner R. C. (2005) Generation of high-affinity human antibodies by combining donor-derived and synthetic complementarity-determining-region diversity. Nat. Biotechnol. 23, 344–348 [DOI] [PubMed] [Google Scholar]
- 20. Raghavan M., Bonagura V. R., Morrison S. L., and Bjorkman P. J. (1995) Analysis of the pH dependence of the neonatal Fc receptor/immunoglobulin G interaction using antibody and receptor variants. Biochemistry 34, 14649–14657 [DOI] [PubMed] [Google Scholar]
- 21. Neuber T., Frese K., Jaehrling J., Jäger S., Daubert D., Felderer K., Linnemann M., Höhne A., Kaden S., Kölln J., Tiller T., Brocks B., Ostendorp R., and Pabst S. (2014) Characterization and screening of IgG binding to the neonatal Fc receptor. MAbs 6, 928–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. West A. P. Jr., and Bjorkman P. J. (2000) Crystal structure and immunoglobulin G binding properties of the human major histocompatibility complex-related Fc receptor. Biochemistry 39, 9698–9708 [DOI] [PubMed] [Google Scholar]
- 23. Oganesyan V., Damschroder M. M., Cook K. E., Li Q., Gao C., Wu H., and Dall'Acqua W. F. (2014) Structural insights into neonatal Fc receptor-based recycling mechanisms. J. Biol. Chem. 289, 7812–7824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schmidt M. M., Townson S. A., Andreucci A. J., King B. M., Schirmer E. B., Murillo A. J., Dombrowski C., Tisdale A. W., Lowden P. A., Masci A. L., Kovalchin J. T., Erbe D. V., Wittrup K. D., Furfine E. S., and Barnes T. M. (2013) Crystal structure of an HSA/FcRn complex reveals recycling by competitive mimicry of HSA ligands at a pH-dependent hydrophobic interface. Structure 21, 1966–1978 [DOI] [PubMed] [Google Scholar]
- 25. Mezo A. R., Sridhar V., Badger J., Sakorafas P., and Nienaber V. (2010) X-ray crystal structures of monomeric and dimeric peptide inhibitors in complex with the human neonatal Fc receptor, FcRn. J. Biol. Chem. 285, 27694–27701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roopenian D. C., Christianson G. J., Proetzel G., and Sproule T. J. (2016) Human FcRn transgenic mice for pharmacokinetic evaluation of therapeutic antibodies. Methods Mol. Biol. 1438, 103–114 [DOI] [PubMed] [Google Scholar]
- 27. Sand K. M., Dalhus B., Christianson G. J., Bern M., Foss S., Cameron J., Sleep D., Bjórås M., Roopenian D. C., Sandlie I., and Andersen J. T. (2014) Dissection of the neonatal Fc receptor (FcRn)-albumin interface using mutagenesis and anti-FcRn albumin-blocking antibodies. J. Biol. Chem. 289, 17228–17239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andersen J. T., Dalhus B., Cameron J., Daba M. B., Plumridge A., Evans L., Brennan S. O., Gunnarsen K. S., Bjórås M., Sleep D., and Sandlie I. (2012) Structure-based mutagenesis reveals the albumin-binding site of the neonatal Fc receptor. Nat. Commun. 3, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McPherson R. A., and Pincus M. R. (2017) Henry's Clinical Diagnosis and Management by Laboratory Methods, 23rd Ed., appendix page e12, Elsevier, St. Louis, MO [Google Scholar]
- 30. Kuo T. T., and Aveson V. G. (2011) Neonatal Fc receptor and IgG-based therapeutics. MAbs 3, 422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dall'Acqua W. F., Woods R. M., Ward E. S., Palaszynski S. R., Patel N. K., Brewah Y. A., Wu H., Kiener P. A., and Langermann S. (2002) Increasing the affinity of a human IgG1 for the neonatal Fc receptor: biological consequences. J. Immunol. 169, 5171–5180 [DOI] [PubMed] [Google Scholar]
- 32. Dall'Acqua W. F., Kiener P. A., and Wu H. (2006) Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J. Biol. Chem. 281, 23514–23524 [DOI] [PubMed] [Google Scholar]
- 33. Vaccaro C., Zhou J., Ober R. J., and Ward E. S. (2005) Engineering the Fc region of immunoglobulin G to modulate in vivo antibody levels. Nat. Biotechnol. 23, 1283–1288 [DOI] [PubMed] [Google Scholar]
- 34. Datta-Mannan A., Witcher D. R., Tang Y., Watkins J., and Wroblewski V. J. (2007) Monoclonal antibody clearance: impact of modulating the interaction of IgG with the neonatal Fc receptor. J. Biol. Chem. 282, 1709–1717 [DOI] [PubMed] [Google Scholar]
- 35. Shields R. L., Namenuk A. K., Hong K., Meng Y. G., Rae J., Briggs J., Xie D., Lai J., Stadlen A., Li B., Fox J. A., and Presta L. G. (2001) High resolution mapping of the binding site on human IgG1 for Fcγ RI, FcγRII, FcγRIII, and FcRn and design of IgG1 variants with improved binding to the FcγR. J. Biol. Chem. 276, 6591–6604 [DOI] [PubMed] [Google Scholar]
- 36. Yu Z., and Lennon V. A. (1999) Mechanism of intravenous immune globulin therapy in antibody-mediated autoimmune diseases. N. Engl. J. Med. 340, 227–228 [DOI] [PubMed] [Google Scholar]
- 37. Bleeker W. K., Teeling J. L., and Hack C. E. (2001) Accelerated autoantibody clearance by intravenous immunoglobulin therapy: studies in experimental models to determine the magnitude and time course of the effect. Blood 98, 3136–3142 [DOI] [PubMed] [Google Scholar]
- 38. Negi V. S., Elluru S., Sibéril S., Graff-Dubois S., Mouthon L., Kazatchkine M. D., Lacroix-Desmazes S., Bayry J., and Kaveri S. V. (2007) Intravenous immunoglobulin: an update on the clinical use and mechanisms of action. J. Clin. Immunol. 27, 233–245 [DOI] [PubMed] [Google Scholar]
- 39. Schwab I., and Nimmerjahn F. (2013) Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat. Rev. Immunol. 13, 176–189 [DOI] [PubMed] [Google Scholar]
- 40. Kazatchkine M. D., and Kaveri S. V. (2001) Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N. Engl. J. Med. 345, 747–755 [DOI] [PubMed] [Google Scholar]
- 41. Petkova S. B., Akilesh S., Sproule T. J., Christianson G. J., Al Khabbaz H., Brown A. C., Presta L. G., Meng Y. G., and Roopenian D. C. (2006) Enhanced half-life of genetically engineered human IgG1 antibodies in a humanized FcRn mouse model: potential application in humorally mediated autoimmune disease. Int. Immunol. 18, 1759–1769 [DOI] [PubMed] [Google Scholar]
- 42. Challa D. K., Bussmeyer U., Khan T., Montoyo H. P., Bansal P., Ober R. J., and Ward E. S. (2013) Autoantibody depletion ameliorates disease in murine experimental autoimmune encephalomyelitis. MAbs 5, 655–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Imbach P., Barandun S., Baumgartner C., Hirt A., Hofer F., and Wagner H. P. (1981) High-dose intravenous gammaglobulin therapy of refractory, in particular idiopathic thrombocytopenia in childhood. Helv. Paediatr. Acta 36, 81–86 [PubMed] [Google Scholar]
- 44. Fehr J., Hofmann V., and Kappeler U. (1982) Transient reversal of thrombocytopenia in idiopathic thrombocytopenic purpura by high-dose intravenous γ globulin. N. Engl. J. Med. 306, 1254–1258 [DOI] [PubMed] [Google Scholar]
- 45. Hansen R. J., and Balthasar J. P. (2002) Effects of intravenous immunoglobulin on platelet count and antiplatelet antibody disposition in a rat model of immune thrombocytopenia. Blood 100, 2087–2093 [PubMed] [Google Scholar]
- 46. Crow A. R., Suppa S. J., Chen X., Mott P. J., and Lazarus A. H. (2011) The neonatal Fc receptor (FcRn) is not required for IVIg or anti-CD44 monoclonal antibody-mediated amelioration of murine immune thrombocytopenia. Blood 118, 6403–6406 [DOI] [PubMed] [Google Scholar]
- 47. Kaneko Y., Nimmerjahn F., and Ravetch J. V. (2006) Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 313, 670–673 [DOI] [PubMed] [Google Scholar]
- 48. Nimmerjahn F., and Ravetch J. V. (2007) The antiinflammatory activity of IgG: the intravenous IgG paradox. J. Exp. Med. 204, 11–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sexton D. J., Chen T., Martik D., Kuzmic P., Kuang G., Chen J., Nixon A. E., Zuraw B. L., Forteza R. M., Abraham W. M., and Wood C. R. (2009) Specific inhibition of tissue kallikrein 1 with a human monoclonal antibody reveals a potential role in airway diseases. Biochem. J. 422, 383–392 [DOI] [PubMed] [Google Scholar]
- 50. Chen L., Kwon Y. D., Zhou T., Wu X., O'Dell S., Cavacini L., Hessell A. J., Pancera M., Tang M., Xu L., Yang Z. Y., Zhang M. Y., Arthos J., Burton D. R., Dimitrov D. S., et al. (2009) Structural basis of immune evasion at the site of CD4 attachment on HIV-1 gp120. Science 326, 1123–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 52. Murshudov G. N., Vagin A. A., and Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 53. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 54. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]