

Abstract
Chemokine-induced directional cell migration is a universal cellular mechanism and plays crucial roles in numerous biological processes, including embryonic development, immune system function, and tissue remodeling and regeneration. During the migration of a stationary cell, the cell polarizes, forms lamellipodia at the leading edge (LE), and triggers the concurrent retraction of the trailing edge (TE). During cell migration governed by inhibitory G protein (Gi)–coupled receptors (GPCRs), G protein βγ (Gβγ) subunits control the LE signaling. Interestingly, TE retraction has been linked to the activation of the small GTPase Ras homolog family member A (RhoA) by the Gα12/13 pathway. However, it is not clear how the activation of Gi-coupled GPCRs at the LE orchestrates the TE retraction in RAW264.7 macrophages. Here, using an optogenetic approach involving an opsin to activate the Gi pathway in defined subcellular regions of RAW cells, we show that in addition to their LE activities, free Gβγ subunits also govern TE retraction by operating two independent, yet synchronized, pathways. The first pathway involves RhoA activation, which prevents dephosphorylation of the myosin light chain, allowing actomyosin contractility to proceed. The second pathway activates phospholipase Cβ and induces myosin light chain phosphorylation to enhance actomyosin contractility through increasing cytosolic calcium. We further show that both of these pathways are essential, and inhibition of either one is sufficient to abolish the Gi-coupled GPCR-governed TE retraction and subsequent migration of RAW cells.
Keywords: calcium, cell migration, G protein, G protein-coupled receptor (GPCR), optogenetics, signal transduction, Retinal, opsin
Introduction
G protein–coupled receptors (GPCRs)2 sense extracellular stimuli and transmit the signal to the cell's interior via activation of heterotrimeric G proteins (1). Heterotrimeric G proteins consist of three subunits: Gα, Gβ, and Gγ. In the inactive state, the Gα subunit is bound to GDP, and all three subunits are bound in a heterotrimeric complex, which is coupled to the cytosolic face of an associated receptor (the GPCR). Upon activation by signal, the GPCR induces a conformational change in its associated G protein, which causes the Gα subunit to exchange GTP for GDP. This in turn induces heterotrimer dissociation into a GαGTP and a free Gβγ subunit (2). These activated G proteins either interact with effectors present on the plasma membrane (PM) or recruit cytosolic effectors to the PM and activate them. Gi-coupled GPCRs control motility in many cell types, in which Gβγ subunits function as the main transducer (3). However, it is unclear whether GαiGTP plays a direct role in Gi pathway–mediated cell migration. Gβγ subunit is an active molecule and does not require additional steps, such as phosphorylation, for activation. This makes it a fast and efficient signal transducer. Gβγ subunits interact with a cohort of downstream effectors, including phosphoinositide 3 kinase (PI3K) (4), adenylyl cyclase (5–8), phospholipase Cβ isoforms (9), and guanine nucleotide exchange factors (RhoGEF) (10, 11). Therefore, Gβγ subunits control a large number of cellular functions, including cell migration (12).
Cell migration is a highly integrated multistep process that plays a fundamental role in a wide array of biological events, including embryonic morphogenesis, wound healing, and immune responses, and also contributes to tissue repair and regeneration (13). Aberrant cell migration is implicated in many pathological conditions, including immune disorders and cancer metastasis (14).
The machinery that governs cell motility can be driven by the mechanisms responsible for intrinsic directionality or by external guidance cues (15). In the absence of external stimuli, certain cell types, including macrophages, exhibit random motility and are driven by excitable-oscillating signaling networks (16). External guidance cues asymmetrically activate cell surface receptors, including receptor-tyrosine kinases (i.e. PDGF receptor) or GPCRs (i.e. CXCR4), thereby inducing either directional migration in stationary cells (RAW264.7) or orienting the migration direction of randomly motile cells (17). Activation of both receptor-tyrosine kinases and GPCRs controls common signaling pathways mediated through serine/threonine-specific protein kinases such as Akt and Raf, Rho GTPases including Ras-related C3 botulinum toxin substrate 1 (Rac1), Ras homolog family member A (RhoA) homolog of cell division control protein (Cdc42), and molecules calcium and diacylglycerol (DAG) as well. Through activation of such pathways, cell surface receptors can govern directional migration by modulating cellular activity at both the leading edge (LE) and trailing edge (TE), as well as by controlling basal motility (18). Non-receptor signaling regulators, including guanine nucleotide exchange factors (GEFs) and guanine nucleotide dissociation inhibitors (GDIs) are involved in cell migration through modulating the activity of heterotrimeric G proteins. Activator of G protein signaling 3 (AGS3) is a GDI and liberates a free Gβγ subunit by binding to GαGDP with nanomolar affinities, whereas AGS1 is a GEF for Gαi and promotes heterotrimer dissociation (19, 20). Similarly, Gα-interacting vesicle-associated protein (GIV) and Dishevelled-binding protein (Daple) also control cell motility through the activation of Gi signaling (21, 22).
Asymmetrically activated GPCRs on the plasma membrane govern migration along the axis of chemokine gradient (23, 24). The region of the cell on which GPCRs are more active becomes the LE. The traction forces generated at the LE pull the cell body toward the gradient, whereas propagating signals to the TE induces its retraction, facilitating the effective relocation of the entire cell (25, 26). This process is termed tread-milling and involves intertwined networks of spatiotemporally coherent as well as segregated yet tightly controlled molecular and cellular events (27). Signaling activities initiated at the LE induce (i) formation of invadopodia and lamellipodia with new focal adhesions, (ii) retraction of the TE accompanied with actomyosin contractility and focal adhesion disassembly, and (iii) active relocation of internal organelles orchestrating directional movement of the entire cell. Whereas the majority of LE activities can be assigned to Gβγ subunit-induced PI3Kγ activation and subsequent phosphatidylinositol 1,4,5-trisphosphate (PIP3) production (28), it is not clear how the activation of Gi-coupled GPCRs at the LE induces the retraction of the TE. The retraction of the cytoskeleton has been attributed to Gα12/13 subunit–mediated RhoA activation through RhoGEFs (29–31). RhoA is initially present in the inactive GDP-bound form. Upon activation, it is converted to the active GTP-bound form. Subsequent activation of Rho family protein kinases results in phosphorylation of myosin light chain phosphatase (MLCP), thereby inhibiting its activity and leading to an increase in MLC phosphorylation. This consequently promotes actomyosin-based contractility (32). During Gαi pathway-directed neutrophil migration, Gβγ subunits in concert with AC9, through DAG and inositol 1,4,5-triphosphate (IP3), activate mTORC2 and PKCβII (33). This suppresses actin remodeling at the LE and phosphorylated myosin II (p-MyoII) activity at the TE. Here, AC9 and protein kinase A (PKA)-mediated inhibition of myosin light chain kinase (MLCK) and RhoA impair the TE retraction (34, 35).
In this study, in addition to its LE functions, we investigated whether Gi pathway–induced generation of free Gβγ subunits also controls TE retraction. We hypothesized that during Gi pathway–controlled cell migration, free Gβγ subunits are primarily responsible for the establishment of front–back polarity, which encapsulates both LE and TE functions and thereby controls the entire process of cell migration. We tested this hypothesis using a mouse macrophage cell line, RAW264.7 (RAW), and blue opsin. Upon optical activation (OA) with 445-nm light (blue light), blue opsin induces precise spatial and temporal control of signaling and allows generation of GαGTP and free Gβγ subunits exclusively at the LE (36, 37). Such GPCR activation mimics the state of a migratory cell experiencing a chemokine gradient. This allows examination of whether the TE retraction signal is generated at the LE. Spectral selectivity of blue opsin to blue light allows for imaging molecular and morphological responses of cells using other wavelengths, including 488 nm (green), 515 nm (yellow), and 595 nm (orange) (37). Our results show that free Gβγ subunits are primarily responsible for controlling the entire migration process in macrophages induced by the activation of the Gi pathway. In addition to its well-known functions in the LE (38), the data demonstrate that Gβγ subunits promote TE retraction through PLCβ-induced calcium mobilization. We show that this essential pathway, together with free Gβγ-RhoGEF–induced phosphorylation of MLCP, promotes actomyosin contractility, resulting in complete TE retraction.
Results
OA of blue opsin for subcellular G protein activation and directional migration of macrophages
To examine whether opsins and G proteins can be activated exclusively at the LE, the Gγ9 assay was used (39). This assay employs the translocation of mCherry Gγ9 (mCh-γ9) from the PM to internal membranes (IMs), generated by activated GPCRs. Global OA of HeLa cells expressing blue opsin and mCh-Gγ9 resulted in the redistribution of mCh-γ9 between the PM (black curve) and IMs (red curve) with a translocation half-time (t½) of ∼7.5 s (Fig. 1, A and B). Termination of OA resulted in complete reversal of the translocation process, whereas continuous OA showed sustained G protein activation, as indicated by the maintenance of accumulated mCh-γ9 in IMs (supplemental Fig. S1A). OA of blue opsin in a subcellular region of interest with an area of 2 × 10 μm2 was activated and resulted in a localized disappearance of mCh-Gγ9 in the PM area exposed and confined accumulation of mCh-Gγ9 at the proximal IMs (Fig. 1C) (yellow arrow). No mCh-Gγ9 localization changes were observed in the distal regions (blue arrow). A plot shows the reduction of mCh intensity on the activated PM regions (solid black) and a corresponding increase in adjacent IMs (solid red) (Fig. 1D). Neither distal IMs nor PMs (dashed lines) showed any fluorescence intensity change. This finding suggests that blue opsin can be used to selectively activate G proteins in a spatially confined region of a cell.
Figure 1.

Adopting optical activation of GPCRs to control the chemokine pathway. A, images of a HeLa cell expressing blue opsin and mCh-γ9. The white box shows the region of OA using 445-nm laser light (1 μW/μm2) pulsed at 0.5 Hz. Before OA, cells were reconstituted with 50 μm 11-cis-retinal. Upon OA of the entire cell, mCh-γ9 on the PM translocates, reducing mCh-γ9 on the PM (yellow arrow) and leading to a gain in IMs (white arrow). Upon termination of OA, mCh-γ9 returns to the PM. B, the plot shows the normalized mCh-γ9 fluorescence intensity changes on the PM and IMs. C, spatially confined OA of blue opsin in the same HeLa cell as in A. Upon OA of the white box area, mCh-γ9 on the activated PM region translocates to IMs, resulting in a localized loss of mCh-γ9. Note the gain of mCh-γ9 in immediate IM regions (yellow arrow). No mCh-γ9 changes were observed in distal regions (blue arrow) of the cell. D, the corresponding plot for C shows the mean fluorescent intensity of mCh-γ9 on the PMs and in IMs (activated and distal). The gray area shows the duration of OA. E, OA (blue box) of Gi-coupled blue opsins with 445-nm light induced RAW cell migration. Cell expresses blue opsin mCh. F, a HeLa cell expressing mCh-γ9, before and after activation of Gi-coupled endogenous CXCR4 receptor with 50 ng/ml SDF-1α. The plot shows mCh intensity on the PM and in IMs. G, images and plots showing translocation of GFP-γ9 in HeLa cells also expressing Gs-coupled β1-AR before and after activation with isoproterenol (10 μm). H, HeLa cells expressing Gq-coupled GRPR and the sensor GFP-γ9 show no translocation of GFP-γ9 after GRPR activation with 1 μm bombesin. I, localized OA (blue box) of Gs-coupled CrBlue in RAW cells failed to induce migration. Blue and green lines indicate the LE and TE of the cell. To the right are the orthogonal slice views of a cross-section of these regions. The arrow (orange) shows mCh intensity changes of this cross-section over time. Scale bar, 10 μm.
Next, an approach was devised to globally image molecular sensors while inducing localized optical activation of G proteins in single cells (supplemental Fig. S2A). This allowed generation of a G protein activity gradient across the cell, segregating the LE and TE. Due to the Gaussian type distribution of intensities of the confined light beam (supplemental Fig. S2A, middle image, blue curve), the 445-nm optical stimulus was placed 1–2 μm away from the periphery of the cell, limiting G protein activity to only the intended region. Using this strategy, the RAW cells expressing blue opsin-mCh were optically activated (blue box) in a confined region (Fig. 1E). The cells showed lamellipodia formation at the activated LE and cell body retraction at the TE. This resulted in an average migration of 35 μm in 20 min. This result suggests that blue opsin can be used to localize G protein activity to the LE, and such activation is sufficient to direct complete migration of RAW cells.
Role of Gα identity in the heterotrimer upon Gβγ subunit generation
If Gβγ subunit is the primary regulator of Gi pathway-induced cell migration, could the free Gβγ subunits generated as a result of Gs- and Gq-coupled GPCR activation also result in cell migration? Activation of Gq and G12/13, as well as Gs pathways, has been shown to control migration in specific cell types, including neutrophils and cancer cells (40–42). Interestingly, the associated migration mechanisms are usually described using the activity of the accompanying Gα subunit. To understand this, generation of Gβγ subunits by Gs- and Gq- coupled GPCR activation was compared with that induced by Gi-coupled endogenous CXCR4 activation by SDF-1α in HeLa cells. HeLa cells expressing green fluorescent protein (GFP)-Gγ9 and Gs-coupled β1-adrenergic receptor (β1-AR) were exposed to 10 μm isoproterenol (Fig. 1G) and showed Gγ9 translocation comparable with that induced by CXCR4 activation (Fig. 1F). In contrast, activation of Gq-coupled gastrin-releasing peptide receptor (GRPR) through exposure to 1 μm bombesin resulted in only a marginally detectable GFP-Gγ9 translocation (Fig. 1H). However, GRPR activation resulted in a robust phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolysis (supplemental Fig. S1B). Activation of GRPR and another Gq-coupled receptor, M3 muscarinic, not only failed to induce Gγ9 translocation, but also failed to induce Gγ1 and Gγ5 translocations (supplemental Fig. S1C). However, they exhibited robust PIP2 hydrolysis (supplemental Fig. S1D). Therefore, regardless of the Gγ subunit in the heterotrimer, activation of Gq-coupled GPCRs results in the generation of fewer free Gβγ subunits.
Effect of Gs pathway activation–induced free Gβγ on RAW cell migration
Because Gs-coupled GPCR activation results in a significant translocation of Gβγ subunits, we examined whether Gs pathway activation also results in RAW cell migration mediated through free Gβγ subunits. An optogenetic chimeric Gs-coupled opsin, CrBlue, was employed to activate the Gs pathway in confined regions of RAW cells (36). CrBlue was engineered by replacing cytosolic loops of blue opsin with that of the Gs-coupled opsin from box jellyfish (Carybdea rastoni) (36). During the initial OA, RAW cells showed minor movement toward the optical input. However, continuous OA of CrBlue did not result in a continuous directional movement along the axis of GPCR activation (Fig. 1I). This may be due to less efficient activation of G proteins by CrBlue or opposing activities of GαsGTP and free Gβγ subunits upon RAW cell migration. Increased cAMP production induced by Gαs activates PKA, and exchange protein directly activated by cAMP (EPAC) controls cell motility (44). Whereas PKA suppresses cell migration, EPAC facilitates it (45, 46). When RAW cells expressing blue opsin-mCh were treated with forskolin and isobutylmethylxanthine to increase cAMP levels, RAW cells failed to migrate upon blue opsin activation (Fig. 2, A and B). However, cells showed unperturbed LE activities, including lamellipodia formation. This finding is in agreement with reports that increasing cAMP and PKA signaling inhibits migration by attenuating RhoA and MLCK activities (8, 47, 48).
Figure 2.

Effect of cAMP/PKA, DAG/PKC, and Gαi-GPCR pathways on RAW cell migration. RAW cells in glass-bottomed dishes expressing blue opsin-mCh and treated with 50 μm 11-cis-retinal were optically stimulated using localized OA (white box) under the following experimental conditions. A, control cells showed complete cell migration with synchronized movement of the LE and the TE. B, 1 μm forskolin and isobutylmethylxanthine prevented the TE retraction, whereas the LE activities remained unperturbed, demonstrating the effect of increased cAMP on cell migration. C, treatment with 10 μm GO6983, a potent PKC inhibitor, did not affect RAW cell migration, suggesting that the DAG/PKC pathway may not have a significant effect on migration. D, incubation of cell with PTx (0.05 μg/ml) for 12 h completely suppressed cell migration. PTx induces ADP-ribosylation and prevents G protein heterotrimer dissociation. E, RAW cells treated with the Gβγ inhibitor, gallein (10 μm, 30 min, 37 °C) also failed to migrate and showed neither LE nor TE activities. F, images of RAW cells transfected with blue opsin GFP, CRY2-mCh-GRK2Ct, and CIBN-CaaX. Local OA resulted in recruitment of CRY2-mCh-GRK2Ct to the PM, near the region of OA. Here, blue opsin activation resulted in release of free Gβγ subunits. However, the recruited CRY2-mCh-GRK2Ct is expected to sequester free Gβγ, preventing activation of its effectors. Even after 20 min of OA, initiation of cell migration was not observed. G, histograms show the extent of movement and migration velocities of the peripheries of the LE and the TE of control and pharmacologically perturbed RAW cells in A–E. *, p < 0.001. (n = 3–10 cells). Scale bar, 10 μm. Error bars, S.E.
Because free Gβγ subunits generated by Gi pathway activation have been shown to regulate neutrophil migration through protein kinase Cβ (PKCβII) (38), we also tested whether PKC influences RAW macrophage migration as well. PKC inhibition with a broad-spectrum PKC inhibitor, GO 6983 (which has a 7 nm IC50 for PKCβ), does not prevent blue opsin-induced RAW cell migration (Fig. 2 and supplemental Movies S1 and S2). Therefore, we anticipate that, during asymmetric activation of the Gs pathway in RAW cells, GαsGTP-induced cAMP reduces the ability of free Gβγ subunits to induce migration.
Roles of Gβγ subunits in Gi pathway-induced RAW cell motility
To isolate the contributions of Gβγ subunits from that of Gαi, RAW cell migration was examined in the presence of a small molecule Gβγ subunit inhibitor, gallein (49, 50). Upon localized OA of blue opsin, control RAW cells migrated with a velocity of ∼1.8 μm/min. Pertussis toxin (PTx) inhibited this migration, indicating that it is governed by the Gi pathway (51) (Fig. 2D). Gallein binds to the effector-binding hot spot in Gβ (52). RAW cells incubated with 10 μm gallein failed to migrate upon localized blue opsin activation, and no LE or TE activities were observed (Fig. 2E and supplemental Movie S3). Plots show the distance traveled by the peripheries of the LE and TE as well as the velocities (Fig. 2G). Fluorescein, a gallein-like compound that does not bind Gβγ, did not show any effect on migration (supplemental Fig. S2, B and C).
To test whether gallein inhibited heterotrimer dissociation or GαGTP signaling, we examined its effect on Gβγ translocation and cAMP responses upon activation of β1-AR. Translocation of GFP-γ9 was unperturbed by 10 μm gallein (supplemental Fig. S3A). Additionally, in the presence of gallein, β1-AR activation in HeLa cells showed an unperturbed cAMP production compared with the untreated cells, as measured using PKA-δRIIβ–PKA-Cα-YFP sensor (supplemental Fig. S3B). Thus, gallein inhibited migration by selectively blocking Gβγ subunit–effector interactions.
We also tested optical inhibition of free Gβγ subunits to examine the effect of localized GαiGTP activity on RAW cell motility. The optogenetic inhibitor is composed of cryptochrome 2 (CRY2) linked to the carboxyl terminus of G protein receptor kinase 2 (GRK2Ct) (53). We expressed PM-targeted cryptochrome-interacting basic-helix-loop-helix (CIB1) and cytosolic CRY2-GRK2Ct together with blue opsin in RAW cells. Using localized OA, CRY2-mCh-GRK2Ct was targeted to predefined LE (Fig. 2F). OA was continued after the addition of retinal to concurrently activate blue opsin at the LE. These cells showed neither lamellipodia formation nor migration, whereas control cells exhibited normal migration. Collectively, these data suggest that whereas free Gβγ subunit is the primary controller, GαGTP may not have a direct role in Gαi-coupled GPCR-mediated RAW cell migration.
Segregating the roles of the Gβγ-PI3K-PIP3 axis on directional motility
Here we examined the significance of PI3K on cell migration relative to other Gβγ subunit effectors. PIP3 has been identified as one of the primary regulators of cytoskeleton remodeling during LE formation (37, 54). Accumulation of PIP3 at the LE sets a feed-forward signal amplification circuit between PI3K and Rac1, and subsequently activates WAVE complex and Arp2/3 to promote actin polymerization, generating traction forces required for cell migration (55, 56). By rapidly reversing the side of OA, we previously showed that the formation of PIP3 at the new LE and its active reduction at the former LE are synchronized (37). This may allow cells to set the migration direction along the axis of the external chemical gradient (36).
To examine whether PIP3 generation at the LE alone can govern both the LE and TE activities, thus supporting complete cell migration, PI3K at the LE was optically activated. The inter-Src homology 2 domain of a p85b-regulatory subunit (iSH2) of PI3K was optically targeted to the user-defined LE on the PM as described previously (Fig. 3A, top) (57). This results in a significant and localized PIP3 production at the LE (Fig. 3A, bottom). A plot shows an increase in PIP3 at the LE. This PIP3 response was similar to that observed in blue opsin activation-induced cell migration. However, optogenetic PI3K activation only resulted in a minor migration response.
Figure 3.

Gβγ governs TE contractility through modulating Ca2+-CaM and MLCK signaling–governed TE contractility. A, a RAW cell–expressing mCh-CRY-iSH2, CIBN- CaaX and AKT-PH-Venus was optically activated to recruit mCh-CRY-iSH2 to a confined region (top). The cell shows localized PIP3 production (bottom images and the plot), similar to that seen with blue opsin activation; however, the optogenetic activation of PIP3 production resulted in only a minor migration response. B, RAW cells transfected with blue opsin mCh were subjected to localized OA in the presence of various inhibitors: 10 μm gallein (for Gβγ), 50 nm wortmannin (PI3K), and 12.5 μm GSK 269962 (ROCK). Cells were incubated with the inhibitor(s) for 30 min at 37 °C before imaging and OA. I, inhibition of Gβγ resulted in a complete suppression of migration with complete cessation of LE and TE activities. II, inhibition of PI3K prevented migration, and a reduction in cell size was also observed. III, ROCK inhibition prevented complete cell migration. Whereas no TE retraction was observed, the LE activities remained unperturbed, resulting in a cell surface area increase. IV, dual inhibition of PI3K and ROCK resulted in neither cell migration nor cell size change. This suggests that ROCK is essential for generation of inward contractile forces. The histogram shows the corresponding percent cell surface changes. C, images of RAW cells transfected with blue opsin mCh and incubated with 10 μm A-7 hydrochloride, a CaM antagonist, and ML-7 hydrochloride, an MLCK antagonist, respectively, for 30 min at 37 °C. I, A-7 hydrochloride inhibited cell migration induced by OA of blue opsin; however, the cell showed intact LE activities. II, MLCK inhibition also prevented RAW cell migration. The histogram shows that compared with the control, both CaM- and MLCK-inhibited cells exhibit a complete lack of TE activities, whereas comparable lamellipodia growth at the LE was observed. **, p < 0.0001 (n = 10). Scale bar, 10 μm. Error bars, S.E.
Next, to examine whether Gβγ subunit-induced PI3K activation/PIP3 production controls TE retraction, blue opsin in RAW cells was activated to induce migration while pharmacologically modulating specific signaling entities in the migration pathway as follows. Inhibition of Gβγ subunits with gallein made RAW cells completely non-responsive to migration induction (Fig. 3, B (I)). Inhibition of PI3K with 50 nm wortmannin also prevented cell migration (Fig. 3B (II)). Wortmannin-treated cells completely ceased LE activities, but they exhibited cell-wide cytoskeleton retraction that was not observed in gallein-treated cells (Fig. 3B (II), yellow arrows). This indicates that Gβγ subunits mediate TE retraction through a PI3K-independent pathway.
Inhibition of Rho kinase (ROCK) with 12.5 μm GSK 269962 inhibited RAW cell migration, whereas LE activities, including lamellipodia formation, remained almost unperturbed (Fig. 3B (III)). Combined inhibition of both PI3K and ROCK also completely eliminated cell migration, with no LE or TE activities observed (Fig. 3B (IV)). A plot shows the percent cell size change, which captures the contractility (positive) in wortmannin-treated cells and the expansion (negative) in ROCK-inhibited cells (Fig. 3B). During migration induction, RAW cells treated with ROCK inhibitor showed a complete lack of TE contractility, whereas LE activities, including lamellipodia formation, remained unchanged. Therefore, we examined whether RAW cells express free Gβγ subunit-controlled signaling components that govern the RhoA pathway. Data from RNA-seq analysis of RAW cells showed expression of multiple types of Rho-GEFs that are potentially involved in activation of RhoA and ROCK (supplemental Fig. S4A). Among them, RAW cells possess Rho114-GEF, which has been identified as a Gβγ subunit-activated RhoGEF (10). Contractility observed at the TE, facilitating its retraction, has been attributed to a reduction in MLCP-induced dephosphorylation of p-MLC (58). ROCK retards this process by deactivating MLCP through its phosphorylation. Therefore, it is likely that Gβγ subunits control the RhoA pathway to maintain a high concentration of p-MLC and facilitate TE retraction during inhibitory GPCR-induced cell migration.
Another RhoGTPase, Cdc42, has been shown to be involved in a number of migration processes and is directly and indirectly controlled by Gβγ subunits (59). Therefore, we also examined whether Cdc42 activity could be detected during blue opsin activation–induced RAW migration. Whereas RAW cells express a significant amount of mRNA for Cdc42 and a Cdc42-GEF, FLJ00018 (ARHGEF42) (supplemental Fig. S4A), RAW cells did not show Cdc42 sensor accumulation at the LE during blue opsin-induced migration (supplemental Fig. S4C).
Calmodulin (CaM) and MLCK increase p-MLC–based contractility
Here we examined the function of the pathway components that link free Gβγ subunits and MLCK in RAW cells. MLCK phosphorylates MLC to generate p-MLC, inducing actomyosin contractility (60). Free Gβγ subunits have been shown to interact with and activate PLC isoforms β1, β2, β3, and δ3, inducing PIP2 hydrolysis. This results in mobilization of stored calcium to the cytosol (61, 62). Ca2+-CaM binds to the M13 catalytic domain and activates MLCK (63). The activated MLCK then induces phosphorylation at Ser-19 on the regulatory light chain of MLC. Alternatively, calcium-stimulated calmodulin-dependent protein kinase kinase 2 (CaMKK2) activates adenosine monophosphate-activated protein kinase (AMPK), resulting in MLCK phosphorylation (64). These phosphorylations increase the Ca2+-CaM concentration required to activate MLCK, setting up a negative feedback loop that suppresses MLCK activity and ultimately leads to down-regulation of MLC phosphorylation (65, 66). To examine the outcome of this complex relationship between cytosolic calcium and MLC phosphorylation in RAW cells, we performed the following experiments.
We first tested whether Gi pathway– induced migration of RAW cells is sensitive to the CaM antagonist A7-hydrochloride (67). The inhibition of CaM suppressed blue opsin activation-induced RAW cell migration (Fig. 3, C–I). However, the cells did produce lamellipodia at the LE, similar to the untreated control cells. Next, the effect of inhibition of MLCK on cell migration was examined using its inhibitor ML-7 hydrochloride. Although cells showed lamellipodia formation at the LE, neither TE retraction nor cell migration was observed (Fig. 3C (II)). A histogram shows the distance traveled by the peripheries of the LE and TE of RAW cells treated with A-7 and ML-7, respectively, compared with control cells (Fig. 3C). Interestingly, inhibition of AMPK did not show a detectable effect on blue opsin–induced RAW cell migration (data not shown). Overall, inhibition of both CaM and MLCK did not have an obvious effect on Gi pathway–induced LE signaling, whereas the cells showed no TE retraction or directional migration (Fig. 3C (I and II)).
Roles of extracellular and cytosolic calcium on TE contractility and cell migration
How cytosolic calcium influences the TE retraction of RAW cells was examined. RAW cells incubated in calcium-free Hanks' balanced salt solution (HBSS) over 30 min were unable to migrate in response to localized blue opsin activation (Fig. 4, A and B). To examine whether this lack of migration is due to reduced cytosolic calcium prior to the migration induction, RAW cells were incubated for 30 min with BAPTA-AM, a cell-permeable calcium chelator. Compared with control RAW cells, BAPTA-AM–treated cells showed no migration, whereas their LE activities remained almost intact (Fig. 4C and supplemental Movie S4). To confirm that the suppression of migration is not due to an unintended effect of BAPTA-AM, the same experiment was performed using thapsigargin, a sarco/endoplasmic reticulum calcium-ATPase pump inhibitor), in place of BAPTA-AM. Before the induction of cell migration, cells were incubated with 0.5 μm thapsigargin to deplete stored ER calcium (68). Analogous to the BAPTA-AM experiment, cells showed no migration, whereas LE activities, including lamellipodia formation, remained unperturbed (Fig. 4D). Histograms show that depletion of cytosolic or sarcoplasmic calcium has the greatest impact on TE retraction, although LE lamellipodia formation and the overall cell migration velocities were also reduced (Fig. 4, E–G).
Figure 4.

Cytosolic calcium regulates TE retraction in RAW cells through a PI3K- independent pathway. A, time lapse images show OA-induced migration of a RAW cell that was transfected with blue opsin mCh. OA-induced lamellipodia formation at the LE and subsequent retraction of the TE results in complete cell migration. B, a RAW cell incubated with calcium-free HBSS for 1 h at 37 °C before the experiment showed no TE retraction, whereas the LE activities remained unchanged. C, before OA, the cell was subjected to chelation of cytosolic calcium by incubation for 30 min with 5 μm BAPTA-AM. D, a RAW cell was incubated with the sarco/endoplasmic reticulum calcium-ATPase blocker 500 nm thapsigargin for 30 min to deplete stored ER calcium. Images show that the cell formed lamellipopdia at the LE on OA but showed a complete lack of TE retraction. E–G, histograms show the extent of movement of the peripheries of the LE and TE and the migration velocities of cells described in A–D. Scale bar, 10 μm. H, RAW cells expressing blue opsin and AKT-PH-mCh were incubated with the calcium chelator BAPTA-AM (5 μm, 30 min at 37 °C). Upon global OA of blue opsin, cells showed robust PIP3 production, similar to control cells. Kymographs of the cellular cross-sections show the reduction of cytosolic and increase in PM-bound AKT-PH-mCh. The plot below was generated using the kymograph and shows the corresponding increase in mCh intensity on the PM. I, upon localized OA, control RAW cells showed complete cell migration with PIP3 production at the LE. Although BAPTA-AM–treated cells also showed PIP3 production at the LE, similar to that seen in control cells, they failed to migrate. The plot shows the corresponding AKT-PH-mCh dynamics at the LE (n = 3). Scale bar, 10 μm. **, p < 0.0001 (n = 10 cells); error bars, S.E.
We then examined the link between cytosolic calcium and Gβγ-governed PI3K activity. Both control and BAPTA-AM–treated cells expressing Akt-PH-mCh (PIP3 sensor) showed a similar and significant PIP3 production upon localized blue opsin activation (Fig. 4H). An orthogonal view shows the mCh dynamics across a section of the cell, exhibiting PIP3 sensor translocation from the cytosol to the PM (Fig. 4H). Nevertheless, only control RAW cells migrated (Fig. 4I and supplemental Movie S5).
Gi-coupled receptor activation induces mobilization of cytosolic calcium
The ability of the Gi-pathway to mobilize calcium to the cytosol through Gβγ subunits was examined. We have previously shown that activation of Gαi-coupled endogenous α2-AR with norepinephrine (NE) induces an increase in cytosolic calcium in HeLa cells (69). Real-time PCR data show that RAW cells possess considerable levels of mRNA for PLCβ2, an enzyme activated by Gβγ subunits (Fig. 5A). To confirm that Gi-coupled GPCR activation induces PLCβ activation and PIP2 hydrolysis through free Gβγ subunits, NE-induced PIP2 hydrolysis in HeLa and RAW cells expressing mCh-PH was examined. OA of blue opsin resulted in significant PIP2 hydrolysis in both cell types (Fig. 5, B and C). PIP2 distribution remained unchanged in PTx- and gallein-treated RAW cells (Fig. 5, D and E). To examine whether the signaling circuits required to mobilize Gβγ subunit-mediated calcium are present in RAW cells, endogenous complement component 5a receptor 1 (c5aR1), a Gi-coupled GPCR, was activated, and the cytosolic calcium was probed using Fluo-4 AM. The addition of 25 μm c5a resulted in a robust increase in cytosolic calcium, which peaked within 20 s (Fig. 6A). RAW cells treated with PTx failed to show c5a-induced calcium mobilization, suggesting that the Gi pathway controls this process (70). To examine whether the ER calcium stores in PTx-treated cells remain intact during the addition of c5a, cells were exposed to thapsigargin. Thapsigargin has been shown to transiently increase cytosolic calcium by blocking calcium entry to the ER (71). PTx-treated RAW cells failed to produce a calcium response upon c5aR activation, but the same cells exhibited a transitory cytosolic calcium increase upon thapsigargin addition (Fig. 6B). To demonstrate that the observed calcium mobilization is independent of Gβγ subunit-mediated PI3K activation, the c5a-induced calcium response was examined in the presence of a PI3K inhibitor, wortmannin. Inhibition of PI3K did not alter the c5a-induced calcium mobilization (Fig. 6C). To directly examine whether the Gαi pathway–mediated cytosolic calcium mobilization is controlled by free Gβγ subunits, c5a receptors were activated in cells incubated with gallein to inhibit Gβγ subunits. As anticipated, cells did not show a calcium increase (Fig. 6D). Cells treated with the gallein-like control compound fluorescein (which does not bind Gβγ) did exhibit calcium mobilization upon c5AR activation (supplemental Fig. S2D).
Figure 5.

Activation of Gαi-GPCR induces PIP2 hydrolysis in RAW cells. A, real-time PCR mapping of genes required for Gβγ to induce MLCK activation in RAW cells. The cDNA of RAW cells was probed using appropriate primers to amplify genes expressed and involved in this pathway. The data show significant expression of PLCβ2, a Gβγ-activated PLC. Data also show considerable expression of CaM isoforms, MLCK and MLCP (MYTP1), suggesting the existence of molecular components that would allow Gβγ to induce MLCK activation. The data are presented as the mean ± S.D. (error bars) (n = 3). B and C, images of a HeLa cell expressing mCh-PH and a RAW cell expressing mCh-PH and blue opsin, respectively, before and after GPCR activation. Activation of endogenous α2-AR with 400 μm NE in HeLa cells and activation of blue opsin using 445-nm light in RAW cells both resulted in PIP2 hydrolysis. The plots show the corresponding mCh-PH changes in the cytosol. D and E, images of RAW cells expressing blue opsin and mCh-PH, treated with PTx (overnight incubation with 0.05 μg/ml PTx at 37 °C) and gallein (30-min incubation with 10 μm at 37 °C), respectively. Both PTx- and gallein-treated cells failed to exhibit PIP2 hydrolysis upon OA of blue opsin. The plots show the corresponding mCh-PH changes in the cytosol. Scale bar, 10 μm.
Figure 6.

Free Gβγ induces mobilization of cytosolic calcium in macrophages. RAW cells were incubated with the probe Fluo-4 AM (1.14 μm) for 25 min at room temperature and washed with HBSS buffer three times before performing the following experiments. A, images of RAW cells before and after activation of endogenous c5aR with 25 μm c5a. Activation of c5aR induced calcium mobilization, and the plot shows calcium response of individual cells. Gray line, time of the agonist addition. B, images of RAW cells incubated with PTx (0.05 μg/ml) to induce ADP-ribosylation and inhibition of Gαi signaling. Inhibition of Gαi signaling prevented c5a-induced calcium mobilization. The same cells exhibited a transient calcium increase upon the addition of 500 nm thapsigargin (Thapsi), which inhibits sarco/endoplasmic reticulum (SR) calcium transport. The plot shows individual cell calcium responses after c5a (black traces) followed by thapsigargin addition (red traces). C, images of RAW cells incubated with 50 nm wortmannin, before and after c5aR activation. The images and plot show that PI3K inhibition has no effect on the c5aR-induced calcium response. D, images of RAW cells before and after exposure to various inhibitors: Gβγ inhibitor, 10 μm gallein; cytosolic calcium chelator, 10 μm BAPTA-AM; IP3R antagonist, 5 μm 2APB. Cells were incubated with inhibitors for 30 min at 37 °C. All of these agents inhibited the c5aR-induced calcium response. E, images of RAW cells incubated in calcium-free buffer containing 5 μm BAPTA, before and after c5AR activation. Lack of extracellular calcium partially reduced the c5aR-induced calcium release (compare with A). F, images of RAW cells expressing blue opsin-mCh before and after opsin activation by 50 μm 11-cis-retinal. Blue opsin induced a robust calcium response only in the presence of retinal (red traces), compared with cells lacking the chromophore (black trace). Scale bar, 10 μm.
The pathway by which free Gβγ subunit–induced calcium mobilization in RAW cells was mapped as follows. Cells were incubated in calcium-free buffer for 1 h. These cells did not exhibit calcium mobilization upon c5aR activation (Fig. 6E). Gβγ subunit-induced PIP2 hydrolysis was examined after blocking inositol triphosphate receptor (IP3R) using 10 μm 2-aminoethoxydiphenyl borate (2APB). In the presence of 2APB, c5a failed to induce calcium mobilization in RAW cells (Fig. 6D). To examine whether calcium enters through an ion channel, c5a-induced calcium was examined in RAW cells after chelating extracellular calcium by adding BAPTA to the cell culture medium (Fig. 6E). Results show that the addition of BAPTA did not perturb (at 150 s) the C5a activation-induced calcium response. Collectively, these data suggest that free Gβγ subunits increase cytosolic calcium through the following pathway: PLCβ activation → PIP2 hydrolysis → IP3 generation → IP3R activation. Because we induced cell migration through the activation of Gi-coupled blue opsin at the LE, blue opsin's ability to induce calcium mobilization in RAW cells was also examined. The cells were preincubated with the calcium indicator Fluo4-AM and continuously imaged using 488-nm light at 1 Hz to capture Fluo-4 fluorescence before and after retinal addition. Upon the addition of retinal to activate opsin, blue opsin–expressing RAW cells showed a calcium response in a similar manner to that induced by the c5a addition (Fig. 6F). Because of the broad absorption spectra of blue opsin that overlaps in its right shoulder with 488 nm (supplemental Fig. S4B), we employed this wavelength for both imaging of Fluo4 and blue opsin activation.
Discussion
We have previously shown that localized OA of blue opsin can activate heterotrimeric G proteins and induce directional cell migration (37, 72). Both LE and TE activities are essential for cell migration. Our optogenetic strategy allowed for delivery of identical optical cues to individual cells, restricting G protein activation only to the LE. This enabled mapping signal propagation from the LE to the TE in real time in living cells. Our data indicate that GαiGTP is not likely to play a direct role in Gi-mediated RAW cell migration. Nevertheless, further investigations are required to confirm this. The lack of RAW cell migration upon localized activation of Gs-coupled CrBlue suggests that GαsGTP signaling may obstruct Gβγ subunit-induced RAW cell migration (Fig. 1F). The loss of the ability of blue opsin to induce RAW cell migration in the presence of elevated cAMP further validates these findings. These observations are also in concert with the previously reported cAMP-mediated inhibition of cell migration (47, 48). Elevated cAMP can prevent contractile force generation through PKA-mediated inhibition of MLC phosphorylation. Although free Gβγ subunits can enhance the activities of AC isoforms 2, 4, and 7 (73, 74), this process requires GαsGTP and therefore eliminates any possibility of cAMP generation during the activation of the Gi pathway. Collectively, these observations suggest that cAMP is likely to negate the ability of free Gβγ subunits from the Gs pathway to induce RAW cell migration. Further, gallein induces complete inhibition of cell migration, demonstrating that free Gβγ subunits play the primary role in controlling the complete cell migration induced by Gi pathway activation.
Free Gβγ subunits interact with and control multiple effectors, many of which can be linked to cell migration. Analysis of RNA-seq data from RAW cells shows the expression of these signaling proteins in RAW cells, validating the existence of the proposed pathway (Fig. 5A). Among them, activation of PI3Kγ, Rac1, and Cdc42 has been shown to be involved in the LE activities (75). Optogenetic activation of Cdc42 in RAW cells resulted in complete cell migration together with accumulation of the Cdc42 sensor at the LE (76). However, with the same sensor, we did not observe detectable Cdc42 activity during blue opsin activation–induced RAW cell migration. This can either be a result of higher affinity of free Gβγ subunits to other effectors or limited sensitivity of the assay to detect a minor Cdc42 activation, or both.
The minor RAW cell migration observed with optogenetic control of PI3K indicates that additional pathways controlled by free Gβγ subunits, other than PI3K and Cdc42, are required for Gi pathway–mediated complete RAW cell migration, especially for the processes involved in TE retraction. The ability of PI3K-inhibited cells to show a cell-wide contractility, while failing to exhibit LE activities, further supports the involvement of additional pathways (Fig. 2F). The RNA-seq data indicates the presence of multiple types of Rho-GEFs in RAW cells that can be involved in the activation of RhoA and subsequently ROCK (supplemental Fig. S4A). Among them, we found Rho114-GEF, which is activated by free Gβγ subunits (10). The deficiency in cell migration observed after inhibition of Gβγ subunits and lack of TE retraction after ROCK inhibition suggest that Gβγ subunits control TE retraction in RAW cells through the activation of RhoGEFs alone or together with other mechanisms.
ROCK promotes p-MLC–mediated contractility to induce TE retraction by deactivating MLCP through its phosphorylation (77). Whereas ROCK can also phosphorylate MLC (78), MLCK is crucial for MLC phosphorylation. Interestingly, real-time PCR data suggest that RAW cells express a considerable amount of PLCβ2 and CaM, obviously linking Gβγ subunits with MLCK though Ca2+-CaM (Fig. 5A). Further, Gi-coupled GPCR activation induces calcium mobilization, which is sensitive to PTx, gallein, and 2APB. This clearly demonstrates the existence of a functional Gβγ-PLCβ-IP3-IP3R-calcium signaling axis. Inhibition of cell motility upon either elimination or reduction of cytosolic calcium using BAPTA-AM and thapsigargin, respectively, indicates that calcium is an essential player in this mechanism. Unperturbed LE activities together with inhibited directional migration in cells lacking cytosolic calcium indicate that the LE and the TE activities are controlled by distinct yet Gβγ subunit–controlled molecular mechanisms. By inactivating MLCP and through direct phosphorylation of MLC, ROCK promotes actomyosin contractility. If the TE retraction is mainly controlled by the RhoA-ROCK pathway, cells should be able to show a significant migratory response in the presence of low concentrations of cytosolic calcium. However, data presented here reject this hypothesis. Regardless of the dual control of cytoskeleton (77, 78), data here show that ROCK is unable to support complete cell migration in the absence of cytosolic calcium. This indicates the existence of a calcium-mediated mechanism that acts as a gatekeeper for the TE retraction. Data from experiments with CaM as well as MLCK inhibition suggest that, independent of PI3K and RhoA pathway activation, Gβγ subunits are likely to control MLC phosphorylation as well. The complete inhibition of cell migration observed upon inhibition of either the Ca2+-CaM or RhoA-ROCK pathway suggests that neither pathway is redundant, and therefore both are mandatory for TE retraction during Gi pathway–governed RAW cell migration.
Overall, our results suggest that Gβγ subunits can solely control complete RAW cell migration through direct regulation of three interconnected yet independently operated mechanisms. The first mechanism includes previously defined Gβγ → PI3Kγ → activation of Rho family GTPases, including Rac1 and Cdc42, which control LE functions. Specific RhoGEFs, including P114-RhoGEF, which controls RhoA → ROCK to increase MLC phosphorylation, come under the second Gβγ subunit–controlled mechanism. From the data presented here, we show a third Gβγ subunit-controlled mechanism that regulates TE retraction through direct activation of PLCβ → calcium → Ca2+-CaM → MLCK activation, subsequently increasing MLC phosphorylation. Additionally, the data also show that, whereas the RhoA-mediated mechanism is essential, it is not sufficient for cells to achieve complete TE retraction (Fig. 7). Therefore, the Gβγ → PLCβ → Ca2+-CaM → MLCK axis is indispensable for MLC phosphorylation-induced actomyosin contractility. Nevertheless, evidence is available for cells diversely utilizing cytosolic calcium to govern cytoskeleton remodeling via mechanisms other than contractility (79, 80). For instance, gonadotropin-releasing hormone–expressing neurons employ calcium and the RhoA/ROCK pathway to promote outward actin in the leading process as a supporting mechanism to migrate (81).
Figure 7.
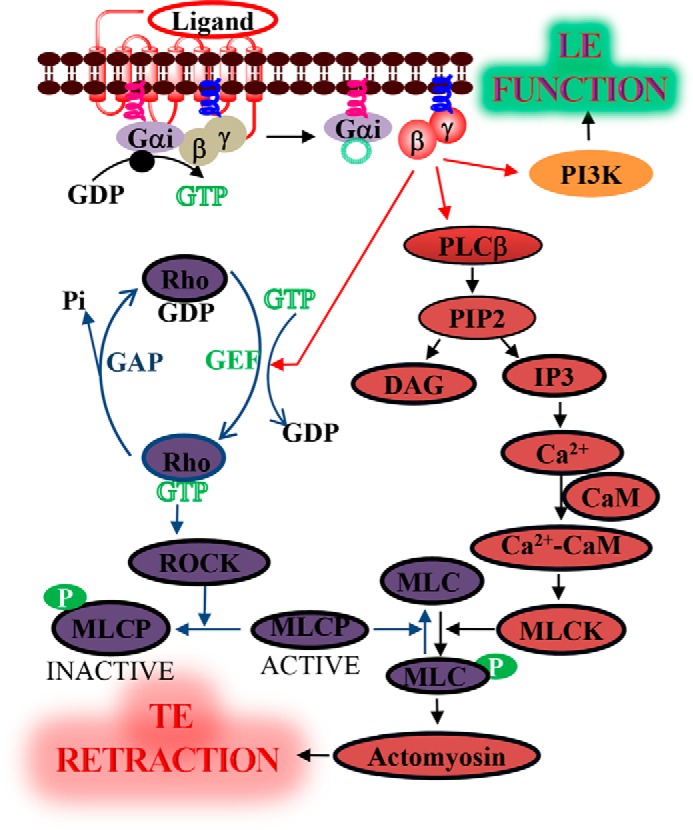
Proposed pathway for Gβγ-induced TE retraction. Activation of Gi-coupled GPCRs induces the heterotrimer dissociation, resulting in GαGTP and free Gβγ. Gβγ simultaneously induces activation of multiple effectors and, thus, several signaling pathways with distinct functionalities. In addition to its roles in the LE, free Gβγ controls TE retraction by deactivating MLCP through the activation of the RhoA pathway and activating MLCK through PLCβ-induced calcium mobilization. Both of these mechanisms result in an increase in phosphorylated MLC, promoting actomyosin contractility and subsequent retraction of the TE.
The local excitation global inhibition (LEGI) model that includes excitable signaling networks has been proposed to examine the differential LE and TE activities during cell migration (37, 82, 83). The present study demonstrates that cytosolic calcium is an integral component of the cell migration signaling networks governed by the Gi pathway. The locally generated calcium at the LE is likely to diffuse through the cytosol to distal regions and promote contractility. Therefore, calcium generated as a result of Gβγ subunit–induced PLCβ activation can be considered as a candidate for the long-sought global inhibitors described in the LEGI model. Although calcium-induced contractility can be global, because of the active cytoskeleton remodeling at the LE, retractions appear to be visible only in distal regions. Considering the conservation of migration mechanisms across cell types, other migratory cells, including cancer cells, may employ similar signaling networks to execute their migration. Therefore, the novel mechanisms as well as the synergistic cross-talk between pathways presented here may not only help to advance understanding of cell migration, but may also identify molecular targets for therapy development.
Experimental procedures
Cell culture and transfections
HeLa cells (ATCC, Manassas, VA) were cultured in minimum essential medium (CellGro) supplemented with 10% dialyzed fetal bovine serum (Atlanta Biologicals) in the presence of 1% penicillin-streptomycin in 60-mm tissue culture dishes at 37 °C in a 5% CO2 humidified incubator and subcultured every 2 or 3 days. At 75% confluence, adherent cells were detached after versene-EDTA (CellGro) and centrifuged at 1000 × g for 3 min; versene-EDTA was aspirated before cells were resuspended in regular culture medium at a cell density of 1 × 106/ml. RAW mouse macrophage cell line (ATCC, Manassas, VA) was cultured in RPMI 1640 (10-041-CV; Corning, Manassas, VA) with 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), l-glutamine, and 1% penicillin-streptomycin in a humidified incubator at 37 °C and 5% CO2. The cells were subcultured every 2–3 days when they achieved 70–80% confluence. For experiments, cells were seeded on 35-mm glass-bottomed dishes (In Vitro Scientific) at 8 × 104/ml cell density and cultured in RPMI 1640 supplemented with 10% dialyzed fetal bovine serum and antibiotics. Cells between passages 3 and 25 were used for experiments. HeLa and RAW cells were transfected using the transfection reagent PolyJetTM (Signagen) according to the manufacturer's protocol. Transfection was performed on cells plated on glass-bottomed dishes. After 5 h of incubation with the transfection reagent, the medium was replaced with 1 ml of fresh culture medium. Cells were imaged 12–24 h after transfection. RAW cells were also transfected by electroporation using the D-032 protocol on an Amaxa Nucleofector device (Lonza, Basel, Switzerland). Each electroporation was performed on 1.25 × 106 cells in 200 μl of Nucleofector solution, followed by the immediate addition of 800 μl of warm culture medium. The cells were then plated on glass-bottomed dishes, placed in an incubator at 37 °C and 5% CO2, and imaged 12–24 h after electroporation.
DNA constructs and reagents
DNA constructs blue opsin-mCh, CrBlue (36), M3 (84), Akt-PH-Venus, CRY2-mCh-GRK2Ct, CIBN-CaaX, mCh-CRY-iSH2, Venus-WGBD (76), and PKA-δRIIβ–PKA-Cα-YFP (85) have been described previously. G protein γ9, γ5, and γ1, Akt-PH-mCh, and mChe-PH constructs were kindly provided by the laboratory of N. Gautam. GRPR-GFP was a kind gift from the laboratory of Zhou-Feng Chen (43). Reagent sources were as follows: PTx, SDF1α, isoproterenol hydrochloride, and norepinephrine from Sigma-Aldrich; gallein from TCI AMERICA; Fluo-4 AM from Molecular Probes, Inc. (Eugene, OR); BAPTA, BAPTA-AM, thapsigargin, A-7 hydrochloride, wortmannin 2APB, and GO 6983 from Cayman Chemical (Ann Arbor, MI); c5a from R&D Systems; GSK 269962 and ML-7 hydrochloride from AdooQ Bioscience (Irvine, CA); Bombesin from Tocris; and 11-cis-retinal from the National Eye Institute. All of the reagents except for PTx (water) and 11-cis-retinal (ethanol) were initially dissolved in DMSO and then diluted in HBSS or cell culture medium before adding to cells.
Live cell imaging and data analysis
Experiments were performed with a ×60, 1.4 numeric aperture oil objective or a ×10, 0.3 numeric aperture objective in a spinning-disk XD confocal TIRF imaging system that is composed of a Nikon Ti-R/B inverted microscope, a Yokogawa CSU-X1 spinning disk unit (5000 rpm), and an Andor FRAPPA unit for photoactivation of manually selected regions of the opsins in real time, all controlled using Andor iQ version 3.1 software (Andor Technologies, Belfast, UK), with 445-, 488-, 515-, and 594-nm solid-state lasers and an iXon ULTRA 897BV back-illuminated deep-cooled EMCCD camera. Blue opsin mCh and Akt-PH-mCh were imaged using 594-nm excitation and 630-nm emission settings, blue opsin-mTurquoise was imaged using 445-nm excitation and 478-nm emission, and Fluo-4 AM was imaged using 488-nm excitation and 515-nm emission, respectively. For OA of blue opsin, cells were reconstituted with 50 μm 11-cis-retinal. OA was then performed with 445-nm blue light (white or blue boxes; 0.8 μW/μm2) pulsed at 0.5 Hz covering the entire cell or user-defined subcellular regions. Iterative OAs were performed at 3-s intervals, adjusting the location of the optical stimuli with respect to the cellular response. Axial focus drifts were circumvented using the Perfect Focus System (Nikon) during cell migration experiments. Mean pixel fluorescence intensity changes in the entire cell or in selected areas of the cell were determined using the Andor iQ version 3.1 software. Time-lapse images were analyzed using the analytical tools accompanying the Andor iQ 3.1 software;, further processing of data was conducted using Origin pro (OriginLab Corp.).
Cytosolic calcium measurements
For intracellular calcium measurements, cells were cultured on cell culture dishes at 37 °C with 5% CO2. Experiments were performed 12–24 h after plating. Cells were washed twice with HBSS with calcium, pH 7.2, and incubated for 50 min at room temperature with the fluorescent calcium indicator Fluo-4 AM (0.28 nm). The fluorescence intensity of Fluo-4 AM (488 nm) was continuously imaged at 1-s intervals using 488-nm excitation and 515-nm emission with confocal microscopy. Fluo-4 AM fluorescence intensities obtained from regions of interest were normalized to initial values.
Real-time quantitative PCR assay
The RAW cells were homogenized, and mRNA was isolated with the GeneJET RNA purification kit according to the manufacturer's protocol. Synthesis of cDNA was performed using the RADIANT cDNA synthesis kit. The gene expression was quantified using real-time PCR using the RADIANT Green LO-ROX quantitative PCR kit (Alkali Scientific, Pompano Beach, FL). DNA amplification was carried out using Icycler (Bio-Rad), and the detection was performed by measuring the binding of the fluorescence dye SYBR Green to double-stranded DNA. All of the primers were ordered from IDT-DNA (supplemental Table S1). The relative quantities of target gene mRNA against an internal control, β-actin, were calculated using the ΔΔCT method. The difference (ΔCT) between the mean values in triplicates of target genes and those of β-actin were calculated using Origin 9 (OriginLab Corp.), and the relative quantified value was expressed as 2−ΔΔCT.
Author contributions
P. S. conducted most of the experiments and analyzed the results. D. K. conducted experiments on optical control of PI3K using iSH2, Gγ9 translocation by SDF1α-induced CXCR4 activation. K. R. conducted experiments on Gγ9 translocation with bombesin, PIP2 hydrolysis, Gαq-induced cell shape change, and CrBlue-induced cell migration. K. S. conducted experiments on the Gγ9 translocation with N. E. A. K. and P. S. conceived the idea for the project and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Anders Tengholm and Dr. Zhou-Feng Chen for the gift of cDNA constructs. We thank Dr. N. Gautam for cDNA constructs for G proteins, GPCRs, and sensors as well as for providing access to RNA-seq data. We thank Dr. Patrick O'Neill for optogenetic constructs and discussions about related experiments. We thank Dr. John L. Payton, Nicole Weiss, Steven Walton, and Saroopa Samaradivakara for experimental assistance, discussions, and comments. We thank Dr. Vanessa Fogg at ScienceDocs Inc. for editing assistance. We also thank Dr. Matthew Toomey for the R script for opsin spectra generation.
This work was supported by the University of Toledo. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S4 and Movies S1–S5.
- GPCR
- G protein–coupled receptor
- PM
- plasma membrane
- GEF
- guanine nucleotide exchange factor
- DAG
- diacylglycerol
- LE
- leading edge
- TE
- trailing edge
- GDI
- guanine nucleotide dissociation inhibitor
- PIP3
- phosphatidylinositol 1,4,5-trisphosphate
- MLCP
- myosin light chain phosphatase
- p-MyoII
- phosphorylated myosin II
- MLC
- myosin light chain
- p-MLC
- phosphorylated MLC
- MLCK
- myosin light chain kinase
- RAW
- RAW264.7
- OA
- optical activation
- β1-AR and α2-AR
- β1- and α2-adrenergic receptor, respectively
- GRPR
- gastrin-releasing peptide receptor
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- EPAC
- exchange protein directly activated by cAMP
- PTx
- pertussis toxin
- iSH2
- inter-Src homology 2 domain
- ROCK
- Rho kinase
- CaM
- calmodulin
- AMPK
- adenosine monophosphate-activated protein kinase
- HBSS
- Hanks' balanced salt solution
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester
- NE
- norepinephrine
- IP3
- inositol 1,4,5-triphosphate
- IP3R
- inositol triphosphate receptor
- 2APB
- 2-aminoethoxydiphenyl borate
- LEGI
- local excitation global inhibition.
References
- 1. Pierce K. L., Premont R. T., and Lefkowitz R. J. (2002) Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 2. Oldham W. M., and Hamm H. E. (2008) Heterotrimeric G protein activation by G-protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 9, 60–71 [DOI] [PubMed] [Google Scholar]
- 3. Swaney K. F., Huang C. H., and Devreotes P. N. (2010) Eukaryotic chemotaxis: a network of signaling pathways controls motility, directional sensing, and polarity. Annu. Rev. Biophys. 39, 265–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brock C., Schaefer M., Reusch H. P., Czupalla C., Michalke M., Spicher K., Schultz G., and Nürnberg B. (2003) Roles of Gβγ in membrane recruitment and activation of p110γ/p101 phosphoinositide 3-kinase γ. J. Cell Biol. 160, 89–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nichols C. G., and Lopatin A. N. (1997) Inward rectifier potassium channels. Annu. Rev. Physiol. 59, 171–191 [DOI] [PubMed] [Google Scholar]
- 6. Choi E. J., Xia Z., Villacres E. C., and Storm D. R. (1993) The regulatory diversity of the mammalian adenylyl cyclases. Curr. Opin. Cell Biol. 5, 269–273 [DOI] [PubMed] [Google Scholar]
- 7. Federman A. D., Conklin B. R., Schrader K. A., Reed R. R., and Bourne H. R. (1992) Hormonal stimulation of adenylyl cyclase through G(i)-protein (βγ) subunits. Nature 356, 159–161 [DOI] [PubMed] [Google Scholar]
- 8. Liu L., Das S., Losert W., and Parent C. A. (2010) mTORC2 regulates neutrophil chemotaxis in a cAMP- and RhoA-dependent fashion. Dev. Cell 19, 845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fogg V. C., Azpiazu I., Linder M. E., Smrcka A., Scarlata S., and Gautam N. (2001) Role of the γ subunit prenyl moiety in G protein βγ complex interaction with phospholipase Cβ. J. Biol. Chem. 276, 41797–41802 [DOI] [PubMed] [Google Scholar]
- 10. Niu J., Profirovic J., Pan H., Vaiskunaite R., and Voyno-Yasenetskaya T. (2003) G protein βγ subunits stimulate p114RhoGEF, a guanine nucleotide exchange factor for RhoA and Rac1: regulation of cell shape and reactive oxygen species production. Circ. Res. 93, 848–856 [DOI] [PubMed] [Google Scholar]
- 11. Ueda H., Nagae R., Kozawa M., Morishita R., Kimura S., Nagase T., Ohara O., Yoshida S., and Asano T. (2008) Heterotrimeric G protein βγ subunits stimulate FLJ00018, a guanine nucleotide exchange factor for Rac1 and Cdc42. J. Biol. Chem. 283, 1946–1953 [DOI] [PubMed] [Google Scholar]
- 12. Clapham D. E., and Neer E. J. (1993) New roles for G-protein βγ-dimers in transmembrane signalling. Nature 365, 403–406 [DOI] [PubMed] [Google Scholar]
- 13. Gether U., Asmar F., Meinild A. K., and Rasmussen S. G. (2002) Structural basis for activation of G-protein-coupled receptors. Pharmacol. Toxicol. 91, 304–312 [DOI] [PubMed] [Google Scholar]
- 14. Jin T., Xu X., and Hereld D. (2008) Chemotaxis, chemokine receptors and human disease. Cytokine 44, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Petrie R. J., Doyle A. D., and Yamada K. M. (2009) Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 10, 538–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huang C.-H., Tang M., Shi C., Iglesias P. A., and Devreotes P. N. (2013) An excitable signal integrator couples to an idling cytoskeletal oscillator to drive cell migration. Nat. Cell Biol. 15, 1307–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sciaccaluga M., D'Alessandro G., Pagani F., Ferrara G., Lopez N., Warr T., Gorello P., Porzia A., Mainiero F., Santoro A., Esposito V., Cantore G., Castigli E., and Limatola C. (2013) Functional cross-talk between CXCR4 and PDGFR on glioblastoma cells is essential for migration. PLoS One 8, e73426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jiménez C., Portela R. A., Mellado M., Rodríguez-Frade J. M., Collard J., Serrano A., Martínez A. C., Avila J., and Carrera A. C. (2000) Role of the PI3K regulatory subunit in the control of actin organization and cell migration. J. Cell Biol. 151, 249–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blumer J. B., and Lanier S. M. (2014) Activators of G protein signaling exhibit broad functionality and define a distinct core signaling triad. Mol. Pharmacol. 85, 388–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peterson Y. K., Bernard M. L., Ma H., Hazard S. 3rd, Graber S. G., and Lanier S. M. (2000) Stabilization of the GDP-bound Conformation of Giα by a peptide derived from the G-protein regulatory motif of AGS3. J. Biol. Chem. 275, 33193–33196 [DOI] [PubMed] [Google Scholar]
- 21. Ghosh P., Garcia-Marcos M., Bornheimer S. J., and Farquhar M. G. (2008) Activation of Gαi3 triggers cell migration via regulation of GIV. J. Cell Biol. 182, 381–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aznar N., Midde K. K., Dunkel Y., Lopez-Sanchez I., Pavlova Y., Marivin A., Barbazán J., Murray F., Nitsche U., Janssen K.-P., Willert K., Goel A., Abal M., Garcia-Marcos M., and Ghosh P. (2015) Daple is a novel non-receptor GEF required for trimeric G protein activation in Wnt signaling. eLife 4, e07091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xie Y., Wolff D. W., Wei T., Wang B., Deng C., Kirui J. K., Jiang H., Qin J., Abel P. W., and Tu Y. (2009) Breast cancer migration and invasion depend on proteasome degradation of regulator of G-protein signaling 4. Cancer Res. 69, 5743–5751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cai H., and Devreotes P. N. (2011) Moving in the right direction: how eukaryotic cells migrate along chemical gradients. Semin. Cell Dev. Biol. 22, 834–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Legler D. F., Uetz-von Allmen E., and Hauser M. A. (2014) CCR7: roles in cancer cell dissemination, migration and metastasis formation. Int. J. Biochem. Cell Biol. 54, 78–82 [DOI] [PubMed] [Google Scholar]
- 26. Kang Y., and Pantel K. (2013) Tumor cell dissemination: emerging biological insights from animal models and cancer patients. Cancer Cell 23, 573–581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ananthakrishnan R., and Ehrlicher A. (2007) The forces behind cell movement. Int. J. Biol. Sci. 3, 303–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kölsch V., Charest P. G., and Firtel R. A. (2008) The regulation of cell motility and chemotaxis by phospholipid signaling. J. Cell Sci. 121, 551–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katoh H., Aoki J., Yamaguchi Y., Kitano Y., Ichikawa A., and Negishi M. (1998) Constitutively active Gα12, Gα13, and Gαq induce Rho-dependent neurite retraction through different signaling pathways. J. Biol. Chem. 273, 28700–28707 [DOI] [PubMed] [Google Scholar]
- 30. Hart M. J., Jiang X., Kozasa T., Roscoe W., Singer W. D., Gilman A. G., Sternweis P. C., and Bollag G. (1998) Direct stimulation of the guanine nucleotide exchange activity of p115 RhoGEF by Gα13. Science 280, 2112–2114 [DOI] [PubMed] [Google Scholar]
- 31. Kranenburg O., Poland M., van Horck F. P., Drechsel D., Hall A., and Moolenaar W. H. (1999) Activation of RhoA by lysophosphatidic acid and Gα12/13 subunits in neuronal cells: induction of neurite retraction. Mol. Biol. Cell 10, 1851–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maekawa M., Ishizaki T., Boku S., Watanabe N., Fujita A., Iwamatsu A., Obinata T., Ohashi K., Mizuno K., and Narumiya S. (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285, 895–898 [DOI] [PubMed] [Google Scholar]
- 33. Geraldes P., and King G. L. (2010) Activation of protein kinase C isoforms and its impact on diabetic complications. Circ. Res. 106, 1319–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mahadeo D. C., Janka-Junttila M., Smoot R. L., Roselova P., and Parent C. A. (2007) A chemoattractant-mediated G(i)-coupled pathway activates adenylyl cyclase in human neutrophils. Mol. Biol. Cell 18, 512–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu L., Gritz D., and Parent C. A. (2014) PKCβII acts downstream of chemoattractant receptors and mTORC2 to regulate cAMP production and myosin II activity in neutrophils. Mol. Biol. Cell 25, 1446–1457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karunarathne W. K., Giri L., Kalyanaraman V., and Gautam N. (2013) Optically triggering spatiotemporally confined GPCR activity in a cell and programming neurite initiation and extension. Proc. Natl. Acad. Sci. U.S.A. 110, E1565–E1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karunarathne W. K., Giri L., Patel A. K., Venkatesh K. V., and Gautam N. (2013) Optical control demonstrates switch-like PIP3 dynamics underlying the initiation of immune cell migration. Proc. Natl. Acad. Sci. U.S.A. 110, E1575–E1583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wu D. (2005) Signaling mechanisms for regulation of chemotaxis. Cell Res. 15, 52–56 [DOI] [PubMed] [Google Scholar]
- 39. Senarath K., Ratnayake K., Siripurapu P., Payton J. L., and Karunarathne A. (2016) Reversible G Protein βγ9 distribution-based assay reveals molecular underpinnings in subcellular, single-cell, and multicellular GPCR and G protein activity. Anal. Chem. 88, 11450–11459 [DOI] [PubMed] [Google Scholar]
- 40. Goulimari P., Kitzing T. M., Knieling H., Brandt D. T., Offermanns S., and Grosse R. (2005) Gα12/13 is essential for directed cell migration and localized Rho-Dia1 function. J. Biol. Chem. 280, 42242–42251 [DOI] [PubMed] [Google Scholar]
- 41. Parent C. A. (2004) Making all the right moves: chemotaxis in neutrophils and Dictyostelium. Curr. Opin Cell Biol. 16, 4–13 [DOI] [PubMed] [Google Scholar]
- 42. Czepielewski R. S., Porto B. N., Rizzo L. B., Roesler R., Abujamra A. L., Pinto L. G., Schwartsmann G., de Queiroz Cunha F., and Bonorino C. (2012) Gastrin-releasing peptide receptor (GRPR) mediates chemotaxis in neutrophils. Proc. Natl. Acad. Sci. U.S.A. 109, 547–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao Z. Q., Liu X. Y., Jeffry J., Karunarathne W. K., Li J. L., Munanairi A., Zhou X. Y., Li H., Sun Y. G., Wan L., Wu Z. Y., Kim S., Huo F. Q., Mo P., Barry D. M., et al. (2014) Descending control of itch transmission by the serotonergic system via 5-HT1A-facilitated GRP-GRPR signaling. Neuron 84, 821–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grandoch M., Roscioni S. S., and Schmidt M. (2010) The role of Epac proteins, novel cAMP mediators, in the regulation of immune, lung and neuronal function. Br. J. Pharmacol. 159, 265–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Almahariq M., Tsalkova T., Mei F. C., Chen H., Zhou J., Sastry S. K., Schwede F., and Cheng X. (2013) A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol. Pharmacol. 83, 122–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yost E. A., Hynes T. R., Hartle C. M., Ott B. J., and Berlot C. H. (2015) Inhibition of G-protein βγ signaling enhances T cell receptor-stimulated interleukin 2 transcription in CD4+ T helper cells. PLoS One 10, e0116575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen L., Zhang J. J., and Huang X. Y. (2008) cAMP inhibits cell migration by interfering with Rac-induced lamellipodium formation. J. Biol. Chem. 283, 13799–13805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Burdyga A., Conant A., Haynes L., Zhang J., Jalink K., Sutton R., Neoptolemos J., Costello E., and Tepikin A. (2013) cAMP inhibits migration, ruffling and paxillin accumulation in focal adhesions of pancreatic ductal adenocarcinoma cells: effects of PKA and EPAC. Biochim. Biophys. Acta 1833, 2664–2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lehmann D. M., Seneviratne A. M., and Smrcka A. V. (2008) Small molecule disruption of G protein βγ subunit signaling inhibits neutrophil chemotaxis and inflammation. Mol. Pharmacol. 73, 410–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Clapham D. E., and Neer E. J. (1997) G protein βγ subunits. Annu. Rev. Pharmacol. Toxicol. 37, 167–203 [DOI] [PubMed] [Google Scholar]
- 51. Ribeiro-Neto F. A., and Rodbell M. (1989) Pertussis toxin induces structural changes in Gα proteins independently of ADP-ribosylation. Proc. Natl. Acad. Sci. U.S.A. 86, 2577–2581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Davis T. L., Bonacci T. M., Sprang S. R., and Smrcka A. V. (2005) Structural and molecular characterization of a preferred protein interaction surface on G protein βγ subunits. Biochemistry 44, 10593–10604 [DOI] [PubMed] [Google Scholar]
- 53. O'Neill P. R., and Gautam N. (2014) Subcellular optogenetic inhibition of G proteins generates signaling gradients and cell migration. Mol. Biol. Cell 25, 2305–2314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chen C. L., Wang Y., Sesaki H., and Iijima M. (2012) Myosin I links PIP3 signaling to remodeling of the actin cytoskeleton in chemotaxis. Sci. Signal. 5, ra10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Machesky L. M., Mullins R. D., Higgs H. N., Kaiser D. A., Blanchoin L., May R. C., Hall M. E., and Pollard T. D. (1999) Scar, a WASp-related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc. Natl. Acad. Sci. U.S.A. 96, 3739–3744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wilson K., Lewalle A., Fritzsche M., Thorogate R., Duke T., and Charras G. (2013) Mechanisms of leading edge protrusion in interstitial migration. Nat. Commun. 4, 2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kakumoto T., and Nakata T. (2013) Optogenetic control of PIP3: PIP3 is sufficient to induce the actin-based active part of growth cones and is regulated via endocytosis. PLoS One 8, e70861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kimura K., Ito M., Amano M., Chihara K., Fukata Y., Nakafuku M., Yamamori B., Feng J., Nakano T., Okawa K., Iwamatsu A., and Kaibuchi K. (1996) Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273, 245–248 [DOI] [PubMed] [Google Scholar]
- 59. Ridley A. J. (2015) Rho GTPase signalling in cell migration. Curr. Opin. Cell Biol. 36, 103–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Takashima S. (2009) Phosphorylation of myosin regulatory light chain by myosin light chain kinase, and muscle contraction. Circ. J. 73, 208–213 [DOI] [PubMed] [Google Scholar]
- 61. Camps M., Hou C., Sidiropoulos D., Stock J. B., Jakobs K. H., and Gierschik P. (1992) Stimulation of phospholipase C by guanine-nucleotide-binding protein βγ subunits. Eur. J. Biochem. 206, 821–831 [DOI] [PubMed] [Google Scholar]
- 62. Smrcka A. V., and Sternweis P. C. (1993) Regulation of purified subtypes of phosphatidylinositol-specific phospholipase Cβ by G protein α and βγ subunits. J. Biol. Chem. 268, 9667–9674 [PubMed] [Google Scholar]
- 63. Somlyo A. P., and Somlyo A. V. (1994) Signal transduction and regulation in smooth muscle. Nature 372, 231–236 [DOI] [PubMed] [Google Scholar]
- 64. Mihaylova M. M., and Shaw R. J. (2011) The AMP-activated protein kinase (AMPK) signaling pathway coordinates cell growth, autophagy, and metabolism. Nat. Cell Biol. 13, 1016–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Horman S., Morel N., Vertommen D., Hussain N., Neumann D., Beauloye C., El Najjar N., Forcet C., Viollet B., Walsh M. P., Hue L., and Rider M. H. (2008) AMP-activated protein kinase phosphorylates and desensitizes smooth muscle myosin light chain kinase. J. Biol. Chem. 283, 18505–18512 [DOI] [PubMed] [Google Scholar]
- 66. Nalli A. D., Kumar D. P., Mahavadi S., Al-Shboul O., Alkahtani R., Kuemmerle J. F., Grider J. R., and Murthy K. S. (2014) Hypercontractility of intestinal longitudinal smooth muscle induced by cytokines is mediated by the nuclear factor-κB/AMP-activated kinase/myosin light chain kinase pathway. J. Pharmacol. Exp. Ther. 350, 89–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Itoh H., and Hidaka H. (1984) Direct interaction of calmodulin antagonists with Ca2+/calmodulin-dependent cyclic nucleotide phosphodiesterase. J. Biochem. 96, 1721–1726 [DOI] [PubMed] [Google Scholar]
- 68. Michelangeli F., and East J. M. (2011) A diversity of SERCA Ca2+ pump inhibitors. Biochem. Soc. Trans. 39, 789–797 [DOI] [PubMed] [Google Scholar]
- 69. Giri L., Patel A. K., Karunarathne W. K., Kalyanaraman V., Venkatesh K. V., and Gautam N. (2014) A G-protein subunit translocation embedded network motif underlies GPCR regulation of calcium oscillations. Biophys. J. 107, 242–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Maurya M. R., and Subramaniam S. (2007) A kinetic model for calcium dynamics in RAW 264.7 cells: 1. mechanisms, parameters, and subpopulational variability. Biophys. J. 93, 709–728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ishikawa S., Fujisawa G., Okada K., and Saito T. (1993) Thapsigargin increases cellular free calcium and intracellular sodium concentrations in cultured rat glomerular mesangial cells. Biochem. Biophys. Res. Commun. 194, 287–293 [DOI] [PubMed] [Google Scholar]
- 72. Karunarathne W. K., O'Neill P. R., and Gautam N. (2015) Subcellular optogenetics: controlling signaling and single-cell behavior. J. Cell Sci. 128, 15–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tang W. J., and Gilman A. G. (1991) Type-specific regulation of adenylyl cyclase by G protein βγ subunits. Science 254, 1500–1503 [DOI] [PubMed] [Google Scholar]
- 74. Sunahara R. K., and Taussig R. (2002) Isoforms of mammalian adenylyl cyclase: multiplicities of signaling. Mol. Interv. 2, 168–184 [DOI] [PubMed] [Google Scholar]
- 75. Srinivasan S., Wang F., Glavas S., Ott A., Hofmann F., Aktories K., Kalman D., and Bourne H. R. (2003) Rac and Cdc42 play distinct roles in regulating PI(3,4,5)P3 and polarity during neutrophil chemotaxis. J. Cell Biol. 160, 375–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. O'Neill P. R., Kalyanaraman V., and Gautam N. (2016) Subcellular optogenetic activation of Cdc42 controls local and distal signaling to drive immune cell migration. Mol. Biol. Cell 27, 1442–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kimura K., Fukata Y., Matsuoka Y., Bennett V., Matsuura Y., Okawa K., Iwamatsu A., and Kaibuchi K. (1998) Regulation of the association of adducin with actin filaments by Rho-associated kinase (Rho-kinase) and myosin phosphatase. J. Biol. Chem. 273, 5542–5548 [DOI] [PubMed] [Google Scholar]
- 78. Amano M., Ito M., Kimura K., Fukata Y., Chihara K., Nakano T., Matsuura Y., and Kaibuchi K. (1996) Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271, 20246–20249 [DOI] [PubMed] [Google Scholar]
- 79. Martini F. J., and Valdeolmillos M. (2010) Actomyosin contraction at the cell rear drives nuclear translocation in migrating cortical interneurons. J. Neurosci. 30, 8660–8670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. He M., Zhang Z. H., Guan C. B., Xia D., and Yuan X. B. (2010) Leading tip drives soma translocation via forward F-actin flow during neuronal migration. J. Neurosci. 30, 10885–10898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Hutchins B. I., Klenke U., and Wray S. (2013) Calcium release-dependent actin flow in the leading process mediates axophilic migration. J. Neurosci. 33, 11361–11371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Xiong Y., Huang C. H., Iglesias P. A., and Devreotes P. N. (2010) Cells navigate with a local-excitation, global-inhibition-biased excitable network. Proc. Natl. Acad. Sci. U.S.A. 107, 17079–17086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ma L., Janetopoulos C., Yang L., Devreotes P. N., and Iglesias P. A. (2004) Two complementary, local excitation, global inhibition mechanisms acting in parallel can explain the chemoattractant-induced regulation of PI(3,4,5)P3 response in dictyostelium cells. Biophys. J. 87, 3764–3774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chisari M., Saini D. K., Cho J.-H., Kalyanaraman V., and Gautam N. (2009) G protein subunit dissociation and translocation regulate cellular response to receptor stimulation. PLoS One 4, e7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Dyachok O., Isakov Y., Sågetorp J., and Tengholm A. (2006) Oscillations of cyclic AMP in hormone-stimulated insulin-secreting β-cells. Nature 439, 349–352 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.