

Abstract
Objective
To provide a review of emerging knowledge from genomics and related basic science, preclinical, and clinical precision medicine studies in head and neck squamous cell carcinoma (HNSCC).
Data Sources
The Cancer Genome Atlas Network (TCGA) publications, PubMed‐based literature review, and ClinicalTrials.gov.
Review Methods
TCGA publications, PubMed, and ClinicalTrials.gov were queried for genomics and related basic science, preclinical, and developmental clinical precision medicine studies in HNSCC.
Results
TCGA reported comprehensive genomic analyses of 279 HNSCC, defining the landscape and frequency of chromosomal copy number alterations, mutations, and expressed genes that contribute to pathogenesis, prognosis, and resistance to therapy. This provides a road map for basic science and preclinical studies to identify key pathways in cancer and cells of the tumor microenvironment affected by these alterations, and candidate targets for new small molecule and biologic therapies.
Conclusion
Recurrent chromosomal abnormalities, mutations, and expression of genes affecting HNSCC subsets are associated with differences in prognosis, and define molecules, pathways, and deregulated immune responses as candidates for therapy. Activity of molecularly targeted agents appears to be enhanced by rational combinations of these agents and standard therapies targeting the complex alterations that affect multiple pathways and mechanisms in HNSCC.
Level of Evidence
NA.
Keywords: Clinical Trials, Genomics, Head and Neck Cancer, Molecular Targeted Therapy
INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) is the most prevalent cancer arising from the epithelium of the upper aerodigestive tract, including the oral cavity, pharynx and larynx, and their anatomic subsites. Each year, HNSCC is diagnosed in over 50,000 Americans and 600,000 people worldwide.1 Nearly half die of their disease, and survivors often suffer significant morbidity affecting voice, speech, taste, dental health, and swallowing. Tobacco, alcohol, betel nut, and other chemical carcinogens are implicated in the etiology of a majority of these cancers, while increasing incidence of human papilloma virus positive (HPV+) HNSCC with a better prognosis has been observed in developing countries. Additionally, a small but important subset of HNSCC arise in younger patients due to hereditary disorders affecting DNA repair.
The Cancer Genome Atlas and other large‐scale genomics studies made possible by recent advances in technology have defined the broader landscape and frequency of chromosomal alterations, mutations, and expressed genes that contribute to pathogenesis, prognosis and resistance of HNSCC to therapy.2, 3, 4, 5, 6, 7 These findings and technologies are increasingly being incorporated in preclinical and clinical studies together with a rapidly expanding armamentarium of molecularly targeted small molecules and biologics such as humanized antibodies. In this review, we summarize salient findings from recent TCGA and other large‐scale genomics studies, and selected preclinical and clinical studies that are exploring the potential of these discoveries to improve therapy of HNSCC. Frequent events affect cell cycle, death, and growth pathways in major subsets. Among important questions being raised, what are the implications and potential of these genomic alterations to define prognosis and strategies for prevention and therapy? Do cancers with gene mutations versus differences in gene dosage (copy number) and expression of a druggable target differ in therapeutic sensitivity? What other concurrent alterations promote resistance, and can combined therapies overcome these mechanisms of resistance?
MATERIALS AND METHODS
Study Methods
TCGA publications, PubMed, and ClinicalTrials.gov were queried for large‐scale genomics and commonly altered target‐related basic science, preclinical, and developmental clinical precision medicine studies in HNSCC. As most clinical studies are phase I safety and pharmacology studies and few were found have reached the stage of phase II/III clinical trial identified through evidence based search methodology using PRISMA guidelines (Preferred Reporting Items for Systematic Reviews and Meta‐Analyses),8 we performed a TCGA publication and PubMed review of the literature for large‐scale genomics studies of HNSCC; preclinical studies targeting corresponding molecules or pathways; and ClinicalTrials.gov review of phase I‐III studies available. Participants, Interventions, Comparisons, Outcomes, and Study design (PICOS) criteria were utilized in weighing studies to include. Studies were prioritized where the population included patients with tumors with histologically confirmed HNSCC, and preclinical studies using genotype confirmed HNSCC‐derived genotyped cell lines, xenografts or patient‐derived tumor xenografts. Genomic tissue and cell lines are predominantly derived from studies where interventions are biased towards surgical patients from whom sufficient tumor was available for macro dissection for enrichment of malignant tumor tissue for genomic studies or cell culture. As a result, TCGA and other studies are enriched and biased towards sampling of HPV(‐) cases from oral cavity and laryngeal sites, relative to HPV‐enriched cases from oropharynx, or nasopharynx often treated by chemoradiotherapy. Comparison for mutations in coding regions of genes, or copy number and expression is possible for studies that included whole exome sequencing, comparative genomic hybridization (CGH), and expression profiling. Statistics‐driven bioinformatic analyses for significant genomic mutations and copy alterations, expression data, and multi‐platform comparisons and integrative analyses of these platforms are limited to TCGA and a smaller number of large‐scale HNSCC studies utilizing common tools. Outcomes data including overall, progression free, or disease‐specific survival for genomics studies is of variable quality, but is included where analyses and statistically significant or near‐significant trends are highlighted.
Search Methods
Articles for TCGA and large‐scale genomic studies, reviews comparing TCGA and prior genomics studies, and related preclinical and clinical studies for HNSCC from January 1, 2010 to April 10, 2017 was performed using PubMed and public databases. Key words included head, neck squamous, genomics, RNA expression, proteomics studies including more than 30 tumors; related terms for basic, preclinical and clinical studies of components or multiple components of pathways altered in ≥5% of tumors.
RESULTS
The Cancer Genome Atlas and Other Large‐Scale Genomics Studies Identify Genes and Chromosomal Regions Linked to Pathogenesis of HNSCC
The advent and improvements in massively parallel (next generation) sequencing and other array based methods during the past decade have made possible the comprehensive multi‐platform TCGA and several large‐scale studies defining the landscape of genomic and epigenetic alterations in HNSCC.2, 3, 4, 5, 6, 9 Globally, most HNSCC harbor complex chromosomal rearrangements, among which there are common and less frequent recurrent alterations that have been associated with HPV status, malignant phenotype, outcome, and certain gene(s) that are of potential clinical significance. More common recurrent chromosomal alterations characterized by both classical karyotyping studies and TCGA, and possible mechanisms for their occurrence and selection during carcinogenesis have been recently reviewed.10 Overall, HNSCC also display relatively frequent DNA mutations, similar to other cancers associated with more mutagenic carcinogens and/or defects in DNA repair.2 Higher mutation frequency is considered a factor in generation of neoantigens necessary for immune recognition and response to new immune checkpoint therapies. In addition to CpG transversions commonly attributed to tobacco carcinogen DNA adducts expected in HPV(‐) tumors, HPV (‐) and (+) HNSCC also display mutations within TpC dinucleotides linked to deregulation of APOBEC cytosine deaminases that repair altered bases.3 These mutations affect certain motifs of relevance to recurrent mutation “hotspots” that can enhance function and potential sensitivity to therapeutics targeting particular oncogenic signal kinases. Rare hereditary mutations or deletions that affect genes involved in DNA repair of chromosomal breakage (Fanconi Anemia‐BRCA pathway genes), telomerase maintenance of chromosomal ends (TERC, TERT genes), and mitotic cell division (MYH9) have been linked to increased risk of HNSCC with chromosomal derangements in younger patients.11, 12, 13
Key to understanding the similarities and differences in pathogenesis between HPV(+) and HPV(‐) HNSCC are the different ways the cell cycle is deregulated to lead to uncontrolled proliferation, accumulation of chromosomal rearrangements and mutations, and malignant progression.3 In HPV(+) HNSCC, early genes E6 and E7 encode oncoproteins that promote degradation of tumor suppressors TP53 and RB1, respectively, unleashing their braking effect that keeps cells from progressing from the quiescent G0 state into G1 and later phases of the cell cycle. HPV(+) cancers also exhibit amplification of E2F1, the transcription factor that promotes G1 cell cycle genes. In HPV(‐) HNSCC, TCGA studies reveal that ∼84% of cancers have mutations of TP53, and a variety of upstream alterations that cumulatively can inhibit RB1 to accomplish a similar result. Essential among these are inactivation of the cyclin dependent kinase CDKN2A, which occurs by chromosome 9p21 deletion, gene mutation, or methylation in nearly all HPV(‐) HNSCC. In contrast to HPV(‐) tumors, HPV(+) HNSCC overexpress the CDKN2A encoded 16 kDa protein p16, which has been found to be a sensitive and relatively specific clinical‐pathologic immunohistochemical marker for HPV status and better prognosis.14 Also common are amplifications or transcriptional activation of cyclin CCND1, CDK6 and MYC that promote proliferation. Thus, virtually 100% of HNSCC have viral or critical endogenous gene alterations affecting the cell cycle.
In addition to proliferation and clonal expansion of cells initiated by these alterations, the inactivation of TP53 by mutation or HPV affects its role in repair of DNA damage and as guardian of genomic integrity. TCGA revealed that most HNSCC harbor complex genomic alterations of varying severity that alone or together with other copy alterations and mutations are emerging as subtypes of potential prognostic and therapeutic significance. Dominating these are concurrent chromosome 3p arm deletions and 3q arm amplifications, which are linked with worse prognosis, and respectively harbor several candidate tumor suppressor and oncogenes.3 Among these, the 3q amplicon includes PI3‐Kinase Catalytic subunit Alpha gene, PIK3CA, which is also the most frequently mutated oncogene in HNSCC. PIK3CA is co‐amplified with and has been linked to enhancing the expression of 3q stemness gene SOX2.15 PI3K also promotes preferential expression of an oncogenic ΔNp63 isoform of TP63 encoded on 3q, involved in squamous differentiation.16 Together, PIK3CA is amplified or mutated in ∼34% of HPV(‐) and 56% of HPV(+) TCGA HNSCC tumors, implicating the PI3K pathway in promoting growth factor dependent or independent growth, and common resistance to EGFR therapies. Consistent with this, smaller subsets harbor mutations or decreased expression of PI3K suppressors (PTEN, PIK3R1), or amplifications or mutations of growth factor receptor tyrosine kinases known to activate PI3K signaling, including EGFR (15%), FGFR1 (10%), ERBB2 (5%), IGF1R (4%), EPHA2 (4%), FGFR2, 3 (2% each), MET (2%).3 Among these, the 8p11 focal amplification harboring FGFR1 also contains WHSC1L1, recently characterized to encode methylase NSD3 that modulates EGFR function and proliferation in HNSCC.17 Overall, over 60% of HPV(‐/+) HNSCC harbored alterations in growth factor receptor and PI3K signaling, making this pathway an important target for developmental therapeutics.
TCGA revealed that ∼30% of HPV(‐) HNSCC display amplification of 11q13, associated with a worse prognosis subtype, that has a median survival of less than 2 years.3 This amplification incorporates several genes of potential biologic and therapeutic relevance, including cyclin D1 (CCND1) and Fas Associated Death Domain (FADD). CCND1 has long been implicated in promoting deregulated G1 cell cycle progression and assumed to be the driver oncogene within this amplicon.18 Consistent with this, most tumors with this amplification lack other alterations in upstream growth factor receptor, RAS or PI3K kinases,3 although some with both may help explain resistance to agents targeting these upstream pathways. Interestingly, while FADD protein was originally shown to mediate cell death as part of Tumor Necrosis Factor Receptor (TNFR) complex, it has also recently been shown to play another role in promoting cell proliferation during G2/M cell division.19 Further, its death function may be blocked by Inhibitor of Apoptosis (IAP) proteins, which are encoded by BIRC2/3 genes, located in an adjacent co‐amplification of 11q22, seen in ∼8% of HNSCC. TCGA and other studies also uncovered inactivating mutations of another TNFR complex cell death mediator, caspase‐8 (CASP8). Mutant CASP8 is found in ∼11% of HPV(‐) tumors, mutually exclusive of amplification of FADD. Tumors with CASP8 mutations often have activating mutations of HRAS or PIK3CA, but few other copy alterations, or TP53 mutations. As such, these tumors appear to represent rarer but predominantly mutation‐type driven subset of HNSCC. Intriguingly, a subset of ∼22% of HPV(+) tumors have 14q32.32 deletions or inactivating mutations of TNFR Associated Factor (TRAF3), implicated in suppressing survival of myeloid cancers and cells infected with other DNA viruses. Recently, the HPV(+) HNSCC subset with loss of TRAF3 or CYLD has been associated with episomal HPV infection and better prognosis, distinguishing these from the subset with predominantly PIK3CA alterations and HPV integration.20 Together, FADD, BIRC2/3, CASP8, TRAF3, and CYLD alter pathways that can lead to activation of proto‐oncogene transcription factors Nuclear Factor‐kappaB/REL that promote genes involved in cell survival, proliferation, angiogenesis, and aberrant inflammation and immunity.21 Overall, ∼44% of HPV(‐) and 31% of HPV(+) HNSCC revealed alterations in cell death/survival and NF‐κB pathways in immune recognition determinants HLA‐A/B an B2M were also seen ∼10% of HNSCC, consistent with mechanisms implicated in escape from immune‐mediated cell death.3
Early exome sequencing and TCGA studies highlighted novel mutations predicted to inactivate NOTCH1,3, 4, 5 encoding a transmembrane signal receptor whose ligand binding and cleavage produces a nuclear transcription factor that promotes a program of genes important in squamous differentiation. Mechanistic studies suggest the overexpression of ΔNp63 enhanced by PI3K on 3q may also suppress NOTCH expression and tumor suppressor function.22, 23 Tumors with loss of NOTCH1 are more poorly differentiated and have been associated with worse prognosis.24 Inactivation of NOTCH and genes FAT1 and AJUBA may also converge on WNT‐β‐catenin signaling to affect cell differentiation. Overall, ∼64% of HPV(‐) and 44% of HPV(+) HNSCC have alterations in differentiation signaling pathway components. Finally, ∼22% of HPV(‐) tumors displayed defects in the KEAP1‐CUL3‐NFE2L2 components of the oxidative stress and damage pathway.
Preclinical and Clinical Studies Elucidating Potential Therapeutic Significance of Genomic Alterations in HNSCC
These studies underscored and clarified the extent to which HPV(‐) and (+) cancers differ in mutational and chromosomal alterations that relate to their pathogenesis, and potential for new therapeutic approaches. Major subsets with alterations in key pathways and frequent mutations have raised interest in the potential of targeting PI3K‐mTOR, MEK‐MAPK, cell cycle, and BIRC/IAPs, in HNSCC (Fig. 1).
Figure 1.
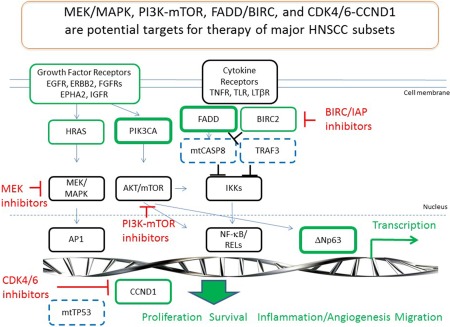
Frequent genomic alterations, pathways and targeted therapeutics under investigation in HNSCC. Genomic amplification or activating mutations of oncogenes (green) and inactivating alterations affecting tumor suppressors (blue dashed) affect key signal pathways and transcription factors that regulate the cancer gene program and phenotype. MEK/MAPK, PI3K‐mTOR, FADD/BIRC and CDK4/6‐CCND1 represent potential targets for therapy.
PI3K‐AKT‐mTOR
The PI3K‐AKT‐mTOR signaling pathway is a vital pathway for growth, survival, and metabolism for which there is evidence of frequent functional signal activation in HNSCC cell lines and tumors.25, 26, 27, 28, 29 Genetic aberrations such as CNAs, mutations, and dysregulation of mRNA are prevalent. A number of first generation PI3K and mTOR inhibitors (Rapamycin, Temsorlimus, Everolimus) have shown activity in in vivo xenograft models.30, 31 Rapamycin has been shown to prevent tumorigenesis and suppress SCC‐15 cells by inhibiting mTOR phosphorylation.32 In a recent study, cell lines with PIK3CA mutations were sensitive to PI3K pathway inhibitors, whereas amplification status or proteomic profiling did not predict sensitivity.29 Our studies showed that cell lines with PIK3CA amplifications or mutations were sensitive to dual PI3K‐mTOR inhibitors, in association with inducible TP53, or AKT phosphorylation.33, 34 Evidence of pharmacodynamic inhibition of signaling and clinical activity was reported in a phase I study of Temsirolimus in newly diagnosed advanced‐stage HNSCC.35 The TEMHEAD trial was a single arm multicenter phase 2 trial of Temsirolimus that looked at refractory/recurrent metastatic squamous cell carcinoma in 40 patients which found that inhibition of the PI3K‐AKT‐mTOR axis was a putative novel treatment paradigm for SCCHN.36 In 33 evaluable patients, disease stabilization occurred in 57.6%, and tumor shrinkage in 39.4%. Neither PIK3CA mutations nor HPV status were predictive for success with Temsirolimus treatment. Fatigue (47.5%), anemia (25.0%), nausea (20.0%), and pneumonia (20.0%) were the most common adverse events. A phase 2 trial evaluating the clinical benefit rate (CBR) of Everolimus in 9 patients was disappointing, with 3 patients discontinued due to toxicity and CBR of 28% showing that Everolimus was inactive as monotherapy in unselected patients with HNSCC.37 The AKT inhibitor MK2206 (Merck, USA) in a phase 2 trial evaluating MK2206 as a single agent in HNSCC has recently been completed and currently awaiting publication of results. Table 1 summarizes trials of PI3K, mTOR and AKT inhibitors and combinations registered in ClinicalTrials.gov.
Table 1.
Trials with Small Molecule Inhibitors Targeting Genomic Pathway Alterations.
| Drug | Combination | Status | Clinical Trial ID |
|---|---|---|---|
| PI3K Inhibitors | |||
| BKM120(Novartis, USA) | Cetuximab | Phase I/II | NCT01816984 |
| Single agent | Phase II | NCT01737450 | |
| Cisplatin | Phase I | NCT02113878 | |
| PX‐866(Oncothyreon, USA) | Cetuximab | Phase I/II | NCT01252628 |
| Docetaxel | Phase I/II | NCT01204099 | |
| BYL719(Novartis, USA) | Cetuximab | Phase I/II | NCT01602315 |
| Single agent | Phase II | NCT02145312 | |
| Paclitaxel | Phase I | NCT02051751 | |
| Cisplatin | Phase I | NCT02537223 | |
| Single agent | Phase II | NCT02145312 | |
| Copanlisib | Cetuximab | Phase I/II | NCT02822482 |
| SF1126 | Single agent | Phase II | NCT02644122 |
| mTOR inhibitors | |||
| Everolimus(Novartis) | Single agent | Phase II | NCT01051791 |
| Single agent | Phase II | NCT01133678 | |
| Carboplatin, Cetuximab | Phase I/II | NCT01283334 | |
| Carboplatin, paclitaxel | Phase I/II | NCT01333085 | |
| Cetuximab | Phase I | NCT01637194 | |
| Erlotinib | Phase II | NCT00942734 | |
| Rapamycin(pfizer,USA) | Single agent | Phase I/II | NCT01195922 |
| Temsirolimus(Pfizer) | Single agent | Phase II | NCT01172769 |
| Cetuximab | Phase II | NCT01256385 | |
| Carboplatin, paclitaxel | Phase I/II | NCT01016769 | |
| Cetuximab | Phase I | NCT02215720 | |
| PI3K/mTOR Dual Inhibitors | |||
| BEZ235(Novartis,USA) | Everolimus | Phase I | NCT01508104 |
| PF04691502(pfizer,USA) | Single agent | Phase I | NCT00927823 |
| PF05212384(Pfizer,USA) | Docetaxel, Cisplatin, Dacomitinib | Phase I | NCT01920061 |
| PD‐901, Irinotecan | Phase I | NCT01347866 | |
| SAR245409(sanofi, USA) | Pimasertib | Phase I | NCT01390818 |
| PQR309 | Single agent | Phase I | NCT02483858 |
| Akt Inhibitors | |||
| MK2206(Merck, USA) | Single agent | Phase II | NCT01349933 |
| MEK inhibitors | |||
| PD‐0325901 | PF‐05212384 | Phase I | NCT01347866 |
| CDK Inhibitor | |||
| Palbociclib | Cetuximab | Phase I/II | NCT03024489 |
| Gedatolisib | Phase I | NCT03065062 | |
| P276–00 | Single agent | Phase I/II | NCT00899054 |
| Single agent | Phase II | NCT00824343 | |
| Smac Mimetics/IAP antagonists | |||
| Debio1143 | Cisplatin | Phase I/II | NCT02022098 |
| Single agent | Phase I | NCT01078649 | |
| GDC0917 | Single agent | Phase I | NCT01908413 |
| Single agent | Phase I | NCT01226277 | |
| LCL‐161 | Paclitaxel | Phase I | NCT01968915 |
| Paclitaxel | Phase I | NCT01240655 | |
| Single agent | Phase I | NCT01098838 | |
| GDC0152 | Single agent | Phase I/II | NCT00977067 |
| Birinapant | Chemotherapy | Phase I/II | NCT01188499 |
| Single agent | Phase I | NCT00993239 | |
| HG5–1029 | Single agent | Phase I | NCT00708006 |
| ASTX‐660 | Single agent | Phase I/II | NCT02503423 |
MEK/MAPK Pathway
The mitogen‐activated protein kinase/extracellular signal‐regulated kinase (MAPK/ERK) pathway is activated in HNSCC by upstream growth factor receptors and HRAS, and may be inhibited by antagonists of these receptors and the intermediate kinase MEK that phosphorylates and activates MAPK/ERK.25, 38, 39 TCGA has identified amplifications of several growth factor receptors including EGFR, ERBB2, IGF1R, FGFR1, 3, EPHA2, MET, and rarer mutations of HRAS, as possible genetic drivers upstream of MAPK/ERK that support MEK kinase as a key signaling intermediate and potential therapeutic target.3 Whilst upstream growth factor receptors have been widely studied, on an individual basis they appear to drive oncogenic signaling and demonstrate clinical activity when targeted in relatively minor HNSCC subsets. However, convergence of these signals and activation of the MEK and ERK effectors of the MAPK pathway is prevalent and has been targeted in preclinical and early clinical studies. Biomarker and clinical responses to MEK inhibitor Trametinib were recently reported in HNSCC in a window of opportunity trial.40 MEK inhibitor PD‐0325901 has shown single‐agent activity and ability to overcome resistance and enhance antitumor effects in vivo with the dual PI3K/mTOR inhibitor PF‐384.34 Disappointingly, phase I studies of MEK and PI3K‐mTOR inhibitors demonstrated enhanced toxicity and narrow therapeutic window of the combination, and eventual progression on or soon after cessation of treatment. Studies in animal models suggest the direct anti‐tumor activity of MEK inhibitors may be offset by suppression of beneficial immune responses elicited by mTOR inhibition or immune checkpoint therapy.41 There are no registered MEK inhibitors in active clinical trial for HNSCC. Table 1 summarizes trials of MEK inhibitor combination registered in ClinicalTrials.gov.
Cell Cycle
The TCGA findings highlighted the potential for investigation of newer CDK4/6 inhibitors due to the near universality of cell cycle dysregulation in HNSCC.3 Consistent with the prevalence of these alterations downstream of growth factor signaling, CDK4/6 blockade was found to enhance the efficacy of EGFR inhibition in SCC.42 There is increasing data that suggests that HPV(+) HNSCC may benefit from CDK inhibition; low‐dose Roscovitine, (CDK inhibitor) significantly inhibited the growth of HPV‐associated xenograft tumors in mice without causing any detectable side effects.43 Of recent interest, high phosphorylation of CDK4/6 target Threonine‐356 on cell cycle regulator RB1 was found to predict survival in HPV(‐) HNSCC, providing a rationale for investigation of these agents.44 In HPV(‐) HNSCC, the CDK4/6 inhibitor LY283519 has been found to be active alone and potent in combination with mTOR inhibitor in HNSCC models in vivo and in vitro.45 A phase 1 study (NCT03065062) is currently recruiting participants looking at Palbociclib in combination with the PI3K/mTOR inhibitor Gedatosilib. Palbociclib(CDK4/6 inhibitor) is also currently being investigated as a single agent in a phase 1 dose escalation study to determine the maximum tolerated dose (MTD) and toxicity (NTC03024489). The MONARCH study was a phase 2 trial assessing the efficacy of a CDK inhibitor P276–00 in HNSCC, which was completed in 2013 but still awaiting publication. With increasing numbers of CDK inhibitors being developed this is an area of future promise.46 Table 1 summarizes trials of CDK inhibitor and combinations registered in ClinicalTrials.gov.
Cell cycle checkpoints allow cells time for DNA repair and maintain genomic integrity before the cell undergoes cell division. Certain signaling cascades due to DNA damage result in the phosphorylation of Wee1 Tyrosine Kinase which is involved in the inhibitory phosphorylation of Cdk1 and Cdk2.47 A number of preclinical studies have elaborated on the function of Wee1 Inhibitors to allow premature entry into cell division with unrepaired DNA resulting in cell death.48, 49 A phase I study (NCT01748825) administered oral 2.5g of AZD1775 (Wee1 inhibitor) for up to 2 weeks in adult patients with refractory tumors, found that partial responses were observed in one patient with head and neck cancer and one patient with ovarian cancer.50
SMAC Mimetics and BIRC/IAP Inhibition
FADD and IAP proteins are amplified and over‐expressed in ∼40% of HNSCC, and can inversely modulate programmed cell death.3, 51 Second Mitochondrial Activator of Caspases (SMAC) mimetics are small molecule inhibitors that mimic smac, an endogenous IAP antagonist. This is a potential therapeutic target that has shown efficacy in preclinical models. Birinapant is a SMAC mimetic that demonstrated activity in FADD and IAP amplified tumor models in combination with death factors TNF and TRAIL, and radiation.52 Seven patients with HNSCC out of a cohort of 50 patients were treated with the bivalent SMAC mimetic with dose range of 0.18 to 63 mg/m3 in a 3 + 3 dose escalation design once weekly, defining a phase II dose.53 Debio 1143 is SMAC mimetic that antagonizes IAP activity (XIAP, cIAP‐1, 2, and ML‐IAP) and promotes apoptosis. Debio 1143 radio‐sensitizing action has also been shown in several in vitro and in vivo HNSCC preclinical models.54 The results of the first‐in‐human oral Debio 1143 trial where 31 patients received oral disease ranging from 5 to 900 mg once daily are encouraging. Optimal treatment response of stable disease was achieved in 5 of the patients and the authors concluded that Debio 1143 should be incorporated with other treatment modalities and sub‐population screening.55 An ongoing Phase I/II trial investigating Debio 1143 is still in the recruitment phase (NCT02022098) that is randomizing 94 participants comparing Debio 1143 to placebo, both with concomitant CRT (cisplatin, radiotherapy). A phase 1 study (NCT01098838) investigating LCL‐161 (SMAC mimetic) had an undisclosed number of HNSCC and showed no objective response to once daily dose ranging of 10 to 3,000 mg, however they concluded that further development of LCL‐161 is warranted given the favorable tolerability and pharmacodynamics activity.56 The use of SMAC mimetics in the treatment of HNSCC is still in its infancy, however the positive results of these early preclinical and phase trials herald hope in the future. Table 1 summarizes trials of CDK inhibitor and combinations registered in ClinicalTrials.gov.
Immunotherapy
Recent exciting advancements of the management of HNSCC have been in the field of immunotherapy. Immune surveillance is an important mechanism to prevent progression of HNSCC. The cancer cells can evade the immune system through multiple mechanisms including T‐cell tolerance, and inhibition of T cell‐related pathways via co‐receptors commonly known as immune checkpoints.57 These checkpoints are negative co‐stimulatory ligands expressed on tumor and infiltrating cells that bind receptors expressed on tumor infiltrating T‐cells that functionally suppress T‐cell function and induce T‐cell apoptosis.58 CTLA‐4 and PD‐1 normally mediate immunological homeostasis by acting as down‐regulators of T‐cell activity after elimination of pathogens, but their expression in HNSCC prematurely stunts effector T‐cell immunity. Checkpoint inhibition primarily via CTLA‐4 and PD‐1 inhibitors is being investigated in a number of phase I/II clinical trials.
Nivolumab is an IgG4 mAb targeting the PD‐1 receptor.59 Since tumors expressing PD‐L1 show the best response to Nivolumab, it is important to understand the proportion of HNSCC that express the PD‐L1 ligand.60 The CheckMate 141 trial (NCT02105636) randomized 361 patients with recurrent HNSCC in a 2:1 ratio to receive Nivolumab every 2 weeks or standard, single‐agent systemic therapy (Ethotrexate, Docetaxel, or Cetuximab).61 In this phase 3 trial, the median overall survival was 7.5 months in the Nivolumab group versus 5.1 months in the standard therapy. The median progression‐free survival (2 months vs 2.3 months) and the response rate (13.3% vs 5.8%) were also better in the Nivolumab group in comparison to the standard therapy. Most significantly perhaps was that treatment‐related adverse events in the Nivolumab group was significantly lower (13.1% vs 35.1%) supporting a more favorable safety profile and quality‐of‐life benefit of immunotherapy. A phase 2 trial (NCT02919683) is specifically looking at Nivolumab in HNSCC of the oral cavity. There is also an ongoing phase 1 study (NCT02124850) assessing whether modulation of biomarkers can predict tumor response in patients administered either Cetuximab and Motolimod (Cohort 1), or Cetuximab, Motolimod and Nivolumab (Cohort 2).
Ipilumumab is an IgG1 mAb targeting the CTLA‐4 receptor.60 Binding to the CTLA‐4 receptors frees the B7 ligand on APCs to bind to the stimulatory CD28 receptor on T cells, resulting in activation.60 Both Nivolumab and Ipilimumab play a role in regulation of T‐cells albeit via different mechanisms and may act synergistically together. A number of trials are still in the recruitment stages with the phase I/2 IMCISION trial (NCT03003637), the phase 2 trial Checkmate 714 (NCT02823574), and the phase 3 Checkmate 651 (NCT02741570) investigating the benefits of immune combination with Ipilimumab and Nivolumab.
Pembrolizumab is a PD‐1 inhibitor that has established safety profile in head and neck cancer patients.7 KEYNOTE‐012 (NCT01848834) is a multicenter phase 1 trial where 104 patients with recurrent or metastatic HNSCC received 10 mg/kg Pembrolizumab intravenously every 2 weeks.7 Of the 104 patients, Pembroluzimab was well tolerated, with 10 (17%) of 60 PDL‐1 positive patients having grade 3 or 4 drug‐related adverse events, the most common of which were increases in the transaminases and hyponatraemia, with 1 patient developing a grade 3 drug‐related rash.7 The study is still under follow‐up analysis but no longer recruiting patients. KEYNOTE‐055 (NCT02255097) is a single‐arm phase 2 study that showed that the response rate was 16% in 171 patients with recurrent/metastic HNSCC who received 200 mg of Pembroluzimab every 3 weeks.62 The study is still ongoing but at the time of analysis 109 (64%) patients reported drug‐related adverse events, with 26 patients (15%) experiencing a grade >3 event.62
Currently, most of the checkpoint inhibitor trials are phase I/II, however there are 8 phase III trials that are currently investigating a checkpoint inhibitor in head and neck cancer: 2 Nivolumab trials (NCT02105636 and NCT02741570); 5 Pembrolizumab (NCT02252042, NCT02358031, NCT03040999, NCT02358031, and NCT02252042); and 1 Tremelimumab (NCT02369874). Table 2 summarizes trials of immune checkpoint and combinations registered in ClinicalTrials.gov.
Table 2.
Trials with Immune Checkpoint Inhibitors.
| Drug | Combination | Status | Clinical Trial ID |
|---|---|---|---|
| Nivolumab | Single agent | Phase III | NCT02105636 |
| Varlilumab | Phase I/II | NCT02335918 | |
| INCB24360 | Phase I/II | NCT02327078 | |
| PLX3397 | Phase I | NCT02526017 | |
| Single agent | Phase I/II | NCT02488759 | |
| Standard therapy | Phase I | NCT02764593 | |
| Ipilimumab | Phase I/II | NCT03003637 | |
| Ipilimumab | Phase II | NCT02823574 | |
| Ipilimumab | Phase III | NCT02741570 | |
| Single agent/ipilumamb | Phase II | NCT02919683 | |
| Epacadostat | Phase I/II | NCT02327078 | |
| Single agent vs Combination(Ipilumamb, BMS‐986016, Daratumamb) | Phase I/II | NCT02488759 | |
| IPI549 | Phase I | NCT02637531 | |
| TAK659 | Phase I | NCT02834247 | |
| Enadenotucirev | Phase I | NCT02636036 | |
| Ipilimumab | Phase II | NCT03097939 | |
| Pembrolizumab | ACP‐196 | Phase II | NCT0454179 |
| Single agent | Phase III | NCT02252042 | |
| Single agent | Phase III | NCT02358031 | |
| INCB24360 | Phase II | NCT02178722 | |
| PLX3397 | Phase I/II | NCT02452424 | |
| Single agent | Phase II | NCT02296684 | |
| MGA271 | Phase I | NCT02475213 | |
| Single agent | Phase II | NCT02255097 | |
| Single agent | Phase II | NCT02769520 | |
| Talimogene Laherparepvec | Phase I | NCT02626000 | |
| Cisplatin | Phase II | NCT02641093 | |
| Single agent | Phase II | NCT02289209 | |
| Cisplatin | Phase II | NCT02777385 | |
| Single agent | Phase II | NCT02609503 | |
| Acalabrutinib | Phase II | NCT02454179 | |
| Cisplatin | Phase III | NCT03040999 | |
| Single agent | Phase II | NCT02841748 | |
| Single agent/cisplatin/carboplatin/5‐FU | Phase III | NCT02358031 | |
| Docetaxel | Phase I/II | NCT02718820 | |
| SD‐101 | Phase I/II | NCT02521870 | |
| Single agent | Phase II | NCT03057613 | |
| Chemoradiotherapy | Phase I | NCT02819752 | |
| Single agent | Phase III | NCT02252042 | |
| Single agent | Phase II | NCT02892201 | |
| Cisplatin, IMRT | Phase I | NCT02775812 | |
| Docetaxel, 5‐FU, Cisplatin | Phase II | NCT03114280 | |
| Single agent | Phase II | NCT03085719 | |
| Vorinostat | Phase I/II | NCT02538510 | |
| Single agent | Phase II | NCT02296684 | |
| Single agent | Phase I | NCT02318771 | |
| Single agent | Phase II | NCT02707588 | |
| Cetuximab | Phase II | NCT03082534 | |
| Cisplatin | Phase I/II | NCT02759575 | |
| RecombinantEphB4‐HSA fusion protein | Phase II | NCT03049618 | |
| Levatinib | Phase I/II | NCT02501096 | |
| PLX3397 | Phase I/II | NCT02452424 | |
| Levatinib | Phase I | NCT03006887 | |
| Single agent | Phase I | NCT01848834 | |
| Single agent | Phase II | NCT02644369 | |
| Atezoluzmab | Obintuzumab | Phase I | NCT02174172 |
| Single agent | Phase I | NCT01375842 | |
| PF‐0502566 | Pembrolizumab | Phase I | NCT02179918 |
| Urelumab | Cetuximab | Phase I | NCT02110082 |
| MEDI4736 | Tremelimumab | Phase III | NCT02369874 |
| AZD9150/AZD5069 | Phase I/II | NCT02499328 | |
| Single agent | Phase II | NCT02207530 | |
| ADXS 11–001 | Phase I/II | NCT02291055 | |
| Mogamulizumab | Phase I | NCT02301130 | |
| Tremelimumab | Mogamulizumab | Phase I | NCT02301130 |
| Ipilumamab | Cetuximab and XRT | Phase I | NCT01935921 |
| MGA271 | Phase I | NCT02381314 | |
| PF‐4518600 | Single agent | Phase I | NCT02315066 |
| AZD17751(Wee1 Inhibitor) | Single agent | Phase I | NCT01748825 |
DISCUSSION AND CONCLUSIONS
TCGA and other large‐scale studies have identified alterations affecting components of several key pathways and functions that provide a roadmap for investigation of developmental therapeutics with potential for precision medicine. The most common include amplifications or mutations of PIK3CA and several growth‐factor receptors, that regulate cell growth and metabolism; a variety of alterations converging on CDK4/6‐RB1‐E2F1, CCND1, and TP53, affecting the cell cycle; FADD and BIRC (IAPs), modulating cell death and survival; and deregulation of immune recognition or activation. New therapeutics targeting these molecules or pathways have shown dramatic single‐agent activity in preclinical models and a few patients, but have mostly demonstrated partial responses or stable disease in clinical trials. This increasingly appears to be due to the fact that most HNSCC tumors harbor complex chromosomal derangements and mutations altering multiple genes and pathways that contribute to the malignant phenotype, whereas relatively fewer are driven primarily by CASP8 with HRAS or HPV with PIK3CA mutations with fewer chromosomal alterations. Recognition of this complexity suggests that investigation of rational combinations targeting several of these key pathways and their interaction with major genomic alterations may reveal wider activity and insight than prematurely focusing on sequence‐based selection of the few patients with predicted activating mutations for single‐agent trials. This includes the potential of marrying treatments that potentiate cell death using chemoradiation or immune checkpoint therapies, inhibitors of PI3K and FADD‐IAP pro‐survival pathways, and MAPK‐cell cycle targets. The number of important genes already found on chromosomes 3 (PIK3CA, SOX2, TP63) and 11 (FADD, BIRC2,3), and others that are implicated in HNSCC pathogenesis and resistance underscore the continuing potential to identify new targets or agents for precision‐medicine‐based investigations.
Financial Disclosure Statement: The NIDCD holds Cooperative Research and Development Agreements with Tetralogic/Medivir and Astex Pharmaceuticals which have provided agents used in preclinical studies cited. Supported by NIDCD Intramural Projects ZIA DC‐000016, 73 and 74.
Conflict of Interest: The authors have no financial conflicts of interest.
BIBLIOGRAPHY
- 1. Rettig EM, D'Souza G. Epidemiology of head and neck cancer. Surg Oncol Clin N Am 2015;24:379–396. [DOI] [PubMed] [Google Scholar]
- 2. Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer‐associated genes. Nature 2013;499:214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517:576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011;333:1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011;333:1154–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pickering CR, Zhang J, Yoo SY, et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov 2013;3:770–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seiwert TY, Burtness B, Mehra R, et al. Safety and clinical activity of pembrolizumab for treatment of recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE‐012): an open‐label, multicentre, phase 1b trial. Lancet Oncol 2016;17:956–965. [DOI] [PubMed] [Google Scholar]
- 8. Stewart LA, Clarke M, Rovers M, et al. Preferred Reporting Items for Systematic Review and Meta‐Analyses of individual participant data: the PRISMA‐IPD Statement. JAMA 2015;313:1657–1665. [DOI] [PubMed] [Google Scholar]
- 9. Seiwert TY, Zuo Z, Keck MK, et al. Integrative and comparative genomic analysis of HPV‐positive and HPV‐negative head and neck squamous cell carcinomas. Clin Cancer Res 2015;21:632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gollin SM. Cytogenetic alterations and their molecular genetic correlates in head and neck squamous cell carcinoma: a next generation window to the biology of disease. Genes Chromosomes Cancer 2014;53:972–990. [DOI] [PubMed] [Google Scholar]
- 11. Boscolo‐Rizzo P, Da Mosto MC, Rampazzo E, et al. Telomeres and telomerase in head and neck squamous cell carcinoma: from pathogenesis to clinical implications. Cancer Metastasis Rev 2016;35:457–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dietz AC, Mehta PA, Vlachos A, et al. Current knowledge and priorities for future research in late effects after hematopoietic cell transplantation for inherited bone marrow failure syndromes: consensus statement from the second Pediatric Blood and Marrow Transplant Consortium International Conference on Late Effects after Pediatric Hematopoietic Cell Transplantation. Biol Blood Marrow Transplant 2017;23:726–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Conti MA, Saleh AD, Brinster LR, et al. Conditional deletion of nonmuscle myosin II‐A in mouse tongue epithelium results in squamous cell carcinoma. Sci Rep 2015;5:14068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hayes DN, Van Waes C, Seiwert TY. Genetic landscape of human papillomavirus‐associated head and neck cancer and comparison to tobacco‐related tumors. J Clin Oncol 2015;33:3227–3234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim BR, Van de Laar E, Cabanero M, et al. SOX2 and PI3K cooperate to induce and stabilize a squamous‐committed stem cell injury state during lung squamous cell carcinoma pathogenesis. PLoS Biol 2016;14:e1002581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barbieri CE, Barton CE, Pietenpol JA. Delta Np63 alpha expression is regulated by the phosphoinositide 3‐kinase pathway. J Biol Chem 2003;278:51408–51414. [DOI] [PubMed] [Google Scholar]
- 17. Hamamoto R, Saloura V, Nakamura Y. Critical roles of non‐histone protein lysine methylation in human tumorigenesis. Nat Rev Cancer 2015;15:110–124. [DOI] [PubMed] [Google Scholar]
- 18. Ramos‐Garcia P, Gil‐Montoya JA, Scully C, et al. An update on the implications of cyclin D1 in oral carcinogenesis. Oral Dis 2016. [DOI] [PubMed] [Google Scholar]
- 19. Bhojani MS, Chen G, Ross BD, Beer DG, Rehemtulla A. Nuclear localized phosphorylated FADD induces cell proliferation and is associated with aggressive lung cancer. Cell Cycle 2005;4:1478–1481. [DOI] [PubMed] [Google Scholar]
- 20. Hajek M, Sewell A, Kaech S, Burtness B, Yarbrough WG, Issaeva N. TRAF3/CYLD mutations identify a distinct subset of human papilloma virus‐associated head and neck squamous cell carcinoma. Cancer 2017;123:1778–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hacker H, Tseng PH, Karin M. Expanding TRAF function: TRAF3 as a tri‐faced immune regulator. Nat Rev Immunol 2011;11:457–468. [DOI] [PubMed] [Google Scholar]
- 22. Dotto GP. Notch tumor suppressor function. Oncogene 2008;27:5115–5123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Okuyama R, Ogawa E, Nagoshi H, et al. p53 homologue, p51/p63, maintains the immaturity of keratinocyte stem cells by inhibiting Notch1 activity. Oncogene 2007;26:4478–4488. [DOI] [PubMed] [Google Scholar]
- 24. Rettig EM, Chung CH, Bishop JA, et al. Cleaved NOTCH1 expression pattern in head and neck squamous cell carcinoma is associated with NOTCH1 mutation, HPV status, and high‐risk features. Cancer Prev Res 2015;8:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bancroft CC, Chen Z, Yeh J, et al. Effects of pharmacologic antagonists of epidermal growth factor receptor, PI3K and MEK signal kinases on NF‐kappaB and AP‐1 activation and IL‐8 and VEGF expression in human head and neck squamous cell carcinoma lines. Int J Cancer 2002;99:538–548. [DOI] [PubMed] [Google Scholar]
- 26. Nathan CO, Amirghahari N, Abreo F, et al. Overexpressed eIF4E is functionally active in surgical margins of head and neck cancer patients via activation of the Akt/mammalian target of rapamycin pathway. Clin Cancer Res 2004;10:5820–5827. [DOI] [PubMed] [Google Scholar]
- 27. Molinolo AA, Hewitt SM, Amornphimoltham P, et al. Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin Cancer Res 2007;13:4964–4973. [DOI] [PubMed] [Google Scholar]
- 28. Vander Broek R, Mohan S, Eytan DF, Chen Z, Van Waes C. The PI3K/Akt/mTOR axis in head and neck cancer: functions, aberrations, cross‐talk, and therapies. Oral Dis 2015;21:815–825. [DOI] [PubMed] [Google Scholar]
- 29. Mazumdar T, Byers LA, Ng PK, et al. A comprehensive evaluation of biomarkers predictive of response to PI3K inhibitors and of resistance mechanisms in head and neck squamous cell carcinoma. Mol Cancer Ther 2014;13:2738–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Amornphimoltham P, Patel V, Sodhi A, et al. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Res 2005;65:9953–9961. [DOI] [PubMed] [Google Scholar]
- 31. Nathan CO, Amirghahari N, Rong X, et al. Mammalian target of rapamycin inhibitors as possible adjuvant therapy for microscopic residual disease in head and neck squamous cell cancer. Cancer Res 2007;67:2160–2168. [DOI] [PubMed] [Google Scholar]
- 32. Brown RE, Zhang PL, Lun M, et al. Morphoproteomic and pharmacoproteomic rationale for mTOR effectors as therapeutic targets in head and neck squamous cell carcinoma. Ann Clin Lab Sci 2006;36:273–282. [PubMed] [Google Scholar]
- 33. Herzog A, Bian Y, Vander Broek R, et al. PI3K/mTOR inhibitor PF‐04691502 antitumor activity is enhanced with induction of wild‐type TP53 in human xenograft and murine knockout models of head and neck cancer. Clin Cancer Res 2013;19:3808–3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mohan S, Vander Broek R, Shah S, et al. MEK inhibitor PD‐0325901 overcomes resistance to PI3K/mTOR inhibitor PF‐5212384 and potentiates antitumor effects in human head and neck squamous cell carcinoma. Clin Cancer Res 2015;21:3946–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ekshyyan O, Mills GM, Lian T, et al. Pharmacodynamic evaluation of temsirolimus in patients with newly diagnosed advanced‐stage head and neck squamous cell carcinoma. Head Neck 2010;32:1619–1628. [DOI] [PubMed] [Google Scholar]
- 36. Grunwald V, Keilholz U, Boehm A, et al. TEMHEAD: a single‐arm multicentre phase II study of temsirolimus in platin‐ and cetuximab refractory recurrent and/or metastatic squamous cell carcinoma of the head and neck (SCCHN) of the German SCCHN Group (AIO). Ann Oncol 2015;26:561–567. [DOI] [PubMed] [Google Scholar]
- 37. Geiger JL, Bauman JE, Gibson MK, et al. Phase II trial of everolimus in patients with previously treated recurrent or metastatic head and neck squamous cell carcinoma. Head Neck 2016;38:1759–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bancroft CC, Chen Z, Dong G, et al. Coexpression of proangiogenic factors IL‐8 and VEGF by human head and neck squamous cell carcinoma involves coactivation by MEK‐MAPK and IKK‐NF‐kappaB signal pathways. Clin Cancer Res 2001;7:435–442. [PubMed] [Google Scholar]
- 39. Dong G, Chen Z, Li ZY, Yeh NT, Bancroft CC, Van Waes C. Hepatocyte growth factor/scatter factor‐induced activation of MEK and PI3K signal pathways contributes to expression of proangiogenic cytokines interleukin‐8 and vascular endothelial growth factor in head and neck squamous cell carcinoma. Cancer Res 2001;61:5911–5918. [PubMed] [Google Scholar]
- 40. Uppaluri R, Winkler AE, Lin T, et al. Biomarker and tumor responses of oral cavity squamous cell carcinoma to trametinib: a phase II neoadjuvant window‐of‐opportunity clinical trial. Clin Cancer Res 2017;23:2186–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cash H, Shah S, Moore E, et al. mTOR and MEK1/2 inhibition differentially modulate tumor growth and the immune microenvironment in syngeneic models of oral cavity cancer. Oncotarget 2015;6:36400–36417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou J, Wu Z, Wong G, et al. CDK4/6 or MAPK blockade enhances efficacy of EGFR inhibition in oesophageal squamous cell carcinoma. Nat Commun 2017;8:13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gary C, Hajek M, Biktasova A, Bellinger G, Yarbrough WG, Issaeva N. Selective antitumor activity of roscovitine in head and neck cancer. Oncotarget 2016;7:38598–38611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Beck TN, Kaczmar J, Handorf E, et al. Phospho‐T356RB1 predicts survival in HPV‐negative squamous cell carcinoma of the head and neck. Oncotarget 2015;6:18863–18874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ku BM, Yi SY, Koh J, et al. The CDK4/6 inhibitor LY2835219 has potent activity in combination with mTOR inhibitor in head and neck squamous cell carcinoma. Oncotarget 2016;7:14803–14813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin‐dependent kinases in cancer therapy. Nat Rev Drug Discov 2015;14:130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dominguez‐Kelly R, Martin Y, Koundrioukoff S, et al. Wee1 controls genomic stability during replication by regulating the Mus81‐Eme1 endonuclease. J Cell Biol 2011;194:567–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hirai H, Arai T, Okada M, et al. MK‐1775, a small molecule Wee1 inhibitor, enhances anti‐tumor efficacy of various DNA‐damaging agents, including 5‐fluorouracil. Cancer Biol Ther 2010;9:514–522. [DOI] [PubMed] [Google Scholar]
- 49. Rajeshkumar NV, De Oliveira E, Ottenhof N, et al. MK‐1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53‐deficient pancreatic cancer xenografts. Clin Cancer Res 2011;17:2799–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Do K, Wilsker D, Ji J, et al. Phase I study of single‐agent AZD1775 (MK‐1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J Clin Oncol 2015;33:3409–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Derakhshan A, Chen Z, Van Waes C. Therapeutic Small Molecules Target Inhibitor of Apoptosis Proteins in Cancers with Deregulation of Extrinsic and Intrinsic Cell Death Pathways. Clin Cancer Res 2017;23:1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Eytan DF, Snow GE, Carlson S, et al. SMAC mimetic birinapant plus radiation eradicates human head and neck cancers with genomic amplifications of cell death genes FADD and BIRC2. Cancer Res 2016;76:5442–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Amaravadi RK, Schilder RJ, Martin LP, et al. A phase I study of the SMAC‐mimetic birinapant in adults with refractory solid tumors or lymphoma. Mol Cancer Ther 2015;14:2569–2575. [DOI] [PubMed] [Google Scholar]
- 54. Matzinger O, Viertl D, Tsoutsou P, et al. The radiosensitizing activity of the SMAC‐mimetic, Debio 1143, is TNFalpha‐mediated in head and neck squamous cell carcinoma. Radiother Oncol 2015;116:495–503. [DOI] [PubMed] [Google Scholar]
- 55. Hurwitz HI, Smith DC, Pitot HC, et al. Safety, pharmacokinetics, and pharmacodynamic properties of oral DEBIO1143 (AT‐406) in patients with advanced cancer: results of a first‐in‐man study. Cancer Chemother Pharmacol 2015;75:851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Infante JR, Dees EC, Olszanski AJ, et al. Phase I dose‐escalation study of LCL161, an oral inhibitor of apoptosis proteins inhibitor, in patients with advanced solid tumors. J Clin Oncol 2014;32:3103–3110. [DOI] [PubMed] [Google Scholar]
- 57. Hashemi‐Sadraei N, Sikora AG, Brizel DM. Immunotherapy and checkpoint inhibitors in recurrent and metastatic head and neck cancer. Am Soc Clin Oncol Educ Book 2016;35:e277–282. [DOI] [PubMed] [Google Scholar]
- 58. Allen CT, Clavijo PE, Van Waes C, Chen Z. Anti‐tumor immunity in head and neck cancer: understanding the evidence, how tumors escape and immunotherapeutic approaches. Cancers 2015;7:2397–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wolchok JD, Kluger H, Callahan MK, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med 2013;369:122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Swanson MS, Sinha UK. Rationale for combined blockade of PD‐1 and CTLA‐4 in advanced head and neck squamous cell cancer‐review of current data. Oral Oncol 2015;51:12–15. [DOI] [PubMed] [Google Scholar]
- 61. Ferris RL, Blumenschein G Jr, Fayette J, et al. Nivolumab for recurrent squamous‐cell carcinoma of the head and neck. N Engl J Med 2016;375:1856–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bauml J, Seiwert TY, Pfister DG, et al. Pembrolizumab for platinum‐ and cetuximab‐refractory head and neck cancer: results from a single‐arm, phase II study. J Clin Oncol 2017:Jco2016701524. [DOI] [PMC free article] [PubMed] [Google Scholar]