

Abstract
Aims
To assess the safety and efficacy of omarigliptin in Japanese patients with type 2 diabetes (T2D).
Methods
In a 24‐week double‐blind trial, 414 patients with T2D were randomized to omarigliptin 25 mg once weekly, sitagliptin 50 mg once daily or placebo. The double‐blind period was followed by a 28‐week open‐label extension during which all patients received omarigliptin 25 mg once weekly. Efficacy endpoints were glycated haemoglobin (HbA1c), 2‐hour postprandial glucose (PPG) and fasting plasma glucose (FPG) levels.
Results
After 24 weeks, the least squares (LS) mean change from baseline in HbA1c was −0.66% for omarigliptin, −0.65% for sitagliptin and 0.13% for placebo. The difference in LS mean for omarigliptin vs placebo was −0.80% (P < .001). The difference in LS mean for omarigliptin vs sitagliptin was −0.02% (95% confidence interval −0.15, 0.12), which met the criterion for non‐inferiority to sitagliptin. Both active treatments provided significant reductions in FPG and 2‐hour PPG compared with placebo (P < .001). Over the 24‐week double‐blind period, there were no clinically meaningful differences in the incidence rates of adverse events among the treatment groups. There was 1 episode of symptomatic hypoglycaemia in the sitagliptin group and none in the omarigliptin or placebo groups. In the 28‐week open‐label period, omarigliptin provided persistent improvements in glycaemic control without notable change in safety profile compared with the double‐blind period. Omarigliptin had no meaningful effect on body weight.
Conclusions
In Japanese patients with T2D, omarigliptin 25 mg once weekly provided significant glucose‐lowering compared with placebo and was non‐inferior to sitagliptin 50 mg once daily. Omarigliptin was generally well tolerated for up to 52 weeks.
Keywords: incretins, oral antihyperglycaemic agent, MK‐3102
1. INTRODUCTION
Omarigliptin (MK‐3102) is a potent, selective, oral dipeptidyl peptidase‐4 (DPP‐4) inhibitor, approved in Japan (MARIZEV), with a half‐life that allows once‐weekly dosing.1 DPP‐4 inhibitors improve glycaemic control in patients with type 2 diabetes (T2D) by prolonging the half‐life of glucagon‐like peptide‐1 (GLP‐1) and glucose‐dependent insulinotropic peptide (GIP), gut‐derived peptides which stimulate insulin secretion and (in the case of GLP‐1) decrease glucagon release in a glucose‐dependent manner.2 Over the past decade, daily‐dosed DPP‐4 inhibitors have become an established part of the therapeutic armamentarium for the treatment of T2D.3
In the present paper, we report the results of a phase III trial conducted in Japanese patients that included a 24‐week double‐blind, placebo‐ and sitagliptin‐controlled period (double‐blind period) and a subsequent 28‐week open‐label extension (open‐label period) in which all patients received omarigliptin. The rationale for the trial, which supported registration in Japan, was to assess the glycaemic efficacy of omarigliptin compared with placebo and a once‐daily DPP‐4 inhibitor.
The objectives of the trial were to assess the safety, tolerability and efficacy of omarigliptin 25 mg once weekly compared with placebo and sitagliptin 50 mg once daily over 24 weeks and to obtain longer‐term (52‐week) data on omarigliptin 25 mg once weekly during the open‐label extension. The hypotheses tested in the present trial were that: (1) after 24 weeks, omarigliptin 25 mg once weekly is superior to placebo in reducing glycated haemoglobin (HbA1c), 2‐hour postprandial glucose (PPG), and fasting plasma glucose (FPG) levels and (2) after 24 weeks, omarigliptin is non‐inferior to sitagliptin 50 mg once daily in reducing HbA1c.
2. RESEARCH DESIGN AND METHODS
2.1. Patients
Trial patients were Japanese men and women, aged ≥20 years, with T2D and a body mass index >18 and <40 kg/m2. At the screening visit, patients who were treatment‐naïve (never on an oral anti‐hyperglycaemic agent [AHA]) or off AHA medication for ≥6 weeks and had National Glycohemoglobin Standardization Program (NGSP) HbA1c ≥7.0% and ≤10.0% were eligible for the study; those on oral AHA medication monotherapy with an NGSP HbA1c ≥6.5% and ≤9.0%, and after a 6‐week AHA wash‐out period, NGSP HbA1c ≥7.0% and ≤10.0%, were also eligible for the study. At week −2, patients were required to have an HbA1c level between ≥7.0% and ≤10.0% and an FPG level ≤12.8 mmol/L.
Patients were excluded from the study if they had type 1 diabetes, a history of ketoacidosis, active liver disease, significant cardiovascular disease, a history of malignancy or haematological disorders, or had been previously treated with sitagliptin or omarigliptin at any time, or with thiazolidinediones or insulin therapy within 12 weeks prior to the screening visit.
Laboratory exclusion criteria included estimated glomerular filtration rate <50 mL/min/1.73 m2, alanine aminotransferase or aspartate aminotransferase >2 times the upper limit of normal, triglycerides >6.78 mmol/L or thyroid‐stimulating hormone outside the central laboratory normal range.
2.2. Study design
The study was a randomized, placebo‐ and sitagliptin‐controlled, parallel‐group, multicentre, double‐blind trial of omarigliptin 25 mg once weekly, with an open‐label extension in which all patients received omarigliptin 25 mg once weekly (Figure S1). The study included a screening period of up to 2 weeks, a wash‐out period of 6 weeks for patients on an oral AHA, a 2‐week single‐blind placebo run‐in period, and a 24‐week double‐blind treatment period followed by a 28‐week open‐label extension period. At randomization, patients were stratified according to their use of oral AHA at screening. Patients were randomized using a double‐dummy design in a 2:2:1 ratio to: omarigliptin 25 mg once weekly/placebo matching sitagliptin 50 mg once daily; sitagliptin 50 mg once daily/placebo matching omarigliptin 25 mg once weekly; or placebo matching omarigliptin 25 mg once weekly/placebo matching sitagliptin 50 mg once daily. Sitagliptin 50 mg once daily was chosen as the relevant dose for use in the study because it is the usual starting dose and the most widely used dose in Japan. An interactive voice response or integrated web response system was used for randomization. Patients not meeting prespecified glycaemic control criteria post‐randomization (from week 4 to week 24, FPG >13.3 mmol/L; after week 24, FPG >11.1 mmol/L) were discontinued from the trial.
A meal tolerance test was conducted at randomization (day 1), and at weeks 24 and 52 (or at discontinuation). Meal tolerance tests were to be performed at trough, 7 days after the previous dose of omarigliptin or placebo matching omarigliptin and prior to the daily sitagliptin or placebo matching sitagliptin dose. The standard meal for the meal tolerance test consisted of ~500 kcal, with 75 g carbohydrate, 14 g fat and 17 g protein. The patient was expected to finish the meal within 15 minutes of beginning to eat. A blood sample for glucose was collected just prior to ingestion of the meal and at 30, 60 and 120 minutes from the start of the meal.
The study (Omarigliptin Protocol 020; ClinicalTrials.gov:NCT01703221) was conducted in accordance with the principles of Good Clinical Practice and was approved by the appropriate institutional review boards.
2.3. Study evaluations
The primary objectives of the present study were to assess the safety and efficacy of omarigliptin for a period of 24 weeks compared with placebo and sitagliptin and to assess the longer‐term safety and tolerability of omarigliptin for up to 52 weeks. The primary hypotheses were that treatment with omarigliptin once weekly for 24 weeks would provide a greater reduction in HbA1c compared with placebo and that treatment with omarigliptin once weekly for 24 weeks would provide a non‐inferior reduction in HbA1c compared with sitagliptin once daily. Secondary hypotheses were that, compared with placebo, 24 weeks of treatment with omarigliptin once weekly would provide greater reductions in 2‐hour PPG and FPG levels. An exploratory objective was the assessment of the effect of 52 weeks of treatment with omarigliptin once weekly on change from baseline in HbA1c, 2‐hour PPG and FPG levels.
2.4. Efficacy endpoints
Changes from baseline in HbA1c, 2‐hour PPG and FPG levels after 24 and 52 weeks of treatment were calculated. The percentages of patients at HbA1c goals of <7.0% and <6.5% at weeks 24 and 52 were also calculated.
2.5. Safety endpoints
Safety endpoints included the incidence rates of adverse events (AEs), percentages of patients meeting predefined limits of change in laboratory variables, change from baseline at weeks 24 and 52 in laboratory variables, ECG, vital signs and body weight. A predefined AE of interest was symptomatic hypoglycaemia.
2.6. Statistical analyses
With 1 exception, the population of all randomized patients who received at least 1 dose of study treatment and had a baseline or a post‐randomization measurement served as the primary population for efficacy analyses. One study patient was randomized to placebo but was incorrectly treated with omarigliptin for 4 once‐weekly doses and then with placebo for approximately 4 weeks and then discontinued by the investigator; prior to unblinding, this patient was excluded from the analyses of efficacy and safety because of the cross‐treatment.
For analyses of the primary efficacy endpoint (change from baseline in HbA1c at week 24), a longitudinal data analysis (LDA) model was used4 with the constraint that true mean at baseline was common to all treatment groups (which was valid as a result of randomization). The analysis model adjusted for treatment, prior AHA therapy status (yes/no), time and the interaction of time by treatment, time by prior AHA therapy status and time by treatment by prior AHA therapy status. The primary hypotheses concerning mean HbA1c change from baseline at week 24 were assessed using the LDA model for comparison of treatment groups. A prespecified, step‐down closed testing procedure was used such that only if the superiority of omarigliptin to placebo was confirmed would the non‐inferiority to sitagliptin be examined. The non‐inferiority of omarigliptin to sitagliptin could be declared if the upper bound of the 2‐sided 95% confidence interval (CI) of least squares (LS) mean between‐group difference was <0.3%.
Using a common standard deviation of 0.658%, and assuming the true mean differences in HbA1c reduction at week 24 between omarigliptin and placebo and between omarigliptin and sitagliptin are 0.5% and 0.0%, respectively, 159, 159 and 79 patients randomized to omarigliptin, sitagliptin and placebo, respectively, would provide >99% power (α = 0.05, 2‐sided test) to show superiority of omarigliptin to placebo and 98% power to demonstrate non‐inferiority of omarigliptin to sitagliptin for the primary hypotheses.
Secondary efficacy endpoints (2‐hour PPG and FPG) were analysed using the LDA model described for HbA1c.
The exploratory endpoint of omarigliptin efficacy up to week 52 was analysed using the LDA model described above (without explicit imputation of missing data) and by calculation of arithmetic means at each time point (without imputation of missing data or adjustment for any factors). Only within‐group analyses were performed because there was no control group in the extension period during which all the patients were treated with omarigliptin.
Analysis of percentages of individuals at the HbA1c goals of <7.0% and <6.5% at weeks 24 and 52 were conducted using the stratified (by prior AHA therapy status) Miettinen and Nurminen method.5 Multiple imputations, based on the LDA model for the analyses of HbA1c, were used to handle missing data.6 Each patient was categorized as a responder (satisfying the HbA1c [NGSP] specific goal of <7.0% or <6.5%) or non‐responder at weeks 24 or 52.
With the exception of the cross‐treated patient noted above, the population of all randomized patients who received at least 1 dose of study treatment was used for the analysis of safety data. Safety and tolerability were assessed by clinical review of all relevant variables including AEs, laboratory tests, ECG, vital signs and body weight. AEs of symptomatic hypoglycaemia were prespecified as events of interest and P values and 95% CIs for between‐treatment group comparisons were calculated using the method of Miettinen and Nurminen.5 For all other endpoints, summary statistics were generated. Change from baseline in body weight at week 24 was analysed using the LDA model described for HbA1c.
To evaluate the longer‐term safety of omarigliptin, the incidence rates (%) of AEs were calculated for the group receiving omarigliptin during the entire duration of the study. Between‐group comparisons and/or estimations of between‐group differences were not performed for the open‐label period. Change from baseline in body weight up to week 52 was analysed as at week 24.
In both phases of this study, potential cases of pancreatitis and prespecified hypersensitivity AEs (anaphylactic reaction, angioedema, asthma‐bronchospasm, erythema multiforme, Stevens–Johnson syndrome, toxic epidermal necrolysis, and drug rash with eosinophilia and systemic symptoms) were evaluated in a blinded manner by external clinical adjudication committees.
3. RESULTS
3.1. Patient disposition and characteristics
A total of 531 patients were screened and 414 patients were randomized at 48 sites in Japan (Figure S2). The most common reason for not randomizing a patient was screen failure (92.3%) and the most common reasons for screen failure were meeting exclusionary laboratory values or not meeting AHA treatment‐related requirements. The first visit of the first patient was on October 26, 2012 and the last visit of the last patient was on April 25, 2014.
Of the 414 randomized patients, 400 (96.6%) completed the 24‐week double‐blind period and 365 (88.2%) completed the 28‐week open‐label period (Figure S2). Baseline demographics and efficacy variables were generally balanced among the groups (Table 1).
Table 1.
Baseline demographic and anthropometric characteristics of randomized patients
| Omarigliptin | Sitagliptin | Placebo | |
|---|---|---|---|
| N = 166 | N = 165 | N = 83 | |
| Age, years | 60 ± 11 | 60 ± 9 | 61 ± 9 |
| Male, n (%) | 104 (62.7) | 115 (69.7) | 57 (68.7) |
| Body Weight, kg | 67 ± 13 | 69 ± 14 | 64 ± 12 |
| BMI, kg/m2 | 25.2 ± 3.7 | 25.4 ± 4.2 | 24.3 ± 3.3 |
| Duration of T2D, years | 7.4 ± 5.5 | 7.4 ± 5.3 | 8.6 ± 5.1 |
| Prior AHA use, n (%) | 65 (39.2) | 61 (37.0) | 32 (38.6) |
| HbA1c, % | 7.9 ± 0.7 | 8.0 ± 0.8 | 8.1 ± 0.7 |
| Range | 6.9‐9.9 | 6.7‐9.9 | 6.9‐10.0 |
| 2‐hour PPG, mmol/L | 13.4 ± 3.5 | 13.4 ± 3.7 | 13.7 ± 3.3 |
| FPG, mmol/L | 9.0 ± 1.7 | 8.8 ± 1.7 | 9.0 ± 1.6 |
Abbreviation: BMI, body mass index.
Values are mean ± standard deviation, unless otherwise indicated.
3.2. Efficacy in the double‐blind period
After 24 weeks of treatment, omarigliptin treatment provided a greater reduction in HbA1c compared with placebo (Table 2; P < .001), and omarigliptin treatment provided non‐inferior reductions in HbA1c compared with sitagliptin treatment (Table 2; LS mean difference in changes from baseline in HbA1c between the omarigliptin and sitagliptin groups: −0.02% [95% CI −0.15, 0.12], with the upper bound of the 2‐sided 95% CI for the between‐group difference being below the prespecified non‐inferiority margin of 0.3%). The profiles for LS mean changes from baseline in HbA1c over time showed near maximal levels of reduction by week 12 with both active treatments, with a continuing modest decrease through to week 24 in both active treatment groups (Figure 1A).
Table 2.
Efficacy endpoints at week 24
| Variable | Omarigliptin | Sitagliptin | Placebo |
|---|---|---|---|
| N = 166 | N = 164 | N = 82 | |
| HbA1c, % | |||
| Change from baselinea | −0.66 (−0.76, −0.57) | −0.65 (−0.74, −0.55) | 0.13 (−0.00, 0.27) |
| vs sitagliptinb | −0.02 (−015, 0.12) | –– | –– |
| vs placebob | −0.80 (−0.96, −0.63)c | −0.78 (−0.94, −0.61)c | –– |
| 2‐hour PPG, mmol/L | |||
| Change from baselinea | −2.35 (−2.75, −1.96) | −2.51 (−2.90, −2.12) | −0.30 (−0.84, 0.23) |
| vs sitagliptinb | 0.16 (−0.37, 0.69) | –– | –– |
| vs placebob | −2.05 (−2.69, −1.41)c | −2.21 (−2.85, −1.57)c | –– |
| FPG, mmol/L | |||
| Change from baselinea | −1.03 (−1.21, −0.84) | −1.15 (−1.34, −0.97) | −0.35 (−0.60, −0.09) |
| vs sitagliptinb | 0.12 (−0.13, 0.37) | –– | –– |
| vs placebob | −0.68 (−0.99, −0.38)c | −0.81 (−1.11, −0.50)c | –– |
LS mean (95% CI) based on an LDA model with terms for treatment, prior AHA therapy status (yes/no), time and the interaction of time by treatment, time by prior AHA therapy status, and time by treatment by prior AHA therapy status with the constraint that the mean baseline is the same for all treatment groups.
Difference in LS means (95% CI).
P < .001.
Figure 1.
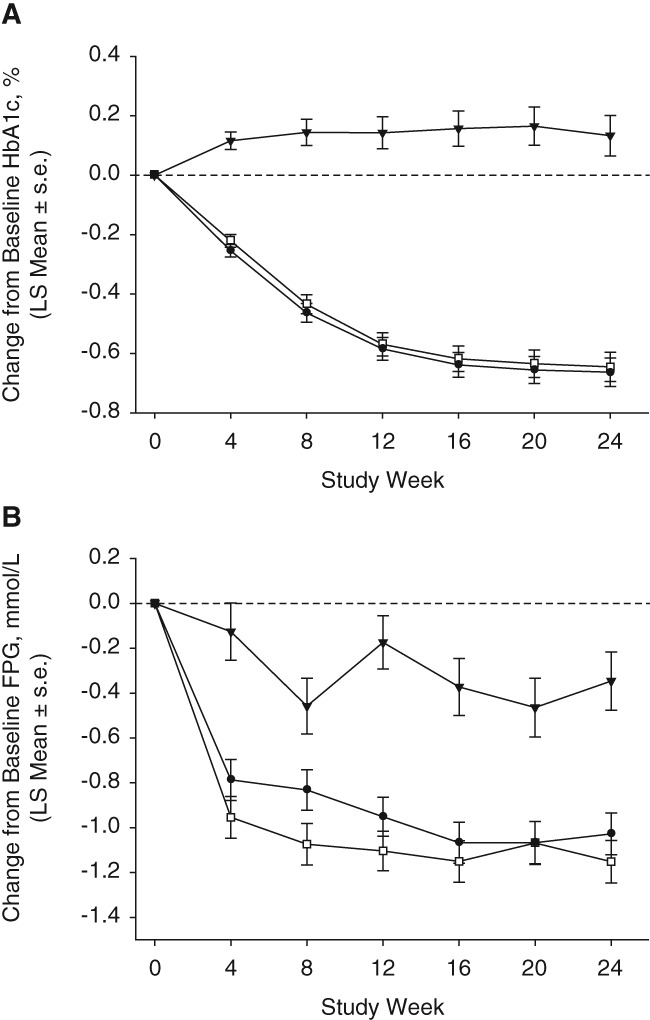
Efficacy measures up to week 24; A, HbA1c; B, FPG; (black triangles, placebo; white squares, sitagliptin; black circles, omarigliptin; based on an LDA model with terms for treatment, prior AHA therapy status (yes/no), time, and interaction of time by treatment, time by prior AHA therapy status, and time by treatment by prior AHA therapy status, with the constraint that the mean baseline was the same for all treatment groups). s.e., standard error
After 24 weeks of treatment, omarigliptin provided a greater reduction in 2‐hour PPG levels compared with placebo (Table 2; P < .001). Sitagliptin and omarigliptin treatment provided similar reductions in 2‐hour PPG (Table 2; LS mean difference in changes from baseline in 2‐hour PPG between the omarigliptin and sitagliptin groups: 0.16 mmol/L [95% CI −0.37, 0.69]).
After 24 weeks of treatment, omarigliptin provided a greater reduction in FPG compared with placebo (Table 2; P < .001). Sitagliptin and omarigliptin treatment provided similar reductions in FPG (Table 2; LS mean difference in changes from baseline in FPG between the omarigliptin and sitagliptin groups: 0.12 mmol/L [95% CI −0.13, 0.37]). The profiles for LS mean changes from baseline in FPG over time showed near maximal levels of reduction by week 4 with both active treatments, with further modest decreases achieved through to week 16 being maintained through to week 24 (Figure 1B). At weeks 4, 8 and 12 the point estimates for FPG were slightly higher in the omarigliptin group compared with the sitagliptin group; however, the standard errors overlapped at weeks 4 and 12 and, at later timepoints, the differences in point estimates for FPG were less notable.
At the end of the double‐blind treatment period, the percentages of patients reaching the HbA1c goals of <7.0% and <6.5% were higher in both active treatment groups compared with placebo (Figure 2). At week 24, 47% and 38% of patients in the omarigliptin and sitagliptin groups, respectively, had HbA1c <7.0% compared with 7% of patients in the placebo group (nominal P < .001 in both cases), while 10% and 9% of patients in the omarigliptin and sitagliptin groups, respectively, had HbA1c <6.5%, compared with 1% of patients in the placebo group (nominal P = .009 and .027 for the omarigliptin and sitagliptin groups, respectively).
Figure 2.
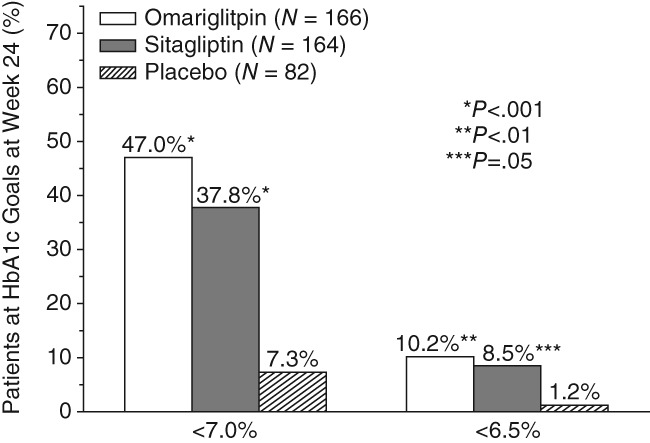
Percentage of patients at HbA1c goals of 7.0% or 6.5% at week 24. P values are associated with differences of active treatment groups from placebo
3.3. Efficacy in the open‐label period
The overall efficacy was derived from both the model‐based LDA method and a simple analysis of mean values (arithmetic mean) from available data without adjustment for any factors (Table S1 and Figures S3A and B). With both analysis methods, treatment with omarigliptin resulted in clinically meaningful reductions from baseline for all glycaemic endpoints.
For HbA1c and FPG, reductions from baseline at week 52 were observed in both statistical approaches, with slightly smaller magnitude using the LDA approach. The reductions in HbA1c and FPG from baseline observed in the omarigliptin/omarigliptin and sitagliptin/omarigliptin groups were similar over time (Figure S3A,B). In the placebo/omarigliptin group improvements in glycaemic variables were observed within 4 weeks of the switch from placebo to omarigliptin and at week 52 the changes were similar to the other treatment groups.
At week 52, the reductions from baseline in 2‐hour PPG were similar in all three treatment groups (Table S1).
At the end of the open‐label period treatment, the percentages of patients reaching the HbA1c goal of <7.0% were 35%, 25% and 32% in the omarigliptin/omarigliptin, sitagliptin/omarigliptin and placebo/omarigliptin groups, respectively. The percentages of patients reaching the HbA1c goal of <6.5% were 7%, 7% and 9%, in the same groups, respectively.
3.4. Safety
3.4.1. Double‐blind period
The incidence rates of AEs, including those that were assessed by the investigator as drug‐related, serious AEs (SAEs), and AEs leading to discontinuation were generally similar between the omarigliptin and sitagliptin groups (Table 3). There were no notable differences in the incidence rates of specific AEs in the omarigliptin and sitagliptin groups compared with the placebo group. The incidence rates of specific AEs with an incidence ≥3% are shown in Table 3. No serious drug‐related AEs or deaths were reported in any treatment group. There were no events of symptomatic hypoglycaemia in the omarigliptin or placebo treatment groups and 1 event in the sitagliptin group (Table 3). No severe hypoglycaemia episodes (any episode of hypoglycaemia for which assistance was required) were reported. There were no investigator‐reported cases or adjudication‐confirmed cases of acute or chronic pancreatitis or serious hypersensitivity. There were no meaningful changes in body weight in the omarigliptin or the sitagliptin treatment groups (LS mean change from baseline 0.04 kg [95% CI −0.25, 0.32] and −0.01 kg [95% CI −0.31, 0.28], respectively) while the placebo group had a small decrease from baseline in body weight (−0.74 kg [95% CI −1.15, −0.32]). There were no meaningful changes in laboratory variables, ECG and vital signs. No notable difference was observed in the percentage of patients that met the QTc predefined limits of change criteria among the treatment groups. As noted in the Statistical Analysis section, 1 patient in the study who was cross‐treated with omarigliptin and then with placebo and then discontinued by the investigator was not included in the overall safety analysis. In this patient, a non‐serious AE of herpes zoster was reported on day 22 (at which time the patient was exposed to incorrectly dispensed omarigliptin). The event was mild in intensity and resolved on day 57. No action was taken with the study medication and the event was assessed by the investigator as not‐related to the study medication.
Table 3.
Adverse events summary: incidences of specific AEsa with incidence ≥3% in ≥1 treatment group and incidence rates for symptomatic hypoglycaemia up to week 24 (double‐blind period)
| Patients, n (%) | Omarigliptin | Sitagliptin | Placebo |
|---|---|---|---|
| N = 166 | N = 164 | N = 82 | |
| With ≥ 1 | |||
| AE | 83 (50.0) | 81 (49.4) | 54 (65.9) |
| Drug‐relatedb AE | 7 (4.2) | 6 (3.7) | 5 (6.1) |
| SAE | 3 (1.8) | 3 (1.8) | 0 (0.0) |
| Drug‐relatedb SAE | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Patient who died | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Who discontinued due to ≥1 | |||
| AE | 1c (0.6) | 2 (1.2) | 0 (0.0) |
| Drug‐relatedb AE | 1 (0.6) | 1 (0.6) | 0 (0.0) |
| SAE | 0c (0.0) | 1 (0.6) | 0 (0.0) |
| Drug‐relatedb SAE | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| With specific AEs with incidence ≥3% in ≥1 treatment group, by SOC | |||
| Gastrointestinal disorders | |||
| Diarrhoea | 2 (1.2) | 3 (1.8) | 3 (3.7) |
| Gastritis | 2 (1.2) | 5 (3.0) | 0 (0.0) |
| Infections and infestations | |||
| Bronchitis | 3 (1.8) | 0 (0.0) | 3 (3.7) |
| Influenza | 3 (1.8) | 5 (3.0) | 4 (4.9) |
| Nasopharyngitis | 21 (12.7) | 18 (11.0) | 25 (30.5) |
| Pharyngitis | 4 (2.4) | 4 (2.4) | 3 (3.7) |
| Respiratory, thoracic and mediastinal disorders | |||
| Upper respiratory tract inflammation | 4 (2.4) | 7 (4.3) | 2 (2.4) |
| With ≥1 AE of symptomatic hypoglycaemiad | 0 (0.0) | 1 (0.6) | 0 (0.0) |
Abbreviation: SOC, system organ class.
According to SOC defined by the Medical Dictionary for Regulatory Activities (MedDRA) classification system.
Assessed by the investigator as related to study drug.
One additional patient, in whom a SAE of “bile duct stone” was reported, discontinued during the double‐blind period of the study. The patient's discontinuation from study was initially reported as attributable to discontinuation criterion related to increased hepatic enzymes (the elevated hepatic enzymes were not reported as an AE). After unblinding this was updated to indicate that the patient's discontinuation from study was attributable to the SAE; therefore, this patient is not included in this table as discontinued because of an AE/SAE, but is included as discontinued because of an AE/SAE in the week 52 summary (Table S2).
Prespecified AE of interest; symptomatic hypoglycaemia: episode with clinical symptoms attributed to hypoglycaemia, without regard to glucose level.
3.4.2. Safety in the open‐label period
Only the group initiating omarigliptin during the double‐blind period could provide safety data for longer‐term (52‐week) omarigliptin treatment. In this group, the overall incidence of AEs was 116/166 patients (69.9%), the incidence of drug‐related AEs was 13/166 patients (7.8%) and the incidence of SAEs was 6/166 patients (3.6%; Table S2). Of these, 3 patients experienced SAEs during the first 24 weeks. One SAE (prostate cancer) was reported in the omarigliptin group during the open‐label extension that was considered by the investigator to be drug‐related. No deaths were reported. Study medication was discontinued as a result of AEs in 4 patients (2.4%) in the 52‐week omarigliptin treatment group (of whom 2 patients discontinued during the first 24 weeks). Three patients (1.8%) discontinued because of drug‐related AEs (double‐blind period: upper respiratory tract infection in 1 patient; open‐label period: drug eruption in 1 patient, and blood glucose increased and HbA1c increased in 1 patient); all of the AEs were mild in intensity. There was no clinically meaningful change in body weight in the 52‐week omarigliptin treatment group (LS mean change from baseline 0.37 kg [95% CI 0.04, 0.71]). No clinically meaningful changes from baseline were seen for laboratory tests, vital signs or ECG variables. No notable safety and tolerability findings were observed in groups switched from sitagliptin or placebo to omarigliptin treatment during the 28‐week open‐label period (Table S2). There were no adjudication‐confirmed cases of acute or chronic pancreatitis or prespecified hypersensitivity adverse reactions.
4. DISCUSSION
In the double‐blind treatment period of this study, 24 weeks of once‐weekly treatment with omarigliptin 25 mg provided greater reductions in HbA1c, 2‐hour PPG and FPG compared with placebo, and provided a reduction in HbA1c that was non‐inferior to daily treatment with sitagliptin 50 mg. Once‐weekly treatment with omarigliptin 25 mg provided reductions in 2‐hour PPG and FPG levels similar to those provided by daily treatment with sitagliptin 50 mg.
The profile of change over time in HbA1c was essentially overlapping for omarigliptin and sitagliptin. While there are apparent differences in the point estimates of FPG between omarigliptin and sitagliptin, these differences are modest and are unlikely to be clinically meaningful. The observation is not related to time required to reach steady‐state. Omarigliptin exhibits negligible accumulation upon repeated dosing and steady‐state is achieved with 1 to 2 once‐weekly doses. In addition, in another trial that compared omarigliptin 25 mg once weekly with sitagliptin 100 mg once daily, no discordance in FPG was observed among the treatment groups at 6 and 12 weeks (visit intervals were different in the 2 studies).7
The exploratory analyses of long‐term efficacy showed that weekly treatment with omarigliptin 25 mg resulted in meaningful reductions from baseline in all glycaemic variables for up to 52 weeks. Modest deterioration of glycaemic control was observed in all 3 groups during the open‐label period. When switched from placebo to omarigliptin 25 mg at week 24, patients in the placebo/omarigliptin group initially demonstrated a magnitude of reduction in HbA1c similar to that achieved by the other treatment groups and, over the remaining treatment period, also exhibited some deterioration in glycaemic control, suggesting the decrease in glycaemic control was not attributable to time‐of‐treatment‐dependent loss of efficacy of DPP‐4 inhibitor therapy with longer‐duration treatment but was more likely attributable to other factors that could occur independently of the treatment, such as loss of trial effect over time. The profile of change in HbA1c over the 52‐week treatment period is generally similar to that reported in daily DPP‐4 inhibitor studies in Japanese patients with T2D.8, 9
The results of the double‐blind and open‐label periods of the study indicated that weekly treatment with omarigliptin 25 mg was well tolerated. During the double‐blind period, there were no notable differences in the overall incidence rates of AEs or drug‐related AEs between the omarigliptin and sitagliptin groups. The overall incidence rates of AEs or drug‐related AEs were not unexpectedly increased during longer‐term treatment with omarigliptin up to 52 weeks. During the double‐blind period, no notable difference was observed between treatment groups in the incidence rates of SAEs or in the incidence of patients discontinued from study medication because of AEs. The incidence rates of SAEs or AEs leading to discontinuation were low up to 52 weeks of treatment.
The absence of symptomatic hypoglycaemia throughout the 52‐week treatment period in the group treated with omarigliptin is consistent with the known low incidence of hypoglycaemia when daily DPP‐4 inhibitors are administered as monotherapy or co‐administered with agents that are not by themselves associated with hypoglycaemia.10
In recent years, a patient‐centred approach has been recommended for the treatment of T2D.3 Available evidence suggests that weekly administered AHAs are viewed positively by patients with T2D11, 12, 13, 14; therefore, a once‐weekly oral AHA may be a therapeutic option when the patient prefers a weekly regimen. Since effective treatment of T2D can be complicated by poor medication adherence,15, 16, 17, 18 a once‐weekly AHA may also have advantages when poor patient adherence to medication has been identified as a barrier to achieving therapeutic goals. A weekly oral AHA may also be useful in other clinical situations (e.g., intermittent assisted healthcare).
The strength of the present trial is that it included both placebo and sitagliptin comparator arms, which allowed an assessment of intrinsic efficacy (placebo comparison) and a direct comparison with the relevant dose of a daily DPP‐4 inhibitor. One limitation of the trial is that patient compliance may have been higher than that which might be observed in real‐world settings. In addition, patient satisfaction with a once‐weekly AHA could not be assessed because to maintain double‐blinding in the placebo‐controlled portion of the trial all treatment groups took once‐weekly (omarigliptin or matching placebo) and once‐daily (sitagliptin or matching placebo) trial medication.
In conclusion, the results of the present study suggest that omarigliptin is an efficacious and generally well‐tolerated, weekly oral AHA for treatment of Japanese patients with T2D. Real‐world studies would be useful to further evaluate the potential benefits of once‐weekly treatment with regard to compliance and satisfaction.
ORCID
Ira Gantz http://orcid.org/0000-0002-6565-7113
Samuel S. Engel http://orcid.org/0000-0002-4439-6356
Supporting information
Table S1. The least squares (LS) means (LDA model analysis) and arithmetic means of the changes from baseline in efficacy endpoints at Week 52.
Table S2. Adverse events (AEs) summary; incidences of specific AEs by MedDRA SOC ‡, with incidence ≥5% in ≥1 treatment group; and incidences of symptomatic hypoglycemia at Week 52 (double‐blind + open‐label period for omarigliptin/omarigliptin group, open‐label period only for sitagliptin/omarigliptin and placebo/omarigliptin groups).
Figure S1. Study design; AHA, anti‐hyperglycemic agent; NGSP, National Glycohemoglobin Standardization Program; R, randomization; q.w., once weekly; q.d., once daily; SCR, screening.
Figure S2. Disposition of patients, double‐blind and open‐label periods. †One patient was initially discontinued in the double‐blind period due to “other” but the reason was updated during the extension, open‐label period, after un‐blinding at Week 24, to “adverse event.” ‡Two patients who experienced adverse events during the placebo run‐in period were later discontinued; one after randomization, but prior to administration of study drug, and the other during the treatment period. §One patient was randomized to placebo but incorrectly treated with omarigliptin and then with placebo and then discontinued by the investigator; prior to unblinding, this patient was excluded from the analyses of efficacy and safety due to the cross‐treatment.
Figure S3. Efficacy measures through Week 52, based on arithmetic means of available data without adjustment by any factors; A, HbA1c; B, FPG; (▼ Placebo/Omarigliptin; □ Sitagliptin/Omarigliptin; ● Omarigliptin/Omarigliptin).
ACKNOWLEDGEMENTS
Editorial assistance was provided by Jennifer Rotonda and Michele McColgan of Merck & Co., Inc., Kenilworth, New Jersey.
Conflict of interest
All authors are employees of MSD K.K. or Merck Sharp & Dohme Corp., subsidiaries of Merck & Co., Inc., Kenilworth, New Jersey, who may own stock and/or hold stock options in the Company.
Author contributions
I.G., T.O., Y.I., K.O., E.A.O., K.D.K., S.S.E. and E.L. are responsible for the work described in this paper. I.G., T.O., Y.I., K.O., K.D.K., S.S.E. and E.L. conceived, designed and/or planned the study. K.O. analysed the data. I.G., T.O., Y.I., K.O., E.A.O., K.D.K., S.S.E. and E.L. interpreted the results. I.G., T.O. and E.A.O. drafted the manuscript. T.O., Y.I., K.O., K.D.K., S.S.E. and E.L. critically reviewed and/or revised the manuscript for important intellectual content. All authors provided final approval of the version to be published and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
APPENDIX 1.
Omarigliptin Study 020 Group
Daishiro Yamada (Jiyugaoka Yamada Clinic of Internal Medicine), Hideki Kuribayashi (Kuribayashi Clinic), Yoshio Kurihara (Kurihara Clinic), Toshiya Okamoto (Okamoto Naika Clinic), Tatsujiro Segawa (Ebetsu Clinic), Kazuhiko Sugiyama (Clinic Sugiyama), Akiko Niiori Onishi (Hitachinaka General Hospital), Hideo Takahashi (Minami Akatsuka Clinic), Hisamoto Kuroda (Green Clinic), Toshio Kawada (Kawada Clinic), Masato Nishiwaki (Keyaki Naika Clinic), Toshiyuki Sugiura (Sugiura Clinic), Shigeo Sawai (Sawai Medical Clinic), Osamu Matsuoka (ToCROM Clinic), Kotaro Shimokawa (Yutenji Medical Clinic), Kazuyuki Mizuyama (Doujin Memorial Meiwa Hospital), Kageki Ito (Ito Internal Medicine & Pediatrics Clinic), Mitsutoshi Kato (Kato Clinic of Internal Medicine), Koki Shin (Shin Clinic), Makiko Suzuki (Koseikai Suzuki Hospital), Yukiko Onishi (The Institute for Adult Diseases, Asahi Life Foundation), Hikaru Ishii (Shin‐Nihonbashi Ishii Clinic), Mitsuru Ohsugi (Toshiba General Hospital), Mikiya Tokui (Tokui Clinic), Hajime Maeda (H.E.C. Science Clinic), Kiyokazu Matoba (Matoba Diabetes Clinic), Akira Yamauchi (Suruga Clinic), Hiroki Yamanoue (Shizuoka Tokushukai Hospital), Hiroshi Hori (Nagoya Kyoritsu Hospital), Satoshi Inoue (OCROM Clinic), Kentaro Ohtoshi (Otoshi Medical Clinic), Yasuro Kumeda (Minamiosaka Hospital), Tetsuro Hiraoka (Hiraoka Naika Clinic), Hiroaki Miyaoka (Saiseikai Matsuyama Hospital), Masamitsu Inoue (Inoue Internal Medical Clinic), Fumihiro Taguchi (Fukuoka Wajiro Hospital), Kensuke Fukuyo (Fukuyo Internal Medicine Clinic), Kiyohide Nunoi (St. Mary's Hospital), Sigeyuki Kouno (Asunaro Naika Clinic), Hiroshi Morinaga (Morinaga Ueno Clinic), Nobuyuki Abe (Abe Diabetes Clinic), Yasuhiro Hashiguchi (Tempozan Naika Medical Clinic, Int. Med.), Hideaki Tanaka (Tanaka Clinic), Hiroaki Tomori (Yaesu Clinic), Koji Nagata (Nagata Naika Clinic), Hiromi Kato (Takaoka Fushiki Hospital), Yoshiyuki Hamamoto (Kitano Hospital), Kunio Hieshima (Jinnouchi Hospital, Diabetes Care Center).
Gantz I, Okamoto T, Ito Y, Okuyama K, O'Neill EA, Kaufman KD, Engel SS, Lai E and the Omarigliptin Study 020 Group . A randomized, placebo‐ and sitagliptin‐controlled trial of the safety and efficacy of omarigliptin, a once‐weekly dipeptidyl peptidase‐4 inhibitor, in Japanese patients with type 2 diabetes. Diabetes Obes Metab. 2017;19:1602–1609. https://doi.org/10.1111/dom.12988
Funding information Funding for this study was provided by MSD K.K., a subsidiary of Merck & Co., Inc., Kenilworth, New Jersey.
REFERENCES
- 1. Biftu T, Sinha‐Roy R, Chen P, et al. Omarigliptin (MK‐3102): a novel long‐acting DPP‐4 inhibitor for once weekly treatment of type 2 diabetes. J Med Chem. 2014;57(8):3205‐3212. [DOI] [PubMed] [Google Scholar]
- 2. Pratley RE, Salsali A. Inhibition of DPP‐4: a new therapeutic approach for the treatment of type 2 diabetes. Curr Med Res Opin. 2007;23(4):919‐931. [DOI] [PubMed] [Google Scholar]
- 3. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes: a patient‐centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care. 2012;35(6):1364‐1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Liang KY, Zeger SL. Longitudinal data analysis of continuous and discrete responses for pre‐post designs. Indian J Stat. 2000;62:134‐148. [Google Scholar]
- 5. Miettinen O, Nurminen M. Comparative analysis of two rates. Stat Med. 1985;4(2):213‐226. [DOI] [PubMed] [Google Scholar]
- 6. Rubin DB. Multiple Imputation for Nonresponse in Surveys. New York: John Wiley & Sons; 1987. [Google Scholar]
- 7. Goldenberg R, Gantz I, Andryuk PJ, et al. Randomized clinical trial comparing the efficacy and safety of treatment with the once‐weekly dipeptidyl peptidase‐4 (DPP‐4) inhibitor omarigliptin or the once‐daily DPP‐4 inhibitor sitagliptin in patients with type 2 diabetes inadequately controlled on metformin monotherapy. Diabetes Obes Metab. 2017;19(3):394‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Araki E, Kawamori R, Inagaki N, et al. Long‐term safety of linagliptin monotherapy in Japanese patients with type 2 diabetes. Diabetes Obes Metab. 2013;15(4):364‐371. [DOI] [PubMed] [Google Scholar]
- 9. Konya H, Yano Y, Matsutani S, et al. Profile of saxagliptin in the treatment of type 2 diabetes: focus on Japanese patients. Ther Clin Risk Manag. 2014;10:547‐558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scheen AJ. Safety of dipeptidyl peptidase‐4 inhibitors for treating type 2 diabetes. Expert Opin Drug Saf. 2015;14(4):505‐524. [DOI] [PubMed] [Google Scholar]
- 11. Polonsky WH, Fisher L, Hessler D, Bruhn D, Best JH. Patient perspectives on once‐weekly medications for diabetes. Diabetes Obes Metab. 2011;13(2):144‐149. [DOI] [PubMed] [Google Scholar]
- 12. Hauber AB, Tunceli K, Yang JC, et al. A survey of patient preferences for oral antihyperglycemic therapy in patients with type 2 diabetes mellitus. Diabetes Ther. 2015;6(1):75‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sen R, Shields AL, Atsuda K. Patient preference for once‐weekly doseing in type 2 diabetes in Japan. JHEOR. 2013;4(1):55‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Suzuki D, Kuroki H, Tsukada A, et al. Do patients choose once weekly DPP‐4 inhibitors? [Japanese]. Progress Med. 2016;36:389‐392. [Google Scholar]
- 15. Cramer JA. A systematic review of adherence with medications for diabetes. Diabetes Care. 2004;27(5):1218‐1224. [DOI] [PubMed] [Google Scholar]
- 16. Tunceli K, Zhao C, Davies MJ, et al. Factors associated with adherence to oral antihyperglycemic monotherapy in patients with type 2 diabetes. Patient Prefer Adherence. 2014;9:191‐197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kirkman MS, Rowan‐Martin MT, Levin R, et al. Determinants of adherence to diabetes medications: findings from a large pharmacy claims database. Diabetes Care. 2015;38(4):604‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Donnan PT, MacDonald TM, Morris AD. Adherence to prescribed oral hypoglycaemic medication in a population of patients with Type 2 diabetes: a retrospective cohort study. Diabet Med. 2002;19(4):279‐284. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The least squares (LS) means (LDA model analysis) and arithmetic means of the changes from baseline in efficacy endpoints at Week 52.
Table S2. Adverse events (AEs) summary; incidences of specific AEs by MedDRA SOC ‡, with incidence ≥5% in ≥1 treatment group; and incidences of symptomatic hypoglycemia at Week 52 (double‐blind + open‐label period for omarigliptin/omarigliptin group, open‐label period only for sitagliptin/omarigliptin and placebo/omarigliptin groups).
Figure S1. Study design; AHA, anti‐hyperglycemic agent; NGSP, National Glycohemoglobin Standardization Program; R, randomization; q.w., once weekly; q.d., once daily; SCR, screening.
Figure S2. Disposition of patients, double‐blind and open‐label periods. †One patient was initially discontinued in the double‐blind period due to “other” but the reason was updated during the extension, open‐label period, after un‐blinding at Week 24, to “adverse event.” ‡Two patients who experienced adverse events during the placebo run‐in period were later discontinued; one after randomization, but prior to administration of study drug, and the other during the treatment period. §One patient was randomized to placebo but incorrectly treated with omarigliptin and then with placebo and then discontinued by the investigator; prior to unblinding, this patient was excluded from the analyses of efficacy and safety due to the cross‐treatment.
Figure S3. Efficacy measures through Week 52, based on arithmetic means of available data without adjustment by any factors; A, HbA1c; B, FPG; (▼ Placebo/Omarigliptin; □ Sitagliptin/Omarigliptin; ● Omarigliptin/Omarigliptin).