

Abstract
Research on chlorine‐free conversions of P4 into organophosphorus compounds (OPCs) has a long track record, but methods that allow desirable, direct P−C bond formations have only recently emerged. These include the use of metal organyls, carbenes, carboradicals, and photochemical approaches. The versatile product scope enables the preparation of both industrially relevant organophosphorus compounds, as well as a broad range of intriguing new compound classes. Herein we provide a concise overview of recent breakthroughs and outline the acquired fundamental insights to aid future developments.
Keywords: main group chemistry, nucleophilic addition, organophosphorus compounds, phosphorus, phosphorus anions
1. Introduction
Organophosphorus compounds (OPCs) are important reagents with widespread use in industry. Especially valuable are the compounds containing P−C bonds, which can be applied as ligands in catalysis or as auxiliaries in C−E coupling reactions (E=C, O, or N).1 The required phosphorus atoms originate from white phosphorus (P4), which is typically converted to PCl3 through large‐scale halogenation and subsequently functionalized by salt elimination reactions (A, Figure 1).2 However, this process generates stoichiometric amounts of halide waste and often involves unselective multi‐step synthetic routes.1 Direct functionalization of P4 could offer an attractive alternative (B), but this strategy is hampered by the unpredictable behavior of the P4 tetrahedron as showcased in the diversity of currently known chemistry. While most of this work has been covered in a number of seminal reviews of the past decade,3, 4, 5 an appealing approach based on direct P−C bond formation, resembling PCl3 substitution,6 has emerged only recently as a promising platform for the selective preparation of OPCs from P4, which is the topic of this review.7
Figure 1.
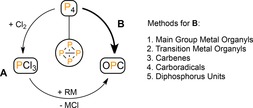
Preparation of organophosphorus compounds (OPCs) from P4.
To understand how P−C bonds can be made using P4 it is instructive to touch on its properties. Most pronounced is its electrophilicity,8 which due to the acute (60ο) bond angles of the P4 cage, is assumed to originate from ring strain (d(P−P)=2.1994(3) Å, gas‐phase electron diffraction),9 even though the expected bending of the P−P bonds (≈5ο) is insignificant according to an AIM analysis (atoms in molecules).10 The bonding in P4 benefits from delocalization of the electrons in s, p, and d cluster orbitals (spherical aromaticity).11 Whereas reduction of P4 by means of cyclic voltammetry (CV) occurs readily, it is irreversible due to bond rupture and polymerization of the formed radical anion (P4 .−).5b, 12 White phosphorus can also be “cracked” both thermally (>1100 K)13 and photochemically (UV irradiation)14 into two transient P2 molecules (P≡P) that polymerize rapidly to the more stable red phosphorus allotrope.15
In this Minireview we highlight recent breakthroughs in P4 chemistry by focusing on reactions that directly create P−C bonds with main group and transition metal organyls, ambiphilic carbenes, and carboradicals as well as on trapping of P2 fragments with organic substrates.
2. Functionalization of P4 Using Main Group Metal Organyls
A common approach to introduce carbon atoms to electrophilic functional groups involves the use of organolithium or Grignard reagents. In 1963, Rauhut and co‐workers were the first to report on the formation of P−C bonds by reaction of either phenyl‐ or n‐butyllithium (or MgBr salts) with ethereal solutions of P4.16 Quenching the resulting deep red suspensions with water or butylhalides afforded mixtures of mostly primary or tertiary phosphanes as detectable products (I, Scheme 1; only Ph shown), but with only low selectivity and poor yields (0–40 %) in addition to large quantities of organopolyphosphines.17 Equally challenging with similar product mixtures are the reactions of P4 with alkynyls (II)18 or with tert‐butyl‐ or methyllithium in combination with Me3SiCl as quenching agent (III).19 The more bulky reagents allowed for formation of cyclotetraphosphanes (e.g. 1; Scheme 1), indicating a more controlled degradation path through steric shielding.20 Using the sterically encumbered Mes*Li (IV; Mes*=2,4,6‐tBu3C6H2), Fluck et al. demonstrated that degradation of P4 is stoppable after a single P−P bond cleavage. They isolated in <10 % yield the first example of a bicyclo[1.1.0]tetraphosphabutane (2) in addition to diphosphene 3.21
Scheme 1.
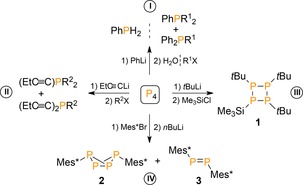
Reactions of P4 with organoalkali reagents. Mes*=2,4,6‐tBu3C6H2, R1=Bu, R2=Et or Pr, X=Br or Cl.
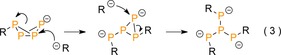
The reactions of P4 with organoalkali reagents proceed through a common highly reactive transient [RP4]− butterfly anion, which is produced after nucleophilic addition and concomitant P−P bond cleavage [Eq. 1].

In 2014, the formation of this elusive intermediate was confirmed by us.22 We used sterically encumbered ArylLi reagents and Lewis acids (LA; B(C6F5)3 or BPh3) to trap the incipient phosphide, thereby selectively obtaining the first examples of LA‐stabilized bicyclo[1.1.0]tetraphosphabutanide anions (4, Scheme 2).23, 24 The HOMO of these species shows a lone pair on the boron‐coordinated wing‐tip P‐atom, which allows for controlled subsequent functionalization of the P4 core. For example, alkylation of 4 b, featuring the strong LA B(C6F5)3, with MeI afforded exclusively endo‐methylated product 5 of which the exo P−B bond could be cleaved to furnish the labile, non‐symmetric organo‐substituted exo,endo‐R2P4 butterfly 6.23 Conversely, alkylation of the more reactive BPh3‐stabilized anion 4 c with Ph3C+PF6 − provided clean and direct access to LA‐free exo,exo‐7, which due to the steric shielding of the bulky trityl group proved kinetically inert.24
Scheme 2.
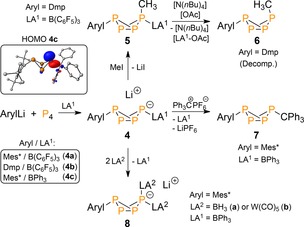
Synthesis of LA‐stabilized [RP4]− butterfly anions and subsequent substitution and transfer reactions. Mes*=2,4,6‐ tBu3C6H2, Dmp=2,6‐dimesitylphenyl.
In contrast to the strongly Lewis acidic B(C6F5)3 group in 4 a, b, the less stabilizing BPh3 in 4 c departs immediately upon endo‐cyclic substitution by BH3 or W(CO)5. In these cases the anionic [RP4]− core transfers to the stronger Lewis acids to give the doubly coordinated anions 8 a and b, respectively (Scheme 2).24 Intriguingly, the high reactivity of 4 c also grants access to OPCs containing P1 and P3 entities through unprecedented [3+1]‐fragmentation reactions using either phenylisocyanate or imidazolium chloride in the presence of an access of the P3‐trapping reagent 1,3‐cyclohexadiene (1,3‐CHD; Scheme 3).25 The reactions proceed through “P” transfer from the LA‐stabilized butterfly anions to the reagents to give spiro‐phosphoranide 9 and carbene–phosphinidene adduct 11, respectively, with concurrent trapping of the liberated diphosphene Mes*P3 by the organic diene that generates in both cases organotriphosphirane 10.
Scheme 3.
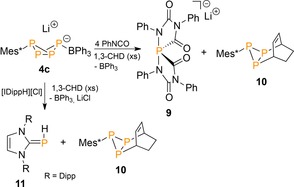
Selective [3+1]‐fragmentation reactions of LA‐stabilized RP4 − butterfly 4 c. Mes*=2,4,6‐tBu3C6H2, IDipp=1,3‐bis(2,6‐diisopropylphenyl)‐imidazol‐2‐ylidene, CHD=cyclohexadiene.
The isolation and versatility of the stabilized [RP4]− anion 4 marks a significant step toward the controlled functionalization of P4 with organolithium reagents. Interestingly, related RP4 derivatives can also be generated from P4 in the coordination sphere of a gold(I) cation, even with unencumbered MesLi,26 which is otherwise precluded due to rapid quenching with boron Lewis acids to Li[MesBAr3]. Exemplary is the reaction of ArylLi with the η2‐P4‐coordinated cationic (NHC)AuI complex 12 (Scheme 4; Aryl=Mes or Dmp, NHC=N‐heterocyclic carbene) that afforded selectively the neutral bicyclic tetraphosphanes 13, which coordinates to an additional gold cation complex with displacement of a P4 molecule to give bimetallic 14.
Scheme 4.
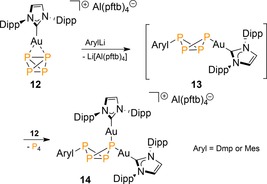
Functionalization of P4 in the coordinated sphere of a coinage metal cation. Dipp=2,6‐diisopropylphenyl, Dmp=2,6‐dimesitylphenyl, pftb=perfluoro‐tert‐butoxy.
In 2015, Hill and co‐workers reported on the selective synthesis of the Mg2+‐stabilized cluster dianion [nBu2P4]2− 15 from P4 and β‐diketiminato n‐butylmagnesium complex [(DippBDI)MgnBu] (DippBDI=HC{C(Me)N(2,6‐iPr2C6H3)}2; Scheme 5).27 The disubstituted P4‐dianion likely results from initial formation of the [nBuP4]− butterfly anion [Eq. (1)] with subsequent nucleophilic addition across the transannular P−P bond [Eq. (2)]. Product 15 reacts with P4 to the polyphosphide cluster [nBu2P8]2− (16), which is isolable due to the stabilizing bulky (DippBDI)Mg2+ centers. These results are in sharp contrast to the noted16 unselective reactions with n‐butylmagnesium bromide and demonstrates the large impact of the employed cation on the outcome of P4 substitution.

Scheme 5.
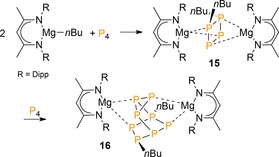
Functionalization of P4 by an n‐butylmagnesium complex. Dipp=2,6‐diisopropylphenyl.
The group of Lerner reported on the synthesis of a trisanionic [R3P4]3− derivative (17, Scheme 6).28 The complex is generated from P4 and mesityllithium in benzene over the course of four days in 60 % yield. Presumably, tetraphosphanetriide 17 results from nucleophilic attack on the common [RP4]− intermediate with simultaneous cleavage of a peripheral P−P bond, followed by a third addition [Eq. 3]. Interestingly, partial protonation of 17 led to lithium diphosphanide [Mes2P2H]− 18, which on quenching with trifluoroacetic acid gave diphosphane Mes(H)P−P(H)Mes. This process hints toward a novel route to prepare lower nuclearity phosphanes.
Scheme 6.
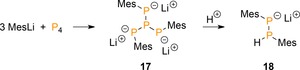
Functionalization of P4 using solvent‐free mesityllithium.
Recently, Zhang functionalized P4 with dianionic 1,4‐dilithio‐1,3‐butadienes to obtain phospholyl lithium derivative 19 in high yield (85 −99 %) and with a broad substrate scope (Scheme 7).29 Computational analysis suggested a cooperative nucleophilic attack of two Csp2−Li bonds on P4 with concurrent cleavage of two P−P bonds and release of Li[P3] to account for the formation of 19. Intriguingly, this mechanism differs from the noted stepwise routes for the direct preparation of the phosphorus anions from P4.
Scheme 7.
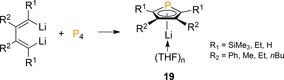
Direct preparation of phospholyl lithium derivatives from P4.
Two examples on p‐block metals complement the work on the s‐block metal organyls. In 1991, Barron reported on the formal insertion of P4 into the Ga−C bond of Ga(tBu)3 to give an endo,exo‐substituted bicyclo tetraphosphorus derivative, which coordinates an additional equivalent of gallium precursor to form the trifunctionalized P4‐butterfly 20 (Scheme 8).30 Subsequently, Power showed related reactivity for the weak thallene dimer TlArDipp2 (ArDipp2=2,6‐(2,6‐iPr2C6H3)C6H3) that yielded instead a linear diaryl‐substituted Ar2P4 chain capped by two thallium centers (21).31 This doubly reduced P4 derivative could be oxidized with iodine to the symmetrically substituted butterfly 22.
Scheme 8.
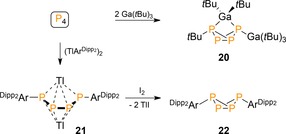
Functionalization of P4 using p‐block organometallic compounds. ArDipp2=2,6‐(2,6‐iPr2C6H3)2C6H3.
3. Functionalization of P4 Using Transition Metal Organyls
Transition metal (TM) complexes have been widely applied for the functionalization of P4,4 but examples that involve direct P−C bond formation are scarce. In 1999, Peruzzini and co‐workers reported on the reaction of rhodium alkyl or aryl ethylene complexes ([(triphos)Rh(R1)(η2‐C2H4)]; R1=Me, Et or Ph, triphos=1,1,1‐tris(diphenylphosphanylmethyl)ethane) with P4 to give the novel complexes [(triphos)Rh(η1:η2‐P4R1)] 23 (Scheme 9).32 Their formation is believed to start by release of ethylene from the rhodium precursor with subsequent oxidative addition of a P4 molecule to give [(triphos)Rh(R1)(η1:η1‐P4)] (IM1) after which migratory insertion of R1 affords the final products. The complexes are thermally labile and allowed extrusion of primary phosphanes by adding molecular hydrogen (PH2R1; albeit in low yield, <15 %) with liberation of cyclo‐P3 complex 24. The reaction illustrates an intriguing stepwise metal‐mediated protocol for the preparation of OPCs from P4. 33 Moreover, 23 (R1 =Me or Ph) reacted with MeOTf (OTf=SO3CF3) or HBF4 to undergo alkylation or protonation at the rhodium‐coordinated PR‐moiety to afford cations of the type [(triphos)Rh(η1:η2‐P4R1R2)][Y] (25, Scheme 9)34 in which the η1:η2‐P4R ligand slowly tumbles with respect to the (triphos)Rh metal center.35
Scheme 9.
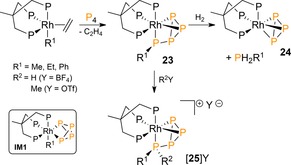
Metal‐mediated direct P−C bond formation using (triphos)rhodium alkyl and aryl ethene complexes. Triphos=1,1,1‐tris(diphenylphosphanylmethyl)ethane, Tf=SO3CF3.
Although not leading directly to P−C bonds from P4, we note that Cummins’ group reported on the use of P4‐derived terminal niobium phosphide [P≡Nb(N(Np)Ar)3]− 26 (Ar=3,5‐Me2C6H3)36 and diniobium octaphosphorus complex (P8)[Nb(OC‐[2Ad]Mes)3]2 27 (Scheme 10),37 which enabled access to a variety of organophosphanes through multistep processes. For example, a phosphalkyne (tBuC≡P) could be prepared by the reaction of 26 with pivaloyl chloride (tBuC(O)Cl) by P for (O)Cl metathesis,38, 39 and a diphosphane in a related fashion by reaction of 26 with chloroiminophosphane ClP=NMes*, which releases transient P2 fragments that are trappable by 1,3‐CHD (see Section 6).40 In addition, the niobium metal centers in 27 could be replaced by organic groups to furnish an organopolyphosphorus framework featuring a rearranged P8 core with four new P−C bonds (see reference 37 for a detailed mechanism).
Scheme 10.
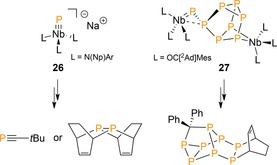
Niobium‐mediated P−C bond formation. Ar=3,5‐Me2C6H3, 2Ad=2‐adamantylidene.
4. Functionalization of P4 Using Carbenes
The ability to stabilize polyphosphorus intermediates along the P4 fragmentation pathway plays an important role in directing its functionalization, which can also be accomplished by ambiphilic carbenes. The first insights were reported by the group of Bertrand, who reacted two equivalents of a cyclic (alkyl)(amino)carbene (CAAC) with P4 to afford the E‐ and Z‐isomers of linear tetraphosphorus chain 28 (Scheme 11; only E‐isomer shown).41 The presumed transient triphosphirene (IM2) reaction intermediate could be trapped with 2,3‐dimethylbutadiene to give 29.42 Likewise, the diphosphene core of 28 underwent a [4+2]‐cycloaddition to yield 30 in which two additional P−C bonds have been introduced.
Scheme 11.
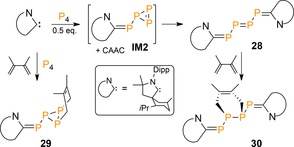
Reactivity of P4 with CAACs. Dipp=2,6‐diisopropylphenyl.
The nature of the carbene influences the fate of the P4 functionalization (Scheme 12). For example, a more electrophilic acyclic (alkyl)(amino)carbene (AAAC) generated bis(carbene) adduct 31 as the cyclopropanation reaction with the initially formed triphosphirene is more favorable compared to ring‐opening (cf. IM2, Scheme 11).43 On the other hand, a less bulky CAAC led instead to trisubstituted P4‐derivative 32 in addition to lower nuclearity P2‐diphosphaalkene 33. Moreover, the small bis(diisopropylamino)cyclopropenylidene fragmented the P4 tetrahedron even further to give P1‐cation 34. These reactions reveal modular reactivity based on both the electronic and steric properties of the carbene.
Scheme 12.
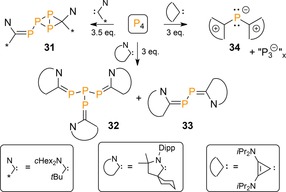
Reactivity of P4 with various carbenes. Dipp=2,6‐diisopropylphenyl.
NHCs less π‐acidic than CAACs react with P4 in a related fashion but with different outcomes. For example, treating P4 with two equivalents of 1,3‐bis(2,6‐diisopropylphenyl)‐imidazolin‐2‐ylidene (SIDipp) gave 35 (Scheme 13), featuring the tetraphosphatriene structural motif, which over time aggregated to the neutral P12 cluster 36.44 The mechanism was postulated to involve a [3+2]‐cycloaddition of 35 with triphosphirene 37 to give intermediate 38, which rearranged to heptaphosphanorbonadiene 39 with loss of two NHCs to afford the final product upon an additional [4+2]‐cycloaddition with 37. As such, the weaker P=C double bonds that are formed by NHCs over CAACs induce aggregation over fragmentation, because the former are better leaving groups.
Scheme 13.
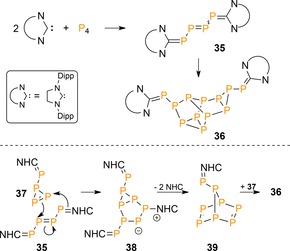
Top: reactivity of P4 with NHCs. Bottom: DFT computed mechanism for the formation of 36. NHC=1,3‐bis(2,6‐diisopropylphenyl)‐imidazolin‐2‐ylidene, Dipp=2,6‐diisopropylphenyl.
Remarkably, reacting P4 with more electrophilic NHCs that bear carbonyl functional groups in the carbon backbones, allows isolation of P8 clusters 40, which are formed by [2+2]‐cycloaddition of the linear R2P4 chains (Scheme 14).45, 46 Furthermore, a highly electrophilic benzamido carbene was shown the insert into one P−P bond to give the expanded five‐membered cage compound 41, which possibly also represents the initial product for other NHC‐induced activations.45, 49
Scheme 14.
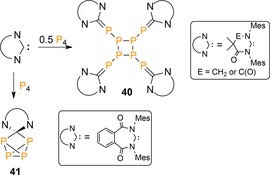
Reactivity of P4 with electrophilic NHCs.
A protocol to furnish OPCs containing P1 units was reported in a joint publication by the groups of Gudat and Grützmacher who treated P4 with equimolar amounts of imidazolium salts and KOtBu.47 The incipient carbene and the tBuOH by‐product react with P4 to generate a phosphaalkene (42, Scheme 15) in addition to a small amount of cation 43 that resembles the CAAC‐initiated fragmentation reported by Bertrand (cf. 34, Scheme 12).46
Scheme 15.
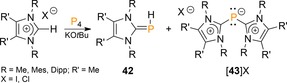
Three‐component reaction of P4 with imidazolium salts and KOtBu. Dipp=2,6‐diisopropylphenyl.
Lastly, a frustrated Lewis pair (FLP) approach, based on the use of carbene ItBu (ItBu=1,3‐bis(tert‐butyl)‐imidazol‐2‐ylidene) and B(C6F5)3, was reported by Tamm et al.48 The NHC was found to bind “abnormally” (mesoionic) in the reaction with P4 and induced a heterolytic P−P bond cleavage [Eq. (1)] to afford the labile zwitterionic exo,exo‐bicyclo[1.1.0]tetraphosphabutane 44 in the presence of the Lewis acid, similar to anion 4 reported recently by our group (Scheme 16).23, 24
Scheme 16.
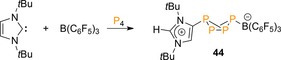
Functionalization of P4 using a carbene–borane Lewis pair.
5. Functionalization of P4 Using Carboradicals
Homolytic cleavage of the P−P bonds with concomitant P−C bond formation, all the way to monophosphanes (PR3), can be accomplished with carboradicals.3 A key challenge is to cleanly generate the radicals in the presence of P4. Illustrative is the early work by the group of Barton, who used P4 as trap in trace oxygen‐initiated radical chain reactions with Barton's PTOC ester‐derived carbon radicals (O‐acyl derivatives of N‐hydroxy‐2‐thiopyridone), which after oxidative work‐up afforded phosphonic acids in high yield (71–86 %; Scheme 17).49
Scheme 17.
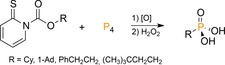
Preparation of phosphonic acids from P4 using Barton esters.
The concept was later extended by the group of Cummins, who synthesized tertiary phosphanes from P4 and radicals generated by halogen (X.) abstraction from haloalkyls or haloaryls with titanium trisanilide Ti(N(tBu)Ar)3 (Scheme 18; Ar=3,5‐Me2C6H3).50 Not only could PPh3 and PCy3 be generated, but also P(SiMe3)3 and P(SnPh3)3 by splitting the higher congener Si−X and Sn−X bonds. Like Barton's radical syntheses, the reactions involve consecutive homolytic P−P bond breaking events and proceed through multiple cyclopolyphosphorus intermediates [Eq. (4)]. This was demonstrated for the more bulky substrates DmpI and MesBr (Dmp=2,6‐dimesitylphenyl), which impede complete substitution to afford bicyclo[1.1.0]tetraphosphabutane exo,endo‐Dmp2P4 (45) and cyclotriphosphirane Mes3P3 (46), respectively. Notably, the oxidized titanium(IV) by‐product X−Ti(N(tBu)Ar)3 can be easily reduced back to the TiIII precursor with sodium amalgam, but due to the strong oxidizing properties of P4 itself (Na/Hg + P4 → Na3P) this process cannot be conducted in situ, which prevents catalytic conversion. It is also of note that electrochemical methods have been employed to furnish related OPCs from P4, which were recently outlined and discussed by Yakhvarov and Budnikova.5
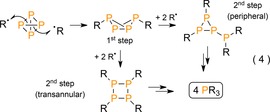
Scheme 18.
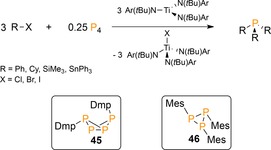
Radical synthesis of tertiary‐ and cyclopolyphosphanes from P4. Dmp=2,6‐dimesitylphenyl, Ar=3,5‐Me2C6H3.
In 2014, the potential of reacting P4 with metal‐mediated radicals was further explored by Scheer and co‐workers.51 They showed that salt elimination from CpBIGNa by CuBr afforded {CpBIG}. radicals that interacted with P4, as observed previously for the bulky {Dmp}. by Cummins,53 to give the exo,exo butterfly CpBIG 2P4 (47, Scheme 19). The presence of free carboradicals was confirmed by EPR, whereas more reactive and less bulky {CpR}. derivatives (CpR=Cp, Cp*, Cp“′ and Cp4iPr) were shown to undergo rapid decomposition, either through radical coupling or forming CpRH. They did react with P4 via an iron‐mediated route (with CpRFeBr2 acting as radical transfer agent) to bicyclic tetraphosphanes 48.
Scheme 19.
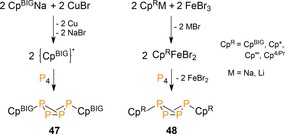
Metal‐mediated radical synthesis of organyl‐substituted P4 butterflies.
6. Functionalization of P4 through P2 Fragments
Cracking white phosphorus into two P2 units has only been explored to a limited extent. The diatomic fragment features a highly reactive P≡P triple bond that allows for Diels–Alder type chemistry. In 2010, the group of Cummins reported on photochemically52 generated P2 that was captured in situ by DA reactions with 1,3‐dienes (Scheme 20).53 The products formed after consecutive [4+2]‐cycloadditions to afford unique organodiphosphanes 49, which have been shown to coordinate to Group 10 metals54 and undergo chalcogenation and alkylation reactions to allow further functionalization of the bicyclic structures.55, 56 Whereas this photochemical protocol transfers cleanly P atoms from P4 into organic frameworks, the isolated yields are only moderate (R=H, 2 %; R=Me, 34 %) due to their lability under the harsh irradiation conditions.
Scheme 20.
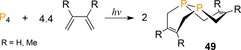
Transfer of photochemically generated P2 to1,3‐dienes.
The transfer of a P2 fragment to an organic substrate was also achieved by Mathey and co‐workers,57 who showed that upon mixing (trimethylsilyl)diazomethanide with P4, a formal [3+2]‐cycloaddition reaction occurs to form diazadiphospholide anion 50 and neutral 51 after protonation (Scheme 21). The product is reminiscent to the recently described all‐inorganic aromatic ion P2N3 −, prepared from reacting azide (N3 −) with a thermally extruded P2 unit from a transannular diphosphorus bisanthracene adduct, P2(C14H10)2.58, 59 Transient P2 may be the intermediate in the formation of 50, but an ionic mechanism related to that observed for the dianions reported by Zhang is also feasible (see Section 2).29
Scheme 21.
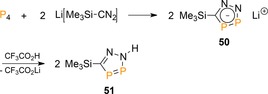
Reaction of P4 with (trimethylsilyl)diazomethanide.
7. Summary and Outlook
The functionalization of P4 through direct P−C bond formation represents a versatile approach for the synthesis of OPCs, and shows potential to circumvent the current use of phosphorus halides. Forming the desired P−C bonds can be accomplished by a number of methods, involving lithium organyls, organometallic complexes, carbenes, carboradicals, and trapping of P2 fragments with dienes. The product scope is varied and includes both industrially relevant phosphanes as well as unique OPCs that are essentially inaccessible through the use of PCl3. These can serve as building blocks to access intriguing additional P‐compound classes, like observed for the LA‐stabilized [RP4]− anions 4 and the R2P4 chains of the type 28, or can be studied as ligands for coordination chemistry as displayed in P4‐butterflies 14 and 20, and explored for organodiphosphanes 49.
The chemistry surveyed reveals substantial progress in controlling P4 functionalization and represents an encouraging entry point for further development. To translate the attained fundamental insights to practical substitution reactions using readily available reagents seems imperative. Exemplary are the protocols reported by Zhang, and Gudat and Grützmacher, using dilithiobutadienes or imidazolium chlorides, respectively, to directly produce phospholide anions 19 and carbene–phosphinidene adducts 42. Also the design of catalytic procedures to facilitate P−C bonding is important. Cummins and Scheer showed Ti‐ and Fe‐mediated radical processes to be potential platforms, and Peruzzini laid the foundation for a rhodium‐assisted cycle. In this regard, photochemistry proves to be an equally promising tool to exploit the underdeveloped P4 → 2 P2 fragmentation. While achieving these goals is ambitious and will require considerable effort, the recent advancements in this field are significant and continue to enable new avenues to be explored, which will hopefully spur the construction of a wealth of valuable new phosphorus products.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Dr. Jaap E. Borger was born in Wageningen, The Netherlands, in 1987 and obtained his B.Sc. in chemistry at the University of Applied Sciences Utrecht in 2009. After working as research associate for Schering‐Plough in Oss (currently MSD), he pursued an M.Sc. in chemistry at the VU University Amsterdam, where he graduated cum laude in 2013. Recently, he completed his doctoral studies under the supervision of Prof. Koop Lammertsma, which focused on the controlled and direct conversion of white phosphorus into organophosphorus compounds. He is now working as a postdoctoral fellow in the group of Prof. Hansjörg Grützmacher at ETH Zürich in Switzerland.

Biographical Information
Dr. A. W. Ehlers obtained his PhD at the Phillipps University of Marburg, Germany on ab initio calculations of Transition Metal‐Ligand bond interactions. He joined the VU University in Amsterdam as Marie Curie Fellow and later as Assistant Professor to study organometallic and main group chemistry for asymmetric homogenous catalysis by DFT. In 2016, he was also appointed as a Visiting Associate Professor in the Department of Chemistry at the University of Johannesburg. Finally, he accepted a position at the University of Amsterdam to study the activity of catalysts in sustainable processes theoretically and by NMR.

Biographical Information
Dr. J. Chris Slootweg obtained his PhD degree at the Vrije Universiteit Amsterdam in 2005. As a post‐doctoral researcher, he studied C−H activation at the ETH Zürich, for which he received a TALENT stipend from NWO. In 2006, he returned as an Assistant Professor to the VU, where he coordinated the Marie Curie Initial Training Network SusPhos on sustainable phosphorus chemistry. In 2013, he received a NWO VIDI grant on main‐group chemistry and catalysis. He was promoted to Associate Professor in May 2014, and moved to the University of Amsterdam in November 2016 to continue exploring his interests in sustainable chemistry.

Biographical Information
Prof. Koop Lammertsma (born in 1949 in Makkum/the Netherlands) was educated at the Universities of Groningen (1974) and Amsterdam (Ph.D. 1979). After postdoctoral work with F. Sondheimer (London), P. v. R. Schleyer (Erlangen‐Nürnberg), and Nobel laureate G. A. Olah (USC) he moved in 1983 to the University of Alabama at Birmingham, USA, to become Full Professor in 1992. In 1996 he moved to the Vrije Universiteit Amsterdam, The Netherlands. Since 2015, he holds a Distinguished Visiting Professor position at the University of Johannesburg, South Africa. His physical organic chemistry has increasingly focused on computationally supported phosphorus chemistry.

Acknowledgements
This work was supported by the Council for Chemical Sciences of the Netherlands Organization for Scientific Research (NWO/CW).
J. E. Borger, A. W. Ehlers, J. C. Slootweg, K. Lammertsma, Chem. Eur. J. 2017, 23, 11738.
References
- 1. Corbridge D. E. C., Phosphorus 2000, Elsevier, Amsterdam, 2000. [Google Scholar]
- 2.Hydrolysis of P4 with NaOH leads to NaH2PO2 and PH3, both of which can also be used to produce phosphorus compounds.
- 3.For reviews on the activation of P4 by main group compounds, see:
- 3a. Scheer M., Balázs G., Seitz A., Chem. Rev. 2010, 110, 4236–4256; [DOI] [PubMed] [Google Scholar]
- 3b. Giffin N. A., Masuda J. D., Coord. Chem. Rev. 2011, 255, 1342–1359. [Google Scholar]
- 4.For reviews on the transition metal-mediated activation of P4, see:
- 4a. Peruzzini M., Gonsalvi L., Romerosa A., Chem. Soc. Rev. 2005, 34, 1038–1047; [DOI] [PubMed] [Google Scholar]
- 4b. Caporali M., Gonsalvi L., Rossin A., Peruzzini M., Chem. Rev. 2010, 110, 4178–4235; [DOI] [PubMed] [Google Scholar]
- 4c. Cossairt B. M., Piro N. A., Cummins C. C., Chem. Rev. 2010, 110, 4164–4177. [DOI] [PubMed] [Google Scholar]
- 5.For reviews on electrocatalytic functionalization of P4, see:
- 5a. Budnikova Y. H., Yakhvarov D. G., Sinyashin O. G., J. Organomet. Chem. 2005, 690, 2416–2425; [Google Scholar]
- 5b. Yakhvarov D. G., Gorbachuk E. V., Sinyashin O. G., Eur. J. Inorg. Chem. 2013, 4709–4726. [Google Scholar]
- 6.For example, PPh3 is produced on an industrial scale by reacting PCl3 with chlorobenzene and sodium. See Ref. [1].
- 7.For a related review, see: Caporali M., Serrano-Ruiz M., Peruzzini M., in Chemistry Beyond Chlorine (Eds.: P. Tundo, L.-N. He, E. Lokteva, C. Mota), Springer International Publishing, Cham, 2016, pp. 97–136. [Google Scholar]
- 8. Fluck E., Pavlidou C. M. E., Janoschek R., Phosphorus Sulfur Relat. Elem. 1979, 6, 469–474. [Google Scholar]
- 9. Cossairt B. M., Cummins C. C., Head A. R., Lichtenberger D. L., Berger R. J. F., Hayes S. A., Mitzel N. W., Wu G., J. Am. Chem. Soc. 2010, 132, 8459–8465. [DOI] [PubMed] [Google Scholar]
- 10. Tsirelson V. G., Tarasova N. P., Bobrov M. F., Smetannikov Y. V., Heteroat. Chem. 2006, 17, 572–578. [Google Scholar]
- 11. Hirsch A., Chen Z., Jiao H., Angew. Chem. Int. Ed. 2001, 40, 2834–2838; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 2916–2920. [Google Scholar]
- 12. Cossairt B. M., Cummins C. C., J. Am. Chem. Soc. 2009, 131, 15501–15511. [DOI] [PubMed] [Google Scholar]
- 13. Bock H., Mueller H., Inorg. Chem. 1984, 23, 4365–4368. [Google Scholar]
- 14. Wang L.-P., Tofan D., Chen J., Voorhis T. V., Cummins C. C., RSC Adv. 2013, 3, 23166–23171. [Google Scholar]
- 15. Rathenau G., Physica 1937, 4, 503–514. [Google Scholar]
- 16.
- 16a. Rauhut M. M., Semsel A. M., J. Org. Chem. 1963, 28, 471–473; [Google Scholar]
- 16b. Rauhut M. M., Semsel A. M., J. Org. Chem. 1963, 28, 473–477. [Google Scholar]
- 17.Similar results were obtained by Yakhvarov and co-workers using Grignard reagents in the presence of ZnBr2, see: Yakhvarov D. G., Ganushevich Y. S., Sinyashin O. G., Mendeleev Commun. 2007, 17, 197–198. [Google Scholar]
- 18. Trofimov B. A., Brandsma L., Arbuzova S. N., Gusarova N. K., Russ. Chem. Bull. 1997, 46, 849–850. [Google Scholar]
- 19.
- 19a. Fritz G., Härer J., Stoll K., Z. Anorg. Allg. Chem. 1983, 504, 47–54; [Google Scholar]
- 19b. Fritz G., Härer J., Z. Anorg. Allg. Chem. 1983, 504, 23–37. [Google Scholar]
- 20.The reaction of potassium cyanide with P4 also proved to be more selective, giving K[P(CN)2] and K[P15] as major products, see: Schmidpeter A., Burget G., Zwaschka F., Sheldrick W. S., Z. Anorg. Allg. Chem. 1985, 527, 17–32; Cummins reported on related reactivity using Na[SnPh3], see: [Google Scholar]; Cummins C. C., Huang C., Miller T. J., Reintinger M. W., Stauber J. M., Tannou I., Tofan D., Toubaei A., Velian A., Wu G., Inorg. Chem. 2014, 53, 3678–3687. [DOI] [PubMed] [Google Scholar]
- 21. Riedel R., Hausen H.-D., Fluck E., Angew. Chem. Int. Ed. Engl. 1985, 24, 1056–1057; [Google Scholar]; Angew. Chem. 1985, 97, 1050–1050. [Google Scholar]
- 22.In 1988, Baudler et al. detected the anion [HP4]− at low temperature by 31P NMR spectroscopy, after reduction of P4 with Na K− naphthalenide, see:
- 22a. Baudler M., Adamek C., Opiela S., Budzikiewicz H., Ouzounis D., Angew. Chem. Int. Ed. Engl. 1988, 27, 1059–1061; [Google Scholar]; Angew. Chem. 1988, 100, 1110–1111; [HP4]− was recently also detected by Mézailles using borohydrides, see: [Google Scholar]
- 22b. Bhattacharyya K. X., Dreyfuss S., Saffon-Merceron N., Mézailles N., Chem. Commun. 2016, 52, 5179–5182. [DOI] [PubMed] [Google Scholar]
- 23. Borger J. E., Ehlers A. W., Lutz M., Slootweg J. C., Lammertsma K., Angew. Chem. Int. Ed. 2014, 53, 12836–12839; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13050–13053. [Google Scholar]
- 24. Borger J. E., Ehlers A. W., Lutz M., Slootweg J. C., Lammertsma K., Angew. Chem. Int. Ed. 2016, 55, 613–617; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 623–627. [Google Scholar]
- 25. Borger J. E., Ehlers A. W., Lutz M., Slootweg J. C., Lammertsma K., Angew. Chem. Int. Ed. 2017, 56, 285–290; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 291–296. [Google Scholar]
- 26. Borger J. E., Bakker M. S., Ehlers A. W., Lutz M., Slootweg J. C., Lammertsma K., Chem. Commun. 2016, 52, 3284–3287. [DOI] [PubMed] [Google Scholar]
- 27. Arrowsmith M., Hill M. S., Johnson A. L., Kociok-Köhn G., Mahon M. F., Angew. Chem. Int. Ed. 2015, 54, 7882–7885; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 7993–7996. [Google Scholar]
- 28. Hübner A., Bernert T., Sänger I., Alig E., Bolte M., Fink L., Wagner M., Lerner H.-W., Dalton Trans. 2010, 39, 7528–7533. [DOI] [PubMed] [Google Scholar]
- 29. Xu L., Chi Y., Du S., Zhang W.-X., Xi Z., Angew. Chem. Int. Ed. 2016, 55, 9187–9190; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 9333–9336. [Google Scholar]
- 30. Power M. B., Barron A. R., Angew. Chem. Int. Ed. Engl. 1991, 30, 1353–1354; [Google Scholar]; Angew. Chem. 1991, 103, 1403–1404. [Google Scholar]
- 31. Fox A. R., Wright R. J., Rivard E., Power P. P., Angew. Chem. Int. Ed. 2005, 44, 7729–7733; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7907–7911. [Google Scholar]
- 32. Barbaro P., Peruzzini M., Ramirez J. A., Vizza F., Organometallics 1999, 18, 4237–4240. [Google Scholar]
- 33.This reactivity could also be achieved by using rhodium or iridium hydrides [(triphos)MH3], but giving PH3 instead of PH2R, see: Peruzzini M., Ramirez J. A., Vizza F., Angew. Chem. Int. Ed. 1998, 37, 2255–2257; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 2376–2378. [Google Scholar]
- 34. Barbaro P., Ienco A., Mealli C., Peruzzini M., Scherer O. J., Schmitt G., Vizza F., Wolmershäuser G., Chem. Eur. J. 2003, 9, 5195–5210. [DOI] [PubMed] [Google Scholar]
- 35. Barbaro P., Caporali M., Ienco A., Mealli C., Peruzzini M., Vizza F., Eur. J. Inorg. Chem. 2008, 1392–1399. [Google Scholar]
- 36.
- 36a. Figueroa J. S., Cummins C. C., J. Am. Chem. Soc. 2003, 125, 4020–4021; [DOI] [PubMed] [Google Scholar]
- 36b. Figueroa J. S., Cummins C. C., Angew. Chem. Int. Ed. 2004, 43, 984–988; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 1002–1006. [Google Scholar]
- 37.
- 37a. Cossairt B. M., Cummins C. C., Angew. Chem. Int. Ed. 2008, 47, 8863–8866; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2008, 120, 8995–8998; [Google Scholar]
- 37b. Cossairt B. M., Cummins C. C., Inorg. Chem. 2008, 47, 9363–9371. [DOI] [PubMed] [Google Scholar]
- 38. Figueroa J. S., Cummins C. C., J. Am. Chem. Soc. 2004, 126, 13916–13917. [DOI] [PubMed] [Google Scholar]
- 39.Similarly, the reaction of 26 with CO2 yields the phosphaethynolate anion, see: Krummenacher I., Cummins C. C., Polyhedron 2012, 32, 10–13. [Google Scholar]
- 40. Piro N. A., Figueroa J. S., McKellar J. T., Cummins C. C., Science 2006, 313, 1276–1279. [DOI] [PubMed] [Google Scholar]
- 41. Masuda J. D., Schoeller W. W., Donnadieu B., Bertrand G., Angew. Chem. Int. Ed. 2007, 46, 7052–7055; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 7182–7185. [Google Scholar]
- 42.Evaluated also computationally, see: Damrauer R., Pusede S. E., Staton G. M., Organometallics 2008, 27, 3399–3402. [Google Scholar]
- 43. Back O., Kuchenbeiser G., Donnadieu B., Bertrand G., Angew. Chem. Int. Ed. 2009, 48, 5530–5533; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5638–5641. [Google Scholar]
- 44. Masuda J. D., Schoeller W. W., Donnadieu B., Bertrand G., J. Am. Chem. Soc. 2007, 129, 14180–14181. [DOI] [PubMed] [Google Scholar]
- 45. Dorsey C. L., Squires B. M., Hudnall T. W., Angew. Chem. Int. Ed. 2013, 52, 4462–4465; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4558–4561. [Google Scholar]
- 46. Martin C. D., Weinstein C. M., Moore C. E., Rheingold A. L., Bertrand G., Chem. Commun. 2013, 49, 4486–4488. [DOI] [PubMed] [Google Scholar]
- 47. Cicač-Hudi M., Bender J., Schlindwein S. H., Bispinghoff M., Nieger M., Grützmacher H., Gudat D., Eur. J. Inorg. Chem. 2016, 649–658. [Google Scholar]
- 48. Holschumacher D., Bannenberg T., Ibrom K., Daniliuc C. G., Jones P. G., Tamm M., Dalton Trans. 2010, 39, 10590–10592. [DOI] [PubMed] [Google Scholar]
- 49.
- 49a. Barton D. H. R., Zhu J., J. Am. Chem. Soc. 1993, 115, 2071–2072; [Google Scholar]
- 49b. Barton D. H. R., Vonder Embse R. A., Tetrahedron 1998, 54, 12475–12496. [Google Scholar]
- 50. Cossairt B. M., Cummins C. C., New J. Chem. 2010, 34, 1533–1536. [Google Scholar]
- 51. Heinl S., Reisinger S., Schwarzmaier C., Bodensteiner M., Scheer M., Angew. Chem. Int. Ed. 2014, 53, 7639–7642; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7769–7773. [Google Scholar]
- 52.For other examples on P4 functionalization using electromagnetic radiation, see: Serrano-Ruiz M., Romerosa A., Lorenzo-Luis P., Eur. J. Inorg. Chem. 2014, 1587–1598. [Google Scholar]
- 53. Tofan D., Cummins C. C., Angew. Chem. Int. Ed. 2010, 49, 7516–7518; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 7678–7680. [Google Scholar]
- 54. Tofan D., Cummins C. C., Chem. Sci. 2012, 3, 2474–2478. [Google Scholar]
- 55. Tofan D., Temprado M., Majumdar S., Hoff C. D., Cummins C. C., Inorg. Chem. 2013, 52, 8851–8864. [DOI] [PubMed] [Google Scholar]
- 56. Knopf I., Tofan D., Beetstra D., Al-Nezari A., Al-Bahily K., Cummins C. C., Chem. Sci. 2017, 8, 1463–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Charrier C., Maigrot N., Ricard L., Floch P. L., Mathey F., Angew. Chem. Int. Ed. Engl. 1996, 35, 2133–2134; [Google Scholar]; Angew. Chem. 1996, 108, 2282–2283. [Google Scholar]
- 58. Velian A., Nava M., Temprado M., Zhou Y., Field R. W., Cummins C. C., J. Am. Chem. Soc. 2014, 136, 13586–13589. [DOI] [PubMed] [Google Scholar]
- 59. Velian A., Cummins C. C., Science 2015, 348, 1001–1004. [DOI] [PubMed] [Google Scholar]