

Abstract
Aims
The MARLINA‐T2D study (ClinicalTrials.gov, NCT01792518) was designed to investigate the glycaemic and renal effects of linagliptin added to standard‐of‐care in individuals with type 2 diabetes and albuminuria.
Methods
A total of 360 individuals with type 2 diabetes, HbA1c 6.5% to 10.0% (48–86 mmol/mol), estimated glomerular filtration rate (eGFR) ≥30 mL/min/1.73 m2 and urinary albumin‐to‐creatinine ratio (UACR) 30–3000 mg/g despite single agent renin‐angiotensin‐system blockade were randomized to double‐blind linagliptin (n = 182) or placebo (n = 178) for 24 weeks. The primary and key secondary endpoints were change from baseline in HbA1c at week 24 and time‐weighted average of percentage change from baseline in UACR over 24 weeks, respectively.
Results
Baseline mean HbA1c and geometric mean (gMean) UACR were 7.8% ± 0.9% (62.2 ± 9.6 mmol/mol) and 126 mg/g, respectively; 73.7% and 20.3% of participants had microalbuminuria or macroalbuminuria, respectively. After 24 weeks, the placebo‐adjusted mean change in HbA1c from baseline was −0.60% (−6.6 mmol/mol) (95% confidence interval [CI], −0.78 to −0.43 [−8.5 to −4.7 mmol/mol]; P < .0001). The placebo‐adjusted gMean for time‐weighted average of percentage change in UACR from baseline was −6.0% (95% CI, −15.0 to 3.0; P = .1954). The adverse‐event profile, including renal safety and change in eGFR, was similar between the linagliptin and placebo groups.
Conclusions
In individuals at early stages of diabetic kidney disease, linagliptin significantly improved glycaemic control but did not significantly lower albuminuria. There was no significant change in placebo‐adjusted eGFR. Detection of clinically relevant renal effects of linagliptin may require longer treatment, as its main experimental effects in animal studies have been to reduce interstitial fibrosis rather than alter glomerular haemodynamics.
Keywords: antidiabetic drug, clinical trial, diabetic nephropathy, DPP‐IV inhibitor, glycaemic control, linagliptin
1. INTRODUCTION
Approximately 35% to 40% of individuals with type 2 diabetes also have chronic kidney disease (CKD),1, 2 defined as albuminuria and/or reduced glomerular filtration rate (GFR). These individuals account for most of the excess risk of premature death seen in the overall type 2 diabetes population.3 Moreover, each renal marker independently predicts the risk of CKD progression, as well as adverse cardiovascular outcomes.4, 5, 6, 7, 8, 9, 10 The current standard of care for individuals with type 2 diabetes and CKD includes individualized glycaemic control and single agent renin‐angiotensin‐aldosterone system (RAAS) blockade with either angiotensin‐converting enzyme (ACE) inhibitors or angiotensin II receptor blockers (ARBs).11, 12 However, individuals with residual albuminuria despite treatment with RAAS blockers still remain at substantial risk for cardio‐renal morbidity and mortality.13 This high residual risk is driving the search for novel therapies to treat diabetic kidney disease.
Dipeptidyl peptidase‐4 (DPP‐4) inhibitors are now embedded in the therapeutic armamentarium as suitable options for managing hyperglycaemia in individuals with type 2 diabetes across the full range of CKD stages.14 DPP‐4 is expressed in many tissues and organs, with the highest levels found in the kidney.15 Several preclinical studies have suggested that targeting kidney DPP‐4 with the high‐affinity inhibitor linagliptin may have direct (ie, non‐glycaemic) renoprotective effects.16, 17, 18, 19 The hypothesis of a direct renal effect of linagliptin was further supported by a pooled analysis of 4 pivotal phase 3 clinical trials. Herein, treatment with linagliptin for 24 weeks was associated with a statistically significant and clinically relevant 28% reduction in albuminuria compared with placebo in type 2 diabetes patients with renal dysfunction who were already receiving ACE inhibitors or ARBs; this effect appeared to be independent of the concomitant improvements in glycaemic control.20 Furthermore, a pooled analysis of 13 randomized clinical trials found that linagliptin treatment of up to 12 to 76 weeks was associated with a statistically significant and clinically relevant 16% reduction in the risk of progression of CKD.21 Based on these observations, 2 independent hypotheses were advanced: first, linagliptin may acutely reduce glomerular damage, thus reducing prevalent albuminuria; second, linagliptin may slow the progression of CKD over the long term.
Two prospective, randomized, controlled studies have been initiated to evaluate these hypotheses: the Efficacy, Safety and Modification of Albuminuria in Type 2 Diabetes Subjects with Renal Disease with LINAgliptin (MARLINA‐T2D) study to investigate potential short‐term albuminuria‐lowering effects of linagliptin; and the CArdiovascular and Renal Microvascular OutcomE Study With LINAgliptin in Patients With Type 2 Diabetes Mellitus (CARMELINA) study to evaluate putative long‐term effects of linagliptin on slowing progression of CKD. MARLINA‐T2D was designed to investigate the glycaemic and renal effects of linagliptin in individuals with type 2 diabetes and residual albuminuria despite single RAAS blockade. Here, the main findings from this study are reported.
2. METHODS
2.1. Study design and participants
The design and methodology of MARLINA‐T2D has been previously reported in detail.22 MARLINA‐T2D was a 24‐week, randomized, double‐blind, placebo‐controlled, phase 3b clinical trial conducted at approximately 80 clinical centres in 12 countries: Canada, Denmark, Finland, France, Germany, Japan, the Philippines, South Korea, Spain, Taiwan, the USA and Vietnam (ClinicalTrials.gov, number NCT01792518). The study protocol was approved by independent ethics committees (IECs)/institutional review boards (IRBs) at each participating centre, and the study was conducted according to the principles of the Declaration of Helsinki and the ICH Harmonised Tripartite Guidelines for Good Clinical Practice.
Eligible individuals were aged 18–80 years with type 2 diabetes, glycated haemoglobin (HbA1c) 6.5% to 10.0% (48‐86 mmol/mol), body‐mass index (BMI) ≤40 kg/m2 and estimated glomerular filtration rate (eGFR) ≥30 mL/min/1.73 m2, based on the Modification of Diet in Renal Disease (MDRD) study equation. To participate in the study, individuals were also required to be either treatment‐naive or receiving ≤2 oral glucose‐lowering drugs (metformin, sulphonylureas, meglitinides or alpha‐glucosidase inhibitors) and/or basal insulin.
To meet the criteria for renal dysfunction, individuals had to have a urinary albumin‐to‐creatinine ratio (UACR) between 30 and 3000 mg/g, or albuminuria >30 mg/L of urine or >30 μg/min clearly documented in the previous 12 months or detected at screening; albuminuria had then to be confirmed with a geometric mean (gMean) UACR value between 30 and 3000 mg/g from 3 consecutive first‐void morning urine samples collected 14 to 16 days before randomization. In addition, each individual was required to be receiving a stable dose of an ACE inhibitor or an ARB but not both (dual or triple blockade of the RAAS was not permitted); additional antihypertensive agents other than RAAS inhibitors were permitted. All antihypertensive agents had to have been administered at the same dose for at least the 10 preceding weeks.
The main exclusion criteria were fasting blood glucose >240 mg/dL (>13.3 mmol/L), history of non‐diabetic kidney disease, renal transplant, presence of urinary tract infection, mean arterial blood pressure >110 mm Hg and/or a cardiovascular event within the previous 3 months; full exclusion criteria have been reported previously.22 All individuals provided written informed consent prior to participation.
2.2. Procedures and endpoints
Following a 2‐week placebo run‐in period, eligible individuals were randomized 1:1 to receive double‐blind, once‐daily oral treatment with linagliptin 5 mg or placebo for 24 weeks. Randomization was stratified by HbA1c value at screening (<8.5% vs ≥8.5% [<69 mmol/mol vs ≥69 mmol/mol]) and gMean of the UACR values measured on the 3 consecutive days leading into the start of placebo run‐in (<300 mg/g vs ≥300 mg/g) and used a block size of 4. The computer‐generated randomization sequence was generated by the study sponsor, and was concealed using a central interactive voice and web response system. Throughout the study, UACR was measured at each visit from 3 morning samples that had been collected on the day of the visit and the 2 preceding days.
The primary efficacy endpoint was change in HbA1c from baseline to week 24, where baseline was defined as the last observation prior to administration of the randomized study drug. The key secondary efficacy endpoint was the time‐weighted average of percentage change from baseline in UACR over the 24 weeks of treatment, where baseline was the gMean of the UACR generated from up to 6 individual measurements at the start and end of the placebo run‐in period. For each of the time points where UACR was assessed during the treatment period, the percentage change from baseline was calculated. The area under the curve (AUC) was then computed for each participant using a trapeze formula by summing the ratios (gMean of UACR measures from each visit/gMean of UACR measures from previous visit) over all days (from first dose of study drug until the scheduled visit date) and dividing by the number of days on treatment at the visit date. AUC per participant was then normalized to 1 day.
Following amendment of the study protocol, the secondary safety endpoint was designated as change from baseline in eGFR after 24 weeks of treatment, as assessed by the Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) cystatin C equation. This amendment was approved by all IECs/IRBs and was made prior to study completion and unblinding. Additional safety endpoints included the incidence and intensity of reported adverse events, which were categorized using the Medical Dictionary for Regulatory Activities (MedDRA), version 18.1. Investigator‐reported hypoglycaemia was defined as an episode of documented blood glucose ≤70 mg/dL (≤3.9 mmol/L) or an episode requiring another person to administer carbohydrate, glucagon or other resuscitative assistance irrespective of a glucose measurement (severe hypoglycaemia). Suspected cardiovascular events were adjudicated by a blinded, external clinical event committee (CEC) comprised of academic cardiologists and neurologists. Similarly, suspected pancreatitis was adjudicated by an external expert CEC. Clinical laboratory measurements were performed by a central laboratory (Quintiles Laboratories).
2.3. Statistical analyses
The required sample size (350 participants, 175 per treatment arm) was calculated as previously described,22 and was intended to provide 99% power to detect a significant difference (α = 0.05, two‐sided), assuming a 0.6% (6.6 mmol/mol) difference, in change in HbA1c from baseline after 24 weeks between treatment groups and 87% power to detect a treatment ratio of 0.79 in the ratio of UACR change from baseline.22 The primary and key secondary endpoints were tested in a hierarchical manner. The primary glycaemic endpoint was analysed with a mixed‐effects model for repeated measures (MMRM) using observed cases, excluding values after glycaemic rescue medication (OC) in the full analysis set (FAS) (all randomized participants who received at least 1 dose of study drug, underwent baseline HbA1c and UACR measurements and at least 1 on‐treatment HbA1c or UACR measurement). The MMRM included baseline HbA1c, baseline log10 (UACR), baseline HbA1c by visit and baseline log10 (UACR) by visit as linear covariates and treatment, visit and visit by treatment interaction as fixed classification effects. Similar MMRM models were fitted for subgroup analyses (Table S1). The percentage of participants with baseline HbA1c ≥7.0% who achieved HbA1c <7.0% at week 24 was analysed post hoc using a logistic regression model in which treatment was a factor and continuous baseline HbA1c and continuous baseline log10 (UACR) were covariates.
The key secondary endpoint was evaluated using an analysis of covariance (ANCOVA) of data from the FAS, with baseline HbA1c and baseline log10 (UACR) as linear covariates and treatment as a fixed classification effect; the last‐observation‐carried‐forward (LOCF) approach was used to replace missing data (including values obtained after glycaemic rescue therapy was started). Because of their non‐normal distribution, UACR data were log10‐transformed prior to ANCOVA analyses. Similar ANCOVA models were performed for subgroup analyses, including a post hoc analysis by background therapy at baseline (ACE inhibitors or ARBs) (Table S2).
The odds of achieving a clinically relevant UACR response with linagliptin compared with placebo at week 24 were analysed post hoc using a logistic regression model. In this analysis, the linagliptin and placebo groups were compared for the proportion of participants with a UACR response at week 24, defined as a reduction in UACR of >20% at week 24 relative to baseline, vs those with no UACR response, defined as no change or an increase in UACR at week 24 compared to baseline. This analysis was performed on the FAS and included eligible participants with a UACR value at week 24, irrespective of introduction of glycaemic rescue therapy (OC‐ROC). The logistic regression model contained treatment as a factor and continuous baseline HbA1c and continuous baseline log10 (UACR) as covariates.
Safety analyses were generally performed using descriptive summaries of adverse events in the treated set (all randomized participants who received at least 1 dose of study drug). Change from baseline in eGFR (CKD‐EPI, cystatin C) in the treated set was analysed using the MMRM for the primary endpoint, with baseline eGFR and baseline eGFR by visit as additional terms.
3. RESULTS
A total of 360 participants were randomized to linagliptin (n = 182) or placebo (n = 178) and comprised the treated set. The FAS consisted of 180 participants in the linagliptin‐treated group and 174 in the placebo‐treated group. Participant disposition is shown in Figure S1.
Baseline demographic and clinical characteristics were generally similar between treatment groups (Table 1). Overall, participants had a mean ± standard deviation (SD) age of 60.6 ± 9.6 years, BMI of 28.4 ± 4.9 kg/m2 and HbA1c of 7.8% ± 0.9% (62.2 ± 9.6 mmol/mol); 63.6% were male, and most were Asian (66.4%) or White (30.3%). Most participants (73.7%) had microalbuminuria (UACR 30‐300 mg/g) and preserved kidney function (eGFR ≥60 mL/min/1.73 m2) (80.0%). All participants were receiving background therapy with either an ACE inhibitor (33.3%) or an ARB (66.7%).
Table 1.
Baseline demographic and clinical characteristics in the treated set
| Linagliptin (n = 182) | Placebo (n = 178) | |
|---|---|---|
| Age, years | 61.0 ± 10.0 | 60.1 ± 9.3 |
| Male, n (%) | 116 (63.7) | 113 (63.5) |
| Race, n (%) | ||
| Asian | 117 (64.3) | 122 (68.5) |
| White | 56 (30.8) | 53 (29.8) |
| Black/African‐American | 8 (4.4) | 3 (1.7) |
| Hawaiian/Pacific Islander | 1 (0.5) | 0 (0.0) |
| BMI, kg/m2 | 28.3 ± 4.8 | 28.6 ± 4.9 |
| Weight, kg | 78.1 ± 18.6 | 77.9 ± 19.3 |
| HbA1ca, % (mmol/mol) | 7.82 ± 0.87 (62.0 ± 9.5) | 7.86 ± 0.89 (62.5 ± 9.7) |
| Time since diagnosis of diabetesa, n (%) | ||
| ≤1 year | 11 (6.1) | 7 (4.0) |
| >1 to 5 years | 25 (13.9) | 40 (23.0) |
| >5 to 10 years | 47 (26.1) | 56 (32.2) |
| >10 years | 97 (53.9) | 71 (40.8) |
| Systolic blood pressurea, mm Hg | 135.2 ± 13.9 | 134.4 ± 13.8 |
| Diastolic blood pressurea, mm Hg | 77.3 ± 9.1 | 78.5 ± 8.3 |
| eGFR (MDRD), mL/min/1.73 m2 | 75.4 ± 23.9 | 72.4 ± 24.4 |
| eGFR (CKD–EPI, cystatin C), mL/min/1.73 m2 | 102.8 ± 49.7 | 94.4 ± 43.3 |
| eGFR (CKD–EPI, cystatin C), mL/min/1.73 m2, n (%) | ||
| ≥90 | 98 (53.8) | 88 (49.4) |
| 60 to <90 | 54 (29.7) | 48 (27.0) |
| 30 to <60 | 25 (13.7) | 39 (21.9) |
| <30 | 5 (2.7) | 3 (1.7) |
| UACRa, mg/g, gMean ± gCV | 120.8 ± 152.9 | 131.9 ± 166.6 |
| UACRa, mg/g, n (%) | ||
| <30 | 11 (6.1)b | 10 (5.7)b |
| 30 to <300 | 134 (74.4) | 127 (73.0) |
| ≥300 | 35 (19.4) | 37 (21.3) |
| Oral antidiabetes monotherapya, n (%) | 64 (35.6) | 65 (37.4) |
| Metformin | 59 (32.8) | 62 (35.6) |
| Oral antidiabetes combination therapy without insulina, n (%) | 42 (23.3) | 47 (27.0) |
| Insulina, n (%) | 64 (35.6) | 48 (27.6) |
| Statins | 109 (59.9) | 107 (60.1) |
| Antihypertensive therapy, n (%) | 182 (100.0) | 178 (100.0) |
| ARBs | 120 (65.9) | 120 (67.4) |
| Calcium antagonists | 79 (43.4) | 88 (49.4) |
| ACE inhibitors | 62 (34.1) | 58 (32.6) |
| Diuretics | 52 (28.6) | 54 (30.3) |
| β‐blockers | 40 (22.0) | 47 (26.4) |
| Other | 11 (6.0) | 15 (8.4) |
Abbreviations: ACE, angiotensin‐converting enzyme; ARBs, angiotensin II receptor blockers; BMI, body mass index; CKD‐EPI, Chronic Kidney Disease Epidemiology Collaboration; eGFR, estimated glomerular filtration rate; FAS, full analysis set; gCV, geometric coefficient of variation; gMean, geometric mean; HbA1c, glycated haemoglobin; MDRD, modification of diet in renal disease; SD, standard deviation; UACR, urinary albumin‐to‐creatinine ratio.
Data are presented as mean ± SD unless otherwise stated.
FAS (linagliptin, n = 180; placebo, n = 174).
Baseline UACR values were defined as the gMean of samples taken on 3 consecutive days immediately before the placebo run‐in and the day before randomization; these patients were eligible based on having UACR >30 mg/g at screening and gMean UACR >30 mg/g for samples taken on 3 consecutive days immediately before the placebo run‐in.
3.1. Efficacy
The adjusted mean ± standard error (SE) difference between linagliptin and placebo in change from baseline in HbA1c after 24 weeks was −0.60% (95% confidence interval [CI], −0.78 to −0.43 [−6.6 mmol/mol, 95% CI, −8.5 to −4.7]; P < .0001) (Figure 1A). The reduction in HbA1c from baseline was consistently larger in the linagliptin group than in the placebo group over time (Figure 1B) and across subgroups (Figure 1C, Figure S2). For participants with HbA1c ≥7.0% at baseline, HbA1c <7.0% at week 24 was achieved by significantly more individuals in the linagliptin group than in the placebo group: 36.2% and 9.3%, respectively (odds ratio, 6.16 [95% CI, 3.13 to 12.15]; P < .0001).
Figure 1.
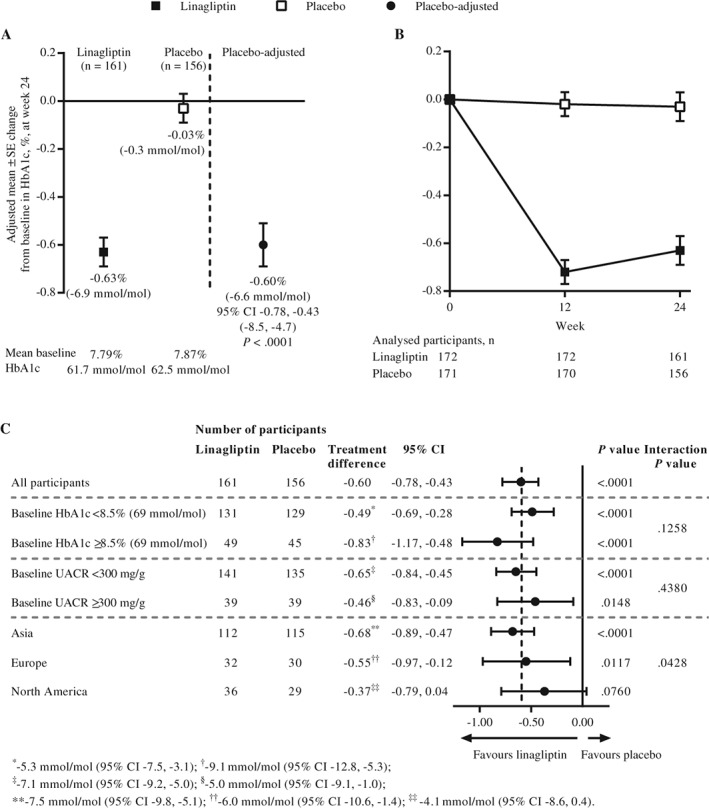
A, Adjusted mean change from baseline in glycated haemoglobin (HbA1c) at week 24 in the full analysis set (FAS) using observed cases excluding values after glycaemic rescue medication (OC). B, Adjusted mean change from baseline in HbA1c over time in the FAS (OC). C, Adjusted mean change from baseline in HbA1c at week 24 in participant subgroups in the FAS (OC). CI, confidence interval; SE, standard error
The time‐weighted average of percentage change from baseline in UACR over 24 weeks was −11.0% (95% CI, −16.8 to −4.7) with linagliptin and −5.1% (95% CI, −11.4 to 1.6) with placebo, a treatment difference of −6.0% (95% CI, −15.0 to 3.0; P = .1954) (Figure 2A). UACR was reduced from baseline over time to a numerically greater extent in the linagliptin group than in the placebo group (Figure 2B). The time‐weighted average of percentage change from baseline in UACR at week 24 for linagliptin compared with placebo was broadly similar across participant subgroups (Figure 2C, Figure S3). Post hoc analysis revealed no significant difference in change from baseline in UACR between participants receiving either an ACE inhibitor or ARB as background therapy: placebo‐corrected adjusted gMean ratios of −14% (95% CI, −28 to 1) and −2% (95% CI, −13 to 10), respectively (P = .1935 for interaction). An additional post hoc analysis comparing clinically relevant UACR responses between the linagliptin and placebo groups suggested that participants treated with linagliptin were approximately 70% more likely to achieve a meaningful response (>20% decrease in UACR at week 24 relative to baseline) than to show no response (odds ratio, 1.67 [95% CI, 1.04 to 2.68]; P = .0351) (Figure 3).
Figure 2.
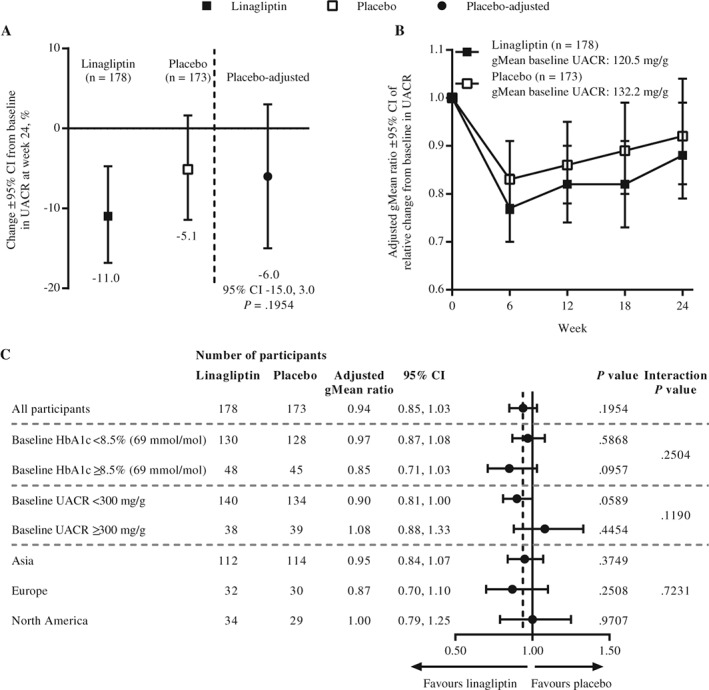
A, Adjusted geometric mean (gMean) for time‐weighted average of percentage change from baseline in urinary albumin‐to‐creatinine ratio (UACR) over 24 weeks in the full analysis set (FAS) (last observation carried forward [LOCF]). B, Adjusted gMean ratio of relative change from baseline in UACR over time in the FAS (LOCF). C, Adjusted gMean ratios for time‐weighted average of percentage change from baseline in UACR at week 24 in participant subgroups in the FAS (LOCF). CI, confidence interval; HbA1c, glycated haemoglobin
Figure 3.
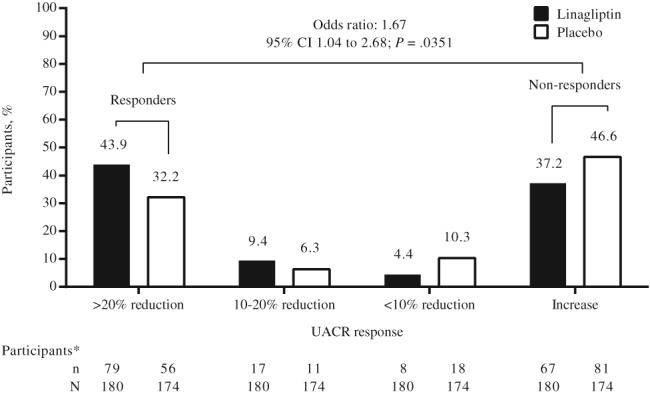
Distribution of urinary albumin‐to‐creatinine ratio (UACR) change from baseline at week 24 by UACR response categories in the full analysis set in eligible participants with a UACR value at week 24 irrespective of glycaemic rescue therapy. *Does not include participants with no UACR data at week 24: linagliptin, n = 9 (5.0%); placebo, n = 8 (4.6%). CI, confidence interval
3.2. Tolerability
A summary of adverse events is shown in Table S3. Adverse events were reported by 107 participants in both the linagliptin and placebo groups (58.8% and 60.1%, respectively), but few were deemed by investigators to be related to the study drug (13 [7.1%] and 11 [6.2%] participants, respectively). Adverse events leading to discontinuation of the study drug occurred in 3 linagliptin‐treated participants (1.6%) and in 2 placebo‐treated participants (1.1%). Serious adverse events occurred in 17 participants treated with linagliptin and in 8 receiving placebo (9.3% and 4.5%, respectively); these included 2 deaths in the linagliptin group (1.1%) and 1 death in the placebo group (0.6%). Serious adverse events reported in linagliptin‐treated participants were related to different acute and chronic medical conditions rather than any single condition.
Investigator‐reported hypoglycaemia occurred in 24 linagliptin‐treated participants (13.2%) and in 10 participants receiving placebo (5.6%), mostly in those receiving concomitant treatment with a sulphonylurea or insulin (Table S3); however, no severe hypoglycaemic episodes occurred. Apart from hypoglycaemia, the most common individual adverse events associated with linagliptin treatment were nasopharyngitis (7.1% and 5.6% of the linagliptin and placebo groups, respectively), hyperglycaemia (4.9% and 4.5%, respectively) and hyperuricaemia (4.4% and 1.7%, respectively) (Table S3).
CEC‐confirmed cardiovascular events occurred in 3 linagliptin‐treated participants (1.6%) and in no participants receiving placebo. CEC‐confirmed pancreatitis occurred in 1 linagliptin‐treated participant (0.5%) and in 2 participants receiving placebo (1.1%). No cases of pancreatic cancer occurred in either treatment group. No participant was hospitalized for heart failure.
No new cases of end‐stage kidney disease occurred during the study. Adjusted mean ± SE difference in change from baseline in eGFR (CKD‐EPI, cystatin C) at week 24 between the linagliptin and placebo groups was −2.63 ± 2.70 mL/min/1.73 m2 (P = .3306). There was also no significant difference in mean change in eGFR between the linagliptin and placebo groups at weeks 6, 12 and 18 (Figure S4).
Ambulatory blood pressure monitoring at baseline and week 24 indicated that blood pressure remained stable during treatment, with no significant difference in mean change between the linagliptin and placebo groups.23
4. DISCUSSION
In the MARLINA‐T2D study reported here, linagliptin significantly improved glycaemic control in type 2 diabetes patients with prevalent renal microvascular complications who were receiving current standard of care for diabetic kidney disease, including ACE inhibitors or ARBs. The glycaemic effects and safety of linagliptin were consistent with previous phase 3 studies of this DPP‐4 inhibitor in individuals with renal impairment.24, 25 However, the albuminuria‐lowering effect of linagliptin over this short‐term treatment period (24 weeks) was not significant.
The discord between the magnitude of the albuminuria‐lowering effect of linagliptin in MARLINA‐T2D (<10%) compared with that seen in a pooled analysis of clinical data (28%)20 may have occurred by chance or may reflect the limitations of the latter type of study. Differences in the characteristics of the study populations and background treatment regimens between the previous pooled analysis and MARLINA‐T2D might explain, at least in part, the divergent findings. Two‐thirds of individuals in the pooled analysis were receiving ACE inhibitors with approximately only one‐third receiving ARBs, whereas the opposite was the case in MARLINA‐T2D. In addition, White patients comprised approximately 70% of the study population of the pooled analysis, with approximately only 25% being Asians, whereas MARLINA‐T2D recruited approximately 66% Asians and 30% Whites. Also, most participants in this trial had modestly elevated UACR levels, where it is possible that the effects might be less, while power in the higher albuminuria subgroup was low and further impacted by high levels of variability. Importantly, UACR was assessed robustly in MARLINA‐T2D (3 samples collected over 3 consecutive days per visit), compared with the pooled analysis (spot urine samples at each visit).
The hypothesis that linagliptin may have nonglycaemic renal effects is based on several lines of evidence. DPP‐4, also known as T‐cell activation antigen CD26, is a ubiquitous glycoprotein with serine exopeptidase activity that exists in both circulating soluble and membrane‐bound forms, with the highest levels for the latter found in the kidney.15 Since the soluble form is believed to exert mainly the anti‐hyperglycaemic effects of DPP‐4 inhibitors, recent research has now focused also on the role of the highly expressed, membrane‐bound form of DPP‐4 found in the kidney. In the healthy human kidney, DPP‐4 appears to be expressed mainly in the tubular compartment, localized at the apical brush boarder of proximal tubular cells.26 Intriguingly, DPP‐4 was observed to be expressed also in glomeruli when individuals developed CKD, suggesting local adaptive mechanisms.27, 28 In addition, preclinical studies have suggested that linagliptin and other DPP‐4 inhibitors may have beneficial pleotropic effects on the kidney, as reviewed by Groop et al.22 Moreover, the improvement in nephropathy and albuminuria observed with the GLP‐1 receptor agonists liraglutide in the LEADER study29 and semaglutide in the SUSTAIN‐6 trial30 suggests at least some involvement of GLP‐1 receptor signalling in mediating the renal effects of DPP‐4 inhibitors.
The MARLINA‐T2D results suggest, however, that linagliptin may have a more modest acute albuminuria‐lowering effect in patients receiving stable RAAS blockade; this was smaller than anticipated based on the pooled analysis. Regardless, these findings do not suggest that linagliptin could impact on kidney function in type 2 diabetes patients through mechanisms associated with reducing albuminuria. In general, it is likely that ubiquitous albuminuria‐lowering effects over the short term may be achieved only by haemodynamic interventions that actively lower glomerular pressure, such as RAAS blockers and, possibly, SGLT2 inhibitors such as empagliflozin,31 dapagliflozin32 or canagliflozin.33 The small mean reduction in albuminuria (−5.1%) in the placebo group probably reflects regression to the mean, given the absence of meaningful changes in blood pressure, kidney function and long‐term glycaemia in this group.
Nevertheless, post hoc analysis of data from MARLINA‐T2D suggests that linagliptin may lower albuminuria to a meaningful extent (>20% in our judgement) in some patients; this cut‐off was based on a meta‐analysis of clinical trials reporting renal outcomes which found that an overall reduction in albuminuria of 19.2% was associated with a statistically significant 17% reduction in the relative risk of end‐stage renal disease (95% CI, 11.4 to 34.2).34 This meta‐analysis also suggested a significant linear correlation between the magnitude of drug‐induced albuminuria reduction and the magnitude of drug effect on risk of end‐stage renal disease.34 As albuminuria may be caused by several different pathophysiological mechanisms, including endothelial dysfunction, podocyte damage and mesangial proliferation,35 responding participants in MARLINA‐T2D may have had one of these underlying pathways activated and targeted by linagliptin.
Recent experimental data point towards linagliptin having anti‐inflammatory,36, 37 anti‐oxidant38 and anti‐fibrotic effects.16, 17, 18, 19 Notably, several studies reported a potential non‐enzymatic, direct effect of linagliptin in the kidney. Linagliptin, by interfering with renal protein–protein interactions of the abundant tubular DPP‐4 protein, down‐regulated pro‐fibrotic pathways and was associated with significant alleviation of tubulo‐interstitial fibrosis.16, 17, 18, 19 These changes would not necessarily be expected to result in lower levels of albuminuria, and the studies suggest that any renoprotective effect of linagliptin might be more likely to prevent progression of CKD over the long term than to have the short‐term UACR‐alleviating effects that MARLINA‐T2D was designed to investigate. This hypothesis is further supported by studies comparing linagliptin and RAAS blockers in diabetic and non‐diabetic animal models of CKD. Results revealed an apparent dissociation between reduction of urinary albumin excretion and reduction of renal oxidative stress, inflammation and fibrosis.19, 27, 39 For example, reduction in kidney fibrosis, a morphological biomarker that is closely correlated to clinical outcome, is much more pronounced in linagliptin‐treated CKD models as compared to the effects of RAAS blockade. Other studies have likewise revealed non‐albuminuric renoprotective effects of linagliptin.16, 40, 41, 42 Although this hypothesis is intriguing, further research is clearly needed to fully elucidate the renal biology and pathophysiology of DPP‐4.
Taken together, these preclinical and clinical study results underline the need for analysis of biomarkers in MARLINA‐T2D to provide information about the effects of linagliptin on kidney fibrosis and oxidative stress. Definition of renal markers for responding patients in this study (eg, biomarkers of activated fibrosis and/or inflammation) could potentially identify personalized treatment opportunities for future research.
The MARLINA‐T2D study has certain strengths and limitations. It is the first randomized clinical study designed to robustly investigate the effects of a DPP‐4 inhibitor, or any incretin therapy, on markers of diabetic kidney disease. As a well‐controlled study, it has high internal validity for inferring the effects of treatment. However, its external validity (generalizability) is limited by the nature of the participants studied (ie, those with early diabetic kidney disease) and it is not known whether the findings can be extrapolated to individuals with more advanced diabetic kidney disease. Of note in this regard, in the SAVOR‐TIMI 53 cardiovascular safety study of saxagliptin, albuminuria decreased more in participants with lower baseline eGFR: −19, −105 and −245 mg/g in those with eGFR >50, 30 to 50 and <30 mL/min, respectively.43 This suggests a possibility that the MARLINA‐T2D study population contained too few individuals with advanced CKD to fully unmask the anti‐albuminuric effect of linagliptin. Finally, and importantly, MARLINA‐T2D assessed a surrogate endpoint rather than actual renal outcomes.
In conclusion, the MARLINA‐T2D study found that linagliptin improved glycaemic control in individuals with type 2 diabetes and early stages of diabetic kidney disease but did not significantly ameliorate acute glomerular damage overall, as estimated using the surrogate endpoint of albuminuria; albeit, significantly more participants in the linagliptin group than in the placebo group experienced a meaningful improvement in albuminuria. The long‐term effect of linagliptin on hard renal outcomes remains to be determined in the ongoing CARMELINA study.
Supporting information
Table S1. Covariates and fixed effects in the mixed‐effect models for repeated measurements used to analyse change from baseline in glycated haemoglobin (HbA1c) in participant subgroups.
Table S2. Covariates and fixed effects in the analysis of covariance models used to analyse time‐weighted average of percentage change from baseline in urinary albumin‐to‐creatinine ratio (UACR) over 24 weeks of treatment in participant subgroups.
Table S3. Summary of adverse events during the 24 weeks of treatment in the treated set of participants
Figure S1. Flow chart of participants. The first participant was enrolled on February 26, 2013 and the last participant completed assessments on December 17, 2015. FAS, full analysis set; qd, once daily.
Figure S2. Adjusted mean change from baseline in glycated haemoglobin at week 24 in race subgroups in the full analysis set using observed cases excluding values after glycaemic rescue medication. Analysis of participants whose race was neither Asian nor white (n = 10) was not conducted as there were fewer observations than the prespecified minimum of five per treatment and visit combination within this category, and pooling with another race category was not appropriate. CI, confidence interval.
Figure S3. Adjusted geometric mean (gMean) ratios for time‐weighted average of percentage change from baseline in urinary albumin‐to‐creatinine ratio (UACR) at week 24 in race subgroups in the full analysis set using last observation carried forward. CI, confidence interval.
Figure S4. Adjusted mean change from baseline in estimated glomerular filtration rate (eGFR) (Chronic Kidney Disease Epidemiology Collaboration [CKD‐EPI], cystatin C) over time in the treated set. SE, standard error.
ACKNOWLEDGEMENTS
The authors thank the participants and staff involved in this study. Data from this study have been presented previously at the American Diabetes Association 76th Scientific Sessions, New Orleans, Louisiana, June 10 to 14, 2016. Medical writing assistance, supported financially by Boehringer Ingelheim, was provided by Giles Brooke, PhD, CMPP, of Envision Scientific Solutions during the preparation of this manuscript.
Conflict of interest
P‐H. G. has been a member of advisory panels for AbbVie, Boehringer Ingelheim, Cebix, Eli Lilly, Janssen, Medscape, Merck Sharp & Dohme Corp., Novartis, Sanofi and AstraZeneca; has received research support from Eli Lilly and Roche; and has been a member of Speaker's Bureaus for AstraZeneca, Boehringer Ingelheim, Eli Lilly, Genzyme, Merck Sharp & Dohme Corp., Novartis, Novo Nordisk and Sanofi. M. E. C., B. H., G. S. and K. S. have received fees for advisory services to Boehringer Ingelheim. V. P. consults for AbbVie, Astellas, Bristol‐Myers Squibb, Boehringer Ingelheim, Eli Lilly, GlaxoSmithKline and Janssen; has received lecture fees or grant support from Baxter, Boehringer Ingelheim, Merck and Pfizer; and his institution has held clinical trial contracts with AbbVie, Roche, Janssen, Servier and Novartis. K. K. has received lecture fees from Boehringer Ingelheim, Eli Lilly and Sanofi. Boehringer Ingelheim, Mitsubishi Tanabe Pharma and Ono Pharmaceutical contributed to establishing the Division of Anticipatory Molecular Food Science and Technology, Medical Research Institute, Kanazawa Medical University. K. K. is under contract for consultancy with Boehringer Ingelheim. M. H. reports consulting fees, lecture fees, moderator fees, supervising fees, payment for manuscript writing, research support or grants from Sanofi, Tanabe Mitsubishi, Takeda, Eli Lilly, Boehringer Ingelheim, Novo Nordisk, Novartis, MSD, Kyowa Hakko Kirin, Daiichi Sankyo, Astellas, Kowa, Asahi Kasei Pharma, Ajinomoto Pharma, Otsuka, Ono, Kaken, Kissei, GlaxoSmithKline, Sanwa Kagaku Kenkyusho, Shionogi, Johnson & Johnson, Sumitomo Dainippon, Chugai, Teijin Pharma, Terumo, Torii, Bayer Yakuhin, Pfizer, Bristol‐Myers Squibb, Mochida, Roche Diagnostics, AstraZeneca, Taisho Toyama and Taisho. R. C. S. has served on the Global Renal Advisory Board for Boehringer Ingelheim. R. T. is a consultant to AbbVie, Amgen, Boehringer Ingelheim, Stealth Peptides, Bristol‐Myers Squibb, Celgene, ZS Pharma and Relypsa and receives grant support from Ardelyx and the NIH. J. C., M. G., T. M., A. K‐W., S. T. and M. vE. are employees of Boehringer Ingelheim.
Author contributions
P‐H. G., M. E. C., V. P., B. H., K. K., M. H., G. S., K. S., R. C. S. and R. T. participated in design of the study, collection and interpretation of data, and drafting and revision of the manuscript. J. C. participated in design of the study, performed the statistical analysis, and participated in interpretation of data and drafting and revision of the manuscript. M. G., T. M., A. K‐W., S. T. and M. vE. participated in design of the study, interpretation of data, and drafting and revision of the manuscript. All authors have approved the final version of the manuscript. P‐H. G. is the guarantor of this work and, as such, had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Groop P‐H, Cooper ME, Perkovic V, et al. Linagliptin and its effects on hyperglycaemia and albuminuria in patients with type 2 diabetes and renal dysfunction: The randomized MARLINA‐T2D trial. Diabetes Obes Metab. 2017;19:1610–1619. https://doi.org/10.1111/dom.13041
Funding information This study was supported by the Boehringer Ingelheim and Eli Lilly and Company Diabetes Alliance
REFERENCES
- 1. Koro CE, Lee BH, Bowlin SJ. Antidiabetic medication use and prevalence of chronic kidney disease among patients with type 2 diabetes mellitus in the United States. Clin Ther. 2009;31:2608–2617. [DOI] [PubMed] [Google Scholar]
- 2. de Boer IH, Rue TC, Hall YN, Heagerty PJ, Weiss NS, Himmelfarb J. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA. 2011;305:2532–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Afkarian M, Sachs MC, Kestenbaum B, et al. Kidney disease and increased mortality risk in type 2 diabetes. J Am Soc Nephrol. 2013;24:302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Zeeuw D, Remuzzi G, Parving HH, et al. Proteinuria, a target for renoprotection in patients with type 2 diabetic nephropathy: lessons from RENAAL. Kidney Int. 2004;65:2309–2320. [DOI] [PubMed] [Google Scholar]
- 5. Eijkelkamp WB, Zhang Z, Remuzzi G, et al. Albuminuria is a target for renoprotective therapy independent from blood pressure in patients with type 2 diabetic nephropathy: post hoc analysis from the Reduction of Endpoints in NIDDM with the Angiotensin II Antagonist Losartan (RENAAL) trial. J Am Soc Nephrol. 2007;18:1540–1546. [DOI] [PubMed] [Google Scholar]
- 6. Ninomiya T, Perkovic V, de Galan BE, et al. Albuminuria and kidney function independently predict cardiovascular and renal outcomes in diabetes. J Am Soc Nephrol. 2009;20:1813–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Zeeuw D, Remuzzi G, Parving HH, et al. Albuminuria, a therapeutic target for cardiovascular protection in type 2 diabetic patients with nephropathy. Circulation. 2004;110:921–927. [DOI] [PubMed] [Google Scholar]
- 8. Holtkamp FA, de Zeeuw D, de Graeff PA, et al. Albuminuria and blood pressure, independent targets for cardioprotective therapy in patients with diabetes and nephropathy: a post hoc analysis of the combined RENAAL and IDNT trials. Eur Heart J. 2011;32:1493–1499. [DOI] [PubMed] [Google Scholar]
- 9. van der Velde M, Matsushita K, Coresh J, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with all‐cause and cardiovascular mortality. A collaborative meta‐analysis of high‐risk population cohorts. Kidney Int. 2011;79:1341–1352. [DOI] [PubMed] [Google Scholar]
- 10. Fox CS, Matsushita K, Woodward M, et al.; Chronic Kidney Disease Prognosis Consortium . Associations of kidney disease measures with mortality and end‐stage renal disease in individuals with and without diabetes: a meta‐analysis. Lancet. 2012;380:1662–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. National Kidney Foundation . KDOQI clinical practice guideline for diabetes and CKD: 2012 update. Am J Kidney Dis. 2012;60:850–886. [DOI] [PubMed] [Google Scholar]
- 12. American Diabetes Association . Standards of medical care in diabetes – 2016. Diabetes Care. 2016;39(suppl 1):S1–S112. [DOI] [PubMed] [Google Scholar]
- 13. de Zeeuw D, Heerspink HJ. Unmet need in diabetic nephropathy: failed drugs or trials? Lancet Diabetes Endocrinol. 2016;4:638–640. [DOI] [PubMed] [Google Scholar]
- 14. Inzucchi SE, Bergenstal RM, Buse JB, et al. Management of hyperglycemia in type 2 diabetes, 2015: a patient‐centered approach: update to a position statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care. 2015;38:140–149. [DOI] [PubMed] [Google Scholar]
- 15. Mentlein R. Dipeptidyl‐peptidase IV (CD26)‐‐role in the inactivation of regulatory peptides. Regul Pept. 1999;85:9–24. [DOI] [PubMed] [Google Scholar]
- 16. Kanasaki K, Shi S, Kanasaki M, et al. Linagliptin‐mediated DPP‐4 inhibition ameliorates kidney fibrosis in streptozotocin‐induced diabetic mice by inhibiting endothelial‐to‐mesenchymal transition in a therapeutic regimen. Diabetes. 2014;63:2120–2131. [DOI] [PubMed] [Google Scholar]
- 17. Shi S, Srivastava SP, Kanasaki M, et al. Interactions of DPP‐4 and integrin beta1 influences endothelial‐to‐mesenchymal transition. Kidney Int. 2015;88:479–489. [DOI] [PubMed] [Google Scholar]
- 18. Gangadharan Komala M, Gross S, Zaky A, Pollock C, Panchapakesan U. Linagliptin limits high glucose induced conversion of latent to active TGFss through interaction with CIM6PR and limits renal tubulointerstitial fibronectin. PLoS ONE. 2015;10:e0141143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsuprykov O, Ando R, Reichetzeder C, et al. The dipeptidyl peptidase inhibitor linagliptin and the angiotensin II receptor blocker telmisartan show renal benefit by different pathways in rats with 5/6 nephrectomy. Kidney Int. 2016;89:1049–1061. [DOI] [PubMed] [Google Scholar]
- 20. Groop PH, Cooper ME, Perkovic V, Emser A, Woerle HJ, von Eynatten M. Linagliptin lowers albuminuria on top of recommended standard treatment in patients with type 2 diabetes and renal dysfunction. Diabetes Care. 2013;36:3460–3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cooper ME, Perkovic V, McGill JB, et al. Kidney disease end points in a pooled analysis of individual patient‐level data from a large clinical trials program of the dipeptidyl peptidase 4 inhibitor linagliptin in type 2 diabetes. Am J Kidney Dis. 2015;66:441–449. [DOI] [PubMed] [Google Scholar]
- 22. Groop PH, Cooper ME, Perkovic V, et al. Dipeptidyl peptidase‐4 inhibition with linagliptin and effects on hyperglycaemia and albuminuria in patients with type 2 diabetes and renal dysfunction: rationale and design of the MARLINA‐T2D trial. Diab Vasc Dis Res. 2015;12:455–462. [DOI] [PubMed] [Google Scholar]
- 23. Cooper ME, Perkovic V, Groop PH, et al. Haemodynamic effects of combination therapy with the DPP‐4 inhibitor linagliptin and renin‐angiotensin system inhibitors in patients with type 2 diabetes. Diabetologia. 2016;56(suppl 1):S366. [Google Scholar]
- 24. McGill JB, Sloan L, Newman J, et al. Long‐term efficacy and safety of linagliptin in patients with type 2 diabetes and severe renal impairment: a 1‐year, randomized, double‐blind, placebo‐controlled study. Diabetes Care. 2013;36:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Laakso M, Rosenstock J, Groop PH, et al. Treatment with the dipeptidyl peptidase‐4 inhibitor linagliptin or placebo followed by glimepiride in patients with type 2 diabetes with moderate to severe renal impairment: a 52‐week, randomized, double‐blind clinical trial. Diabetes Care. 2015;38:e15–e17. [DOI] [PubMed] [Google Scholar]
- 26. Schlatter P, Beglinger C, Drewe J, Gutmann H. Glucagon‐like peptide 1 receptor expression in primary porcine proximal tubular cells. Regul Pept. 2007;141:120–128. [DOI] [PubMed] [Google Scholar]
- 27. Sharkovska Y, Reichetzeder C, Alter M, et al. Blood pressure and glucose independent renoprotective effects of dipeptidyl peptidase‐4 inhibition in a mouse model of type‐2 diabetic nephropathy. J Hypertens. 2014;32:2211–2223. [DOI] [PubMed] [Google Scholar]
- 28. Stiller D, Bahn H, August C. Demonstration of glomerular DPP IV activity in kidney diseases. Acta Histochem. 1991;91:105–109. [DOI] [PubMed] [Google Scholar]
- 29. Marso SP, Daniels GH, Brown‐Frandsen K, et al.; LEADER Steering Committee , LEADER Trial Investigators . Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2016;375:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Marso SP, Bain SC, Consoli A, et al.; SUSTAIN‐6 Investigators . Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. 2016;375:1834–1844. [DOI] [PubMed] [Google Scholar]
- 31. Cherney D, Lund SS, Perkins BA, et al. The effect of sodium glucose cotransporter 2 inhibition with empagliflozin on microalbuminuria and macroalbuminuria in patients with type 2 diabetes. Diabetologia. 2016;59:1860–1870. [DOI] [PubMed] [Google Scholar]
- 32. Heerspink HJ, Johnsson E, Gause‐Nilsson I, Cain VA, Sjostrom CD. Dapagliflozin reduces albuminuria in patients with diabetes and hypertension receiving renin‐angiotensin blockers. Diabetes Obes Metab. 2016;18:590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heerspink HJ, Desai M, Jardine M, Balis D, Meininger G, Perkovic V. Canagliflozin slows progression of renal function decline independently of glycemic effects. J Am Soc Nephrol. 2017;28:368–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Heerspink HJ, Kropelin TF, Hoekman J, de Zeeuw D, on behalf of the Reducing Albuminuria as Surrogate Endpoint (REASSURE) Consortium . Drug‐induced reduction in albuminuria is associated with subsequent renoprotection: a meta‐analysis. J Am Soc Nephrol. 2015;26:2055–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Satchell SC, Tooke JE. What is the mechanism of microalbuminuria in diabetes: a role for the glomerular endothelium? Diabetologia. 2008;51:714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fadini GP, Bonora BM, Cappellari R, et al. Acute effects of linagliptin on progenitor cells, monocyte phenotypes, and soluble mediators in type 2 diabetes. J Clin Endocrinol Metab. 2016;101:748–756. [DOI] [PubMed] [Google Scholar]
- 37. Zhuge F, Ni Y, Nagashimada M, et al. DPP‐4 inhibition by linagliptin attenuates obesity‐related inflammation and insulin resistance by regulating M1/M2 macrophage polarization. Diabetes. 2016;65:2966–2979. [DOI] [PubMed] [Google Scholar]
- 38. Takashima S, Fujita H, Fujishima H, et al. Stromal cell‐derived factor‐1 is upregulated by dipeptidyl peptidase‐4 inhibition and has protective roles in progressive diabetic nephropathy. Kidney Int. 2016;90:783–796. [DOI] [PubMed] [Google Scholar]
- 39. Chaykovska L, Alter ML, von Websky K, et al. Effects of telmisartan and linagliptin when used in combination on blood pressure and oxidative stress in rats with 2‐kidney‐1‐clip hypertension. J Hypertens. 2013;31:2290–2299. [DOI] [PubMed] [Google Scholar]
- 40. Ishibashi Y, Matsui T, Maeda S, Higashimoto Y, Yamagishi S. Advanced glycation end products evoke endothelial cell damage by stimulating soluble dipeptidyl peptidase‐4 production and its interaction with mannose 6‐phosphate/insulin‐like growth factor II receptor. Cardiovasc Diabetol. 2013;12:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakashima S, Matsui T, Takeuchi M, Yamagishi SI. Linagliptin blocks renal damage in type 1 diabetic rats by suppressing advanced glycation end products‐receptor axis. Horm Metab Res. 2014;46:717–721. [DOI] [PubMed] [Google Scholar]
- 42. Nistala R, Habibi J, Aroor A, et al. DPP4 inhibition attenuates filtration barrier injury and oxidant stress in the zucker obese rat. Obesity. 2014;22:2172–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mosenzon O, Leibowitz G, Bhatt DL, et al. Effect of saxagliptin on renal outcomes in the SAVOR‐TIMI 53 trial. Diabetes Care. 2017;40:69–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Covariates and fixed effects in the mixed‐effect models for repeated measurements used to analyse change from baseline in glycated haemoglobin (HbA1c) in participant subgroups.
Table S2. Covariates and fixed effects in the analysis of covariance models used to analyse time‐weighted average of percentage change from baseline in urinary albumin‐to‐creatinine ratio (UACR) over 24 weeks of treatment in participant subgroups.
Table S3. Summary of adverse events during the 24 weeks of treatment in the treated set of participants
Figure S1. Flow chart of participants. The first participant was enrolled on February 26, 2013 and the last participant completed assessments on December 17, 2015. FAS, full analysis set; qd, once daily.
Figure S2. Adjusted mean change from baseline in glycated haemoglobin at week 24 in race subgroups in the full analysis set using observed cases excluding values after glycaemic rescue medication. Analysis of participants whose race was neither Asian nor white (n = 10) was not conducted as there were fewer observations than the prespecified minimum of five per treatment and visit combination within this category, and pooling with another race category was not appropriate. CI, confidence interval.
Figure S3. Adjusted geometric mean (gMean) ratios for time‐weighted average of percentage change from baseline in urinary albumin‐to‐creatinine ratio (UACR) at week 24 in race subgroups in the full analysis set using last observation carried forward. CI, confidence interval.
Figure S4. Adjusted mean change from baseline in estimated glomerular filtration rate (eGFR) (Chronic Kidney Disease Epidemiology Collaboration [CKD‐EPI], cystatin C) over time in the treated set. SE, standard error.