

 The decomposition of formic acid to H2 and CO2 under metal-free conditions has been unveiled using dialkylborane derivatives as catalysts.
The decomposition of formic acid to H2 and CO2 under metal-free conditions has been unveiled using dialkylborane derivatives as catalysts.
Abstract
Formic acid is at the crossroads of novel sustainable energy strategies because it is an efficient H2 carrier. Yet, to date, its decomposition to H2 relies on metal-based catalysts. Herein, we describe the first metal-free catalysts able to promote the dehydrogenation of formic acid. Using dialkylborane derivatives, HCOOH is decomposed to H2 and CO2 in the presence of a base with high selectivity. Experimental and computational results point to the involvement of bis(formyloxy)borates as key intermediates in the C–H bond activation of a formate ligand.
Introduction
Besides its utilization as a preservative and antibacterial agent in the agriculture industry,1 formic acid (FA) is at the crossroads of novel sustainable energy strategies. With a high energy density of 2104 W h L–1, FA is indeed investigated as a potential energy vector either directly as a fuel in electrochemical fuel cells2 or as an intermediate in the formation of methanol3 (4900 W h L–1). Additionally, FA is an attractive hydrogen carrier because of its relatively high hydrogen content (4.4 wt%), and “hydrogen batteries” have been proposed,4 which rely on the reversible hydrogenation of CO2 and the dehydrogenation of formic acid in basic media, for H2 storage. Importantly, efficient catalysts are required to promote the rapid and selective dehydrogenation of FA to CO2 and molecular H2 and their design has been the focus of many efforts within the last two decades.5 While early studies focused on the development of ruthenium6 and iridium complexes,7 the current state of the art now also includes efficient iron catalysts8 and the search for noble-metal free catalysts is an active research field. Notably, Berben et al. have disclosed in 2014 that molecular aluminium-based complexes with a bis(imino)pyridine ligand promote the selective dehydrogenation of formic acid in the presence of triethylamine with TOFs of up to 5200 h–1.9 More recently, Maschmeyer et al. 10 have shown that sodium germanate heterogeneously promotes H2 formation from formic acid vapors at 300 °C. So far, a metal-free catalyst able to convert HCOOH to H2 and CO2 has yet remained elusive. The main challenge of this endeavour lies in the necessity of a double, antagonistic, activation of formic acid. First, the acidic O–H proton must be abstracted with the aid of a Brønsted base to yield a formate anion. The C–H bond cleavage then requires a Lewis-acidic species, able to promote hydride abstraction despite the presence of a strongly coordinating formate medium.
Herein, we disclose that dialkylborane derivatives are unprecedented organocatalysts for the selective dehydrogenation of FA to molecular hydrogen and CO2 in the presence of a basic additive. Mechanistic insights are provided based on experimental observations and DFT calculations and they highlight the role of bis(formyloxy)borates in this catalytic transformation.
Results and discussion
While the deprotonation of the O–H bond in FA is easily promoted by a Brønsted base having a pK a > 3.7, the activation of the C–H bond in the resulting formate anion is anticipated to be a rate-limiting step and it necessitates a Lewis acid able to abstract a hydride anion and release CO2. For the design of an efficient catalyst, it is thus mandatory to seek a Lewis acid able to interconvert a formate anion and CO2, with a low activation energy and negligible exergonicity. In this context, we have shown recently that guanidines are highly efficient organocatalysts in the hydroboration of CO2 at 25 °C.11 In particular, experimental and computational results pointed to a mechanism where adduct 1, formed from MTBD (7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene) and the stoichiometric BBN–H hydroborane, promotes the reduction of CO2 to the formate salt [2+ , HCOO–] with a modest activation barrier of 23.8 kcal mol–1 and a nearly athermic balance (ΔG = –3.0 kcal mol–1) (Scheme 1). This result suggests that intermediate 2+ could serve as a catalyst to promote the reverse transformation, namely the dehydrogenation of formate anions, and catalyze H2 generation by quenching of the formed B–H linkage with HCOOH.
Scheme 1. Computed pathway (M05-2X/6-31+G*) in THF for the rate-determining step in the MTBD-catalyzed hydroboration of CO2 to formate.

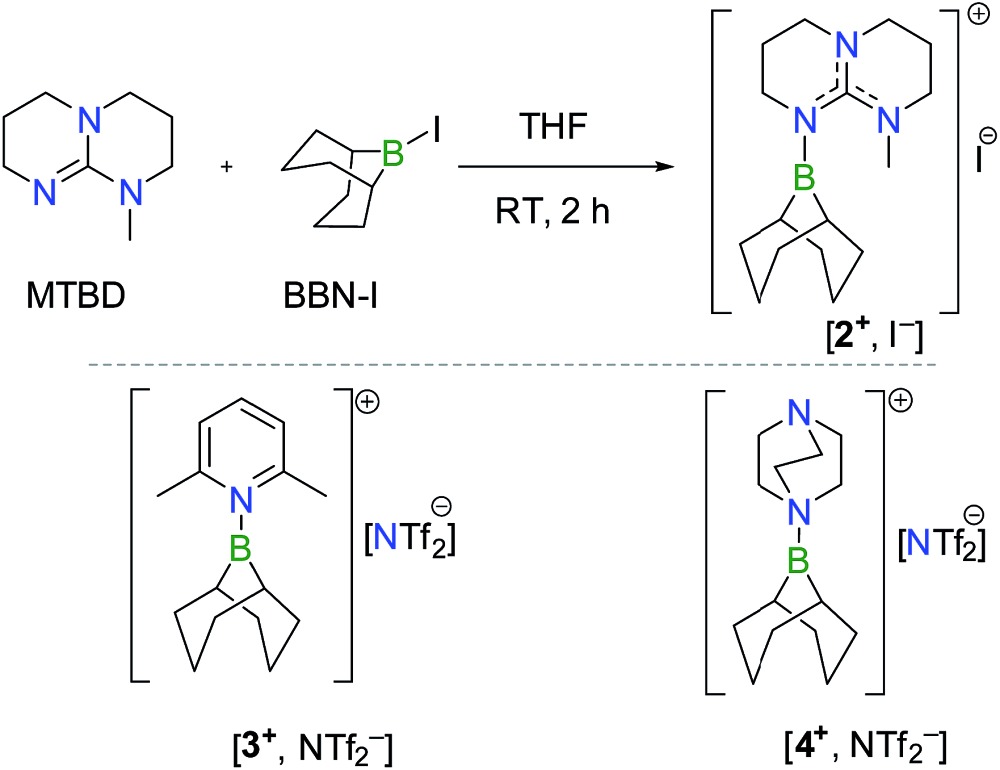
To test this hypothesis, [2+ , I–] was synthesized in 81% yield by nucleophilic displacement of the iodine atom in BBN–I by MTBD in THF (eqn (1)). The 11B{1H} NMR spectrum of 2+ displays a resonance at +57.2 ppm, consistent with the structure of an electrophilic tricoordinate boron center. This 11B chemical shift is similar to that found in the borenium salts [3+ , NTf2 –] (+59.2 ppm) and [4+ , NTf2 –] (+56.9 ppm).12 The description of 2+ as a borenium cation stabilized by a π-donor nitrogen ligand was further confirmed by X-ray diffraction (Fig. 1a). In the solid state, the boron atom adopts a trigonal planar geometry with a N–B bond length of 1.428(2) Å, close to the N–B distance found in aminoboranes, suggesting π-donation from the nitrogen atom of the guanidine.
Fig. 1. (a) ORTEP view of 2+ in [2+ , I–]. Displacement ellipsoids are drawn at the 50% probability level. Counterion and hydrogen atoms are omitted. (b) ORTEP view of compound 5– in [TBDH+, 5– ]. Displacement ellipsoids are drawn at the 30% probability level. TBDH+ and hydrogen atoms on the BBN bicycle have been removed for clarity.

 |
1 |
Using 5 mol% [2+ , I–] as the catalyst, the dehydrogenation of 13C-labelled FA was undertaken in d 8-THF at 130 °C. Monitoring the reaction mixture by 1H and 13C NMR demonstrated the complete consumption of FA and the clean formation of H2 (4.55 ppm) and 13CO2 (126 ppm) as the only 13C-enriched product after 4 days. In contrast, a blank experiment, carried out in the absence of the catalyst, showed no decomposition of FA under identical conditions. Nonetheless, a more careful insight into the proceedings of the catalytic reaction at shorter times revealed a lengthy initiation period, hence a lack of reproducibility (vide infra).
In order to increase both the rate and the reproducibility of the reaction, the dehydrogenation of FA was investigated in the presence of triethylamine (TEA) as an external base. The positive influence of bases on the kinetics of FA conversion to H2 and CO2 is indeed well established with metal catalysts. 6b,9 To our delight, the combination of 5 mol% [2+ , I–] with triethylammonium formate (5FA/2TEA) in THF leads reproducibly to the clean formation of H2 and CO2 in ca. 26% yield (±5%) after 19 h (entry 1, Table 1) and >99% yield after 45 h, while no H2 evolution was observed in the absence of the catalyst (see ESI†). To the best of our knowledge, this result represents the first example of an organocatalytic dehydrogenation of FA.
Table 1. Metal-free catalytic dehydrogenation of HCOOH/NEt3 (5 : 2) a .
| ||||
| Entry | Catalyst (amount, mol%) | Solvent | Conv. b [%] | TON (time, h) |
| 1 | [2+ , I–] (5.0) | TDF | 26 | 5.2 (19) |
| 2 | BBN–I (5.0) | TDF | 48 | 9.6 (19) |
| 3 | BBN–I (5.0) | C6D6 | <5 | — |
| 4 | BBN–I (5.0) | Tol-d 8 | <5 | — |
| 5 | BBN–I (5.0) | CD3OD | <5 | — |
| 6 | BBN–I (5.0) | CD3CN | 84 | 16.8 (19) |
| 7 | BBN–H (5.0) | CD3CN | 52 | 10.4 (19) |
| 8 | [TBDH+, 5– ] (5.0) | CD3CN | 67 c | 13.4 (19) |
| 9 | Cy2B–I (10.0) | CD3CN | >99 d | 10 (4.5) |
| 10 | Cy2B–I (5.0) | CD3CN | >99 | 20 (19) |
| 11 | Cy2B–I (1.0) | CD3CN | 79 | 79 (19), 88 (23), 100 (40) |
| 12 | [Et3NH+, 6– ] (5.0) | CD3CN | >99 c | 20 (8) |
| 13 | [Et3NH+, 6– ] (1.0) | CD3CN | 78 c | 78 (19), 94 (23), 100 (26) |
aReaction conditions: 0.2 mmol FA, 0.08 mmol TEA, 0.2 mL deuterated solvent.
bConversions determined by 1H NMR using mesitylene (10 μL) as an internal standard; mean values over at least 2 runs.
cCatalyst was recovered unchanged.
dFull conversion reached within 4.5 h.
Interestingly, in the presence of TEA, commercially available BBN–I proves to be an efficient catalyst and within 19 h at 130 °C it affords H2 and CO2 in 48% yield from FA (entry 2, Table 1). In comparison, as noted above, only 26% yield is observed with 5.0 mol% [2+ , I–], suggesting that the Lewis basicity of MTBD might be detrimental. Importantly, the solvent has a dramatic effect on the H2 generation with BBN–I in the presence of TEA. Whereas no reaction occurs in non-polar solvents such as benzene or toluene (entries 3 and 4, Table 1), 84% FA is dehydrogenated after 19 h at 130 °C in MeCN with 5.0 mol% BBN–I (entry 6, Table 1). Nonetheless, the catalytic system is inactive in the presence of polar and protic solvents such as methanol, presumably because of the competitive complexation of methanol to boron (entry 5, Table 1). The reaction temperature also plays an important role in the outcome of the reaction and the yield plummets from 84 to 18% after 19 h, when the reaction temperature is lowered from 130 to 120 °C. Decreasing the temperature below 120 °C results in the complete deactivation of the catalytic system, even after prolonged time (ca. 30 h). Finally, the amount of base present in the reaction medium strongly controls the productivity and, using 5.0 mol% BBN–I, no reaction occurs in pure FA with 0.0 or 5.0 mol% MTBD, while 49% dehydrogenation is measured after 19 h with 10.0 mol% MTBD (see ESI†).
 |
2 |
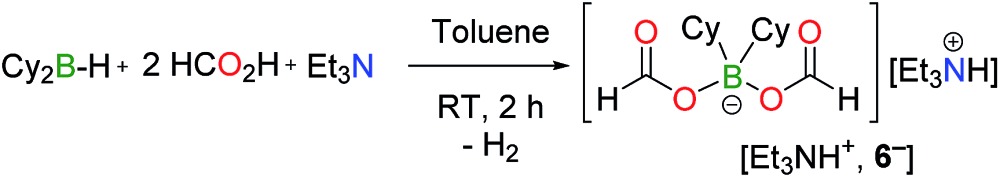
Monitoring the product distribution by 1H and 13C NMR reveals the formation of a common catalytic intermediate in the dehydrogenation of 13C-labelled FA with 2/5 TEA when [2+ , I–] or BBN–I are used as catalysts. This intermediate is the only boron species detected in solution at room temperature within the reaction mixture and it can be formulated as a bis(formyloxy)borate anion (5– ). 5– features two formate moieties characterized by a doublet at 8.41 ppm (1 J CH = 203 Hz) in the 1H NMR spectrum and a 13C{1H} resonance at 167 ppm. The 11B{1H} NMR spectrum presents a single peak at 8.98 ppm in d 3-MeCN, consistent with an electron-poor tetra-coordinated boron center. In fact, 5– is formed quantitatively when [2+ , I–] is reacted with 2 equiv. FA and 1 equiv. TEA at 25 °C (eqn (2)). 5– can be isolated as a well-defined salt via the dehydrogenation of hydroborane BBN–H with 2 equiv. FA and a stoichiometric amount of a base, namely 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), MTBD or TEA (eqn (3)). While [Et3NH+, 5– ] is a colorless oil, [TBDH+, 5– ] was isolated as a crystalline solid enabling its structure determination by X-ray diffraction (Fig. 1b). To date, [TBDH+, 5– ] is the first example of a bis(formyloxy)borate characterized and it displays B–O and C–O bond lengths (B–O: 1.542(3) and 1.528(3) Å, C–O: 1.279(3) and 1.293(3) Å, C O: 1.217(3) and 1.218(3) Å) similar to those encountered in anionic mono(formyloxy)borates derived from B(C6F5)3 (ref. 13) or HBAr2 (ref. 14) (Ar = 2,4,6-tris(trifluoromethyl)phenyl).
The ability of [TBDH+, 5– ] to promote the decarboxylation of formate ions was further probed using this salt as a catalyst in the dehydrogenation of FA. With 5.0 mol% [TBDH+, 5– ], 67% CO2/H2 are produced from a 5FA/2TEA mixture after 19 h at 130 °C and [Et3NH+, 5– ] could be recovered at the end of the reaction (entry 8, Table 1).15 In agreement with eqn (3), BBN–H is also catalytically active (entry 7, Table 1).
 |
3 |
 |
4 |
Overall, the experimental findings gathered with BBN–I as a catalyst reveal the importance of bis(formyloxy)borate 5– as a catalytically active species. R2B–X (X = Cl, Br, I, OMe, OTf) precursors were thus screened to determine the electronic and/or steric influence of the R group on the catalytic activity.
Catechol borane and pinacol borane derivatives (catB–Cl, catB–Br or pinB–OMe) are completely inactive in the dehydrogenation of FA, even in the presence of TEA (entries 25, 26 and 27, Table S1†). In contrast, dicyclohexylborane derivatives exhibit a three-fold increase in activity, compared to the BBN system; and the quantitative dehydrogenation of 5FA/2TEA is achieved within only 4.5 h at 130 °C with 10 mol% Cy2B–I (entry 9, Table 1). Interestingly, the reaction still proceeds smoothly with 1.0 mol% Cy2B–I and 79% conversion is observed after 19 h (TON19h = 79, TOF = 4.1 h–1) and 88% after 23 h (TON23h = 88, TOF = 3.8 h–1). A TON of 100 requires a longer reaction time of 43 h. Importantly, with either Cy2B- or BBN-based catalysts only <0.2% CO was detected by GC (see ESI†). Similar to BBN–I, Cy2B–I is readily converted in the reaction medium to a catalytically active bis(formyloxy)borate anion (6– ) in the presence of FA and TEA (eqn (4) and entries 12 and 13, Table 1).
The pivotal role of the bis(formyloxy)borates 5– and 6– was further probed using DFT techniques (at the M06-2X/6-311+G(d,p) level of theory), so as to account for their observation throughout the catalysis (Scheme 2). In agreement with the experimental observations, 5– is found as the lowest energy species on the potential energy surface. No direct transition state (TS) could be localized to directly connect the saturated bis(formyloxy)borate 5– with CO2 and the hydridoborate intermediate 9– , suggesting that the decarboxylation of 5– requires the decoordination of one formate ligand to restore a vacant orbital on the boron center. The trigonal formyloxyborane 7 is indeed engaged in an unfavorable equilibrium with 5– (ΔG = +11.1 kcal mol–1). Formation of 9– from 7 can then proceed via a β-hydride elimination (TS2 ), similar to ruthenium catalysts.6 In addition, Berben et al. proposed that aluminium complexes of the bis(imino)pyridine ligand can promote the decarboxylation of a formate ligand via a β-hydride abstraction assisted by the ancillary nitrogen ligand.9 Nonetheless, the corresponding activation energy of 51.9 kcal mol–1 is incompatible with the applied reaction conditions. In addition, a lower energy pathway was identified that corresponds to the direct hydride abstraction of an external formate anion by the neutral boron electrophile 7 via TS1 (26.6 kcal mol–1). This result confirms that, with a base/boron catalyst molar ratio of 1, no free formate anion is available and the decarboxylation of 7 can only proceed via TS2 , therefore inhibiting the catalytic dehydrogenation of pure FA with [2+ , I–].
Scheme 2. Computed pathways in acetonitrile (at the M06-2X/6-311+G(d,p) level of theory) and proposed catalytic cycle for the dehydrogenation of formic acid catalyzed by BBN–H derivatives.

Formed by the decarboxylation of 7, the hydridoborate intermediate 9– lies 12.9 kcal mol–1 above the starting materials, accounting for our unsuccessful attempts to observe a B–H functionality.16 9– is prone to protonolysis and two different pathways have been computationally unveiled for the dehydrogenation of FA with 9– . H2 release by quenching of the B–H group with an external FA molecule proceeds via TS4 , with an activation barrier of 15.9 kcal mol–1 from 9– . Interestingly, a pathway involving the displacement of the formate ligand in 9– by HCOOH and subsequent intramolecular dehydrogenation (via TS3 ) cannot be ruled out, as it involves a low activation barrier of 18.7 kcal mol–1 from 9– . The dehydrogenation step reforms 7, which is ultimately converted to the more stable borate 5– . Overall, the decomposition of FA involves two steps, namely the decarboxylation of a trigonal boron formate intermediate (via TS1 , 26.6 kcal mol–1) and the subsequent deprotonation of FA by the reactive hydridoborate intermediate (via TS3 or TS4 , 31.6 and 28.8 kcal mol–1, respectively) (Scheme 2). Both steps thus have similar energy demands and no rate-determining step (RDS) can be unequivocally proposed. Nevertheless, the consistency of the latter barriers with regard to the applied reaction conditions was further established using the energetic span model.17 With an energetic span of δE = 28.8 kcal mol–1, the calculated TOF at 130 °C (403 K) is 1.0 h–1 which is in the same order of magnitude as the experimental TOF (0.6–0.9 h–1 from Table 1, entries 6–8).
The influence of replacing the BBN scaffold with Cy2B- or catB-backbones has been investigated computationally (Schemes S2 and S3†). TS1 , TS3 and TS4 are very close in energy for the catalytically active BBN and Cy2B systems and the mechanistic model does not account for the greater catalytic activity of Cy2B–I. Nonetheless, the energy surface is significantly modified when catechol borane derivatives are used as catalysts (Scheme S3†). Indeed, the coordination of the π-donor catecholate ligand to the boron atom lowers the electrophilicity of the catalyst and hampers the activation of the C–H bond by hydride abstraction, which necessitates an activation energy of 38.8 kcal mol–1. Finally, the mechanism depicted in Scheme 2 also stresses that a bis(formyloxy)borate species (such as 5– or 6– ) is the resting state of the catalyst and must dissociate to a catalytically active trigonal formyloxyborane intermediate. As a consequence, a highly electrophilic borane derivative might prevent the generation of the active species, and, experimentally, the potent electrophile B(C6F5)3 was found inactive in the dehydrogenation of 5FA/2TEA, even after 48 h at 130 °C (entry 28, Table S1†). This result suggests that the catalyst should bear at least two formate ions to balance the electrophilicity of the boron catalyst. In that respect, BCl3 enables the slow dehydrogenation of formic acid with a TON of 9 and a TOF of 0.5 h–1 after 19 h at 130 °C (entry 31, Table S1†).
Conclusions
In conclusion, the first organocatalysts able to promote the dehydrogenation of formic acid have been unveiled with TONs of up to 100. Using dialkylborane derivatives and their corresponding bis(formyloxy)borates, HCOOH is decomposed to H2 and CO2 in the presence of a base with high selectivity. Capitalizing on the mechanistic insights, current efforts are devoted to increasing the catalytic activity of these new boron catalysts and exploring the reduction chemistry of bis(formyloxy)borates.
Acknowledgments
For financial support of this work, we acknowledge CEA, CNRS, Labex CHARMMMAT, CINES and ERC (Starting Grant Agreement n.336467). T.C. thanks the Foundation Louis D. – Institut de France for its support. The authors thank Dr Cédric Tard (Université Paris Diderot, France) for the assistance with GC analyses.
Footnotes
References
- Reutemann W. and Kieczka H., in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000. [Google Scholar]
- Uhm S., Lee H. J., Lee J. Phys. Chem. Chem. Phys. 2009;11:9326–9336. doi: 10.1039/b909525j. [DOI] [PubMed] [Google Scholar]
- (a) Miller A. J., Heinekey D. M., Mayer J. M., Goldberg K. I. Angew. Chem., Int. Ed. 2013;52:3981–3984. doi: 10.1002/anie.201208470. [DOI] [PubMed] [Google Scholar]; (b) Savourey S., Lefevre G., Berthet J. C., Thuery P., Genre C., Cantat T. Angew. Chem., Int. Ed. 2014;53:10466–10470. doi: 10.1002/anie.201405457. [DOI] [PubMed] [Google Scholar]; (c) Wesselbaum S., Moha V., Meuresch M., Brosinski S., Thenert K. M., Kothe J., Stein T. V., Englert U., Hölscher M., Klankermayer J., Leitner W. Chem. Sci. 2015;6:693–704. doi: 10.1039/c4sc02087a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddien A., Federsel C., Sponholz P., Mellmann D., Jackstell R., Junge H., Laurenczy G., Beller M. Energy Environ. Sci. 2012;5:8907–8911. [Google Scholar]
- (a) Johnson T. C., Morris D. J., Wills M. Chem. Soc. Rev. 2010;39:81–88. doi: 10.1039/b904495g. [DOI] [PubMed] [Google Scholar]; (b) Grasemann M., Laurenczy G. Energy Environ. Sci. 2012;5:8171–8181. [Google Scholar]; (c) Laurenczy G., Dyson P. J. J. Braz. Chem. Soc. 2014;25:2157–2163. [Google Scholar]
- (a) Fellay C., Dyson P. J., Laurenczy G. Angew. Chem., Int. Ed. 2008;47:3966–3968. doi: 10.1002/anie.200800320. [DOI] [PubMed] [Google Scholar]; (b) Loges B., Boddien A., Junge H., Beller M. Angew. Chem., Int. Ed. 2008;47:3962–3965. doi: 10.1002/anie.200705972. [DOI] [PubMed] [Google Scholar]; (c) Alsabeh P. G., Mellmann D., Junge H. and Beller M., in Ruthenium in Catalysis, Springer International Publishing, 2014, vol. 48, pp. 45–79. [Google Scholar]
- (a) Hull J. F., Himeda Y., Wang W. H., Hashiguchi B., Periana R., Szalda D. J., Muckerman J. T., Fujita E. Nat. Chem. 2012;4:383–388. doi: 10.1038/nchem.1295. [DOI] [PubMed] [Google Scholar]; (b) Barnard J. H., Wang C., Berry N. G., Xiao J. Chem. Sci. 2013;4:1234–1244. [Google Scholar]; (c) Wang W. H., Xu S., Manaka Y., Suna Y., Kambayashi H., Muckerman J. T., Fujita E., Himeda Y. ChemSusChem. 2014;7:1976–1983. doi: 10.1002/cssc.201301414. [DOI] [PubMed] [Google Scholar]
- (a) Bielinski E. A., Lagaditis P. O., Zhang Y., Mercado B. Q., Wurtele C., Bernskoetter W. H., Hazari N., Schneider S. J. Am. Chem. Soc. 2014;136:10234–10237. doi: 10.1021/ja505241x. [DOI] [PubMed] [Google Scholar]; (b) Boddien A., Loges B., Gartner F., Torborg C., Fumino K., Junge H., Ludwig R., Beller M. J. Am. Chem. Soc. 2010;132:8924–8934. doi: 10.1021/ja100925n. [DOI] [PubMed] [Google Scholar]; (c) Boddien A., Mellmann D., Gartner F., Jackstell R., Junge H., Dyson P. J., Laurenczy G., Ludwig R., Beller M. Science. 2011;333:1733–1736. doi: 10.1126/science.1206613. [DOI] [PubMed] [Google Scholar]
- Myers T. W., Berben L. A. Chem. Sci. 2014;5:2771–2777. [Google Scholar]
- Amos R. I., Heinroth F., Chan B., Zheng S., Haynes B. S., Easton C. J., Masters A. F., Radom L., Maschmeyer T. Angew. Chem., Int. Ed. 2014;53:11275–11279. doi: 10.1002/anie.201405360. [DOI] [PubMed] [Google Scholar]
- Das Neves Gomes C., Blondiaux E., Thuery P., Cantat T. Chem.–Eur. J. 2014;20:7098–7106. doi: 10.1002/chem.201400349. [DOI] [PubMed] [Google Scholar]
- (a) Narula H. N. C. K. Inorg. Chem. 1985;24:2532–2539. [Google Scholar]; (b) Denmark S. E., Ueki Y. Organometallics. 2013;32:6631–6634. doi: 10.1021/om400582k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley A. E., Thompson A. L., O'Hare D. Angew. Chem., Int. Ed. 2009;48:9839–9843. doi: 10.1002/anie.200905466. [DOI] [PubMed] [Google Scholar]
- Lu Z., Wang Y., Liu J., Lin Y.-J., Li Z. H., Wang H. Organometallics. 2013;32:6753–6758. [Google Scholar]
- Contrary to trialkyboranes, 5– was found insensitive to acidic protonolysis during the catalysis and the cleavage of one of the Csp3 –B bonds within the BBN-backbone was never observed
- Although there is no experimental evidence for the formation of 9– , isolation of the related [HC(O)OB(H)Ar2]– anion has been reported by Wang et al., see ref. 14
- (a) Kozuch S., Shaik S. Acc. Chem. Res. 2011;44:101–110. doi: 10.1021/ar1000956. [DOI] [PubMed] [Google Scholar]; (b) Kozuch S. WIREs Comput. Mol. Sci. 2012;2:795–815. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.