

Summary
Rare inherited bleeding disorders (IBD) are a common cause of bleeding tendency. To ensure a correct diagnosis, specialized laboratory analyses are necessary. This study reports the results of an upfront diagnostic strategy using targeted whole exome sequencing. In total, 156 patients with a significant bleeding assessment tool score participated in the study, of which a third had thrombocytopenia. Eighty‐seven genes specifically associated with genetic predisposition to bleeding were analysed by whole exome sequencing. Variants were classified according to the five‐tier scheme. We identified 353 germline variants. Eight patients (5%) harboured a known pathogenic variant. Of the 345 previously unknown variants, computational analyses predicted 99 to be significant. Further filtration according to the Mendelian inheritance pattern, resulted in 59 variants being predicted to be clinically significant. Moreover, 34% (20/59) were assigned as novel class 4 or 5 variants upon targeted functional testing. A class 4 or 5 variant was identified in 30% of patients with thrombocytopenia (14/47) versus 11% of patients with a normal platelet count (12/109) (P < 0·01). An IBD diagnosis has a major clinical impact. The genetic investigations detailed here extricated our patients from a diagnostic conundrum, thus demonstrating that continuous optimization of the diagnostic work‐up of IBD is of great benefit.
Keywords: bleeding disorders, genetic analysis, platelet disorders
Rare inherited bleeding disorders (IBD) are a heterogeneous group of diseases that are very complex to diagnose. The work‐up of patients with mild bleeding disorders is hampered by the fact that the number of possible causes is extensive (>51 different inherited platelet abnormalities have been genetically described) but the incidence of each defect is very low (<1/100 000) (Nurden & Nurden, 2013; Lentaigne et al, 2016). Some patients may have several defects working in concert to produce a mild bleeding disorder (Stockley et al, 2015). Bleeding tendency may also be caused by vascular defects, such as Ehlers–Danlos syndrome (EDS) or hereditary telangiectasia (HT). Thus, a correct diagnosis requires not only a careful interview on symptoms and family history but also functional and, in many cases, genetic testing. Advanced functional platelet assays are only available at specialized coagulation centres and do not cover vascular or connective tissue disorders. In the pre‐molecular genetics era, the mere complexity (and cost) of these investigations rendered a significant proportion of patients, between 47–67%, without a proper diagnosis (Quiroga & Mezzano, 2012).
Advances in gene sequencing techniques, enforced by the Human Genome Project, have made the rapid sequencing of multiple genes a promising tool in the diagnostic work‐up of these patients. A remarkable effort has been made by the ThromboGenomics consortium that has designed an next generation sequencing (NGS)‐based panel targeting 63 genes relevant for inherited bleeding and thrombotic disorders, allowing for a molecular diagnosis in cases where an IBD or thrombotic disorder is suspected on laboratory or clinical findings (Simeoni et al, 2016). Another approach has been taken by the UK Genotyping and Phenotyping of Platelets (GAPP) study that enrols patients with increased bleeding tendency of unknown aetiology. In this study, the presence of known genetic causes is ruled out by functional and genetic testing. The GAPP study recently showed that a platelet defect was identified in approximately 60% of the investigated participants (>500 patients) by using whole exome sequencing (WES) techniques (Fletcher et al, 2015; Watson et al, 2013).
The advantage of WES compared to gene panels is the option to consecutively expand the number of genes analysed as information on the underlying genetic disorders increases. As for all new screening methods, caution must be taken when interpreting the results, especially in regard to variant filtering. Hence, WES identifies tens of thousands of variants in each patient, of which the majority are common benign variants that are filtered out by bioinformatics tools. However, a large number of variants need interpretation and subsequent classification. Classification of the clinical significance of a genetic variant ranges from benign to pathogenic and, to ensure a consistent and standardized output, variant classification must follow international consensus‐based guidelines (Amendola et al, 2016; Rehm et al, 2013; Richards et al, 2015; Matthijs et al, 2016).
In the present study, to improve the diagnostic outcome in patients referred for an IBD, we applied WES to identify variants in 87 preselected, already known disease‐associated genes. Variants were classified according to the five‐tier scheme. Variants of unknown significance (VUS) were further filtered according to their Mendelian inheritance patterns as well as in silico analyses. Prior to initiation of the WES strategy, only patients with an established diagnosis of haemophillia or Von Willebrand syndrome type 2 or suspected carriers had been offered classic genetic testing at our institutions. Ultimately, targeted functional tests were performed, allowing patients suffering from excessive bleeding to be diagnosed with IBD. The results obtained by implementation of WES at an early stage of the diagnostic work‐up in patients suspected of an IBD are presented here.
Materials and methods
Enrolment of patients
We performed the International Society on Thrombosis and Haemostasis (ISTH) bleeding assessment tool (BAT) score (Elbatarny et al, 2014) in all patients referred to the Department of Haematology, Rigshospitalet (RH), Copenhagen University Hospital, Denmark, and the Coagulation Unit, Skaane University Hospital, Malmö, Sweden, between April 2013 and January 2016 on the basis of a suspected IBD. Patients without evidence of a coagulation factor deficiency [i.e. normal levels of activated partial thromboplastin time (APTT), International normalised ratio, prothrombin time, fibrinogen, FVIII, von Willebrand factor antigen (VWF:Ag) and VWF activity measured with ristocetin cofactor activity or the glycoprotein (GP) Ib enzyme‐linked immunosorbent assay] and a significant BAT score (>3 in men, >5 in women, >2 in children), as well as patients suspected of an inherited thrombocytopenia, were included in the study. Prior to WES analysis, all patients from RH received oral and written information about WES and also the possible occurrence of incidental findings. All patients signed informed consent to publication of their data in concordance with the Helsinki Declaration, and permission for data publication was granted by the local ethics committee (H‐15006580) and the data registry (30‐1470). Patients from Malmö fulfilling the same inclusion‐ and exclusion criteria were included in the study approved by the local ethics committee (Dnr 2014/209). After oral and written informed consent and in concordance with the Helsinki Declaration, further blood samples were obtained for genetic sequencing. The diagnostic flow is illustrated in Fig 1.
Figure 1.
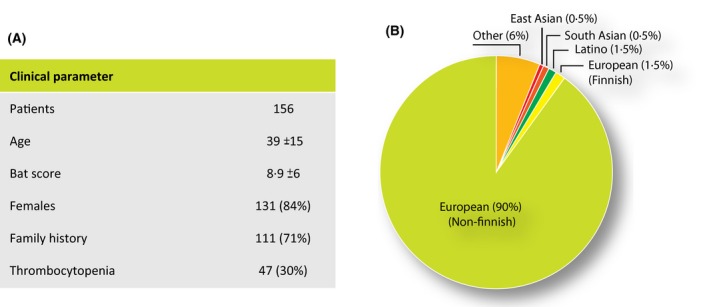
Patient characteristics. A) Clinical parameter. BAT (bleeding assessment tool). B) Patient ethnicity
Clinical evaluation
A family history including information on relatives with increased bleeding tendency or thrombocytopenia and consanguinity was obtained for all patients. Patient ethnicity was registered. A physical examination was performed, focussing on skin bruising, petechiae, skin elasticity and telangiectasia. In patients suspected of a collagen disorder, the Beighton joint hypermobility score was performed (Zeitoun et al, 2013). In patients with symptoms of HT, the oral cavity was thoroughly examined.
Selection of predisposing genes
Based on a continuous literature search between 2013 and 2016, 87 genes predisposing to bleeding disorders were selected, 63 of which were platelet related and 24 were non‐platelet related (Table 1 and Table S1). The non‐platelet related genes included coagulation factors, ADAMTS13, plasminogen activator inhibitor and alpha1‐antiplasmin. We also examined EDS and HT predisposing genes. As protein disulphide isomerase (PDI) was recently described to play a key role in clot formation (Jasuja et al, 2010), the P4HB gene was included to examine whether loss‐of‐function in P4HB could be a previously undiscovered cause of bleeding tendency. Finally, the search was expanded to genes associated with specific phenotypic (bleeding, thrombocytopenia, thrombocytopathy or symptomatic (hypermobility) search terms.
Table 1.
Genes predisposing to bleeding disorders
| Gene | Inheritance | Associated features | Blood smear | |
|---|---|---|---|---|
| Adhesion receptors | ||||
| Bernard‐Soulier syndrome biallelic | GP1BA; GP1BB; GP9 | AR | Severe thrombocytopenia | Giant platelets |
| Bernard‐Soulier syndrome monoallelic | GP1BA; GP1BB | AD | Mild thrombocytopenia | Large platelets |
| Platelet‐type von Willebrand disease | GPIBA gain‐of‐function | AD | Thrombocytopenia worse during pregnancy, infections, stress | Normal |
| Glycoprotein VI deficiency | GP6 | AR | Normal platelet count | Normal |
| Receptors for soluble agonists | ||||
| ADP receptor deficiency | P2RY12 | AR | Reduced response to ADP | Normal |
| Thromboxane A2 receptor | TBXA2R | AD | Reduced response to AA | Normal |
| Signalling pathways | ||||
| Cytosolic phospholipase A2 | PLA2G4A | AR | GI ulcers, low TBXB2 levels, reduced ADP and collagen response | Normal |
| G‐protein coupled receptors | P2RY1; F2R; F2RL3; ADRA2A | AR | Reduced response to ADP, TRAP | Normal |
| Ghosal syndrome | TBXAS1 | AR | Anaemia, increased bone density, reduced response to AA | Normal |
| Platelet secretion | ||||
| Hermansky‐Pudlak | HPS1; HPS3; HPS4; HPS5; HPS6; AP3B1; DTNBP1; BLOC1S3; BLOC1S6 | AR | Albinism, visual loss, immunodeficiency, neurologic symptoms | Normal |
| Chediak‐Higashi | LYST | AR | Infections, albinism, neurologic deterioration | Azurophil granules leukoc |
| Familial haemophagocytic lymphohistiocytosis, type 5 | STXBP2; UNC13D; STX11 | AR | Low fibrinogen levels, hearing loss, diarrhoea, infections | Normal |
| Grey platelet syndrome | NBEAL2 | AR | Splenomegaly, bone marrow fibrosis, high vitamin B12 | Grey platelets |
| Platelet‐type bleeding disorder 17 | GFI1B | AD | Dysplastic megakaryocytes | Grey platelets |
| ARC syndrome | VPS33B; VIPAS39 | AR | Developmental retardation | Absent alpha granules |
| Quebec platelet disorder | PLAU (gain‐of‐function) | AD | Increased fibrinolysis | Normal |
| Dense granule abnormalities | NBEA | AD | Autism | Reduced dense granules |
| SRC‐related grey platelet syndrome | SRC E527K (gain‐of‐function) | AD | Bone defects, facial dysmorphia | Reduced alpha granules |
| Platelet‐type bleeding disorder 12 | PTGS1 | AR | Reduced AA response, reduced TBXB2 level | Normal |
| Platelet aggregation | ||||
| Platelet‐type bleeding disorder 18 | RASGRP2 | AR | Reduced response to ADP, TRAP | Normal |
| LAD type III | FERMT3 | AR | Glanzmann‐like, immunodeficiency | Normal |
| Glanzmann thrombasthenia | ITGA2B; ITGB3; ITGA2 | AR | Severe bleeding | Normal |
| Procoagulant activity | ||||
| Scott syndrome | ANO6 | AR | Phosphatidylserine exposure deficiency | Normal |
| Small platelets thrombocytopenia | ||||
| Wiskott‐Aldrich syndrome; X‐linked thrombocytopenia | WAS | XL | Immunodeficiency, eczema | Small platelets |
| CARST syndrome | FYB | AR | Petechial rash | Small platelets |
| Large platelets thrombocytopenia | ||||
| MYH9‐related disorder | MYH9 | AD | Hearing loss, cataract, nephropathy | Giant platelets, Döhle bodies |
| GATA1 disorder | GATA‐1 | XL | Dyserythropoiesis, thalassaemia, leukaemia | Grey platelets |
| Sitosterolaemia | ABCG5; ABCG8 | AR | High cholesterol, tendon xanthomas, anaemia | Stomatocytes |
| Filamin related | FLNA | XL | Periventricular nodular heterotopia, epilepsy | Macrothrombocytes |
| TUBB1 related | TUBB1 | AD | Defective platelet microtubules | Macrothrombocytes |
| ACTN1 related | ACTN1 | AD | Impaired cytoskeleton | Macrothrombocytes |
| Platelet‐type bleeding disorder 19 | PRKACG | AR | Impaired cytoskeleton | Macrothrombocytes |
| Normal size thrombocytopenia | ||||
| Congenital amegakaryocytic | MPL | AR | Severe thrombocytopenia, bone marrow aplasia | Normal |
| THC2‐related | ANKRD26 5’ UTR; MASTL; ACBD5 | AD | Propensity to leukaemia, dysmegakarypoiesis | Normal |
| FDP/acute myeloid leukaemia | RUNX | AD | Propensity to leukaemia, aspirin‐like platelet defect | Abnormal alpha granules |
| THC4 | CYCS | AD | Cytochrome C gain‐of‐function, premature platelet release | Normal |
| FLI1‐related | FLI1 | AD | Abnormal alpha granules | Grey platelets |
| ETV6‐related | ETV6 (ETS binding site mutation) | AD | Red cell macrocytosis, haematological malignancies | Elongated alpha granules |
| CTRUS‐related | HOXA11 | AD | Radioulnar synostosis, deafness, skeletal abnormal | Normal |
| Stormorken syndrome | STIM1; ORAI1 | AD | Anaemia, asplenia, myopathy, headache, ichthyosis | Normal |
| SLFN14‐related | SLFN14 (GTP/ATP binding domain) | AD | Reduced dense granules | Normal |
| ANKRD18A‐related | ANKRD18A | AR | – | Normal |
| Tangier disease | ABCA1 | AR | Lack of HDL cholesterol, heart disease, splenomegaly | Normal |
| Collagen disorder | ||||
| Ehlers‐Danlos syndrome | COL5A1; COL3A1; COL1A1; COL5A2 | AD | Hypermobility, abnormal skin and scarring | Normal |
| Blood vessel abnormality | ||||
| Hereditary telangiectasia | ACVRL1; ENG; SMAD4 | AD | Epistaxis, GI bleeding, arteriovenous malformations | Normal |
| Rare coagulation factor deficiencies | ||||
| PAI1 deficiency | SERPINE1 | AR | Increased fibrinolysis | Normal |
| Alpha‐2 plasmin inhibitor deficiency | SERPINF2 | AR | Increased fibrinolysis | Normal |
| α2‐Antiplasmin deficiency | SERPINA2 | AR/AD | Increased fibrinolysis | Normal |
| Fibrinogen | FGA; FGB; FGG | AR | Dysfibrinogenaemia, abnormal fib‐TEG | Normal |
| FV | F5 | AR | Factor V deficiency | Normal |
| FVII | F7 | AD/AR | Factor VII deficiency | Normal |
| FX | F10 | AD/AR | Factor X deficiency | Normal |
| FXI | F11 | AD/AR | Delayed bleeding following trauma and surgery | Normal |
| FXIII | F13A1; F13B | AD/AR | Umbilical bleeding, ICH, abortions, late bleeding | Normal |
| FVIII | F8 | XL | Haemophilia A | Normal |
| FIX | F9 | XL | Haemophilia B | Normal |
| FII | F2 | AR | Prothrombin <10%, high INR | Normal |
| von Willebrand disease | VWF | AD | Von Willebrand syndrome | Normal |
| Inherited TTP | ADAMTS13 | AR | Thrombocytopenia, haemolysis, neurological symptoms | Normal |
| Protein disulphide isomerase | P4HB | ? | Defects in initiation of thrombus formation | Normal |
AA, arachidonic acid; AD, autosomal dominant; AR, autosomal recessive; ARC, Arthrogryposis‐Renal dysfunction‐Cholestasis; CARST, congenital autosomal recessive small‐platelet thrombocytopenia; CTRUS, Congenital thrombocytopenia and radio‐ulnar synostosis:GI, gastrointestinal; ICH, intracranial haemorrhage; INR, International Normalised Ratio; LAD, Leucocyte adhesion deficiency; THC, thrombocytopenia;XL, X‐linked.
Whole exome sequencing
The exon coverage of all 87 genes was evaluated from in‐house validation exome sequencing data with a minimum coverage of 50 × to ensure sufficient coverage of targeted genes. Genomic DNA was purified from whole blood using a QIAamp DNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. For enrichment of the exome either SeqCap EZ Exome v3 (Roche Nimblegen, Madison, WI, USA) or SureSelect All Exon Kit v4 or v5 (Agilent Technologies, Santa Clara, CA, USA) was used. Sequencing was conducted using the HiSeq2500 and NextSeq500 platforms from Illumina (San Diego, CA, USA). In brief, 1 μg of genomic DNA was fragmented on a Covaris S2 (Woburn, MA, USA) to an average size of 250 bp. Up until September 2015, the trimming, 3’‐adenylation and ligation of Illumina adaptors were performed on an SPRI‐TE nucleic acid extractor using SPRIworks Fragment Library Cartridges I (Beckman Coulter, Brea, CA, USA) with a size selection of 200–400 bp. From September 2015, the trimming, 3’‐adenylation and adaptor ligations were done on a Sciclone G3 robot (Perkin Elmer, Waltham, MA, USA) using Illumina‐compatible KAPA library DNA adaptors (Roche Diagnostics, Basel, Switzerland). Sequencing was performed as paired‐end sequencing, 2 × 101 bases or 2 × 151 bp, resulting in approximately 100 mol/l paired‐end reads. Sequencing data were processed using Consensus Assessment of. Sequence And Variation (CASAVA) v1.8.2 (Illumina, San Diego, CA, USA).
Data processing
Generated fastq files were uploaded to CLC bio (Genomics Workbench v2.5.1; Qiagen) for trimming of the last 3’ base, mapping and variant calling. All the algorithms applied to the samples were custom made by CLC bio. The reads were mapped to the reference human genome hg19/GRCh37 with custom algorithm CLC4 (https://www.qiagenbioinformatics.com/products/clc-genomics-workbench/) for a comparison between the CLC4 algorithm and the most commonly known aligners. Quality control of the target sequencing was performed on the read mappings. The variant calling was performed by a Maximum Likelihood approach on a Bayesian model. Variants were called with a minimum of 9 × coverage, 3 counts and 25% frequency. Variants were further filtered using the Ingenuity Variant Analysis tool (Qiagen), excluding the low quality scores and variants (>1% in background population), and including variants found within the 87‐gene list and variants associated with bleeding, thrombocytopenia and thrombocytopathy as well as specific symptomatic associations.
Variant classification and in silico analysis
The initial classification of identified variants was performed according to the five‐tier scheme as recommended by The American College of Medical Genetics and Genomics (Richards et al, 2015): class 5 is pathogenic; class 4 probably pathogenic; class 3 uncertain significance; class 2 probably benign; and class 1 benign. To select VUS for specific functional testing, we developed the Öresund clinical score, which combines the Mendelian inheritance pattern with the results of four computational analyses (Table 2). We filtered out heterozygous variants in genes known to have a recessive Mendelian inheritance pattern. For further filtering, all the class 3 variants were subjected to computational predictive analyses. Given that the algorithms for missense variants are not entirely identical and guidelines recommend a combination of the in silico tools (Richards et al, 2015), we applied four different predictive tools: Align GVGD (A‐GVGD); PolyPhen‐2; MutationTaster; and SIFT (Tavtigian et al, 2006; Adzhubei et al, 2010; Kumar et al, 2009; Schwarz et al, 2010). If a VUS scored as ‘damaging’ in three of the four predictive tools, we considered the computational analysis as significant and thus viewed the VUS as a possible candidate variant for downstream functional testing. The frequency of the variants was obtained from the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) or the Exome Sequencing Project (ESP, https://embedthis.com/esp/doc/users/database.html) databases. Candidate variants with an Öresund clinical score >2, selected as described above, were examined by available specific functional tests [such as flow cytometry (FC), transmission electron microscopy (TEM), immunohistochemistry and plasma‐thromboxane B2]. Variants of unknown significance with Öresund score 1‐2 were left as class 3.
Table 2.
Öresund clinical score
| Score | Inheritance | Variants of unknown significance (VUS) | In silico analysis |
|---|---|---|---|
| 4 | AR | Homozygous;a | Damaging |
| 4 | AD | Heterozygous;a | Damaging |
| 4 | XL | Hemizygous;a | Damaging |
| 3b | AR | Homozygous;b | Damaging |
| 3a | AD | Heterozygous;b | Damaging |
| 3a | XL | Hemizygous;b | Damaging |
| 2b | AD | Heterozygous;b | Benign |
| 2b | XL | Hemizygous;b | Benign |
| 2a | AR | Heterozygous;b | Damaging |
| 1 | AR | Heterozygous;b | Benign |
AD, autosomal dominant; AR, autosomal recessive; XL, X‐linked.
Previously reported or supported by screening tests.
Not previously reported or validated by functional tests.
Initial functional testing
A peripheral blood smear was performed in all patients with thrombocytopenia. These smears were evaluated by the CellaVision™ software (CellaVision‐DM1200; Sysmex, Kobe, Japan). Giant platelets were defined as abnormal platelets when the size of erythrocytes or larger. Macrothrombocytes were defined as abnormal platelets when the size was between 66% and 100% that of erythrocytes. Multiplate analyses were performed on whole blood following stimulation by four different agonists on a multiplate analyser (Verum Diagnostica GmbH, Munich, Germany) (Michelson, 2009; Albanyan et al, 2015). Light transmission aggregometry (LTA) was performed according to routine clinical protocols (Cattaneo et al, 2013; Pai et al, 2011). Subjects with a platelet count <100 × 109/l were excluded from the LTA and multiplate analyses.
Specific functional testing
Assessment of platelet activation by FC was performed as previously described (Nishibori et al, 1993; Stenberg et al, 1985; Shattil et al, 1985). Briefly, platelet activation and granule release were investigated by FC using markers for CD63, CD62P and activated GPIIb/IIIa. For each patient, aliquots of whole blood (20 μl) were incubated with 20 μl of CD42b‐APC antibodies (Cat. No. 551061; Becton Dickinson [BD], Stockholm, Sweden) for 15 min at room temperature. Subsequently, 40 μl of diluted blood was incubated with 40 μl of platelet‐activating agonists and a 20‐μl mix of markers for degranulation and activation. The following agonists and concentrations were used: ADP (Cat. No. 58‐64‐01; Sigma‐Aldrich, Stockholm, Sweden) final concentration (CF) 10 μmol/l and 1 μmol/l; CRP‐XL (from Dr. Richard W. Farndale, University of Cambridge, UK) CF 20 μg/ml and 2 μg/ml; collagen (CHRONO‐PAR Cat. No. N‐385) 1 mg/ml and TRAP‐6 (Cat. No. N‐8365; Bachem, Bubendorf, Switzerland) CF 25 μmol/l and 5 μmol/l. The following activation markers were used: 5 μl PAC‐1‐FITC (Cat. No. 340507; BD); 5 μl CD62P‐PE (Cat. No. IM1759u; Beckman Coulter, Bromma, Sweden); 2·5 μl CD63‐V450 (Cat. No. 561984; BD). The samples were incubated at room temperature for 15 min and fixed through incubation with 0·6% CellFIX (BD) for 10 min. The samples were diluted in Isoflow (Beckman Coulter) before being analysed using a Gallios or Navios flow cytometer (Beckman Coulter). In the FC, the laser setting W2 was used and data were analysed using Kaluza Flow Cytometry Software (Beckman Coulter). Platelet activation and degranulation were expressed as percentage of cells positive for each activation marker or mean fluorescence intensity. LTA and FC analyses of platelet receptors were performed before genetic analysis in patients from Malmö.
Transmission electron microscopy
Platelet‐rich plasma for TEM was prepared using the following protocol. Blood was collected into 2 × 5 ml sodium citrate 3·2% tubes and centrifuged at 200 g for 10 min at 20°C. The plasma fraction was transferred to a plastic tube with 2% glutaraldehyde in 0·05 mol/l phosphate buffer. The sample was fixed for 1 h before being centrifuged at 200 g for 10 min at 20°C. Samples were further processed by the Core Facility for Integrated Microscopy (CFIM), University of Copenhagen, Denmark, where they were fixed with 2% glutaraldehyde in 0·05 mol/l phosphate buffer for at least 24 h at 4°C. They were then washed three times in 0·12 mol/l cacodylate buffer before being post‐fixed in 1% OsO4 and 0·05 mol/l (1·5%) K3Fe(CN)6 in 0·12 mol/l cacodylate buffer for 2 h at room temperature. Samples were washed three times with water before being incubated with 1% uranyl acetate for 1 h at 4°C. Following another three washes with water, they were dehydrated through a graded series of alcohol. Samples were infiltrated in a resin and propylene oxide gradient before being infiltrated with pure resin. They were then embedded in 100% resin for 24 h at 60°C. Ultra‐thin sections (50 nm) were cut with a Leica EM UC7 microtome and mounted for examination at 80 kV using a Philips CM100 transmission electron microscope. Images were obtained using an Olympus Veleta camera system and iTEM FEI software. Patient samples were documented by images from 50 sections, where each image contained at least one platelet. Platelet sections from 10 healthy controls were also assessed in the same manner. The sections were visually assessed with respect to platelet size as well as the presence of alpha granules and dense bodies.
Whole‐mount electron microscopy with thrombin stimulation
For evaluation of dense granule secretion in patients with missense variants in familial haemophagocytic lymphohistiocytosis (FHL)‐associated genes (UNC13D, STX11, STXBP2), platelet‐rich plasma (prepared as described above) was stimulated with thrombin (0·1 iu/ml for 3 min) and examined by whole‐mount TEM (White, 2004; Al et al, 2012). The numbers of dense granules per platelet before and after thrombin stimulation were counted in patients and in a control group (20 healthy adults). Fifty images were evaluated from each patient and control. Results were interpreted as the ratio of dense granule numbers before and after thrombin stimulation. An aberrant result of >3 standard deviations (SD) of the control group was considered to be an indication of decreased dense granule secretion (Fager et al, 2017).
Immunohistochemistry
To assess platelets and granulocytes, blood smears were either stained with May‐Grünwald‐Giemsa stain or subjected to immunofluorescence analysis using a rabbit polyclonal antibody against non‐muscle myosin IIA (Sigma‐Aldrich Chemie GmbH, Steinheim, Germany).
Multidisciplinary team conference
Each patient's genetic and functional test results in conjunction with their clinical phenotype were discussed amongst the referring clinicians, a genomic biochemist, a clinical biochemist and a clinical geneticist at a multidisciplinary team (MDT) conference.
Results
Patient characteristics
In total, 156 patients (120 from RH and 36 from Malmö) were included in the study. The mean age was 39 ± 15 years (mean ± 1SD) and 131 (84%) were female. The majority of patients (71%, 111/156) had a positive family history of increased bleeding tendency and 30% (47/156) had thrombocytopenia (defined as a platelet count below the normal range). The majority of patients were of European ethnicity (90%). The mean BAT score was 8·9 ± 6 (1SD) (Fig 1).
Efficacy of WES in diagnostics of IBD
The mean coverage of the exomes of the samples was 84 × , where 96·96% of the regions were covered with at least 9 × . In total, 353 predominantly missense variants (2·3 variants per patient) were detected in 80 of the 87 genes (Table S2), and at least one variant within the selected 87 genes was identified in 95% (148/156) of the patients. We found that 5% (8/156 patients) harboured a known pathogenic variant and were diagnosed with a bleeding disorder as a direct consequence of WES. Of the 345 previously unreported variants, 29% (99) were predicted to be significant by computational analyses. According to the known Mendelian inheritance pattern of each gene, 17% (59 variants) in 38 patients were predicted as being significant VUS and assigned an Öresund clinical score >2, thus designating them as relevant candidates for further investigations. Variants of unknown significance with Öresund score 1–2 were left as class 3. Considering patients diagnosed as a consequence of WES, as well as those harbouring variants selected for further investigations, 29% (46 patients) of our cohort harboured variants or VUS that could explain or contribute to IBD. As a result of targeted functional testing and clinical EDS evaluation, 20/59 (34%) VUS with an Öresund clinical score >2 were assigned as novel class 4 or 5 variants. Furthermore, a class 4 or 5 variant was identified in 30% (14/47) of patients with thrombocytopenia versus 11% (12/109) of patients with a normal platelet count (P < 0·01).
Known pathogenic variants
Table 3 lists all the known pathogenic variants. The patient characteristics were as follows.
Table 3.
Known pathogenic variants
| Patient | Platelet count (× 109/l) | Gene | Zygosity | Variant | AA change | Type | References |
|---|---|---|---|---|---|---|---|
| 68 | 131 | F8 | Het | c.3637delA | p.Ile1213Phefs*5 | fs | Lin et al (1993) |
| 1 | 127 | F11 | Het | c.1288G>A | p.Ala430Thr | m | Liu et al (2015) |
| 46 | 106 | GATA1 | Hemi | c.647G>A | p.Arg216Gln | m | Tubman et al (2007) |
| 117 | 245 | GATA1 | Het | c.647G>A | p.Arg216Gln | m | Tubman et al (2007) |
| 1 | 127 | ITGB3 | Hom | c.31T>C | p.Trp11Arg | m | Nurden et al (2015) |
| 60 | 43 | MYH9 | Het | c.5521G>A | p.Glu1841Lys | m | Saposnik et al (2014) |
| 64 | 21 | RUNX1 | Het | c.602G>A | p.Arg201Gln | m | Latger‐Cannard et al (2016) |
| 102 | 84 | RUNX1 | Het | c.602G>A | p.Arg201Gln | m | Latger‐Cannard et al (2016) |
| 23 | 292 | VWF | Het | c.2435delC | p.Pro812Argfs*31 | fs | Zhang et al (1992) |
Het= heterozygous, Hom=homozygous, Hemi= hemizygous, AA=amino acid, fs=frames‐shift variant, m=missense variant.
Patient 1
We identified a diagnosis of Glanzmann thrombasthenia in this patient with mild thrombocytopenia (homozygosity of ITGB3 (c.31T>C, p.Trp11Arg)) in whom multiplate analysis demonstrated absent aggregation with all agonists employed. This patient was also diagnosed with a mild factor XI deficiency (heterozygosity of F11 (c.1288G>A, p.Ala430Thr)) with reduced levels of FXI (0·49 iu/l) (Ref.: 0·67–1·27 iu/l). For severe bleeding and major surgery, activated factor VII would be the choice for haemostatic treatment in this patient.
Patients 46 and 117
An X‐linked variant in transcription factor GATA‐1 (c.647G>A, p.Arg216Gln) was found in Patient 117, a 32‐year‐old male with macrothrombocytopenia and severe bleeding tendency. TEM demonstrated clearly reduced numbers of alpha granules (Fig 2). Red blood cell morphology was normal. The same variant was found in his mother, who had a normal platelet count but a severe lifelong bleeding tendency. Patient 46 was diagnosed with X‐linked macrothrombocytopenia without dyserythropoiesis. He has two daughters and his family will undergo genetic counselling.
Figure 2.

Transmission Electron Microscopy images. (A) Control: Normal size platelet with average alpha granule content. (B) Image from patient with a GATA1 variant demonstrating a representative macrothrombocyte and empty alpha granules (E). C and D: Representative platelets from a patient with an FLI1 variant showing a reduced amount of alpha granules, a giant alpha granule (G) and open platelet structures filled with glycogen particles [“scrolls” (S)] in the cytoplasm. M, mitochondria.
Patients 64 and 102
Two unrelated patients with normal size thrombocytopenia harboured a RUNX1 (c.602G>A, p.Arg201Gln) variant. The older patient (Patient 64) had a lifelong history of severe thrombocytopenia and bleeding tendency and she developed myelodysplasia when aged 65 years. She had no children and did not wish to undergo genetic counselling. However, she had always speculated on the cause of her illness, which now appeared well resolved. The younger patient (Patient 102) had a brother and father with chronic thrombocytopenia who both harboured the same RUNX1 variant found by polymerase chain reaction analysis and Sanger sequencing. Neither the younger proband (Patient 102) nor her RUNX1 variant‐positive relatives have been diagnosed with myelodysplastic syndromes, but they will undergo genetic counselling and haematological vigilance.
Patient 23
In this patient, with previously unresolved IBD (BAT score 7) and grey zone levels of VWF [VWF:Ag 0·39 iu/l (Ref.: 0·58–1·69 iu/l) and Ristocetin cofactor (VWF:RCo) 0·43 iu/l (Ref.: 0·52–1·58 iu/l)], a frameshift variant in VWF (c.2435delC, p.Pro812Argfs*31) was detected, and she was consequently diagnosed with von Willebrand disease (VWD) type 1. She now receives appropriate desmopressin treatment and genetic counselling and carries a von VWD bleeder ID card.
Patient 60
A patient with inherited blindness (Leber congenital amaurosis), thrombocytopenia, giant platelets and leucocyte inclusion bodies in the peripheral blood smear harboured an MYH9 (c.5521G>A, p.Glu1841Lys) variant. This patient was referred for WES as part of a family investigation, because three of her first and second degree relatives had severe bleeding tendency. None of her referred relatives had thrombocytopenia or MYH9 variants and their bleeding cause remains unsolved. Patient 60 did not complain of hearing loss and no proteinuria was detected. The 1841 variant is only rarely associated with renal problems; however, it is recommended to test renal function about once a year and to perform a hearing examination every 5 years in patients with MYH9‐associated thrombocytopenia (Althaus & Greinacher, 2010).
Patient 68
An FVIII variant (c.3637delA, p.Ile1213Pfs*5) was found in Patient 68 who had slight thrombocytopenia (131 × 109/l) and grey platelets in the peripheral blood smear. Her brother had severe haemophilia A, however Patient 68 had never undergone genetic testing prior to WES. Thus her carrier status was established, facilitating pregnancy‐related genetic counselling. However, no genetic cause of the thrombocytopenia was found. The FVIII level in Patient 68 was 0·56 iu/l (Ref.: 0·50–1·5 iu/l).
Novel pathogenic variants (class 5)
As a result of further variant filtering by the Öresund clinical score and targeted functional testing, 20 variants were assigned as novel class 4 or 5 variants (Table 4).
Table 4.
Novel class 4 and 5 variants
| Patient | Platelet count (×109/l) | BAT score | Gene | Variant | Frequency (%) | In silico prediction | Result of functional tests | Diagnosis | Variant class |
|---|---|---|---|---|---|---|---|---|---|
| 127 | 284 | 8 | COL1A1 | c.4018G>A, p.Gly1340Ser | – | Damaging | EDS classical phenotype | Ehlers‐Danlos syndrome | 4 |
| 123 | 282 | 5 | COL1A1 | c.436C>A, p.Pro146Thr | 0 | Damaging | EDS classical phenotype | Ehlers‐Danlos syndrome | 4 |
| 54 | 224 | 17 | COL5A1 | c.193C>T, p.Arg65Trp | 0·19 | Damaging | EDS classical phenotype | Ehlers‐Danlos syndrome | 4 |
| 33 | 334 | 4 | F11 | c.599G>A, p.Cys200Tyr | 0·0082 | Damaging | FXI 0·44 × 103 iu/l (Ref.: 0·67–1·27) | Mild FXI deficiency | 5 |
| 106 | 131 | 10 | FLI1 | c.1019G>C, p.Arg340Pro | – | Damaging | Macrothrombocytes; TEM: abnormal alpha granules; family segregation studies confirmative | FLI1‐related thrombocytopathy | 4 |
| 13 | 44 | 5 | GP1BA | c.247C>T, p.Leu83Phe | 0·00083 | Damaging |
Giant platelets FC: reduced expression of GP1BA (CD42b) |
Monoallelic Bernard‐Soulier syndrome | 4 |
| 49 | 68 | 2 | GP1BA | c.58T>G, p.Cys20Gly | – | Damaging | Macrothrombocytes; 50% giant platelets; FC: reduced expression of GP1BA (CD42b) | Monoallelic Bernard‐Soulier syndrome | 4 |
| 3 | 200 | 7 |
ITGB3
ITGB3 |
c.1192delG, p.Ala398Profsa24 c.1639T>G, p.Cys547Gly |
– – |
Damaging Damaging |
LTA: absent aggregation following stimulation with all agonists except ristocetin | Glanzmann thrombasthenia |
5 5 |
| 21 | 143 | 14 |
ITGB3
ITGB3 |
c.567delA, p.Tyr190Thrfsa17 c.1525C>T, p.Gln509a |
– – |
Damaging Damaging |
LTA: absent aggregation FC: low PAC‐1 expression |
Glanzmann thrombasthenia |
5 5 |
| 133 | 106 | 2 | MYH9 | c.3493C>T, p.Arg1165Cys | – | Damaging | Immunohistochemistry: leucocyte inclusion bodies present | MYH9‐related macrothrombocytopenia | 5 |
| 16 | 183 | 10 | NBEA | c.8350G>T, p.Val2784Phe | 0·61 | Damaging |
TEM: reduced dense granules Multiplate: reduced response to AA |
Delta storage pool deficiency | 4 |
| 92 | 347 | 11 | NBEA | c.8350G>T, p.Val2784Phe | 0·61 | Damaging | TEM: reduced, atypical dense granules Decreased ATP release by LTA | Delta storage pool deficiency | 4 |
| 151 | 219 | 11 | NBEA | c.1185A>C, p.Glu395Asp | 0·15 | Damaging | FC: reduced expression of CD63 after stimulation with ADP, TRAP and collagen | Delta storage pool deficiency | 4 |
| 76 | 59 | 8 | NBEAL2 a | c.4928_4929delAT, p.Asp1643Glyfsa34 | – | No data |
FC: reduced CD62P expression Large grey platelets in blood smear High B12 levels |
Grey platelet syndrome | 5 |
| 20 | 382 | PTGS1 | c.337C>T, p.Arg113Cys | 0·05 | Damaging | ELISA: reduced TBXB2 levels | Prostaglandin synthase‐1 deficiency | 4 | |
| 136 | 61 | 4 | RUNX1 | c.308C>T, p.Pro103Leu | – | Damaging |
FC: reduced CD63 expression Family segregation studies confirmative |
RUNX1‐related thrombocytopenia | 4 |
| 62 | 215 | 9 | UNC13D | c.466C>T, p.Arg156Trp | 0·016 | Damaging | WM TEM: reduced secretion of dense granules upon thrombin stimulation LTA: abnormal aggregation with ADP | Dense granule secretion deficiency | 4 |
| 47 | 130 | 3 | WAS b | c.1208C>T, p.Pro403Leu | 0·023 | Damaging | Small platelets; immune deficiency: reduced IgA and IgM | X‐linked thrombocytopenia | 4 |
AA, arachidonic acid; EDS,; ELISA, enzyme‐linked immunosorbent assay; FC, flow cytometry; LTA, light transmission aggregometry; TEM, transmission electron microscopy; WM TEM, whole mount transmission electron microscopy.
Homozygous.
Hemizygous.
Patient 76
A homozygous frameshift variant in NBEAL2 (c.4928_4929delAT, p.Asp1643Glyfs*34) was concluded to cause grey platelet syndrome in this 32‐year‐old female with a platelet count of 59 × 109/l and grey macrothrombocytes in the peripheral blood smear. She had previously been misdiagnosed with idiopathic thrombocytopenic purpura and treated with intravenous immunoglobulin and prednisone during pregnancy without response. FC analysis confirmed that the CD62P expression on activated platelets was reduced. Her B12 levels were supranormal (>1200 μmol/l). Patient 76 will receive platelet transfusions and tranexamic acid as haemostatic treatment related to severe bleeding and surgery, genetic counselling and haematological surveillance for development of splenomegaly and bone marrow fibrosis.
Patient 133
A novel MYH9 (c.3493C>T, p.Arg1165Cys) variant was identified in Patient 133 with macrothrombocytopenia. Leucocyte inclusion bodies were not detected in the peripheral blood smear but were demonstrated by immunohistochemistry, thus revealing the morphology of an MYH9‐associated syndrome. Consequently, this patient will be examined for proteinuria, cataract and hearing loss, while severe bleeding and surgery will be treated with desmopressin and tranexamic acid as prophylactic platelet transfusions are rarely indicated in MYH9‐related thrombocytopenia.
Patient 21
Compound heterozygosity of two variants in ITGB3 (c.567delA, p.Tyr190Thrfs*17 and c.1525C>T, p.Gln509*) was discovered in this 71‐year‐old male with a lifelong bleeding tendency, slightly reduced platelet count (143 × 109/l) and a previously unsettled diagnosis, with the presence of post‐platelet transfusion glycoprotein antibodies. Platelet aggregation was absent by multiplate evaluation. FC demonstrated <5% GPIIb/IIIa expression and thus confirmed a suspected diagnosis of Glanzmann thrombasthenia. Therefore, activated factor VII will be the choice of haemostatic treatment for this patient for major bleeding and surgery.
Patient 3
Compound heterozygosity of two ITGB3 variants (c.1192delG, p.Ala398Profs*24 and c.1639T>G, p.Cys547Gly) and absent platelet aggregation by LTA with all agonists employed except ristocetin also confirmed a suspected diagnosis of Glanzmann thrombasthenia in this patient with severe bleeding tendency and a normal platelet count.
Patient 33
A patient with a normal APTT (33 s; Ref.: 28–35 s) harboured a variant in F11 (c.599G>A, p.Cys200Tyr). The bleeding tendency in Patient 33 was characterized by bruising and oozing following surgical procedures. Specific testing of the FXI level showed a reduced level (0·44 iu/l; Ref.: 0·62–1·0 iu/l). No other cause of bleeding was found. Consequently, this patient was concluded to have mild FXI deficiency, thus indicating treatment with tranexamic acid and fresh frozen plasma during episodes of severe bleeding, trauma or surgery.
Novel class 4 variants
Patient 47
A WAS variant (c.1208C>T, p.Pro403Leu) was detected in a young male suffering from a syndrome with muscle dystonia, mild immune deficiency (reduced IgM and IgA), infections of the skin and a slightly reduced platelet count (130 × 109/l). Neither of his parents had thrombocytopenia. The peripheral blood smear demonstrated small platelets and his phenotype was identical to an X‐linked thrombocytopenia. The variant was designated as a class 4 because Wiskott‐Aldrich Syndrome protein expression was not analysed. Thus, the cause of immunodeficiency in this patient was probably resolved by WES.
Patient 106
A variant in the gene encoding the transcription factor friend leukaemia integration 1 (FLI1; c.1019G>C, p.Arg340Pro), located in a highly conserved DNA‐binding domain, was found in this male patient with macrothrombocytopenia (platelet count 131 × 109/l) and severe bleeding tendency (BAT score 10). TEM demonstrated abnormal alpha granules with rare scrolls resembling the open platelet structures filled with glycogen particles previously described in Medich giant platelet syndrome (Gunning et al, 2013) (Fig 2C and D). Family linkage studies showed that the mother of Patient 106 and her two sisters with thrombocytopenia harboured the same FLI1 variant. The platelet counts of one of his mother`s sisters and of his half‐brother were normal and they both carried an FLI1 wildtype.
Patients 16, 92 and 151
A recurrent variant in NBEA (c.8350G>T, p.Val2784Phe) was identified in two unrelated patients with normal platelet counts. They both exhibited a reduced number of dense granules on TEM examination. Decreased ATP release by LTA was demonstrated in one patient (Patient 92) and the other patient (Patient 16) had a reduced response to arachidonic acid and collagen by multiplate analysis. Hence, the variant was concluded to be probably pathogenic. Given that the brother of Patient 16 required bone marrow transplantation, a search for an unrelated donor was initiated and identified due to the suspicion of an inherited platelet disorder. Another NBEA variant (c.1185A>C, p.Glu395Asp) was identified in Patient 151, who had a family history, lifelong bleeding tendency (BAT score 11) and a recent diagnosis of antiphospholipid antibody syndrome (APAS) with multiple arterial thromboses. She therefore received warfarin treatment and experienced prolonged and transfusion‐demanding bleeding following surgery, even though warfarin was appropriately paused preoperatively and she was not treated with antiplatelet agents. FC demonstrated impaired expression of CD63 and TEM showed a reduced amount of dense granules. A probable diagnosis of delta storage pool deficiency was concluded. Due to APAS and risk of thrombosis, Patient 151 cannot be treated with tranexamic acid or desmopressin for future surgeries, and thus enhanced surgical haemostatic procedures should be ensured.
Patients 49 and 13
A previously unreported heterozygous GP1BA variant (c.58T>G, p.Cys20Gly) was identified in a patient (Patient 49) without significant bleeding tendency but with chronic thrombocytopenia (68 × 109/l) and 50% giant platelets in the peripheral blood smear,. FC showed reduced expression of GP1BA and normal expression of platelet activation markers (CD62P, PAC‐1, CD63). A probable diagnosis of monoallelic Bernard‐Soulier syndrome (BSS) was therefore concluded. Three more patients with low platelet counts (44–94 × 109/l) and giant platelets in the peripheral blood smear harboured a shared rare heterozygous variant in GP1BA (c.247C>T, p.Leu83Phe). In one of these patients (49), FC showing reduced levels of GP1BA was again diagnostic of monoallelic BSS. The two other patients are pending further functional testing.
Patient 136
A RUNX1 (c.308C>T, p.Pro103Leu) variant was detected in this 38‐year‐old woman with thrombocytopenia. FC analysis of her platelet function showed impaired CD63 expression, indicating dense granule deficiency. Moreover, her son, father, grandfather and cousin suffered from bleeding symptoms and thrombocytopenia, and her sister was diagnosed with myelodysplastic syndrome. An identical RUNX1 variant was found in her six‐year‐old son, who had a platelet count of 79 × 109/l. The family will receive haematological surveillance and genetic counselling.
Patient 20
A thromboxane synthase (PTGS1) variant (c.337C>T, p.Arg113Cys) was identified with another PTGS1 variant (c.1003G>A, p.Val481Ile) with a relatively high frequency in the background population (0·77%) in Patient 20, who had a normal platelet count and severe bleeding tendency. Her levels of thromboxane B2 (TBXB2) in plasma were reduced compared to controls and a probable diagnosis of prostaglandin synthase‐1 deficiency was concluded. Patient 20 has been advised to avoid antiplatelet agents and non‐steroidal anti‐inflammatory drugs and receives tranexamic acid treatment for minor bleeding. A family investigation of her two sons is ongoing, as they both present with bleeding problems.
Patients 127, 123 and 54
Three patients harboured collagen variants [Patient 127: COL1A1 (c.4018G>A, p.Gly1340Ser), Patient 123: COL1A1 (c.436C>A, p.Pro146Thr) and Patient 54: COL5A1 (c.193C>T, p.Arg65Trp)] that were classified as probable pathogenic class 4 variants because the patients fulfilled the clinical criteria of classical type of EDS based on a clinical evaluation performed by a rheumatologist expert in EDS. Patient 127 also harboured a damaging variant in COL3A1 but Patient 127 did not fulfill the clinical criteria of vascular of type EDS, thus the COL3A1 variant was left as a class 3. The treatment of EDS requires an MDT and Patients 127, 123 and 54 will be referred to rheumatologist care and physical therapy. Haemostatic treatment includes tranexamic acid, desmopressin and meticulous stitching following invasive procedures.
Patient 62
Studies in rodents have indicated an increased bleeding tendency in heterozygous carriers of variants in genes associated with FHL due to the impaired release of procoagulant mediators by platelet degranulation (Ren et al, 2010). These findings prompted us to conduct specific functional testing in 12 patients with heterozygous VUS with an Öresund clinical score of 2a in the FHL‐associated genes UNC13D, STX11 and STXBP2. Impaired secretion from platelet dense granules by at least one of the assays (whole‐mount TEM, LTA and FC) was confirmed in eight of the patients as compared to healthy controls. In patient 62, harbouring UNC13D (c.466C>T, p.Arg156Trp), this variant was strongly associated with impaired dense granule secretion following thrombin stimulation. Consequently, Patient 62 was assigned a probable diagnosis of impaired platelet dense granule secretion linked to a heterozygous UNC13D variant (Fager et al, 2017).
Discussion
Whole exome sequencing is rapidly being implemented as a diagnostic tool for a wide spectrum of haematological diseases (Camps et al, 2016; Hansen et al, 2016; Leinoe et al, 2016; Zhang et al, 2016). Prompted by the diagnostic conundrum for the majority of IBD ‐ suspected patients, we adopted WES into the diagnostic strategy at our institutions. This approach was made possible due to a pre ‐ established in‐house genomic centre and an existing close collaboration between the two coagulation units localized in the Öresund Region. To date, in the diagnostic work‐up of rare IBD, NGS‐based gene panels have been implemented by two large research collaborations: the ThromboGenomics platform (Simeoni et al, 2016) and the GAPP study group (Johnson et al, 2016). We implemented WES in our clinics, allowing consecutive expansion of the number of genes analysed. We included both platelet‐ and non‐platelet‐related genes in the IBD gene panel and report a higher number of class 4 and 5 variants in patients with thrombocytopenia compared to those with normal platelet counts. The genetic diagnosis of rare IBD with normal platelet counts is hampered by a knowledge gap, while it is estimated that the genetic causes of thrombocytopenia have been identified in approximately 50% of patients (Johnson et al, 2016). One of the greater challenges we encountered in the process was the classification of VUS in the many relatively new genes and their subsequent translation into clinical practice. Hence, we developed the Öresund clinical score as a simple pragmatic approach, incorporating in silico predictions with inheritance patterns to assess the possible clinical relevance of the identified variants and to select for downstream specific functional testing. The Öresund clinical score led to 59 possible clinically relevant VUS in 38 patients, of which 20 (34%) were assigned as novel class 4 or 5 variants. To address the constraint that in silico analyses are never 100% accurate (Thusberg et al, 2011), we combined four in silico methods for variant filtering to direct the specific functional studies. Nevertheless, the selection of variants for downstream functional testing using the Öresund clinical score may have caused false negative results. The clinical usability of the Öresund clinical score needs to be evaluated in an independent patient cohort, where functional testing of all identified VUS is performed.
Eight patients (5%) were diagnosed as a direct consequence of WES, and 20 previously unreported class 4 or 5 variants were identified in 12% (18/156) of patients. An IBD diagnosis has a major clinical impact. The genetic investigations detailed here extricated our patients from a diagnostic conundrum. The remaining 83% (130 patients) rendered without a proper diagnosis may harbour pathogenic variants in unknown genes or regulatory regions or their IBD might result from a multigenic inheritance mode. Exome sequencing carries the fundamental limitation of not being able to assess the impact of regulatory and non‐coding regions. The genetic background of IBD is likely to be heterogeneous with contributions from multiple genetic alterations, which may also interact with acquired factors such as comorbidity and medications (Watson et al, 2013). Combined heterozygous variants, particularly in genes involved in signalling pathways, platelet granule trafficking or granule biogenesis, may result in a severe bleeding phenotype. Thus, extended functional testing of compound heterozygous variants in the same biological pathways warrants further investigations.
Fourteen patients (9%) had a VUS in collagen genes (COL5A1, COL5A2, COL3A1 and COL1A1) predicted to be damaging by in silico analyses. However, only three collagen variants were classified as probable pathogenic class 4 variants in patients fulfilling the clinical criteria of autosomal dominant inherited EDS. Collagen mediates platelet adhesion and activation following injury to the vascular wall and is essential for the maintenance of haemostasis (Manon‐Jensen et al, 2016). It is currently unclear whether collagen missense variants are causal of the bleeding tendency in patients not fulfilling the clinical EDS criteria. Of particular interest, only one of our patients had a (benign) variant in the gene (P4HB) encoding PDI, indicating that PDI variants were not a common cause of bleeding disorders in our patient cohort. We anticipate that an additional number of our patients will be diagnosed by WES, especially those patients with a normal platelet count, given the considerable efforts that are currently being directed towards the discovery of new causal variants in IBD patients (Bariana et al, 2017; Johnson et al, 2016; Simeoni et al, 2016).
It is highly probable that numerous bleeding‐associated genes will be discovered in the coming years. These efforts will inevitably lead to an optimized annotation of novel variants in the increasing number of genes found to predispose to bleeding, and will therefore allow early genetic analysis to be more efficiently translated into clinical practice. The advantage of WES, in comparison to custom capture gene panels, is that it enables expansion by analysing newly assigned IBD‐predisposing genes. The disadvantages of WES analysis in a diagnostic clinical setting are that it requires a genomic sequencing laboratory in combination with data scientist with NGS‐data analysis expertise. Once an atomized pipeline has been established, the manual labour of a WES analysis is in line with a single gene analysis. For example, in our setting, a WES analysis including diagnostic report can be ready within three weeks from blood sample submission. The expenses of a WES analysis are cost effective provided that the diagnosis was not established by the primary genetic analysis (Stark et al, 2017). Implementing WES at an early phase of the work‐up of IBD may eliminate the need for complex and expensive laboratory analyses. However, translating the comprehensive results of genetic analysis into haematological practice requires an efficient multidisciplinary pipeline represented by a number of specialized centres, e.g. genomic medicine, integrated microscopy and FC, coagulation laboratory, clinical genetics and rheumatology. The multidisciplinary efforts are particularly relevant when the examined genes include syndromes with bleeding phenotypes not associated with a platelet disorder.
In conclusion, we found that WES of predefined genes is a powerful tool for elucidating potential pathogenic variants in patients with IBD and a feasible method to direct specific functional testing by FC and TEM. Looking forward, we intend to proceed with this approach, relying on up‐front WES to guide downstream personalised laboratory diagnostic procedures and to expand the exciting functional tests. Patients with thrombocytopenia are often misdiagnosed with idiopathic thrombocytopenic purpura, which may prompt ineffective and potentially harmful treatment with immunosuppressive therapy, intravenous immunoglobulin and splenectomy. Hence, a diagnosis of IBD can have important clinical implications beyond specific haemostatic treatment and genetic counselling. Our study supports the view that continuously aiming to improve the diagnostic work‐up of IBD is of great benefit.
Author contributions
EL, EZ, FCN and MR designed the project; EL, EZ, PK and NA provided and examined the patients; SK, OØ and MR conducted the NGS‐data analysis; EL, EZ, OØ and MR interpreted the results; EN and KQ were responsible for the functional analyses; EL, EZ, MR, FCN, PK, NA, SK, OØ, KQ and JK were involved in writing the draft of the manuscript and approved the final manuscript.
Conflict of Interest
All the authors declare no conflict of interest.
Supporting information
Table S1. List of 87 predisposing genes including their NM accession no. and reference of associated inherited bleeding disorder.
Table S2. All class 3, 4 and 5 variants (total = 353) identified in the patients (n = 156).
Acknowledgements
We acknowledge Lars Kjeldsen, Department of Haematology, Rigshospitalet, for his visionary support of this study. We thank Miriam Yan Juk Guo, Aseeba Ayub and Cecilia Brunhoff Håkansson for excellent laboratory assistance. We are also indebted to Dr. Albert Huisman, Department of Clinical Chemistry and Haematology, The University Medical Centre Utrecht, The Nederlands, for assistance with the thromboxane analysis. Moreover, we are very grateful to Prof. Dr. med. Andreas Greinacher and his team, Institute of Immunology and Transfusion Medicine, University Hospital Greifswald, Greifswald, Germany for immunofluorescence analysis. For funding of analysis of Swedish patients, we are grateful to Jon Persson Foundation and the Royal Physiografic Society, Lund, Sweden.
References
- Adzhubei, I.A. , Schmidt, S. , Peshkin, L. , Ramensky, V.E. , Gerasimova, A. , Bork, P. , Kondrashov, A.S. & Sunyaev, S.R. (2010) A method and server for predicting damaging missense mutations. Nature Methods, 7, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al, H.R. , Ren, Q. , Ye, S. , Karim, Z.A. , Filipovich, A.H. & Whiteheart, S.W. (2012) Munc18b/STXBP2 is required for platelet secretion. Blood, 120, 2493–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanyan, A. , Al‐Musa, A. , AlNounou, R. , Al, Z.H. , Nasr, R. , AlJefri, A. , Saleh, M. , Malik, A. , Masmali, H. & Owaidah, T. (2015) Diagnosis of Glanzmann thrombasthenia by whole blood impedance analyzer (MEA) vs. light transmission aggregometry. International Journal of Laboratory Hematology, 37, 503–508. [DOI] [PubMed] [Google Scholar]
- Althaus, K. & Greinacher, A. (2010) MYH‐9 Related platelet disorders: strategies for management and diagnosis. Transfusion Medicine and Hemotherapy, 37, 260–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amendola, L.M. , Jarvik, G.P. , Leo, M.C. , McLaughlin, H.M. , Akkari, Y. , Amaral, M.D. , Berg, J.S. , Biswas, S. , Bowling, K.M. , Conlin, L.K. , Cooper, G.M. , Dorschner, M.O. , Dulik, M.C. , Ghazani, A.A. , Ghosh, R. , Green, R.C. , Hart, R. , Horton, C. , Johnston, J.J. , Lebo, M.S. , Milosavljevic, A. , Ou, J. , Pak, C.M. , Patel, R.Y. , Punj, S. , Richards, C.S. , Salama, J. , Strande, N.T. , Yang, Y. , Plon, S.E. , Biesecker, L.G. & Rehm, H.L. (2016) Performance of ACMG‐AMP variant‐interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. American Journal of Human Genetics, 99, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bariana, T. K. , Ouwehand, W. H. , Guerrero, J. A. & Gomez, K. ; the BRIDGE Bleeding, Thrombotic and Platelet Disorders and ThromboGenomics Consortia (2017), Dawning of the age of genomics for platelet granule disorders: improving insight, diagnosis and management. British Journal of Haematology, 176: 705–720. [DOI] [PubMed] [Google Scholar]
- Camps, C. , Petousi, N. , Bento, C. , Cario, H. , Copley, R.R. , McMullin, M.F. , van Wijk, R. , Ratcliffe, P.J. , Robbins, P.A. & Taylor, J.C. (2016) Gene panel sequencing improves the diagnostic work‐up of patients with idiopathic erythrocytosis and identifies new mutations. Haematologica, 101, 1306–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo, M. , Cerletti, C. , Harrison, P. , Hayward, C.P. , Kenny, D. , Nugent, D. , Nurden, P. , Rao, A.K. , Schmaier, A.H. , Watson, S.P. , Lussana, F. , Pugliano, M.T. & Michelson, A.D. (2013) Recommendations for the standardization of light transmission aggregometry: a consensus of the Working Party from the Platelet Physiology Subcommittee of SSC/ISTH. Journal of Thrombosis and Haemostasis, 11, 1183–1189. [DOI] [PubMed] [Google Scholar]
- Elbatarny, M. , Mollah, S. , Grabell, J. , Bae, S. , Deforest, M. , Tuttle, A. , Hopman, W. , Clark, D.S. , Mauer, A.C. , Bowman, M. , Riddel, J. , Christopherson, P.A. , Montgomery, R.R. , Rand, M.L. , Coller, B. & James, P.D. (2014) Normal range of bleeding scores for the ISTH‐BAT: adult and pediatric data from the merging project. Haemophilia, 20, 831–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fager, F.M. , Leinoe, E. , Rossing, M. , Norstrom, E. , Strandberg, K. , Steen, S.T. , Qvortrup, K. & Zetterberg, E. (2017) Germline heterozygous variants in genes associated with familial hemophagocytic lymphohistiocytosis as a cause of increased bleeding. Platelets, 11, 1–9. [DOI] [PubMed] [Google Scholar]
- Fletcher, S.J. , Johnson, B. , Lowe, G.C. , Bem, D. , Drake, S. , Lordkipanidze, M. , Guiu, I.S. , Dawood, B. , Rivera, J. , Simpson, M.A. , Daly, M.E. , Motwani, J. , Collins, P.W. , Watson, S.P. & Morgan, N.V. (2015) SLFN14 mutations underlie thrombocytopenia with excessive bleeding and platelet secretion defects. The Journal of Clinical Investigation, 125, 3600–3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunning, W. , Dole, M. , Brecher, M. & White, J.G. (2013) The Medich giant platelet syndrome: two new cases. Platelets, 24, 107–112. [DOI] [PubMed] [Google Scholar]
- Hansen, J.W. , Westman, M.K. , Sjo, L.D. , Saft, L. , Kristensen, L.S. , Orskov, A.D. , Treppendahl, M. , Andersen, M.K. & Gronbaek, K. (2016) Mutations in idiopathic cytopenia of undetermined significance assist diagnostics and correlate to dysplastic changes. American Journal of Hematology, 91, 1234–1238. [DOI] [PubMed] [Google Scholar]
- Jasuja, R. , Furie, B. & Furie, B.C. (2010) Endothelium‐derived but not platelet‐derived protein disulfide isomerase is required for thrombus formation in vivo. Blood, 116, 4665–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, B. , Lowe, G.C. , Futterer, J. , Lordkipanidze, M. , MacDonald, D. , Simpson, M.A. , Sanchez‐Guiu, I. , Drake, S. , Bem, D. , Leo, V. , Fletcher, S.J. , Dawood, B. , Rivera, J. , Allsup, D. , Biss, T. , Bolton‐Maggs, P.H. , Collins, P. , Curry, N. , Grimley, C. , James, B. , Makris, M. , Motwani, J. , Pavord, S. , Talks, K. , Thachil, J. , Wilde, J. , Williams, M. , Harrison, P. , Gissen, P. , Mundell, S. , Mumford, A. , Daly, M.E. , Watson, S.P. & Morgan, N.V. (2016) Whole exome sequencing identifies genetic variants in inherited thrombocytopenia with secondary qualitative function defects. Haematologica, 101, 1170–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, P. , Henikoff, S. & Ng, P.C. (2009) Predicting the effects of coding non‐synonymous variants on protein function using the SIFT algorithm. Nature Protocols, 4, 1073–1081. [DOI] [PubMed] [Google Scholar]
- Latger‐Cannard, V. , Philippe, C. , Bouquet, A. , Baccini, V. , Alessi, M.C. , Ankri, A. , Bauters, A. , Bayart, S. , Cornillet‐Lefebvre, P. , Daliphard, S. , Mozziconacci, M.J. , Renneville, A. , Ballerini, P. , Leverger, G. , Sobol, H. , Jonveaux, P. , Preudhomme, C. , Nurden, P. , Lecompte, T. & Favier, R. (2016) Haematological spectrum and genotype‐phenotype correlations in nine unrelated families with RUNX1 mutations from the French network on inherited platelet disorders. Orphanet Journal of Rare Diseases, 11, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinoe, E. , Nielsen, O.J. , Jonson, L. & Rossing, M. (2016) Whole‐exome sequencing of a patient with severe and complex hemostatic abnormalities reveals a possible contributing frameshift mutation in C3AR1. Cold Spring Harbor Molecular Case Studies, 2, a000828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentaigne, C. , Freson, K. , Laffan, M.A. , Turro, E. & Ouwehand, W.H. (2016) Inherited platelet disorders: toward DNA‐based diagnosis. Blood, 127, 2814–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S.W. , Lin, S.R. & Shen, M.C. (1993) Characterization of genetic defects of hemophilia A in patients of Chinese origin. Genomics, 18, 496–504. [PubMed] [Google Scholar]
- Liu, H. , Wang, H.F. , Tang, L. , Yang, Y. , Wang, Q.Y. , Zeng, W. , Wu, Y.Y. , Cheng, Z.P. , Hu, B. , Guo, T. & Hu, Y. (2015) Genetic analysis in Factor XI deficient patients from central China: identification of one novel and seven recurrent mutations. Gene, 561, 101–106. [DOI] [PubMed] [Google Scholar]
- Manon‐Jensen, T. , Kjeld, N.G. & Karsdal, M.A. (2016) Collagen‐mediated hemostasis. Journal of Thrombosis and Haemostasis, 14, 438–448. [DOI] [PubMed] [Google Scholar]
- Matthijs, G. , Souche, E. , Alders, M. , Corveleyn, A. , Eck, S. , Feenstra, I. , Race, V. , Sistermans, E. , Sturm, M. , Weiss, M. , Yntema, H. , Bakker, E. , Scheffer, H. & Bauer, P. (2016) Guidelines for diagnostic next‐generation sequencing. European Journal of Human Genetics, 24, 1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelson, A.D. (2009) Methods for the measurement of platelet function. American Journal of Cardiology, 103, 20A–26A. [DOI] [PubMed] [Google Scholar]
- Nishibori, M. , Cham, B. , McNicol, A. , Shalev, A. , Jain, N. & Gerrard, J.M. (1993) The protein CD63 is in platelet dense granules, is deficient in a patient with Hermansky‐Pudlak syndrome, and appears identical to granulophysin. The Journal of Clinical Investigation, 91, 1775–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurden, A.T. & Nurden, P. (2013) Glycoproteins, inherited diseases of platelets, and the role of platelets in wound healing. Bulletin de L'Académie Nationale de Médecine, 197, 349–358. [PubMed] [Google Scholar]
- Nurden, A.T. , Pillois, X. , Fiore, M. , Alessi, M.C. , Bonduel, M. , Dreyfus, M. , Goudemand, J. , Gruel, Y. , Benabdallah‐Guerida, S. , Latger‐Cannard, V. , Négrier, C. , Nugent, D. , Oiron, R.D. , Rand, M.L. , Sié, P. , Trossaert, M. , Alberio, L. , Martins, N. , Sirvain‐Trukniewicz, P. , Couloux, A. , Canault, M. , Fronthroth, J.P. , Fretigny, M. , Nurden, P. , Heilig, R. & Vinciguerra, C. (2015) Expanding the mutation spectrum affecting αIIbβ3 integrin in glanzmann thrombasthenia: screening of the ITGA2B and ITGB3 genes in a large International Cohort. Human Mutation, 36, 548–561. [DOI] [PubMed] [Google Scholar]
- Pai, M. , Wang, G. , Moffat, K.A. , Liu, Y. , Seecharan, J. , Webert, K. , Heddle, N. & Hayward, C. (2011) Diagnostic usefulness of a lumi‐aggregometer adenosine triphosphate release assay for the assessment of platelet function disorders. American Journal of Clinical Pathology, 136, 350–358. [DOI] [PubMed] [Google Scholar]
- Quiroga, T. & Mezzano, D. (2012) Is my patient a bleeder? A diagnostic framework for mild bleeding disorders. Hematology/ the Education Program of the American Society of Hematology, 2012, 466–474. [DOI] [PubMed] [Google Scholar]
- Rehm, H.L. , Bale, S.J. , Bayrak‐Toydemir, P. , Berg, J.S. , Brown, K.K. , Deignan, J.L. , Friez, M.J. , Funke, B.H. , Hegde, M.R. & Lyon, E. (2013) ACMG clinical laboratory standards for next‐generation sequencing. Genetics in Medicine, 15, 733–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren, Q. , Wimmer, C. , Chicka, M.C. , Ye, S. , Ren, Y. , Hughson, F.M. & Whiteheart, S.W. (2010) Munc13‐4 is a limiting factor in the pathway required for platelet granule release and hemostasis. Blood, 116, 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W.W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. & Rehm, H.L. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saposnik, B. , Binard, S. , Fenneteau, O. , Nurden, A. , Nurden, P. , Hurtaud‐Roux, M.F. & Schlegel, N. ; French MYH9 network . (2014) Mutation spectrum and genotype‐phenotype correlations in a large French cohort of MYH9‐Related Disorders. Molecular Genetics & Genomic Medicine, 2, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz, J.M. , Rodelsperger, C. , Schuelke, M. & Seelow, D. (2010) MutationTaster evaluates disease‐causing potential of sequence alterations. Nature Methods, 7, 575–576. [DOI] [PubMed] [Google Scholar]
- Shattil, S.J. , Hoxie, J.A. , Cunningham, M. & Brass, L.F. (1985) Changes in the platelet membrane glycoprotein IIb.IIIa complex during platelet activation. Journal of Biological Chemistry, 260, 11107–11114. [PubMed] [Google Scholar]
- Simeoni, I. , Stephens, J.C. , Hu, F. , Deevi, S.V. , Megy, K. , Bariana, T.K. , Lentaigne, C. , Schulman, S. , Sivapalaratnam, S. , Vries, M.J. , Westbury, S.K. , Greene, D. , Papadia, S. , Alessi, M.C. , Attwood, A.P. , Ballmaier, M. , Baynam, G. , Bermejo, E. , Bertoli, M. , Bray, P.F. , Bury, L. , Cattaneo, M. , Collins, P. , Daugherty, L.C. , Favier, R. , French, D.L. , Furie, B. , Gattens, M. , Germeshausen, M. , Ghevaert, C. , Goodeve, A.C. , Guerrero, J.A. , Hampshire, D.J. , Hart, D.P. , Heemskerk, J.W. , Henskens, Y.M. , Hill, M. , Hogg, N. , Jolley, J.D. , Kahr, W.H. , Kelly, A.M. , Kerr, R. , Kostadima, M. , Kunishima, S. , Lambert, M.P. , Liesner, R. , Lopez, J.A. , Mapeta, R.P. , Mathias, M. , Millar, C.M. , Nathwani, A. , Neerman‐Arbez, M. , Nurden, A.T. , Nurden, P. , Othman, M. , Peerlinck, K. , Perry, D.J. , Poudel, P. , Reitsma, P. , Rondina, M.T. , Smethurst, P.A. , Stevenson, W. , Szkotak, A. , Tuna, S. , van Geet, C. , Whitehorn, D. , Wilcox, D.A. , Zhang, B. , Revel‐Vilk, S. , Gresele, P. , Bellissimo, D.B. , Penkett, C.J. , Laffan, M.A. , Mumford, A.D. , Rendon, A. , Gomez, K. , Freson, K. , Ouwehand, W.H. & Turro, E. (2016) A high‐throughput sequencing test for diagnosing inherited bleeding, thrombotic, and platelet disorders. Blood, 127, 2791–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark, Z. , Schofield, D. , Alam, K. , Wilson, W. , Mupfeki, N. , Macciocca, I. , Shrestha, R. , White, S.M. & Gaff, C. (2017) Prospective comparison of the cost‐effectiveness of clinical whole‐exome sequencing with that of usual care overwhelmingly supports early use and reimbursement. Genetics in Medicine: Official Journal of the American College of Medical Genetics. 2017 Jan 26. doi: 10.1038/gim.2016.221. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- Stenberg, P.E. , McEver, R.P. , Shuman, M.A. , Jacques, Y.V. & Bainton, D.F. (1985) A platelet alpha‐granule membrane protein (GMP‐140) is expressed on the plasma membrane after activation. Journal of Cell Biology, 101, 880–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockley, J. , Nisar, S.P. , Leo, V.C. , Sabi, E. , Cunningham, M.R. , Eikenboom, J.C. , Lethagen, S. , Schneppenheim, R. , Goodeve, A.C. , Watson, S.P. , Mundell, S.J. & Daly, M.E. (2015) Identification and characterization of novel variations in platelet G‐Protein Coupled Receptor (GPCR) genes in patients historically diagnosed with type 1 von Willebrand Disease. PLoS ONE, 10, e0143913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian, S.V. , Deffenbaugh, A.M. , Yin, L. , Judkins, T. , Scholl, T. , Samollow, P.B. , de Silva, D. , Zharkikh, A. & Thomas, A. (2006) Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. Journal of Medical Genetics, 43, 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thusberg, J. , Olatubosun, A. & Vihinen, M. (2011) Performance of mutation pathogenicity prediction methods on missense variants. Human Mutation, 32, 358–368. [DOI] [PubMed] [Google Scholar]
- Tubman, V.N. , Levine, J.E. , Campagna, D.R. , Monahan‐Earley, R. , Dvorak, A.M. , Neufeld, E.J. & Fleming, M.D. (2007) X‐linked gray platelet syndrome due to a GATA1 Arg216Gln mutation. Blood, 109, 3297–3299. [DOI] [PubMed] [Google Scholar]
- Watson, S.P. , Lowe, G.C. , Lordkipanidze, M. & Morgan, N.V. (2013) Genotyping and phenotyping of platelet function disorders. Journal of Thrombosis and Haemostasis, 11, 351–363. [DOI] [PubMed] [Google Scholar]
- White, J.G. (2004) Electron microscopy methods for studying platelet structure and function. Methods in Molecular Biology, 272, 47–63. [DOI] [PubMed] [Google Scholar]
- Zeitoun, J.D. , Lefevre, J.H. , deParades, V , Sejourne, C. , Sobhani, I. , Coffin, B. & Hamonet, C. (2013) Functional digestive symptoms and quality of life in patients with Ehlers‐Danlos syndromes: results of a national cohort study on 134 patients. PLoS ONE, 8, e80321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Z.P. , Falk, G. , Blombäck, M. , Egberg, N. & Anvret, M. (1992) Identification of a new nonsense mutation in the von Willebrand factor gene in patients with von Willebrand disease type III. Human Molecular Genetics, 1, 61–62. [DOI] [PubMed] [Google Scholar]
- Zhang, J. , Barbaro, P. , Guo, Y. , Alodaib, A. , Li, J. , Gold, W. , Ades, L. , Keating, B.J. , Xu, X. , Teo, J. , Hakonarson, H. & Christodoulou, J. (2016) Utility of next‐generation sequencing technologies for the efficient genetic resolution of haematological disorders. Clinical Genetics, 89, 163–172. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of 87 predisposing genes including their NM accession no. and reference of associated inherited bleeding disorder.
Table S2. All class 3, 4 and 5 variants (total = 353) identified in the patients (n = 156).