

Summary
Microbial utilization of complex polysaccharides is a major driving force in shaping the composition of the human gut microbiota. There is a growing appreciation that finely tuned polysaccharide utilization loci enable ubiquitous gut Bacteroidetes to thrive on the plethora of complex polysaccharides that constitute “dietary fiber”. Mixed-linkage β(1,3)/β(1,4)-glucans (MLGs) are a key family of plant cell wall polysaccharides with recognized health benefits, but whose mechanism of utilization has remained unclear. Here, we provide molecular insight into the function of an archetypal MLG utilization locus (MLGUL) through a combination of biochemistry, enzymology, structural biology, and microbiology. Comparative genomics coupled with growth studies demonstrated further that syntenic MLGULs serve as genetic markers for MLG catabolism across commensal gut bacteria. In turn, we surveyed human gut metagenomes to reveal that MLGULs are ubiquitous in human populations globally, which underscores the importance of gut microbial metabolism of MLG as a common cereal polysaccharide.
eTOC Blurb
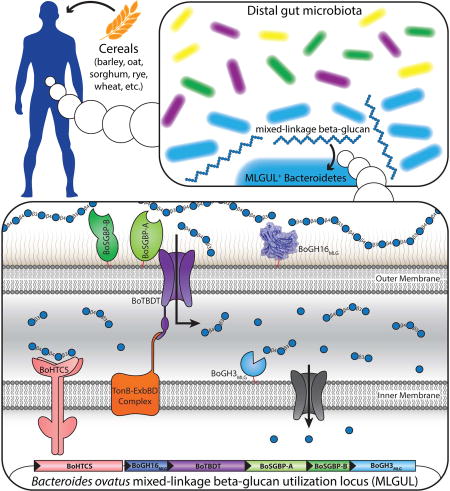
Mixed-linkage β(1,3)/β(1,4)-glucan (MLG) is an important complex dietary polysaccharide (dietary fiber), the degradation of which in the human gut depends on the resident microbiota. Tamura et al. outline the molecular mechanism of MLG utilization by Bacteroides ovatus and reveal that the majority of surveyed humans possess MLG-utilizing Bacteroidetes.
Introduction
The composition and homeostasis of the human gut microbiota has a profound and intimate connection to various aspects of our physiology, health and wellbeing (Littman and Pamer, 2011). Indeed, a multitude of diseases such as type 2 diabetes, inflammatory bowel diseases (IBDs), and cancer have been linked to alterations in the population and proportion of microbes in this highly complex and dynamic ecosystem that exists in our large intestine (Biedermann and Rogler, 2015; Fujimura et al., 2010; Kau et al., 2011; Schwabe and Jobin, 2013). The molecular mechanisms by which the microbiota exerts influence on human health are largely unresolved and undoubtedly complex, yet may hold the key to personalized medicine through therapeutics that target the gut microbial ecosystem (Blanton et al., 2016; Haak et al., 2017; Kootte et al., 2012; Subramanian et al., 2015).
A major factor in shaping the composition and physiology of the gut microbiota is the influx of complex glycans – popularly known as “dietary fibre” – that evade degradation by the limited set of human genome-encoded glycoside hydrolases (Hamaker and Tuncil, 2014; El Kaoutari et al., 2013; Koropatkin et al., 2012). Indeed, regular ingestion of plant polysaccharides is integral to maintaining a healthy balance of microbes in our lower gastrointestinal tract (De Filippo et al., 2010; Sonnenburg and Sonnenburg, 2014). Members of the Bacteroidetes, a dominant phylum in the human gut, possess an arsenal of Polysaccharide Utilization Loci (PUL) to target a wide range of complex glycans (El Kaoutari et al., 2013). Analogous to the archetypal Bacteroides thetaiotaomicron starch utilization system (Sus), a hallmark of all Bacteroidetes PULs is the organization of genes clustered around tandem susC/susD homologs (encoding a TonB dependent transporter, TBDT, and a cell-surface glycan-binding protein, SGBP, respectively) (Martens et al., 2009). Additional co-localized and co-regulated SGBP(s), a cohort of enzymes, and a transcriptional regulator typically make up a machinery that acts in concert to sense, break down, and import complex glycans (Grondin et al., 2017; Hemsworth et al., 2016). Many such PULs, each targeting specific glycan structures, have been identified by genomics and transcriptomics (see, e.g., the seminal study by (Martens et al., 2011), but detailed functional characterization lags severely behind (reviewed in (Grondin et al., 2017; Martens et al., 2014)). Developing a precise understanding of the molecular details of complex glycan utilization by individual members of the microbiota is essential to designing targeted therapies based on prebiotics, probiotics, and synbiotics (Ciorba, 2012; Slavin, 2013), as well as novel therapeutic interventions.
Recently, comprehensive functional analysis has revealed the detailed molecular mechanisms by which individual PULs enable human gut Bacteroidetes to utilize predominant plant polysaccharides, including the matrix glycans, xyloglucan (Hemsworth et al., 2016; Larsbrink et al., 2014; Tauzin et al., 2016), xylan (Rogowski et al., 2015), β-mannan (Bågenholm et al., 2017), and rhamnogalacturonan II (Ndeh et al., 2017). Mixed-linkage β(1,3)/β(1,4)-glucans (MLGs, Fig. 1A) from cereal grains constitute an additional key group of dietary glycans, whose utilization by gut microbes was previously unresolved at the molecular level. MLGs are abundant in the aleurone layer of common cereals, including oats (3–8 % dry weight) and barley (2–20 % dry weight) (El Khoury et al., 2012). Beyond their obvious potential to contribute to energy intake (Cummings and Macfarlane, 1997; McNeil, 1984), MLGs have been linked to a range of health benefits, e.g. promoting healthy cholesterol and blood glucose levels, ameliorating insulin resistance, and mitigating metabolic syndrome (El Khoury et al., 2012). In particular, the cholesterol lowering effect of oat MLG has long been recognized by the United States Food and Drug Administration (FDA) as well as the United Kingdom Joint Health Claims Initiative (JHCI), and been confirmed by subsequent studies (Othman et al., 2011).
Figure 1. Cereal mixed-linkage β(1,3)/β(1,4)-glucan (MLG) and MLG Utilization Locus (MLGUL) structures.

A: Chemical structure of MLG, consisting of a linear glucan chain of β(1,4)-linked cellotriosyl and cellotetraosyl units linked by β(1,3) bonds. MLGs from various sources (barley, oat, lichenin, etc) vary in the ratio of cellotriose to cellotetraose units (Lazaridou et al., 2004). Arrows indicate the specific site of hydrolysis by the vanguard endo-glucanase of the MLGUL, BoGH16MLG. B: Genetic organization of the B. ovatus MLGUL and syntenic loci in select Bacteroidetes species. Homologous genes are connected by colored bars and the locus tag of the TBDT of each syntenic MLGUL is given on the right. See also Figure S1; Table S1.
The mechanisms behind these systemic benefits of MLG are, however, incompletely understood, in part due to a lack of understanding of MLG metabolism by individual members of the human gut microbiota. Thus, we report here the molecular characterization of a mixed-linkage glucan utilization locus (MLGUL) in the common symbiont B. ovatus. Identifying syntenic MLGUL in other Bacteroidetes revealed that as the archetype, this MLGUL serves as a molecular marker for MLG utilization across the Bacteroidetes phylum, thereby enabling future functional prediction across species.
Results
Identification of a multi-gene locus responsible for MLG utilization by B. ovatus
A putative MLGUL was previously identified in B. ovatus (Fig. 1B) based on the presence of a tandem susC/susD homolog signature (Martens et al., 2009) and high-level expression of select genes in the presence of bMLG (Martens et al., 2011). Individual genes in the locus, BACOVA_02741-02745, were all substantially upgregulated (125 to 298-fold) during growth on bMLG vs. glucose as sole carbon sources (Table S1). BACOVA_02742 and BACOVA_02743 encode the signature TBDT/SGBP pair with 28% and 19% protein sequence identity to SusC and SusD, respectively. The putative MLGUL was additionally predicted to encode a second, non-homologous SGBP (BACOVA_02744), a hybrid two-component sensor/transcriptional regulator (HTCS, BACOVA_02740), and up to three glycoside hydrolases (GHs).
BoGH16MLG (BACOVA_02741) is a member of Glycoside Hydrolase Family 16 (GH16), a family of endo-β-glucanases in the Carbohydrate Active Enzymes (CAZy) classification (Cantarel et al., 2009). GH16 notably includes canonical bacterial MLG endo-glucanases (endo-MLGase) (Planas, 2000), along with a diversity of endo-glucanases and endo-galactanases (Eklof and Hehemann). BoGH3MLG (BACOVA_02745) is classified into Glycoside Hydrolase Family 3 (GH3), whose members include exo-β-glucosidases (Fincher et al.). Notably, we have determined that BACOVA_02738, which is predicted to encode a second GH3 exo-β-glucosidase, is unlikely to be part of the MLGUL for three reasons: (1) BACOVA_02738 was not significantly upregulated on MLG (1.6-fold vs. glucose control, Table S1), (2) a corresponding gene is not found among syntenic loci (Fig. 1B), and (3) the encoded enzyme was catalytically feeble compared to BoGH3MLG on β-glucosides relevant to MLG saccharification (vide infra).
To determine the correlation between the presence of the predicted MLGUL and growth of B. ovatus on MLG, we constructed an isogenic mutant of B. ovatus Δtdk (Larsbrink et al., 2014) in which a contiguous region of DNA encoding genes BACOVA_02738-02745 was deleted (B. ovatus ΔMLGUL). Vis-à-vis the parent strain, the B. ovatus ΔMLGUL was able to grow equally well on glucose as the sole carbon source, however the ability to grow on bMLG was completely abolished (Fig. S1). Thus, the putative MLGUL is necessary to confer B. ovatus the ability to utilize MLG.
Enzymology and structural biology of BoGH16MLG, the vanguard MLGase
Cellular localization
The GH family membership of BoGH16MLG suggested a potential role as the vanguard enzyme catalyzing polysaccharide backbone cleavage at the cell surface as the essential first step in MLG utilization. Indeed, the presence of a predicted Type II signal sequence (determined with LipoP 1.0 (Juncker and Willenbrock, 2003)) suggested that the protein is membrane-anchored via lipidation of the N-terminal cysteine residue (Paetzel et al., 2002). To validate this prediction, B. ovatus Δtdk was grown on minimal medium with either glucose or bMLG as a sole carbon source prior to immunolocalization of BoGH16MLG. As shown in Fig. 2A, BoGH16MLG was clearly visualized on the outer surface of cells in which the presence of the polysaccharide induced MLGUL expression, but was absent from cells grown on glucose (Fig. S2C, S2F). Further analysis of cellular fractions by Western blotting revealed the presence of BoGH16MLG in the membrane fraction, corroborating its attachment to the outer membrane (Fig. 2C). Interestingly, BoGH16MLG was also detected in the lysate supernatant (soluble periplasmic or cytoplasmic proteins) and in the culture supernatant (secreted protein) (Fig. 2C). While the former may represent anchored protein released into the soluble fraction during cell lysis, detection in the culture supernatant could result from packaging and release in outer membrane vesicles, which has previously been observed for other Bacteroidetes glycoside hydrolases (Elhenawy et al., 2014).
Figure 2. Enzyme localization analysis.

Phase contrast microscopy and corresponding fluorescence microscopy images of B. ovatus Δtdk cells grown in minimal medium with bMLG as the sole carbon source probed with custom polyclonal antibodies against recBoGH16MLG (A) and recBoGH3MLG (B). C: Western blots of protein collected from the culture supernatant, cell lysate supernatant, and cell lysate membrane fraction of B. ovatus Δtdk cells grown in minimal medium with glucose or bMLG as a sole carbon source. See also Figure S2.
Substrate and product specificity
To verify the leading catalytic role of BoGH16MLG in MLG utilization and determine the specificity of the enzyme for individual β-glucans, recombinant BoGH16MLG produced in E. coli (recBoGH16MLG, Fig. S3A, S3B) was screened for hydrolytic activity against a library of polysaccharides. No activity was observed on tamarind xyloglucan, beechwood xylan, wheat arabinoxylan, carob galactomannan, konjac glucomannan, synthetic carboxymethylcellulose, synthetic hydroxyethylcellulose, Xanthomonas campestris xanthan gum, or Ulva sp. ulvan. In this initial screen, BoGH16MLG was minimally active on all-β(1,3)-glucans, including Laminaria digitata laminarin, yeast β-glucan, and Alcaligenes faecalis curdlan, but exhibited high specific activity on bMLG. The optimum pH of 6.5 (consistent with function in the distal human colon) and maximum temperature range of 45 to 55 °C was determined using bMLG as substrate (data not shown).
Subsequent Michaelis-Menten kinetic analysis at the pH optimum and 37 °C demonstrated that BoGH16MLG is a highly predominant mixed-linkage β(1,3)/β(1,4)-glucanase (MLGase), with a 33-fold higher specificity constant, kcat/Km, for bMLG (Fig. 1A) over laminarin, an all-β(1,3)-glucan with single β(1,6)-linked glucosyl branches (Fig. 3A, Table S2) (Martin et al., 2007). BoGH16MLG was even less efficient on the other two all-β(1,3)-glucans for which activity was initially observed: The kcat/Km, was 147-fold higher for bMLG than yeast β-glucan (similar in structure to laminarin but with longer β(1,6)-linked glucose branches (Lowman et al., 2011)) and nearly four orders of magnitude higher than that of high curdlan, a 22 kDa, non-branched β(1,3)-glucan (Harada et al., 1968) (Fig. 3A, Table S2).
Figure 3. BoGH16MLG kinetics and MLGUL GHs product analysis.

A: BoGH16MLG initial-rate kinetics curves fitted to the Michaelis-Menten equation for β-glucan polysaccharide substrates on which it is active. Laminarin was reduced to laminaritol by sodium borohydride reduction to reduce background in the BCA assay. Curve fitting was done on OriginPro 2015 and error bars represent standard deviations from the mean. B: Chromatograms of bMLG and its hydrolysis products by BoGH16MLG and BoGH3MLG separated by HPAEC-PAD. Red: full length bMLG polysaccharide. Dark blue: reaction progress time course and limit digest of bMLG hydrolysis by 10 nM BoGH16MLG. Cyan: reaction progress time course and limit digest of BoGH16MLG products hydrolysis by 25 nM BoGH3MLG. Standards are shown below in black: solid lines are those corresponding to limit digest products and dotted line to intermediate products. See also Figures S3, S4 and S5; Tables S2 and S3.
Detailed product analysis was employed to determine the mode of hydrolysis, endo vs. exo, and linkage specificity of recBoGH16MLG to gain information on the nature of the MLG cleavage products at the B. ovatus cell surface. HPLC analysis at selected time points in the hydrolysis showed the initial production of very large oligosaccharide fragments, which were progressively converted into shorter species and ultimately to two distinct oligosaccharides in the limit-digest (Fig. 3B). This product evolution indicates that BoGH16MLG operates through an endo-dissociative mode of action in which the MLG polysaccharide is stochastically cleaved along the backbone.
Comparison with oligosaccharide standards (Fig. 3B) and additional LC-MS analysis (data not shown) revealed that the limit-digest products were the mixed-linkage trisaccharide, G4G3G [Glcβ(1–4) Glcβ(1–3) Glc], and the mixed-linkage tetrasaccharide, G4G4G3G [Glcβ(1–4) Glcβ(1–4) Glcβ(1–3) Glc]. Thus, BoGH16MLG specifically hydrolyzes β(1,4)-linkages of glycosyl residues that are immediately preceded by a β(1,3)-linked glucosyl residue (toward the non-reducing chain end). This specificity is typical of bacterial endo-MLGases within GH16 (Gaiser et al., 2006; Mcgregor et al., 2017; Planas, 2000).
To provide more refined insight into BoGH16MLG substrate specificity, Michaelis-Menten kinetics were determined for a series of chromogenic glycosides (Fig. S4A, S4B; Table S3). recBoGH16MLG had no activity on the ortho-chloro-para-nitrophenyl (CNP) β-glycosides of glucose (G-CNP), cellobiose (G4G-CNP), cellotriose (G4G4G-CNP), nor on para-nitrophenyl (pNP) β-glucoside (G-pNP). Weak activity was observed on the pNP and CNP β-glycosides of laminaribiose (G3G), consistent with a requirement for a β(1,3) linkage spanning the -2 to -1 active-site subsites (GH subsite nomenclature according to (Davies et al., 1997)), as was indicated by the bMLG limit-digest analysis (vide supra). Likewise, G4G3G-CNP and G4G4G3G-CNP were specifically and efficiently hydrolyzed to release the aglycone, with no cleavage of the internal glycosidic bonds. The specificity constants (kcat/Km values) for CNP release from these mixed-linkage tri- and tetrasaccharides were 800- and 1500-fold greater than that of G3G-CNP, respectively, which indicate that potential -3 and -4 subsites contribute 17 kJ/mol and 1.6 kJ/mol to transition state stabilization (ΔΔG‡). Indeed, a very significant contribution from the -3 subsite is a common feature of GH16 endo-MLGases (Gaiser et al., 2006; McGregor et al., 2016; Planas, 2000).
BoGH16MLG tertiary structure
Three-dimensional structures of recBoGH16MLG were solved by X-ray crystallography to reveal the molecular basis for MLG recognition and hydrolysis. The apo structure of recBoGH16MLG was determined to a resolution of 1.8 Å by molecular replacement using the structure of Zobellia galactanivorans laminarinase ZgLamCGH16-E142S (PDB code 4CRQ) (Labourel et al., 2015) as a search model (See Table S4 for processing and refinement statistics). The crystal contained two polypeptide chains in the asymmetric unit corresponding to residues I35-L271 of wild-type BoGH16MLG for both chains (residue numbering is from transcriptional start site according to the genomic sequence). No electron density was observed for the N-terminal His6-tag and subsequent 15 amino acids in either chain of the recombinant protein, which suggests that residues C19-D34 of the wild-type enzyme constitute a flexible linker sequence to distance the catalytic module from the outer membrane surface (residues M1-S18 comprise the predicted signal peptide); the sidechain of C19 would constitute the site of N-terminal lipidation (Paetzel et al., 2002). The overall fold of BoGH16MLG consists of a β-jelly roll architecture typical of other GH16 members (Davies and Sinnot, 2008): Two antiparallel β-sheets stack on top of each other with the concave face forming the polysaccharide substrate binding cleft. The end-on arrangement of the two chains in the asymmetric unit hinted at the possibility of the formation of a dimer (Fig 4A). Size-exclusion chromatography, however, indicated that BoGH16MLG exists as a monomer in solution (data not shown) which, together with steric considerations of polysaccharide binding through the active-site cleft, indicates that end-on contacts observed between Chains A and B are artifacts of crystal packing.
Figure 4. BoGH16MLG structural biology.

A: the overall structure of the BoGH16MLG:G4G4G3G asymmetric unit containing two polypeptide chains shown from orthogonal views with the bound oligosaccharides in yellow and the transparent surface representation in white. Chain A cartoon is shown in cyan, and chain B cartoon is shown in slate blue throughout the figure. B: Mixed-linkage tetrasaccharide ligand modelled into chain A of BoGH16MLG with the opaque surface representation in gray and the oligosaccharide colored according to B-factors. The glucose in subsite -4 is outside of the active site cleft and has significantly higher B factor than the glucose units in subsites -1 to -3. C: Tyr-181 rotamers observed in the complex structure with the 2Fo-Fc map of the tyrosines shown contoured at 0.5σ in grey. D: Tyr-181 residues observed in the apo structure with the 2Fo-Fc map of the tyrosines shown contoured at 0.5σ in grey. E: Wall-eyed stero view of the active site of chain A of the BoGH16MLG:G4G4G3G complex. Hydrogen bonding interactions are shown as dashed black lines, sugars are shown in yellow with its 2Fo-Fc map contoured at 1σ in orange, and the conserved GH16 active site residues shown in purple. Hydrophobic stacking interactions in addition to hydrogen bonds position the mixed-linkage oligosaccharide in the negative subsite of BoGH16MLG. See also Figures S5 and S6; Tables S4 and S5.
The sidechains of the conserved GH16 catalytic residues (Planas, 2000), comprising Glu-143 (nucleophile), Asp-145 (electrostatic “helper”) and Glu-148 (acid/base) are presented on the same face of one β-strand (β8), pointing into the active-site cleft. Notably, these residues are contained in a EXDXXE consensus sequence that is typical of bacterial GH16 laminarinases (β(1,3)-specific endo-glucanases). The insertion of an extra amino acid (underlined), typically methionine, results in a so-called “β-bulge” secondary structural motif that is not found among canonical bacterial MLGases, which instead possess a regular β-strand (Barbeyron et al., 1998; Michel et al., 2001).
Commensurate with this observation, the closest eight structural homologs identified using the Dali server (Holm and Rosenstrom, 2010) feature a β-bulge active-site motif (Table S5). Specifically, the top match (Z-score = 29.3) was the structure of laminarinase “ZgLamCGH16-E142S“ from Zobellia galactanivorans (PDB code 4CTE) (Labourel et al., 2015), which has 38% amino acid identity and superimposed with BoGH16MLG with a root mean square deviation (RMSD) value of 2.0 Å over 211 out of 231 Cα pairs. In comparison, the closest GH16 homolog with a regular active-site β-strand was the lichenase (MLGase) from Paenibacillus macerans (PDB code 1MAC) (Hahn et al., 1995), which has a comparable Z-score of 25.1 and an RMSD value of also 2.0 Å over 200 out of 212 Cα pairs, despite having only 22% amino acid identity with BoGH16MLG.
Soaking crystals of the wild-type enzyme with G4G4G3G yielded a product complex with 1.8 Å resolution (Table S4). The complete tetrasaccharide was modelled in electron density spanning subsites -1 to -4 in the active-site cleft of Chain A, while the electron density for the fourth glucosyl residue in subsite -4 was not resolved in chain B. This is most likely due to disorder of this residue since the corresponding -4 Glc in Chain A is fully solvent exposed, makes no contact with the enzyme, and has significantly higher B-factors (Fig. 4B). These structural observations are consistent with kinetic data for chromogenic MLG oligosaccharides (Table 1), which likewise suggest the existence of three primary negative subsites, -1 to -3, and a weakly interacting -4 subsite.
Table 1.
| Enzyme | Substrate | kcat (s−1) | Km (mM) | kcat/Km (s−1 mM−1) | Assay |
|---|---|---|---|---|---|
| BoGH3MLG | β-Glc-pNP | 59.5 ± 1.46 | 2.95 ± 0.14 | 20.2 | pNP |
|
|
|||||
| gentiobiose (G6G) | ND | ND | 0.0571 | HK/G6PDH | |
|
|
|||||
| cellobiose | 5.52 ± 0.19 | 7.47 ± 0.48 | 0.739 | HK/G6PDH | |
| cellotriose | 22.1 ± 0.3 | 0.859 ± 0.033 | 25.7 | HK/G6PDH | |
| cellotetraose | 17.3 ± 0.5 | 0.687 ± 0.044 | 25.2 | HK/G6PDH | |
| cellopentaose | 19.4 ± 0.8 | 0.777 ± 0.060 | 25.0 | HK/G6PDH | |
| cellohexaose | 17.4 ± 0.4 | 0.747 ± 0.041 | 23.3 | HK/G6PDH | |
|
|
|||||
| laminaribiose | 28.0 ± 1.1 | 1.90 ± 0.12 | 14.7 | HK/G6PDH | |
| laminaritriose | 34.2 ± 1.0 | 0.911 ± 0.052 | 37.5 | HK/G6PDH | |
| laminaritetraose | 31.3 ± 2.3 | 0.898 ± 0.135 | 34.9 | HK/G6PDH | |
| laminaripentaose | 39.5 ± 3.4 | 1.27 ± 0.20 | 31.1 | HK/G6PDH | |
|
|
|||||
| MLGO3 A (G3G4G) | 61.6 ± 1.6 | 0.997 ± 0.040 | 61.8 | HK/G6PDH | |
| MLGO3 B (G4G3G) | 24.7 ± 1.3 | 0.521 ± 0.064 | 47.4 | HK/G6PDH | |
| MLGO4 A (G3G4G4G) | 55.7 ± 2.7 | 1.33 ± 0.12 | 41.9 | HK/G6PDH | |
| MLGO4 B (G4G4G3G) | 30.8 ± 2.0 | 0.736 ± 0.106 | 41.8 | HK/G6PDH | |
| MLGO4 C (G4G3G4G) | 15.7 ± 0.3 | 0.601 ± 0.031 | 26.1 | HK/G6PDH | |
|
| |||||
| BACOVA_02738 (GH3) | β-Glc-pNP | 0.212 ± 0.004 | 2.53 ± 0.13 | 0.0838 | pNP |
ND: not determined (in cases where Michealis-Menten curve fitting was not feasible, individual kcat and Km values are not reported and kcat/Km value was determined from linear curve fit to initial-rate data in the [S] ≪ Km(apparent) range). Data are represented as the fitted parameters ± standard deviation. Highlighted in bold are the biologically relevant substrates that BoGH3MLG encounters in the periplasmic space. See also Figure S4.
In both Chain A and B, the three glucosyl residues spanning subsites -1 to -3 are well defined and virtually identical. The reducing-end glucosyl residue in the -1 subsite is in the β-conformation, with the C1 hydroxyl hydrogen bonded to Tyr-181, which is observed in a dual conformation in both chains of the G4G4G3G-complex (Fig. 4C). Interestingly, this dual conformation is not observed in the apo-form of the enzyme; Tyr-181 is “swung in” to the active site in chain B, while it is “swung out” in chain A, the sidechain from chain A stacking on top of the chain B sidechain (Fig. 4D). The conformation of this sidechain will be key to determining the nature of the positive substrate binding subsites, indeed, comparison with other GH16 endo-glucanases clearly suggests the presence of two positive subsites (Gaiser et al., 2006; Planas, 2000). Whether the dynamics observed for Tyr-181 are an artefact of crystallisation, or perhaps play a role in substrate binding and product release is unclear in the absence of an enzyme-substrate complex spanning the positive subsites.
With regard to specific interactions in the negative subsites, subsite -1 is further characterized by hydrogen bonds between Glu-143 and the C2 hydroxyl, Trp-125 and the C6 hydroxyl, as well as Glu-148 and the ring oxygen and the C1 hydroxyl. This glucose is also positioned by a stacking interaction with Trp-125 and Trp-129 (Fig. 4E), both of which are conserved across all GH16 laminarinases. At subsite -2, highly conserved Arg-97 forms a hydrogen bond with the C6 hydroxyl, and Asn-60 hydrogen bonds to the C2 hydroxyl as well as to the glucosidic bond oxygen between the -1 and -2 sugars. Another conserved residue, Trp-138, serves as a platform that stacks with the subsite -2 glucose. In subsite -3, the main interaction is stacking against Trp-58, which also forms a hydrogen bond to the glucosidic bond oxygen between the -3 and -4 sugars (Fig. 4E). Together, these interactions in subsite -3 are responsible for 17 kJ/mol of transition-state stabilization (vide supra).
Downstream saccharificaiton of mixed-linkage oligosaccharides produced by BoGH16MLG
To elucidate the mechanism for the downstream conversion of the oligosaccharide products of BoGH16MLG to glucose for primary metabolism, we examined the biochemistry of the two predicted exo-β-glucosidases, BoGH3MLG and BACOVA_02738(GH3) associated with the MLGUL.
Cellular localization of BoGH3MLG and the BACOVA_02738(GH3) gene product
BoGH3MLG and BACOVA_02738(GH3) were unambiguously predicted by SignalP 4.0 (Petersen et al., 2011) to contain a secretion signal peptide, while LipoP 1.0 (Juncker and Willenbrock, 2003) additionally indicated a Type II lipoprotein signal sequence (Paetzel et al., 2002) in BoGH3MLG only. The same B. ovatus _tdk cultures used for BoGH16MLG localization, containing glucose or bMLG as the sole carbon source, were probed using polyclonal antibodies independently raised against recombinant BoGH3MLG and the BACOVA_02738(GH3) gene product. Neither protein was detected on the cell surface by fluorescence microscopy, especially in the presence of bMLG which induces BoGH16MLG production (Fig. 2B, Fig. S2A). BoGH3MLG induction by bMLG was confirmed by a Western blot of cellular fractions, which also confirmed that this enzyme is membrane anchored (Fig. 2C).
In contrast, the BACOVA_02738(GH3) gene product was detected to a higher degree in B. ovatus cells grown in minimal medium with glucose as a sole carbon source compared to cells induced with bMLG (Fig. S2B). The lack of upregulation by bMLG is consistent with transcriptional analysis which showed a limited change in expression in bMLG vs. glucose (1.6-fold), which was two orders of magnitude lower than definitive MLGUL genes (Table S1). The higher detection in uninduced cells is explained by the high basal expression of BACOVA_02738(GH3) (more than an order of magnitude greater than all MLGUL members; Table S1). The lack of detection in minimal medium containing bMLG is due to high amounts of induced MLGUL proteins diminishing the presence of the BACOVA_02738(GH3) gene product when normalized to total protein (Fig. S2B).
Substrate product specificity of BoGH3MLG and BACOVA_02738(GH3)
Initial activity screening on chromogenic pNP glycosides (see Experimental Procedures) revealed that both recBoGH3MLG and recBACOVA_02738(GH3) are specific exo-β-glucosidases (activity on other pNP glycosides was undetectable at micromolar enzyme concentrations). However, recBACOVA_02738(GH3) is strikingly feeble compared to recBoGH3MLG on G-β-pNP (kcat/Km values of 0.084 mM−1 s−1 versus 20 mM−1 s−1; Fig. S4C, S4D, Table 1). Further, measuring Michealis-Menten kinetic parameters on cello- and laminari-oligosaccharides was not feasible due to overall poor activity and low production yields (data not shown). These kinetic results corroborate the above comparative genetic and transcriptional analyses, collectively suggesting BACOVA_02738(GH3) is not integral to the MLGUL. Hence, this enzyme was not characterized further.
To investigate oligosaccharide substrate preference of the BoGH3MLG, we conducted initial-rate kinetics analyses on a series of gluco-oligosaccharides of distinct linkage composition and degrees of polymerization (d.p.). The non-reducing-end glucose was hydrolyzed from all-β(1,4)-linked cello-oligosaccharides (d.p. 2–6), all-β(1,3)-linked laminari-oligosaccharides (d.p. 2–5), and mixed-linkage β(1,3)/β(1,4)-gluco-oligosaccharides (d.p. 3–4, 5 examples) with comparable efficiencies, according to classic Michaelis-Menten saturation kinetics (Fig. S4E, S4F; Table 1). In this series, only cellobiose (G4G) was poorly hydrolyzed by BoGH3MLG vis-à-vis the β(1,3)-linked congener laminaribiose (G3G) and all other gluco-oligosaccharides (e.g., G4G has a kcat/Km value 20-fold lower than G3G, Table 1). The β(1,6)-linked diglucoside gentiobiose (G6G) was also a very poor substrate, with a kcat/Km value 260-fold lower than that of G3G. Gluco-oligosaccharides with a β (1,3)-linked glucosyl unit at the non-reducing end all have slightly higher kcat values compared to those with a β(1,4)-linkage in this position, which typically contributes to higher kcat/Km values for the former, when substrates of equal d.p. are compared. However, the magnitude of these differences, which are often less than 2-fold, indicate that BoGH3MLG is essentially agnostic to β(1,3) versus β(1,4) linkages. These results also suggest that in addition to a single negative subsite (-1) commensurate with its exo-activity, BoGH3MLG has only two positive subsites that contribute to catalysis: in each gluco-oligosaccharide series, tetrasaccharides and larger are hydrolyzed with identical kcat/Km values to the corresponding trisaccharides.
Product analysis following extended incubation of BoGH3MLG with G4G4G3G and G4G3G demonstrated that BoGH3MLG completely degrades the BoGH16MLG limit-digest products to glucose. HPLC also revealed that laminaribiose (G3G) is the only new intermediate formed during the course of hydrolysis (Fig. 3B). This demonstrates that BoGH3MLG sequentially hydrolyzes one glucose unit at a time from the non-reducing end of MLG oligosaccharides, viz.: G4G4G3G → G + G4G3G (also present in the starting mixture) → G+ G3G → G + G. Notably, the individual kcat and Km values for each step are nearly identical (Table 1).
BoGH3MLG and BACOVA_02738(GH3) primary structures
Despite extensive efforts, we were unable to crystallize the key β-glucosidase BoGH3AMLG for experimental tertiary structure determination. However, BoGH3MLG has 63% sequence identity to a B. ovatus β-glucosidase (BoGH3B, PDB code 5JP0 (Hemsworth et al., 2016)), from the xyloglucan utilization locus (Fig. S5A) and, as such, was amenable to tertiary structure homology modelling. Phyre2 (Kelley et al., 2015) utilized PDB code 5JP0 as the sole template and 728 out of 742 residues (98% of the sequence, excluding the signal peptide) were modelled with 100 % confidence. The model suggests that BoGH3MLG possesses a homologous three-domain architecture with the active site being formed at the interface of the (α/β)8 TIM barrel and α/β sandwich domains (Fig. S5B). The predicted catalytic nucleophile and acid/base residues are Asp-309 and Glu-453, respectively, based on primary and tertiary alignment (Fig. S5A, S5C). Two tryptophan residues were modelled on opposite sides of the entrance to the active site pocket (Fig. S5D), forming a narrow “coin slot”, which may effect a preference towards β(1,3)- and β(1,4)-linked glucans and poor activity against β(1,6)-linked gentiobiose (Table 1). In constrast, enzymes that lack a homologous Trp-453 have a more open entrance to the active site and show broad activity against β(1,2)- and β(1,6)-linked glucans in addition to β(1,3)- and β(1,4)-linked glucans (Karkehabadi et al., 2014; Pozzo et al., 2010).
In comparison, BACOVA_02738(GH3) possess catalytic residues homologous to BoGH3MLG and similar GH3 members, despite having only 31% sequence identity to BoGH3MLG (Fig. S5A). The most similar characterized GH3 member to BACOVA_02738(GH3) among ca. 300 members identified in the CAZy is a Chrysosporium lucknowense β-glucosidase with 39% sequence identity (Dotsenko et al., 2012).
Syntenic MLGUL are molecular markers of MLG utilization across the Bacteroidetes
Refined functional characterization of the catalytic specificity of GH components significantly increases confidence in the use of individual PULs as genetic markers of complex carbohydrate metabolism among Bacteroidetes (Cuskin et al., 2015; Larsbrink et al., 2014; Rogowski et al., 2015; Sonnenburg et al., 2010). The MLGUL characterized here represents the only PUL in B. ovatus that confers growth on MLG from cereals. To understand the wider distribution of MLG metabolic capacity among symbiotic Bacteroidetes in the human gut, we correlated the presence of a syntenic MLGUL with growth on bMLG for 354 individual Bacteroidetes strains representing 29 different species.
A total of 121 strains across just 7 of the species were able to grow on bMLG (Fig. 5). In particular, 33 of 33 B. ovatus strains (including the type strain ATCC 8483) grew well on bMLG, as well as 44 of 45 strains of the closely related B. xylanisolvens. The majority of B. uniformis strains tested (33 out of 35) were also competent bMLG utilizers. The limited penetrance of the MLGUL across the genus clearly demonstrates nutrient-niche specialization among individual Bacteroides species.
Figure 5. Penetrance map of MLG utilization ability across diverse human gut Bacteroidetes.

The phylogenetic tree was constructed from fully sequenced strains of the species shown. The number of strains of each species tested for growth is depicted to scale as a black circle at each leaf. The number of those strains that grew on bMLG as a sole carbon source is shown to scale in red within the black circle.
Comparative analysis of available genomic sequences revealed that strains able to grow on bMLG as the sole carbon source harbor a syntenic MLGUL (Fig. 1B). Previous transcriptional analysis demonstrated that the syntenic MLGUL found in B. cellulosilyticus is also activated during growth on bMLG (McNulty et al., 2013). Concordance between the presence of a syntenic MLGUL and the ability to utilize MLG is further highlighted by the lack of a MLGUL in the B. uniformis ATCC 8492, one of only two strains of B. uniformis that could not grow on bMLG. Based on this analysis, we can also predict MLG utilization ability in two sequenced species of Prevotella, Pr. copri DSM 18205 and Pr. multiformis DSM16608, important members of the Bacteroidetes from the human gut and oral cavity, respectively (Fig. 1B).
Discussion
A molecular model for MLG utilization by B. ovatus
Our current suite of data suggests a model by which the MLGUL gene products work in concert to enable the utilization of MLG (Fig. 6), analogous to that of other PUL-encoded systems (Grondin et al., 2017). Thus, BoGH16MLG is anchored to the outer membrane where it plays a leading role in fragmenting large MLG polysaccharide chains (typical d.p. 700 – 5000, depending upon the plant species of origin (Lazaridou et al., 2004; Zheng et al., 2011)) into oligosaccharides that can be imported into the periplasm via the TBDT. Notably, the specific limit-digest products of BoGH16MLG endo-hydrolysis identified here, viz. the trisaccharide G4G3G and the tetrasaccharide G4G4G3G (Fig. 3B), have been shown previously to bind the periplasmic sensor domain of the HTCS encoded by BACOVA_02740 (KD 300 µM and 400 µM, respectively), while the intact MLG polysaccharide does not (Martens et al., 2011). Monomeric glucose, all-β(1,4)-linked cello-oligosaccharides, and all-β(1,3)-linked laminari-oligosaccharides are also not bound by the HTCS (Martens et al., 2011), indicating that the unique linkages present in MLG are central to inducing the MLGUL system. Thus, there is an essential, yet distant, interplay between the outer-membrane localized MLGase and the HTCS in specific nutrient sensing.
Figure 6. Model of mixed-linkage β-glucan saccharification by the concerted action of the MLGUL machinery.

Gene products are colored analogously to the gene locus in Fig. 1. The cell surface localized endo-MLGase BoGH16MLG cleave large mixed-linkage β-glucan polysaccharides into shorter fragments which are imported into the periplasm via the TonB dependent transporter, BoTBDT. This glycan capture and transport process at the cell surface is aided by the two surface glycan binding proteins BoSGBP-A and BoSGBP-B. The smaller mixed-linkage β-glucan fragments in the periplasm bind the sensor domain of the hybrid two-component sensor BoHTCS to induce upregulation of the system. Periplasmic exo-β-glucosidases BoGH3MLG and BACOVA_02738(GH3) act from the non-reducing ends to liberate individual glucose monomers which are imported into the cell and metabolized.
It is therefore likely that the BoGH16MLG limit-digest products, or minimal repeats of these structures [(G4G4G3G)m(G4G3G)n], comprise the main products transported through the TBDT in vivo. Recent studies on inulin (β(2,1)-fructan) utilization suggest that some TBDTs are able to import longer polysaccharide chains (e.g. d.p. >20) (Rakoff-Nahoum et al., 2016). Regardless of length, the efficient exo-hydrolytic activity of BoGH3MLG in the periplasm is sufficient to completely saccharify all imported oligosaccharides to glucose (Fig. 3B), to feed primary metabolism in the cytosol. In this process, the trisaccharide substrate of the HTCS, G4G3G, will always be generated regardless of the imported saccharide chain length, ensuring continual production of the MLGUL up-regulation signal until substrate is exhausted. Interestingly, BoGH3MLG will never encounter cellobiose (G4G), towards which it has relatively weak activity (Fig. S4F; Table 1), in this process; the final step of saccharification of MLGOs is the hydrolysis of the competent substrate laminaribiose (G3G).
Structural enzymology reveals complex trajectories for the evolution of MLG activity in GH16
Previous phylogenetic analyses of GH16 have suggested that the evolution of the active-site β-bulge motif EXDXXE, which is widespread among Clan GH-B (comprising GH16 and GH7), to a regular β-strand motif EXDXE is a defining feature that delineates endo-β(1,3)-glucanases (laminarinases, EC 3.2.1.39 and EC 3.2.1.6) from mixed-linkage endo-β(1,3)/β(1,4)-glucanases (licheninases, EC 3.2.1.73), respectively (Barbeyron et al., 1998; Michel et al., 2001). In this context, the observation that BoGH16MLG is highly specific for MLG, despite having a β-bulge motif, was surprising.
Using the CAZy Database as a starting point (http://www.cazy.org/GH16_characterized.html) together with mining of the primary literature, we generated a contemporary maximum-likelihood phylogeny of all biochemically characterized GH16 members (Fig. S6). This analysis indicates that although canonical, normal β-strand MLGases do form a monophyletic group as previously observed, MLGase activity is in fact widespread among the historical “laminarinase” group, in which BoGH16MLG is itself positioned. Despite currently limited and disparate kinetic data for individual enzymes, it also appears that it is not possible to define further substrate-specific clades within this group based on molecular phylogeny alone, in light of weak bootstrap support. This precludes defining any single evolutionary event giving rise to unique trajectories for the further diversification of extant laminarinases and MLGases in this clade. Instead, it appears that diverse, subtle mutations have allowed the independent evolution of predominant laminarinase or MLGase activity numerous times across a flat evolutionary landscape. As such, we suggest that this GH16 subgroup should be more generally referred to as the “laminarinase/MLGase group” going forward.
Detailed tertiary structural comparison of 10 β-bulge-containing members of this laminarinase/MLGase group revealed, however, that predominant laminarinases harbor a significantly more protruding loop (which is often, but not always, longer) connecting strands β2 and β3 than predominant MLGases (Fig. S7A, S7B). Structural superposition with the BoGH16MLG:G4G4G3G complex indicates that this loop in predominant laminarinases would clash with MLG in the negative subsites, instead favoring binding of an all-β(1,3)-glucan that curves away from this loop. Such curvature is exemplified by the ZgLamCGH16-E142S:thio-β-1,3-trisaccharide structure (Fig. S7A, PDB code 4CTE) (Labourel et al., 2015). Indeed, Ilari et al. observed that shortening the homologous loop in LamA from the archeon Pyrococcus furiosus (Fig. S7A, PDB code 2VY0) by 4 amino acids increased the activity towards MLG by 10-fold (Ilari et al., 2009). Likewise, BglF from Nocardiopsis sp. F96 (Fig. S6B, PDB code 2HYK) and LamR from Rhodothermus marinus (Fig. S7B, PDB code 3ILN), which have a 3.3- and 8.5-fold greater specificity constant and specific activity, respectively, toward MLG than laminarin, also have a smaller loop, similar to BoGH16MLG, in this position. The canonical, regular-β-strand MLGase from Paenibacillus macerans (Fig. S7C, PDB code 1MAC) and Bacillus licheniformis (Fig. S7C, PDB code 1GBG), similarly have a small loop at this position.
Taken together, these analyses reveal a complex evolutionary landscape that computational phylogenetic analysis fails to resolve. Despite using a manually curated, structure-based sequence alignment as input, the maximum-likelihood numerical approach did not delineate the members of the laminarinase/MLGase group on the basis of the distinct active-site loop differences observed in tertiary structures (Fig. S7). Instead, the phylogeny was likely obfuscated by diverse, random variations in amino acid composition across the entire β-sandwich domain, which clearly limits large-scale, unsupervised phylogenetic analysis of these GH16 members. Moreover, analysis of both MLG and laminarin specificity (as a minimum) for individual members of this group, in light of their tertiary structures, is essential to avoid potential mis-annotation of these enzymes.
Mining metagenomic data reveals the ubiquity of MLG utilization in the human gut and beyond
Using syntenic MLGULs as genetic markers, we surveyed the publicly available metagenome data of 426 adults to understand the capacity of human populations to derive nutrition from cereal MLGs. We were able to distinguish the species of origin based on nucleotide sequence except for MLGULs from B. ovatus and B. xylanisolvens, which were strikingly similar at 97% nucleotide identity. The B. ovatus/B. xylanisolvens and B. uniformis MLGULs are the most prevalent; both are observed in about 70% of the total human cohort (Fig. 7). The Pr. copri MLGUL is more often the sole MLGUL of an individual than that of B. cellulosilyticus when only one is present (Fig. 7, cyan lines), despite the latter being more frequent in total. Overall, 92.5% of the subjects harbor at least one of the five different MLGULs identified in this study, irrespective of nationality or whether they have been diagnosed with IBD. MLGULs are ubiquitously detectable despite variability in sampling depth across different metagenomics sequencing projects (Fig. 7). The prevalence of MLGULs across different nationalities is consistent with MLG from cereal grains being a ubiquitous component of the human diet. Indeed, the importance of MLG utilization is underscored by the upregulation of the MLGUL in the ceca of mice fed a complex plant cell wall diet (Martens et al., 2011). Similar widespread global distribution in human populations has been observed for xyloglucan utilization loci (Larsbrink et al., 2014). These observations are sharply contrasted by the PUL that mediates utilization of the red algal polysaccharide porphyran, which is essentially confined to subjects from Japan, where seaweed is a common part of the diet (Hehemann et al., 2010; Larsbrink et al., 2014). Interestingly, we were unable to detect MLGULs in four unweaned infants sampled in the Japanese metagenome project (data not included in our analysis of adult metagenomes). This is consistent with a dearth of Bacteroidetes in infants who receive the bulk of their nutrition from milk and are not yet consuming plant polysaccharides (Urokawa et al., 2007)
Figure 7. Bacteroidetes MLGULs from a survey of 426 adult human gut metagenomes.

Vertical lines represent the presence (cyan when unique, blue when one of multiple) or absence (black) of a corresponding species-related MLGUL in a single individual. The total number of MLGULs observed in an individual is shown in the bottom row, colored according to the legend in the top left corner. The frequency of MLGUL occurrence across all 426 individuals is shown on the right. Variation in sequencing depth in megabase pair is illustrated in the graph below: grey lines show the depth for individual subjects and black lines show the average depth of each metagenomics project.
Moving beyond the human microbiota, we can likewise predict MLG utilization ability in Dysgonomonas gadei and Pr. oryzae (formerly Xylanibacter oryzae) based on the presence of a syntenic MLGUL. These species are commonly found in the termite hindgut and decomposing rice straw, respectively. This provides direct evidence that an analogous MLG utilization system is employed by Bacteroidetes operating in environments beyond the human gut.
Conclusion
Complex carbohydrates that promote the growth of beneficial microbes in our distal large intestine are a cornerstone of a healthy diet. MLGs in particular have long been known to impart healthful effects (Othman et al., 2011), yet its mechanism of utilization for fermentation by gut microbes was unknown. Our work here sheds light on the fine-tuned mechanism that B. ovatus and other Bacteroidetes has evolved to efficiently utilize MLGs in the highly competitive environment of the human gut microbiota. The finding that a majority of humans possess microbes that can utilize this ubiquitous cereal polysaccharide highlights the relevance of potential therapeutic interventions based on MLG utilization to the general population. The present study also sets the stage for future work to understand the quantitative contributions of individual members of the microbiota and their cognate enzymes to MLG utilization in the human gut (Patrascu et al., 2017; Zhong et al., 2015).
Experimental Procedures
Microbiology
B. ovatus gene deletions were constructed using allelic exchange as previously described (Koropatkin et al., 2008). Anaerobic growth profiles were measured as previously described (Martens et al., 2011). Details of localization analysis by immunofluorescence microscopy and immunoblotting are provided in the Supplemental Experimental Procedures.
Cloning, expression, and purification of recombinant enzymes
The genes encoding BoGH16MLG, BoGH3MLG, and BACOVA_02738(GH3) were cloned into expression vectors for recombinant production in E. coli. Details of cloning strategies, production, and purification are provided in the Supplemental Experimental Procedures.
Enzyme kinetics and product analysis
Thorough kinetic analysis was conducted on a panel of polysaccharides, oligosaccharides, and chromogenic substrates. Products of enzymatic reactions were analyzed by HPAEC-PAD and HILIC-MS. Details of enzymatic assays, analytical methods, as well as sources of commercial substrates are provided in the Supplemental Experimental Procedures.
X-ray crystallography
Crystals of BoGH16MLG were screened and optimized by sitting drop vapor diffusion method. The structures of the apo- and G4G4G3G-BoGH16MLG were solved by molecular replacement. Details of crystallization, data collection, and structure solution are provided in the Supplemental Experimental Procedures.
Bioinformatics
Phylogenetic anlaysis of select GH16 and GH3 sequences was conducted based on structure-guided alignment. Metagenomic survey was carried out by nucleotide BLAST of MLGUL sequences against various metagenome sequence data. Details are provided in the Supplemental Experimental Procedures.
Statistical analysis
All kinetic assays were done in triplicate. Michaelis-Menten parameters are reported as fitted values ± standard deviation throughout the article. All growth experiment results are averages of two biological replicates.
Supplementary Material
Highlights.
The molecular mechanism of MLG utilization by Bacteroides ovatus is presented.
BoGH16MLG possesses structural features suited for efficient MLG degradation.
MLG utilization loci (MLGULs) serve as genetic markers for MLG catabolism.
Bacteroidetes MLGULs are ubiquitous in the gut microbiota of human populations.
Note: MLG may be expanded to “mixed-linkage beta-glucan” for clarity, if the character count restriction can be relaxed.
Acknowledgments
We thank Diamond Light Source (Harwell, UK) for access to beamlines I02 and I03 (proposal mx9948) that contributed to the results presented here. Work in Vancouver was supported by Operating Grants from the Canadian Institutes for Health Research (MOP-137134 and MOP-142472) and infrastructure support from the Canadian Foundation for Innovation and the British Columbia Knowledge Development Fund. We thank Constance M. Bahr (Koropatkin group, U. Michigan) for invaluable assistance with microscopy. We thank Adriana Cabrera (Brumer group) for preparing laminaritol by sodium borohydride reduction of laminarin. We thank Nicholas McGregor (Brumer group) for assistance with LC-MS. We thank Alexander H. Viborg (http://research.ahv.dk/) for access to CAZy database tools. We thank Hila Behar (Brumer group) for assistance with BLAST analysis of human metagenome sequences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author contributions
KT cloned, expressed, and purified recombinant enzymes, conducted and analyzed kinetics for hydrolysis of polysaccharides, oligosaccharides, and chromogenic substrates, determined hydrolysis products, conducted phylogenetic and structural analyses, carried out metagenomics survey, and wrote the article. GRH determined the crystal structures of apo- and G4G4G3G-complexed BoGH16MLG. GD conducted enzyme localization analyses. TR, NP, and KU conducted reverse genetics and growth analyses. NJ synthesized G3G-CNP. GJD, ECM, and HB designed and directed research and co-wrote the article with input from all authors.
Accession numbers
The atomic coordinates and structure factors of apo- and G4G4G3G-complexed BoGH16MLG reported in this paper can be accessed through the PDB with identifiers 5NBO and 5NBP, respectively.
References
- Bågenholm V, Reddy SK, Bouraoui H, Morrill J, Kulcinskaja E, Bahr CM, Aurelius O, Rogers T, Xiao Y, Logan DT, et al. Galactomannan Catabolism Conferred by a Polysaccharide Utilization Locus of Bacteroides ovatus. 2017;292:229–243. doi: 10.1074/jbc.M116.746438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbeyron T, Gerard A, Potin P, Henrissat B, Kloareg B. The Kappa-Carrageenase of the Marine Bacterium Cytophaga drobachiensis. Structural and Phylogenetic Relationships Within Family-16 Glycoside Hydrolases. Mol. Biol. Evol. 1998;15:528–537. doi: 10.1093/oxfordjournals.molbev.a025952. [DOI] [PubMed] [Google Scholar]
- Biedermann L, Rogler G. The intestinal microbiota: its role in health and disease. Eur. J. Pediatr. 2015;174:151–167. doi: 10.1007/s00431-014-2476-2. [DOI] [PubMed] [Google Scholar]
- Blanton LV, Barratt MJ, Charbonneau MR, Ahmed T, Gordon JI. Childhood undernutrition, the gut microbiota, and microbiota-directed therapeutics. Science. 2016;352:1533–1533. doi: 10.1126/science.aad9359. [DOI] [PubMed] [Google Scholar]
- Cantarel BI, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009;37:233–238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciorba M. A Gastroenterologist’s Guide to Probiotics. Clin. Gastroenterol. Hepatol. 2012;10:960–968. doi: 10.1016/j.cgh.2012.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JH, Macfarlane GT. Role of intestinal bacteria in nutrient metabolism. JPEN. J. Parenter. Enteral Nutr. 1997;21:357–365. doi: 10.1177/0148607197021006357. [DOI] [PubMed] [Google Scholar]
- Cuskin F, Lowe EC, Temple MJ, Zhu Y, Cameron EA, Pudlo NA, Porter NT, Urs K, Thompson AJ, Cartmell A, et al. Human gut Bacteroidetes can utilize yeast mannan through a selfish mechanism. Nature. 2015;517:165–169. doi: 10.1038/nature13995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies GJ, Sinnot ML. Sorting the diverse. Biochem. J. 2008;30:26–32. [Google Scholar]
- Davies GJ, Wilson KS, Henrissat B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem. J. 1997;321:557–559. doi: 10.1042/bj3210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dotsenko GS, Sinitsyna OA, Hinz SWA, Wery J, Sinitsyn AP. Bioresource Technology Characterization of a GH family 3 β-glycoside hydrolase from Chrysosporium lucknowense and its application to the hydrolysis of β-glucan and xylan. Bioresour. Technol. 2012;112:345–349. doi: 10.1016/j.biortech.2012.02.105. [DOI] [PubMed] [Google Scholar]
- Eklof J, Hehemann J-H. Glycoside Hydrolase Family 16. [accessed 10 February 2017];CAZypedia. available at URL http://www.cazypedia.org/
- Elhenawy W, Debelyy MO, Feldman MF. Preferential packing of acidic glycosidases and proteases into Bacteroides outer membrane vesicles. mBio. 2014;5:1–12. doi: 10.1128/mBio.00909-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, Collini S, Pieraccini G, Lionetti P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. U.S.A. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fincher G, Mark B, Brumer H. Glycoside Hydrolase Family 3. [accessed 10 February, 2017];CAZypedia. available at URL http://www.cazypedia.org/
- Fujimura K, Slusher N, Cabana M, Lynch S. Role of the gut microbiota in defining human health. Expert Rev Anti Infect Ther. 2010;8:435–454. doi: 10.1586/eri.10.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiser OJ, Piotukh K, Ponnuswamy MN, Planas A, Borriss R, Heinemann U. Structural basis for the substrate specificity of a Bacillus 1,3-1,4-β-glucanase. J. Mol. Biol. 2006;357:1211–1225. doi: 10.1016/j.jmb.2006.01.014. [DOI] [PubMed] [Google Scholar]
- Grondin JM, Tamura K, Déjean G, Abbott DW. Polysaccharide Utilization Loci: Fueling Microbial Communities. J. Bacteriol. 2017;199:1–15. doi: 10.1128/JB.00860-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haak BW, Levi M, Wiersinga WJ. Microbiota-targeted therapies on the intensive care unit. Curr. Opin. Crit. Care. 2017;23:1–8. doi: 10.1097/MCC.0000000000000389. [DOI] [PubMed] [Google Scholar]
- Hahn M, Pons J, Planas a, Querol E, Heinemann U. Crystal structure of Bacillus licheniformis 1,3-1,4-beta-D-glucan 4-glucanohydrolase at 1.8 A resolution. FEBS Lett. 1995;374:221–224. doi: 10.1016/0014-5793(95)01111-q. [DOI] [PubMed] [Google Scholar]
- Hamaker BR, Tuncil YE. A perspective on the complexity of dietary fiber structures and their potential effect on the gut microbiota. J. Mol. Biol. 2014;426:3838–3850. doi: 10.1016/j.jmb.2014.07.028. [DOI] [PubMed] [Google Scholar]
- Harada T, Misaki A, Saito H. Curdlan: A bacterial gel-forming β-1-3-glucan. Arch. Biochem. Biophys. 1968;124:292–298. doi: 10.1016/0003-9861(68)90330-5. [DOI] [PubMed] [Google Scholar]
- Hehemann J-H, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature. 2010;464:908–912. doi: 10.1038/nature08937. [DOI] [PubMed] [Google Scholar]
- Hemsworth GR, Thompson AJ, Stepper J, Sobala ŁF, Coyle T, Larsbrink J, Spadiut O, Goddard-Borger ED, Stubbs KA, Brumer H, et al. Structural dissection of a complex Bacteroides ovatus gene locus conferring xyloglucan metabolism in the human gut. Open Biol. 2016;6:1–14. doi: 10.1098/rsob.160142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L, Rosenstrom P. Dali server: Conservation mapping in 3D. Nucleic Acids Res. 2010;38:545–549. doi: 10.1093/nar/gkq366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilari A, Fiorillo A, Angelaccio S, Florio R, Chiaraluce R, Van Der Oost J, Consalvi V. Crystal structure of a family 16 endoglucanase from the hyperthermophile Pyrococcus furiosus - Structural basis of substrate recognition. FEBS J. 2009;276:1048–1058. doi: 10.1111/j.1742-4658.2008.06848.x. [DOI] [PubMed] [Google Scholar]
- Juncker A, Willenbrock H. Prediction of lipoprotein signal peptides in Gram negative bacteria. Protein Sci. 2003;12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kaoutari A, Armougom F, Gordon JI, Raoult D, Henrissat B. The abundance and variety of carbohydrate-active enzymes in the human gut microbiota. Nat. Rev. Microbiol. 2013;11:497–504. doi: 10.1038/nrmicro3050. [DOI] [PubMed] [Google Scholar]
- Karkehabadi S, Helmich KE, Kaper T, Hansson H, Mikkelsen N, Gudmundsson M, Piens K, Fujdala M, Banerjee G, Scott-craig JS, et al. Biochemical Characterization and Crystal Structures of a Fungal Family 3 β-Glucosidase, Cel3A from Hypocrea jecorina. 2014;289:31624–31637. doi: 10.1074/jbc.M114.587766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature. 2011;474:327–336. doi: 10.1038/nature10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Khoury D, Cuda C, Luhovyy BL, Anderson GH. Beta glucan: Health benefits in obesity and metabolic syndrome. J. Nutr. Metab. 2012;2012:1–28. doi: 10.1155/2012/851362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kootte RS, Vrieze A, Holleman F, Dallinga-Thie GM, Zoetendal EG, de Vos WM, Groen AK, Hoekstra JBL, Stroes ES, Nieuwdorp M. The therapeutic potential of manipulating gut microbiota in obesity and type 2 diabetes mellitus. Diabetes, Obes. Metab. 2012;14:112–120. doi: 10.1111/j.1463-1326.2011.01483.x. [DOI] [PubMed] [Google Scholar]
- Koropatkin NM, Martens EC, Gordon JI, Smith TJ. Starch Catabolism by a Prominent Human Gut Symbiont Is Directed by the Recognition of Amylose Helices. Structure. 2008;16:1105–1115. doi: 10.1016/j.str.2008.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koropatkin NM, Cameron Ea, Martens EC. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 2012;10:323–335. doi: 10.1038/nrmicro2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labourel A, Jam M, Legentil L, Sylla B, Hehemann JH, Ferrières V, Czjzek M, Michel G. Structural and biochemical characterization of the laminarinase ZgLamCGH16 from Zobellia galactanivorans suggests preferred recognition of branched laminarin. Acta Crystallogr. Sect. D Biol. Crystallogr. 2015;71:173–184. doi: 10.1107/S139900471402450X. [DOI] [PubMed] [Google Scholar]
- Larsbrink J, Rogers TE, Hemsworth GR, McKee LS, Tauzin AS, Spadiut O, Klinter S, Pudlo Na, Urs K, Koropatkin NM, et al. A discrete genetic locus confers xyloglucan metabolism in select human gut Bacteroidetes. Nature. 2014;506:498–502. doi: 10.1038/nature12907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaridou A, Biliaderis CG, Micha-Screttas M, Steele BR. A comparative study on structure-function relations of mixed-linkage (1→3), (1→4) linear β-D-glucans. Food Hydrocoll. 2004;18:837–855. [Google Scholar]
- Littman DR, Pamer EG. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe. 2011;10:311–323. doi: 10.1016/j.chom.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowman DW, West LJ, Bearden DW, Wempe MF, Power TD, Ensley HE, Haynes K, Williams DL, Kruppa MD. New insights into the structure of (1-3,1-6)-β-D-glucan side chains in the Candida glabrata cell wall. PLoS One. 2011;6:1–10. doi: 10.1371/journal.pone.0027614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC, Koropatkin NM, Smith TJ, Gordon JI. Complex glycan catabolism by the human gut microbiota: The bacteroidetes sus-like paradigm. J. Biol Chem. 2009;284:24673–24677. doi: 10.1074/jbc.R109.022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC, Lowe EC, Chiang H, Pudlo NA, Wu M, McNulty NP, Abbott DW, Henrissat B, Gilbert HJ, Bolam DN, et al. Recognition and degradation of plant cell wall polysaccharides by two human gut symbionts. PLoS Biol. 2011;9:1–16. doi: 10.1371/journal.pbio.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC, Kelly AG, Tauzin AS, Brumer H. The devil lies in the details: How variations in polysaccharide fine-structure impact the physiology and evolution of gut microbes. J. Mol. Biol. 2014;426:3851–3865. doi: 10.1016/j.jmb.2014.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K, McDougall BM, McIlroy S, Jayus Chen J, Seviour RJ. Biochemistry and molecular biology of exocellular fungal β-(1,3)- and β-(1,6)-glucanases. FEMS Microbiol. Rev. 2007;31:168–192. doi: 10.1111/j.1574-6976.2006.00055.x. [DOI] [PubMed] [Google Scholar]
- Mcgregor N, Yin V, Tung C, Petegem F Van, Brumer H. Crystallographic insight into the evolutionary origins of xyloglucan endotransglycosylases and endohydrolases. Plant J. 2017;89:651–670. doi: 10.1111/tpj.13421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor N, Morar M, Fenger TH, Stogios P, Lenfant N, Yin V, Xu X, Evdokimova E, Cui H, Henrissat B, et al. Structure-function analysis of a mixed-linkage β-glucanase/xyloglucanase from the key ruminal bacteroidetes prevotella bryantii B14. J. Biol. Chem. 2016;291:1175–1197. doi: 10.1074/jbc.M115.691659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeil NI. The contribution of the large-intestine to energy supplies in man. Am. J. Clin. Nutr. 1984;39:338–342. doi: 10.1093/ajcn/39.2.338. [DOI] [PubMed] [Google Scholar]
- McNulty NP, Wu M, Erickson AR, Pan C, Erickson BK, Martens EC, Pudlo NA, Muegge BD, Henrissat B, Hettich RL, et al. Effects of Diet on Resource Utilization by a Model Human Gut Microbiota Containing Bacteroides cellulosilyticus WH2, a Symbiont with an Extensive Glycobiome. PLoS Biol. 2013;11:1–20. doi: 10.1371/journal.pbio.1001637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel G, Chantalat L, Duee E, Barbeyron T, Henrissat B, Kloareg B, Dideberg O. The κ-carrageenase of P. carrageenovora features a tunnel-shaped active site: A novel insight in the evolution of clan-B glycoside hydrolases. Structure. 2001;9:513–525. doi: 10.1016/s0969-2126(01)00612-8. [DOI] [PubMed] [Google Scholar]
- Ndeh D, Rogowski A, Cartmell A, Luis AS, Baslé A, Gray J, Venditto I, Briggs J, Zhang X, Labourel A, et al. Complex pectin metabolism by gut bacteria reveals novel catalytic functions. Nature. 2017;544:65–70. doi: 10.1038/nature21725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Othman RA, Moghadasian MH, Jones PJH. Cholesterol-lowering effects of oat β-glucan. Nutr. Rev. 2011;69:299–309. doi: 10.1111/j.1753-4887.2011.00401.x. [DOI] [PubMed] [Google Scholar]
- Paetzel M, Karla A, Strynadka NCJ, Dalbey RE. Signal peptidases. Chem. Rev. 2002;102:4549–4579. doi: 10.1021/cr010166y. [DOI] [PubMed] [Google Scholar]
- Patrascu O, Béguet-Crespel F, Marinelli L, Chatelier E Le, Abraham A, Leclerc M, Klopp C, Terrapon N, Henrissat B, Blottière HM, et al. A fibrolytic potential in the human ileum mucosal microbiota revealed by functional metagenomic. Sci. Rep. 2017;7:1–15. doi: 10.1038/srep40248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Planas A. Bacterial 1,3-1,4-β-glucanases: structure, function and protein engineering. Biochim. Biophys. Acta. 2000;1543:361–382. doi: 10.1016/s0167-4838(00)00231-4. [DOI] [PubMed] [Google Scholar]
- Pozzo T, Pasten JL, Karlsson EN, Logan DT. Structural and Functional Analyses of β-Glucosidase 3B from Thermotoga neapolitana: A Thermostable Three-Domain Representative of Glycoside Hydrolase 3. J. Mol. Biol. 2010;397:724–739. doi: 10.1016/j.jmb.2010.01.072. [DOI] [PubMed] [Google Scholar]
- Rakoff-Nahoum S, Foster KR, Comstock LE. The evolution of cooperation within the gut microbiota. Nature. 2016;533:255–259. doi: 10.1038/nature17626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogowski A, Briggs JA, Mortimer JC, Tryfona T, Terrapon N, Lowe EC, Baslé A, Morland C, Day AM, Zheng H, et al. Glycan complexity dictates microbial resource allocation in the large intestine. Nat. Commun. 2015;6:1–15. doi: 10.1038/ncomms8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwabe RF, Jobin C. The microbiome and cancer. Nat. Rev. Cancer. 2013;13:800–812. doi: 10.1038/nrc3610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slavin J. Fiber and prebiotics: Mechanisms and health benefits. Nutrients. 2013;5:1417–1435. doi: 10.3390/nu5041417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Sonnenburg JL. Starving our microbial self: The deleterious consequences of a diet deficient in microbiota-accessible carbohydrates. Cell Metab. 2014;20:779–786. doi: 10.1016/j.cmet.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Zheng H, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, Sonnenburg JL. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141:1241–1252. doi: 10.1016/j.cell.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian S, Blanton LV, Frese SA, Charbonneau M, Mills DA, Gordon JI. Cultivating healthy growth and nutrition through the gut microbiota. Cell. 2015;161:36–48. doi: 10.1016/j.cell.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauzin AS, Kwiatkowski KJ, Orlovsky NI, Smith CJ, Creagh AL, Haynes CA, Wawrzak Z, Brumer H, Koropatkin NM. Molecular Dissection of Xyloglucan Recognition in a Prominent Human Gut Symbiont. mBio. 2016;7:e02134–15. doi: 10.1128/mBio.02134-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urokawa KK, Toh TI, Uwahara TK, Shima KO, Oh HT, Oyoda AT, Ori HM, Gura YO, Hrlich DSE, Toh KI, et al. Comparative Metagenomics Revealed Commonly Enriched Gene Sets in Human Gut Microbiomes. DNA Res. 2007;14:169–181. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Li L, Wang Q. Distribution and molecular characterization of β-glucans from hull-less barley bran, shorts and flour. Int. J. Mol. Sci. 2011;12:1563–1574. doi: 10.3390/ijms12031563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Marungruang N, Fak F, Nyman M. Effects of two whole-grain barley varieties on caecal SCFA, gut microbiota and plasma inflammatory markers in rats consuming low- and high-fat diets. Br. J. Nutr. 2015;113:1558–1570. doi: 10.1017/S0007114515000793. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.