

Abstract
Colorectal cancer (CRC) is a leading cause of cancer mortality. Diet has a significant influence on colon cancer risk. Identifying chemopreventive agents, dietary constituents, practices and/or diet supplements that promote gut health and reduce the incidence of intestinal neoplasias and CRC could significantly impact public health. Sphingadienes (SDs) are dietary sphingolipids found in plant-based food products. SDs are cytotoxic to colon cancer cells and exhibit chemopreventive properties. The aim of the present study was to develop a sensitive and robust ultra-high performance liquid chromatography tandem mass spectrometry (UHPLC-MS/MS) method for quantifying SDs in food products and biological samples. The assay was linear over a concentration range of 80 nM to 50 μM and was sensitive to a detection limit of 3.3 nM. Post-extraction stability was 100% at 24h. SD content in soy oils was approximately 10 nM. SDs were detected transiently in the plasma of adult mice 10 minutes after gavage delivery of a 25 mg/kg bolus and declined to baseline by 1 hour. SD uptake in the gut was maximal in the duodenum and peaked 1 hour after gavage delivery. Disappearance of SDs in the lower gastrointestinal tract suggests either rapid metabolism to yet unidentified products or potentially luminal export.
Keywords: sphingolipids, sphingadienes, mass spectrometry, soy, chemoprevention, colon cancer
1.0. Introduction
Colorectal cancer (CRC) is diagnosed in over a million people each year (1). Worldwide, CRC represents 10% of all cancers (2). In the US, it is the 3rd leading cancer diagnosis and cause of cancer death. CRC develops slowly from pre-malignant colon adenomas and usually occurs in those over 50. The long latency between development of colon adenomas and progression to CRC creates a window of opportunity for chemoprevention in patients with CRC risk factors (3).
Epidemiological and preclinical evidence suggest that dietary habits affect CRC incidence (4-10). CRC rates rise in developing nations as they adopt “western” diets high in fat and low in antioxidants (11). Sphingolipids are ubiquitous membrane lipids found in our bodies and many constituents of the human diet, including meat, dairy and whole soy products, which contain approximately 600 pmol sphingolipids/ gram soy (12-14). The average consumption of sphingolipids is about 0.3-0.4 grams per day. All sphingolipids contain a long chain amino base backbone, which in mammalian cells is sphingosine. Sphingolipid metabolism is a major activity of gut enterocytes (15). Sphingolipids in mammalian food products are degraded in the gut lumen by enzymes located in the brush border, resulting in the release of sphingosine, which is actively taken up by enterocytes. There, it is either utilized in lipid biosynthesis or converted to the bioactive sphingolipid, sphingosine-1 -phosphate (S1P)(16). S1P is mitogenic and promotes colitis and CAC via STAT3-dependent mechanisms that promote cell transformation, an early step in carcinogenesis (17-19). Further, human colitic bowel exhibits genetic changes that favor S1P formation and reduce its degradation by S1P lyase (SPL), leading to S1P accumulation (17).
Soy and other plants such as rice and wheat brans contain a unique family of sphingolipids called sphingadienes (SDs) that contain two double bonds at C4 and C8-positions (16). Desaturation of SD at the C8-position are synthesized by Δ8-sphingolipid desaturase, which leads to the formation of cis- and trans- isomers C18-sphinga-(4E, 8Z)-dienes (20). Higher order animals can incorporate SDs from plants by direct absorption as the free sphingoid base or by liberating SDs from intestinal breakdown of glucosylceramides that contain SD as a constituent (20,21).
Metabolic fates of SD remain incompletely understood. The presence of SD-derived sphingoglycolipids, sphingomyelin (SM) and ceramides in human breast milk (22), plasma (23,24), animal meats (25), and mouse brain (26) suggest that SDs may be in part metabolized to more complex sphingolipids. However, little is known regarding metabolic role(s) of SD and their downstream metabolic products.
We and others showed in cells and in vivo that phytosphingolipids such as SDs exert chemopreventive effects in the gut and attenuate tumorigenesis initiated by chronic inflammation (17,27-30). We discovered that oral administration of SDs at a dose of 25 mg/kg bw reduces colon cytokine levels, upregulates SPL expression, reduces S1P levels in colon tissues, and suppresses spontaneous intestinal tumor formation, colitis and CAC in mouse models of CRC (17,31). We have also shown that SDs reduce cell proliferation and induce autophagy and apoptosis in CRC cells through inhibition of AKT, WNT and STAT3 signaling pathways (27,31).
Although SDs hold promise as chemopreventive agents, we are unaware of any reports describing detailed methodology for their quantitation in food products and biological samples. We have developed a facile assay to quantitate SDs with a single-phase liquid extraction system which is followed by a rapid UHPLC-MS/MS analysis. We show the utility of our assay for quantifying soy-type C18 (4E, 8Z) SDs in soy oils, tofu and human plasma and characterize the uptake of SDs into murine intestinal tissues and plasma over time after administration of an oral bolus.
2.0. Materials and methods
2.1. Chemicals
4E,8Z-Sphingadiene (SD), and sphingosine (d17:1; C17-SO) were purchased from Avanti Polar Lipids Inc (AL, USA). Purity of these standards were greater than 98%. Ammonium formate, sucrose, potassium chloride, Tris-Cl, Ethylenediaminetetraacetic acid (EDTA), phosphate-buffered saline tablets (pH 7.4), diethylenetriaminepentaacetic acid (DTPA) and formic acid were purchased from Sigma Aldrich and were of the highest reagent grade (MO, USA). Isopropanol (HPLC grade), ethyl acetate (reagent grade), chloroform (reagent grade) and methanol (LCMS grade) were purchased from Thermo Fisher Scientific (MA, USA). Chelex 100 Resin was purchased from BioRad.
2.2. Sample preparation
2.2.1. Standard stock preparations
Stock solutions of SD were prepared by dissolving 1 mg SDs in 1 ml of methanol and diluting to desired working concentrations with methanol. C17-SO internal standard was prepared by dissolving 2.86 mg in 10 ml of methanol. Solutions were aliquoted into amber glass vials and kept in -20°C until analysis.
2.2.2. Calibration curve
The calibration curve of SD consisted of eleven levels of calibrators (0.08, 0.25, 0.6, 1.5, 2.5, 5, 10, 20, 30, 40, 50 μM). The calibration curves were prepared in either PBS or in human plasma. Metal free PBS buffer was prepared by dissolving five PBS tablets (Sigma Aldrich) in 1L of milliQ water. To this solution, 3 g of Chelex-100 resin was added and stirred overnight and filtered using a 0.45 μm filter. DTPA (100 μM final) was added to filtered PBS solution. Briefly, 100 μl of the calibrator solutions were added to either 100 μl of PBS or human plasma and were extracted using the protocols below:
2.2.3. Extraction and homogenization buffers
Extraction buffer for liquid samples (biological fluids, medium or buffer) consisted of Isopranol:ethyl acetate solutions (15:85 v/v). Extraction buffer for tissues or other solid samples consisted of a mixture of isopropanol:water:ethyl acetate (30:10:60 v/v/v). Homogenization buffer for tissues consisted of 50 mM Tris-HCl (pH 7.4) buffer containing 0.25 M sucrose, 25 mM potassium chloride and 0.5 mM EDTA.
2.2.4. SD extraction from biological fluids or buffers
Samples or standard solutions (200 μl) were mixed with 1.8 ml of PBS in borosilicate glass tubes (13 × 100 mm). To these solutions, 10 μl of internal standard (C17-SO, 5 μM) was added and briefly vortexed. Appropriate extraction buffer (2 ml) was added and briefly vortexed. These samples were centrifuged at 3000 RPM at 4°C for 10 min. The upper organic phase was transferred into a new glass tube with a glass Pasteur pipette. Concentrated formic acid (3 μl) was added to the bottom layer and briefly vortexed. Acidified samples were re-extracted with the addition of 2 ml of extraction buffer followed by brief vortexing and centrifugation at 3000 RPM at 4°C for 10 min. The upper organic phase from the second extraction was combined with the previously collected samples and vortexed. A two-ml aliquot of the samples was transferred into a new glass tube and dried down completely either under a constant stream of nitrogen or by using the speedvac under ambient temperature (Thermo Savant SpeedVac system SPD1000). Dried samples were reconstituted with 200 μl of Mobile Phase B (described in section 2.3). and transferred into a HPLC vial with glass inserts for injection.
2.2.5. SD extraction from solid tissues, cells or tofu samples
Frozen tissue sections (∼20 mg) were weighed and transferred into a 2 ml tube containing 1 ml of pre-chilled homogenization buffer (described in section 2.3.3). To these samples, glass beads were added to each tube and homogenized using the FastPrep FP120 tissue disrupter system (Thermo Fisher Inc.) with a setting of 5 for 30 sec. Samples were taken out and vortexed, and the FastPrep homogenization step was repeated for an additional 30 sec. Homogenized samples were centrifuged at 20K RPM at 4°C for 5 min. Supernatant was transferred to a new 1.5 ml tube, and a 30 μl aliquot was taken for protein measurements.
For extraction of SD, 200 μl of tissue extracts were mixed with 1.8 ml of PBS in borosilicate glass tubes (10 × 13 mm). To these samples, 10 μl of internal standard (C17-SO, 5 μM) were added and briefly vortexed. Two ml of extraction buffer (described in section 2.3) were added and briefly vortexed. These samples were sonicated in an ice chilled water bath for 30 sec. This sonication step was repeated 3 times. Following sonication, samples were centrifuged at 3000 RPM for 10 min at 4°C. The upper organic phase was transferred to a new glass tube. To the bottom aqueous phase, 3 μl of concentrated formic acid were added and briefly vortexed. These samples were re-extracted with the addition of 2 ml of extraction buffer, vortexed and centrifuged at 3000 RPM for 10 min at 4°C. The upper phase from the second extraction was combined together with the first, and a 2 ml aliquot was dried down to completion using a speedvac. Dried samples were reconstituted with 200 μl of Mobile phase B and transferred into HPLC vials with glass inserts for storage and injection.
2.2.6. Extraction of SD from soy oil samples
For extraction of SD from oil samples, 200 μl of oil samples were mixed with 3 ml of chloroform:methanol (2:1 v/v) in boroscilicate glass tubes. Internal standard (C17-SO) was added (3 μl). To these samples, 1ml of PBS was added and briefly vortexed, followed by centrifugation at 2000 RPM for 5 min. The bottom chloroform layer was transferred to a new glass tubes. To the remaining top layer, 3 μl of concentrated formic acid were added, and samples were re-extracted with the addition of 2 ml of chloroform. Samples were vortexed and centrifuged at 2000 RPM for 5 min. Bottom fractions were collected and combined and dried in a speedvac under ambient temperature. Dried samples were reconstituted in 200 μl of mobile phase B and transferred into a HPLC vial with glass inserts for storage and analysis.
2.3. LC-MS/MS settings
A 1290 ultra-high performance LC (UHPLC) system coupled to an Agilent 6490 Triple Quadrupole (QqQ) MS equipped with Agilent Jet Stream (AJS)-electrospray ionization interface was used. The instrument was operated by Mass Hunter Workstation software. Precursor and production ion selection and optimization of collision energies were performed manually by flow injection of analytical standards. A Zorbax SB C18 UHPLC column (5 μm 2.1 × 50 mm; PN 860975-902; Agilent, CA, USA) maintained at 50°C was used. A binary gradient of mobile phase A (1 mM ammonium formate in water containing 0.2% formic acid) and B (1 mM ammonium formate in methanol with 0.2% formic acid) was delivered at a constant flow rate of 1 ml/min. The total run time was 4 min. Initial gradient condition was maintained at 80% B and a linear gradient to 100% B within 2 min and was returned to baseline condition at 2.1 min to allow for ∼ 2 min column re-equilibration.
Mass detection was used in the positive electrospray ionization (ESI+) mode with multiple reaction monitoring (MRM). The MRM transitions used for SD detection were 298.2→280.2 [collision energy (CE); 8 V], and the transitions for C17-SO were 286.5→268.3 (CE 4 V). The general source settings in the positive ionization modes were as follows: gas temperature 200°C; gas flow, 14 Lmin-1; nebulizer 20 psi; sheath gas temperature 250°C; sheath gas flow 11 L min-1; capillary voltage 3000 V; and nozzle voltage; 0V. The fragmentor voltage of 380V and a dwell time of 15 ms were used for all mass transitions, and both Q1 and Q3 resolutions were set to nominal mass unit resolution.
2.4. Human plasma
Human plasma was obtained by percutaneous phlebotomy of pediatric patients undergoing endoscopy to rule out inflammatory bowel disease. All samples were obtained from patients with no evidence of gross pathology on endoscopic inspection and no evidence of microscopic pathology on biopsy samples. Blood was collected in EDTA tubes, and plasma was isolated by low speed centrifugation at 4°C. Samples remained in -80°C until use. These studies were conducted in accordance with an approved Institutional Review Board (IRB) protocol. Human plasma was used for testing matrix effects, recovery and precision of our SD assay.
2.5. Oral SD absorption time course study
To determine the time course of SD uptake in the gut tissues and plasma, SD solutions (4mg/ml) were prepared in 0.5% methylcellulose (w/v) dissolved in pH 4.0 water. Solutions were kept in 4°C for 2 hours to allow SD to be solubilized in methylcelluose solution. This dose of SD was used based on our prior evidence of efficacy in lowering gut inflammation in mice (27). C57/BL6 wild type mice of 8-12 weeks of age were weighed, and SD was delivered by oral gavage at a dose of 25 mg/kg body weight. Baseline samples were obtained from mice (n = 3) administered vehicle (0.5% methylcellulose solution without SD). Additional groups of mice were euthanized at each specified time point by CO2 asphyxiation (n = 3 per time point), followed by exsanguination with cardiac puncture to obtain blood. Duodenum, jejunum, ileum, and colon sections were separated, flushed with cold PBS, and flash-frozen in liquid nitrogen. Samples were stored in -80°C until analysis. All animal care, husbandry, euthanasia and tissue harvest in this study were performed in accordance with approved Institutional Animal Care and Use Committee protocols.
2.6. Data analysis
2.6.1. Matrix effect
MS/MS areas generated from known amounts of standards (A) in PBS were compared with those obtained from plasma samples spiked with the same amount of analyte (B). The ratio (B/A × 100) is defined as the absolute matrix effect (ME%). A value of 100% indicates that there is no absolute matrix effect. There is signal enhancement if the value is >100% and signal suppression if the value is <100%. For comparisons of ME across different biological backgrounds, signals generated from C17-SO was used to quantify ME.
2.6.2. Precision and accuracy
The precision and accuracy studies were performed by calculating the relative standard deviation (RSD) and recoveries obtained from the QC samples. Intra-day precision (each n = 3) was evaluated by analysis of QC samples at different times of the same day. Inter-day precision (n = 3) was determined by repeated analysis of QC samples three times over a 2-day period.
2.6.3 Linearity, limits of detection and limits of quantification calculations
A 13-point calibration curve was generated for SD within the concentration ranges of 0.08 μmol/L to 50 μmol/L. Linearity was assessed by plotting the area ratios of SD over the internal standard against the known concentrations of SD spiked into the plasma or PBS prior to extraction. Linear regression analysis was performed to calculate the slope and the intercept of the best-fit line. Limits of detection were estimated by: LOD = 3Sa/slope of curve. Limits of quantification were estimated by: LOQ=10Sa/slope of curve. Sa is defined as standard deviation of response estimated by Y-residuals.
2.6.4. Time course of SD absorption
SD concentrations quantified at each of the time points were compared by one-way ANOVA, with Turkey's post-hoc tests. P<0.05 was considered statistically significant. Differences between the total amount of SD absorbed in different gut sections were determined by one-way ANOVA with Tukey's post-hoc analysis.
3.0. Results and Discussion
3.1. Optimization of detection parameters
A few mass spectrometry-based profiling methods to identify SDs as part of structural components of complex sphingolipids in plants, animal tissues and biological fluids have been reported (20,22,26). However, quantitative validation and optimization of these methods for detecting SDs in food matrixes and in human biological samples have not been described. As reviewed by (32), extraction methods using classic Bligh and Dyer protocol for traditional lipidomic analysis are inefficient in recovering more polar O-phosphorylated species such as sphingosine1-phosphate and possibly SD-1-phosphate compounds. An alternative single step liquid-liquid extraction procedure that preserves polar O-acylated SPLs and phosphorylated bases have been described (33). In this paper, we describe an adaptation of this procedure for quantitative assay for SD screening.
Extraction efficiency is critical for detecting SD which is a low abundant compound in plasma and tissues. We initially compared extraction efficiencies of traditional two step Bligh-Dyer and a single phase extraction with ethyl-acetate:isopropanol:water system that was described previously (33). Relative to chloform:methanol:water system, the ethyl acetate:isopropol:water system afforded higher and more reproducible recoveries of SD, other sphingoid bases (e.g. sphingosine) and internal standards (data not shown). Extraction efficiencies, recovery and SD and other sphingoid bases (SB)
To optimize a facile methodology for SD detection, a combination of formic acid (FA) and ammonium formate (AF) were used as mobile phase modifiers to improve separation and sensitivity of detection. Figure 1A shows the structure of SD with the dotted line showing the fragmentation that yields loss of –OH group. Figure 1B represents the product ion scan of SD, and it shows the protonated quasi-molecular parent ion [M+H+] of SD at m/z of 298.4 and a pre-dominant product ion m/z of 280.4, which is generated by the loss of OH group. We also observed the presence of m/z 262.2 fragment specific for the d18:2 backbone of SD, as reported by others (22). However, as shown in Figure 1B, the signal intensity for m/z of 280.4 was much higher and therefore, we chose m/z 298.4 → 280.4 transition for quantifying SD. We did not observe any significant in-source fragmentation of SD (data not shown), and the fragmentor and collision energies were optimized to yield highest intensity for detecting m/z 298.4 -→ 280.4 transition. For the quantitative assay of SD, d17:1 sphingosine (C17-SO) was chosen as an internal standard, as it is not a naturally occurring sphingolipid, and it shares similar chemical structure and chromatographic behaviors as SD.
Figure 1. Optimization of SD detection.
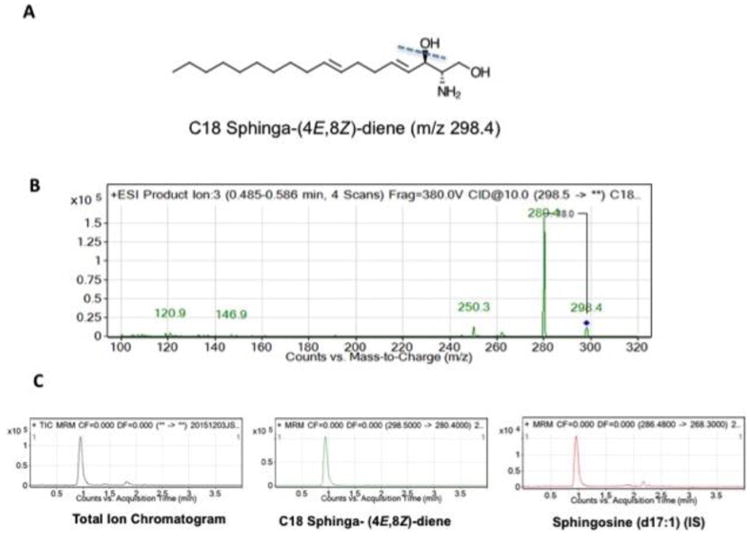
Panel 1A shows the structure of C18 sphinga-(4E,8Z)-diene (m/z 298.4), the SD used in the development of the assay. Panel 1B shows the mass spectrum of collision-induced dissociation fragmentation pattern obtained with C18 (4E, 8Z)-sphingadiene. Please note the precursor ion m/z of 298.4 for the parent. Loss of water yields a major product ion of m/z 280.4. For detection and quantification of SD, mass transition of 298.4 →280.4 was used. Panel 1C show the total ion chromatogram and extracted ion chromatograms for SD and C17-SO, which is used as internal standard for quantification.
For chromatographic separation of SD, a Zorbax SB C18 UHPLC column was chosen as a stationary phase. We initially compared the performance of this column versus Phenomenex Luna 3μ C18(2) HPLC column but found that higher efficiency of UHPLC separation method allowed rapid separation of solvent front and our analytes of interests. As shown in Figure 1C, SD and C17-SO co-elute at a retention time of 0.9 ± 0.07 min (mean ± SD). Chromatographic gradient conditions were optimized to detect SD and C17-SO internal standard within a 4-min run time. Additional equilibration time afforded greater reproducibility in peak retention time when analyzing > 40 samples in a single batch run (data not shown).
3.2. Determination of absolute matrix effect (ME)
Matrix effects, or ion suppression, can be defined as the change in response observed for a given concentration of target analyte in the presence of other sample components. To determine whether our internal standardization was effective in compensating for the matrix effects, we determined the matrix effects of plasma using the strategy applied by Matuszewski et al (34). As shown in Table 1, ME% of plasma was approximately 85.5 % across concentration ranges of 5-40 μmol/L tested. Coefficient of variances of signal intensities in plasma was in general higher than that observed in PBS (Table 1). Variances observed in SD were closely matched with C17-SO internal standard (Table 1). Matrix effects in other complex sample backgrounds such as tissues and tofu homogenates were also tested (Figure 1). Results showed similar MEs in the different matrix tested, indicating that the extraction procedures removed potential ion suppressing agents across the different biological backgrounds with similar efficiency (Figure 2). Because the general recovery and signal intensities of our internal standards were closely matched with SD, the mean accuracy after adjusting for internal standard was similar in both PBS and plasma matrix. These results suggest that our choice of internal standard optimized the detection accuracy of SD in complex matrices.
Table 1. Matrix effects and Coefficient of Variance of Peak Areas for SD and SO.
| Conc. (μmol/L) | ME (%) | Coefficient of Variance | |||
|---|---|---|---|---|---|
| SD in PBS | SD in Plasma | Int. Std. in PBS | Int. Std. in Plasma | ||
| 5 | 82.1 ± 11.7 | 3.3 | 16.6 | 2.4 | 14.2 |
| 10 | 87.5 ± 13.7 | 7.6 | 15.5 | 5.5 | 12.8 |
| 20 | 88.1 ± 13.4 | 3.8 | 15.4 | 3.5 | 18.1 |
| 30 | 87.4 ± 5.7 | 4.8 | 9.0 | 3.0 | 11.1 |
| 40 | 85.8 ± 6.4 | 1.0 | 7.5 | 3.1 | 6.1 |
ME – Matrix Effect
Figure 2. Comparisons of matrix effects of different biological backgrounds.
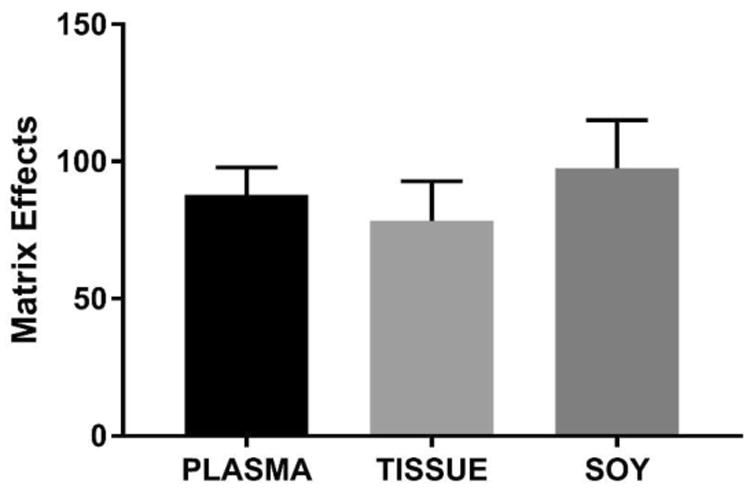
ME of plasma, tissue and soy homogenates were calculated as described in methods. Average matrix effects were 87.8 ± 10.0, 78.3 ± 14.6, 97.6 ± 17.5% in plasma, tissue and soy, respectively (Average of 6 replicates) and were similar across the different bioological matrices.
To test for the linearity of the assay, a 13-point calibration curve was generated with SD over the concentration ranges of 0.08 to 50 μM in PBS and in plasma. As shown in Figure 3, the linearity of SD standard curve was excellent with r2 >0.99 for both PBS and plasma-spiked standards. The slope of the linear fit line for PBS was 0.36 ± 0.003 (mean ± SD) and was similar to the slope calculated for plasma-spiked standards (0.35 ± 0.004; mean ± SD), suggesting that the correction of recovery variance made from the C17-SO internal standard largely corrected the higher variances seen in plasma spiked samples.
Figure 3. 13 point-calibration curve for SDs in PBS and plasma.
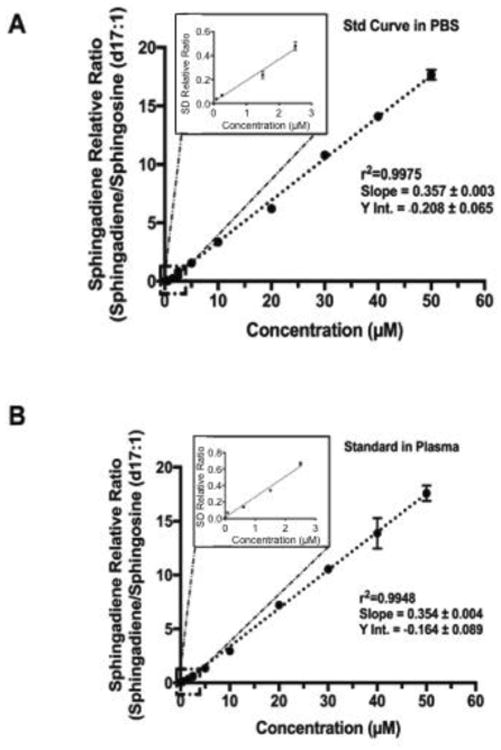
The linearity of SD assay over the concentration ranges of 80 nM to 50 μM are shown. In panel A, the standard curve was created in PBS. Panel B shows the identical curve generated in human plasma matrix. Extraction procedure involves a simple liquid extraction with ethyl-acetate:isopropanol:water mixture followed by collection and drying down of the organic phase. Dried samples are simply reconstituted in methanol. Excellent linearity and similar slope and intercept was observed for both conditions, indicating that the matrix effects of plasma was negligible. The Inset shows the linearity of the assay over the lowest concentration ranges tested.
To estimate the limits of detection, a separate 4-point curve was generated within the ranges of 5 nmol/L to 100 nmol/L. Slope of the curve at these lower ranges were 0.18 ±0.005 and Y-intercept of 0.11 ± 0.0002 (r2=0.9987; p<0.0001). Using the slope and the standard deviation of this Y-intercept, we estimate the limits of detection to be 3.3 nmol/L and limits of quantification to be 11 nmol/L.
3.3. Precision, accuracy and stability
Table 2 shows the precision, accuracy, intra- and inter-day variances of SD quantification in plasma matrix. Both intra- and inter-day measurements show excellent concordance. Precision, expressed as relative standard deviations of measurements, were between 4.2 to 11% for intra-day and 4.3 to 15.3% for the inter-day measurements (Table 2). Accuracy for the lowest amount of SD spiked (0.2 μmol/L) was 81.8% and 86.4 for intra- and inter-day measurements. Post-extraction stability of SD was tested by re-analyzing the freshly extracted samples following 24 hrs in autosampler set at 4°C. As shown in Figure 4, no loss in signal intensity was observed for SD or C17-SO under these analytical conditions.
Table 2. Precision and Accuracy of SD Quantification in Plasma.
| Calc. Conc. | Precision RSD (%) | Accuracy (%) | ||||
|---|---|---|---|---|---|---|
| Conc. (μmol/L) | Intra-Day | Inter-Day | Intra-Day | Inter-Day | Intra-Day | Inter-Day |
| 0.2 | 0.2 ± 0.03 | 0.18 ± 0.02 | 11 | 15.3 | 81.8 | 86.4 |
| 2.0 | 1.8 ± 0.2 | 1.75 ± 0.2 | 10.0 | 11.4 | 96.2 | 88.5 |
| 10 | 8.1 ± 0.50 | 8.07 ±0.6 | 4.7 | 4.9 | 97.8 | 98.8 |
| 20 | 19.7 ± 1.2 | 19.8 ± 1.6 | 6.0 | 8.1 | 98.5 | 92.6 |
| 40 | 37.9 ± 3.7 | 37.9 ± 3.8 | 9.8 | 10.0 | 98.3 | 99.8 |
| 50 | 48.1 ± 2.0 | 49.2 ± 2.05 | 4.2 | 4.3 | 97.8 | 97.9 |
Figure 4. Post-extraction stability of SDs in plasma.
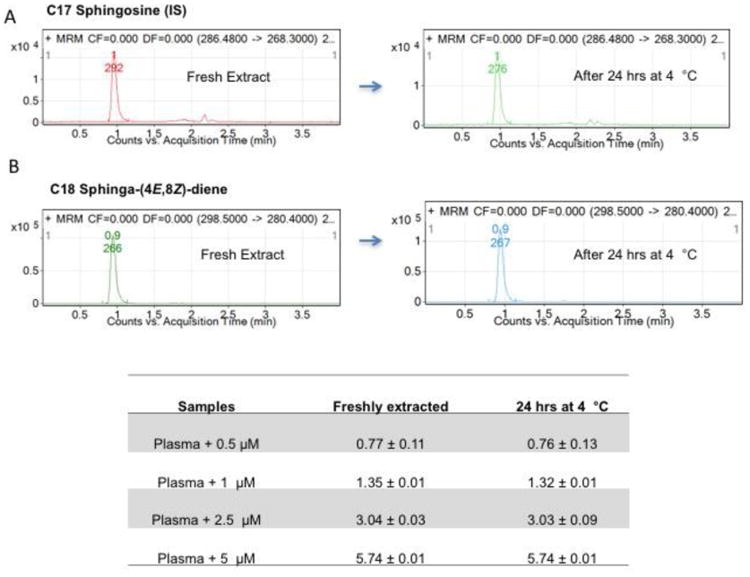
To determine the post-extraction stability of SDs, analyzed samples were kept in 4°C for 24 hours and reinjected. Panel A shows the extracted ion chromatograph of SDs and C17-SO internal standards. Table shows the stability of SD compounds spiked in plasma and as shown, minimal loss is detected at 24 hrs.
3.4. SD quantification in soy oil and tofu
Soy oil and tofu products are physiological relevant dietary sources of SD. To quantify amounts of SD that may available from these products, three different soy oils and three different tofu products were analyzed for SD content. Oil matrix required modification of extraction procedure, and SDs were extracted from soy oil using the standard chloroform:methanol extraction procedure as described in Methods. Comparisons of three soy oil products showed a range of values from 4.2 ± 0.5 nmol/L to 18.7 ± 3.5 nmol/L of SD (Figure 5A). Analysis of three readily available commercial tofu products showed similar values across all products tested with average values of 10.2 ± 3.5 mg of SD per kg wet weight. In human breast milk, approximately 7% of SM contains SD bases, and we estimate that ∼ 1.8 mg of SD can be derived from 1 Kg of dried breast milk (22). Others have reported that approximately 5% to 7% of SM from beef and pork is composed of SD. Based on the total amount of 470 mg/Kg wet weight, we estimate that meat-derived SM may supply ∼24 mg of SD per kg (25). Based on our calculations, the amount of SD in tofu is comparable to amounts found in meat and therefore represent an excellent source of SD.
Figure 5. SD concentrations in common soy-based food products.
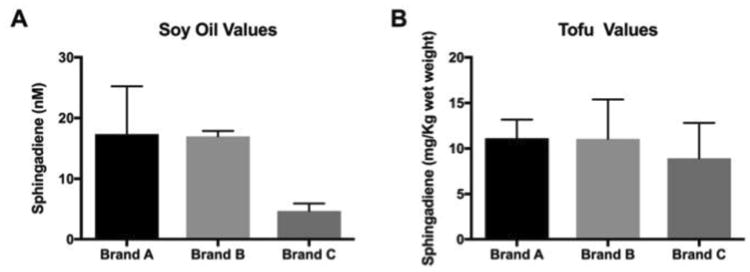
Panel A shows ranges of SDs quantified in soy oil products ranged from 4.7 to 17.4 μM. Each brand was analyzed in duplicate. Panel B shows ranges of SDs detected in tofu products.
3.5. Time-course of SD absorption in mice
Thus far, only two studies have examined the relative uptake efficiencies of SD versus SO in cells and in mice (20). In human Caco-2 cells, accumulation of SO is preferred over that of SDs and was found to be limited by the higher rate of apical SD excretion by MDR1 encoded P-glycoprotein (35). In mice, SDs supplied as a component of maize-derived glucosylceramide were absorbed into the lymphatic fluids at a level that is approximately ten-fold less than SO (20). Although these two studies establish the potential dietary absorption of SD, in vivo kinetic parameters of SD uptake, including specific regions where gut-absorption may occur and the rates of plasma absorption have not been directly addressed. To fill this gap, a time course analysis of SD absorption in the gut and plasma was performed following oral gavage with SDs (25 mg/kg body wt.). Absorption determination in the gut tissue necessitated use of independent sets of mice at each time point to ascertain approximate rates of absorption of SDs. Because of limitation of this study design, fundamental pharmacokinetic parameters such as AUC, T1/2, Tmax and Cmax were not assessed.
To determine, relative absorption across the small intestine, temporal changes in SD content in duodenum, jejunum, and ileum were measured. Baseline concentration of SD in the, duodenum, jejunum, and ileum were 230 ± 64, 130 ± 18 and 269 ± 66 pmol/mg proteins, respectively. Following oral gavage, the highest increases in SD were observed in the duodenum section at the 1 hr time point (38.8 ± 20.8 nmol/mg protein; p<0.001) and subsequently decreased to 1.3 ± 0.7 nmol/mg protein at 24 hrs. Second highest accumulation was observed in the jejunum section. In this section, the highest accumulation of SD was detected at 2 hrs, but the absolute concentration was significantly less than that observed in the duodenum (0.3 ± 0.08 nmol/mg protein; p<0.001). Interestingly, concentrations of SD in the ileum section remained unchanged following oral gavage, suggesting that most SD absorption occurs in the duodenum. Measurement of SD in the colon also showed lack of significant uptake over time (Figure 6D).
Figure 6. Time course of SD absorption in the small intestine and colon sections.
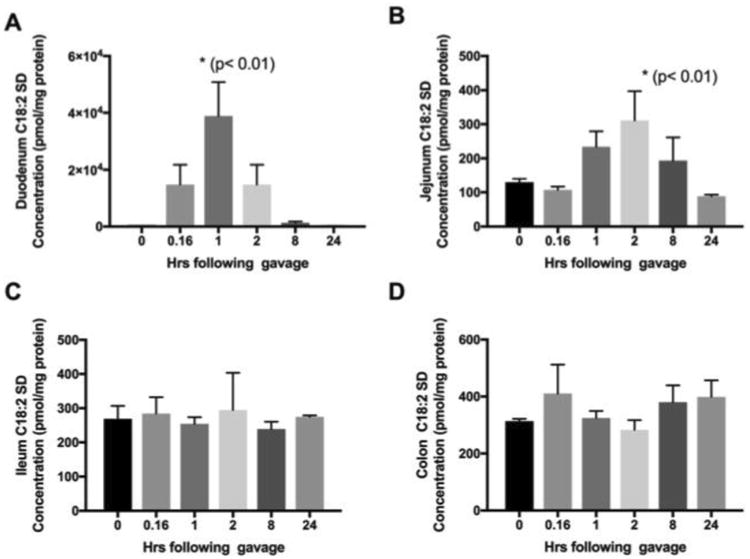
To determine the uptake characteristics along the small intestine and to locate major sites of absorption, intestinal segments were obtained from mice euthanized at specified times following oral gavage with 25 mg/kg SDs. Time-dependent SD uptake in the duodenum (panel A), jejunum (panel B), ileum (panel C), and colon (panel D) are shown. Results are mean and standard deviation of three mice per time point.
Uptake of SDs as a free base may be different than SDs absorbed as components of more complex sphingolipids, such as SM and Cer. Others have reported the presence of alkaline sphingomyelinases and ceramidases in the gut that can hydrolyze and release SDs from more complex sphingolipids (36-38). Interestingly, the expression levels of both sphingomyelinase and ceramidase have been shown to be highest in the duodenum and jejunum sections of the small intestines (36-38). Our data along with these previous observations suggest that duodenum and jejunum may be significant sites of sphingoid base absorption in vivo. It is interesting to note that the stable accumulation of SD was not observed in any sections of the gut. This may be due to rapid apical excretion of SD by P-glycoporin, as observed in studies with human intestinal cells (39). Small amounts of SD may be metabolized to other complex sphingolipids.
The time course of SD absorption is shown in Figure 7. Baseline plasma concentration of SD was 0.30 ± 0.001 μM (mean ± SD). Following oral gavage with SD, plasma SD increased to 0.57 ± 0.06 μM within 10 min (0.16 hr; p<0.0001) and decreased to baseline levels within 1 hr (Figure 7). Considering the temporal patterns of SD uptake we observed in the different sections of the small intestine, the acute increase in plasma SD at 10 min may be more related to para-cellular leakage due to bolus delivery of SD solution by gavage.
Figure 7. Time course of SD appearance in mice plasma.
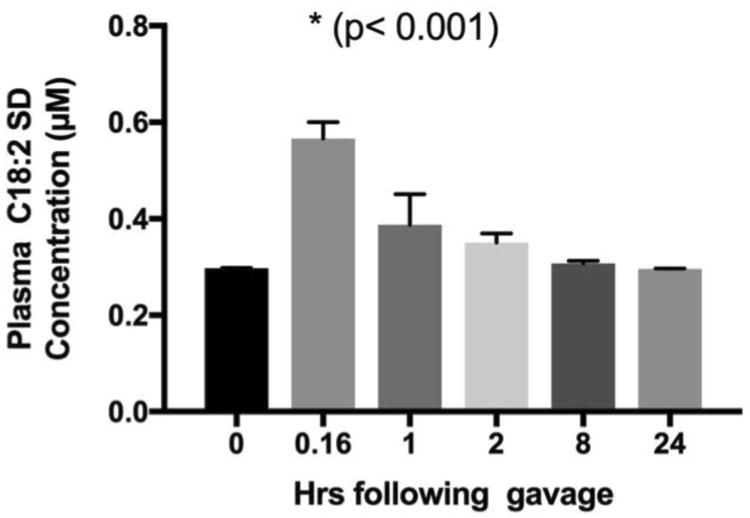
To determine oral bioavailability of SDs in vivo, C57/B6 mice were administered 25 mg/kg SDs by gavage, and 3 mice were sacrificed at each of the specified time points to obtain blood and tissue samples. A small but significant increase in plasma SD concentration was observed within 10 min following oral gavage and levels returned to baseline within an hour post gavage.
Anti-inflammatory and chemopreventive effects of SDs are garnering interest in them as potential preventative agents against the development of colon cancer. Despite promising data showing their anti- inflammatory and carcinogenic effects, the exact mechanism of action of cytotoxic SDs are incompletely understood. The bioactivity of SDs may be due to direct interactions with unknown cellular targets. It is also possible that SDs incorporated into more complex sphingolipids including Cer or SM may exert their own independent anti-inflammatory effects. Evidence for apical secretion of SDs in the Caco-2 cell system also suggests a possibility that SDs may modulate gut mucosal cell populations. Apical excretion and accumulation of SDs in the luminal side of ileum and colon or retention of SDs within the luminal contents of the ileum, cecum and colon may allow for interactions with the gut microbiome that may be protective against the development of inflammation in these regions. In support of that notion, a spate of recent studies provides evidence that long chain sphingoid bases and other sphingolipids may provide protection against bacterial invasion and biofilm formation in vivo (40-43). In the future, it will be interesting to establish whether SDs act at least in part by impacting and/or remodeling the gut microbiota.
4.0. Conclusions
We have developed a facile method for SD quantification that can be used for food and biological sample analysis. Our SD extraction procedure and the use of a C17-SO internal standard minimize the matrix effects of complex tissues samples. Excellent recovery, precision and accuracy were observed. Linearity is preserved over nM to high μM ranges of expected physiological levels of SDs. Availability of this method will be helpful to other investigators seeking to quantify SDs for nutritional chemoprevention studies.
Acknowledgments
This work was supported by NIH (National Cancer Institute) grant R01CA129438, S10OD018070, and a supplement from the Office of Dietary Supplements (all to JDS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schoen RE, Pinsky PF, Weissfeld JL, Yokochi LA, Church T, Laiyemo AO, Bresalier R, Andriole GL, Buys SS, Crawford ED, Fouad MN, Isaacs C, Johnson CC, Reding DJ, O'Brien B, Carrick DM, Wright P, Riley TL, Purdue MP, Izmirlian G, Kramer BS, Miller AB, Gohagan JK, Prorok PC, Berg CD Team, P.P. Colorectal-cancer incidence and mortality with screening flexible sigmoidoscopy. N Engl J Med. 2012;366:2345–2357. doi: 10.1056/NEJMoa1114635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Centelles JJ. General aspects of colorectal cancer. ISRN Oncol. 2012;2012:139268. doi: 10.5402/2012/139268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Winawer SJ, Zauber AG. The advanced adenoma as the primary target of screening. Gastrointest Endosc Clin N Am. 2002;12:1–9. v. doi: 10.1016/s1052-5157(03)00053-9. [DOI] [PubMed] [Google Scholar]
- 4.Giovannucci E, Willett W. Dietary factors and risk of colon cancer. Ann Med. 1994;26:443–452. doi: 10.3109/07853899409148367. [DOI] [PubMed] [Google Scholar]
- 5.Itano O, Fan K, Yang K, Suzuki K, Quimby F, Dong Z, Jin B, Edelmann W, Lipkin M. Effect of caloric intake on Western-style diet-induced intestinal tumors in a mouse model for hereditary colon cancer. Nutr Cancer. 2012;64:401–408. doi: 10.1080/01635581.2012.660672. [DOI] [PubMed] [Google Scholar]
- 6.Mazzei JC, Zhou H, Brayfield BP, Hontecillas R, Bassaganya-Riera J, Schmelz EM. Suppression of intestinal inflammation and inflammation-driven colon cancer in mice by dietary sphingomyelin: importance of peroxisome proliferator-activated receptor γ expression. J Nutr Biochem. 2011;22:1160–1171. doi: 10.1016/j.jnutbio.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mazzei JC, Zhou H, Brayfield BP, Hontecillas R, Bassaganya-Riera J, Schmelz EM. Suppression of intestinal inflammation and inflammation-driven colon cancer in mice by dietary sphingomyelin: importance of peroxisome proliferator-activated receptor gamma expression. J Nutr Biochem. 2011;22:1160–1171. doi: 10.1016/j.jnutbio.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Newmark HL, Yang K, Kurihara N, Fan K, Augenlicht LH, Lipkin M. Western-style diet-induced colonic tumors and their modulation by calcium and vitamin D in C57Bl/6 mice: a preclinical model for human sporadic colon cancer. Carcinogenesis. 2009;30:88–92. doi: 10.1093/carcin/bgn229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pabla B, Bissonnette M, Konda VJ. Colon cancer and the epidermal growth factor receptor: Current treatment paradigms, the importance of diet, and the role of chemoprevention. World J Clin Oncol. 2015;6:133–141. doi: 10.5306/wjco.v6.i5.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang K, Kurihara N, Fan K, Newmark H, Rigas B, Bancroft L, Corner G, Livote E, Lesser M, Edelmann W, Velcich A, Lipkin M, Augenlicht L. Dietary induction of colonic tumors in a mouse model of sporadic colon cancer. Cancer Res. 2008;68:7803–7810. doi: 10.1158/0008-5472.CAN-08-1209. [DOI] [PubMed] [Google Scholar]
- 11.Rasool S, Kadla SA, Rasool V, Ganai BA. A comparative overview of general risk factors associated with the incidence of colorectal cancer. Tumour Biol. 2013;34:2469–2476. doi: 10.1007/s13277-013-0876-y. [DOI] [PubMed] [Google Scholar]
- 12.Sullards MC, Lynch DV, Merrill AH, Jr, Adams J. Structure determination of soybean and wheat glucosylceramides by tandem mass spectrometry. J Mass Spectrom. 2000;35:347–353. doi: 10.1002/(SICI)1096-9888(200003)35:3<347::AID-JMS941>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 13.Oskouian B, Saba JD. Death and taxis: what non-mammalian models tell us about sphingosine-1-phosphate. Semin Cell Dev Biol. 2004;15:529–540. doi: 10.1016/j.semcdb.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 14.Abnet C, Borkowf C, Qiao Y, Albert P, Wang E, Merrill A, Jr, Mark S, Dong Z, Taylor P, Dawsey S. A cross-sectional study of human serum sphingolipids, diet and physiological parameters. J Nutr. 2001;131:2748–2752. doi: 10.1093/jn/131.10.2748. [DOI] [PubMed] [Google Scholar]
- 15.Nilsson A, Duan RD. Absorption and lipoprotein transport of sphingomyelin. J Lipid Res. 2006;47:154–171. doi: 10.1194/jlr.M500357-JLR200. [DOI] [PubMed] [Google Scholar]
- 16.Fyrst H, Saba JD. An update on sphingosine-1-phosphate and other sphingolipid mediators. Nat Chem Biol. 2010;6:489–497. doi: 10.1038/nchembio.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Degagné E, Pandurangan A, Bandhuvula A, Kumar A, Eltanawy A, Zhang M, Yoshinaga Y, Nefedov M, de Jong P, Fong L, Young S, Bittman R, Ahmedi Y, Saba J. Sphingosine-1-phosphate lyase downregulation promotes colon carcinogenesis through STAT3-activated microRNAs. J Clin Invest. 2014;124:5368–5384. doi: 10.1172/JCI74188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Degagne E, Saba JD. S1pping fire: Sphingosine-1-phosphate signaling as an emerging target in inflammatory bowel disease and colitis-associated cancer. Clinical and experimental gastroenterology. 2014;7:205–214. doi: 10.2147/CEG.S43453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang J, Nagahashi M, Kim EY, Harikumar KB, Yamada A, Huang WC, Hait NC, Allegood JC, Price MM, Avni D, Takabe K, Kordula T, Milstien S, Spiegel S. Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell. 2013;23:107–120. doi: 10.1016/j.ccr.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sugawara T, Tsuduki T, Yano S, Hirose M, Duan J, Aida K, Ikeda I, Hirata T. Intestinal absorption of dietary maize glucosylceramide in lymphatic duct cannulated rats. J Lipid Res. 2010;51:1761–1769. doi: 10.1194/jlr.M002204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishikawa J, Takada S, Hashizume K, Takagi Y, Hotta M, Masukawa Y, Kitahara T, Mizutani Y, Igarashi Y. Dietary glucosylceramide is absorbed into the lymph and increases levels of epidermal sphingolipids. J Dermatol Sci. 2009;56:220–222. doi: 10.1016/j.jdermsci.2009.09.002. [DOI] [PubMed] [Google Scholar]
- 22.Blaas N, Schuurmann C, Bartke N, Stahl B, Humpf HU. Structural profiling and quantification of sphingomyelin in human breast milk by HPLC-MS/MS. J Agric Food Chem. 2011;59:6018–6024. doi: 10.1021/jf200943n. [DOI] [PubMed] [Google Scholar]
- 23.Renkonen O. Presence of sphingadienine and trans-monoenoic fatty acids in ceramide monohexosides of human plasma. Biochim Biophys Acta. 1970;210:190–192. doi: 10.1016/0005-2760(70)90078-0. [DOI] [PubMed] [Google Scholar]
- 24.Renkonen O, Hirvisalo E. Structure of plasma sphingadienine. J Lipid Res. 1969;10:687–693. [PubMed] [Google Scholar]
- 25.Fischbeck A, Kruger M, Blaas N, Humpf HU. Analysis of sphingomyelin in meat based on hydrophilic interaction liquid chromatography coupled to electrospray ionization-tandem mass spectrometry (HILIC-HPLC-ESI-MS/MS) J Agric Food Chem. 2009;57:9469–9474. doi: 10.1021/jf9025376. [DOI] [PubMed] [Google Scholar]
- 26.Colsch B, Afonso C, Popa I, Portoukalian J, Fournier F, Tabet JC, Baumann N. Characterization of the ceramide moieties of sphingoglycolipids from mouse brain by ESI-MS/MS: identification of ceramides containing sphingadienine. J Lipid Res. 2004;45:281–286. doi: 10.1194/jlr.M300331-JLR200. [DOI] [PubMed] [Google Scholar]
- 27.Kumar A, Pandurangan AK, Lu F, Fyrst H, Zhang M, Byun HS, Bittman R, Saba JD. Chemopreventive sphingadienes downregulate Wnt signaling via a PP2A/Akt/GSK3beta pathway in colon cancer. Carcinogenesis. 2012;33:1726–1735. doi: 10.1093/carcin/bgs174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Inamine M, Suzui M, Morioka T, Kinjo T, Kaneshiro T, Sugishita T, Okada T, Yoshimi N. Inhibitory effect of dietary monoglucosylceramide 1-O-beta-glucosyl-N-2′-hydroxyarachidoyl-4,8-sphingadienine on two different categories of colon preneoplastic lesions induced by 1,2-dimethylhydrazine in F344 rats. Cancer Sci. 2005;96:876–881. doi: 10.1111/j.1349-7006.2005.00127.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hossain Z, Sugawara T, Hirata T. Sphingoid bases from sea cucumber induce apoptosis in human hepatoma HepG2 cells through p-AKT and DR5. Oncol Rep. 2013;29:1201–1207. doi: 10.3892/or.2013.2223. [DOI] [PubMed] [Google Scholar]
- 30.Rozema E, Binder M, Bulusu M, Bochkov V, Krupitza G, Kopp B. Effects on inflammatory responses by the sphingoid base 4,8-sphingadienine. Int J Mol Med. 2012;30:703–707. doi: 10.3892/ijmm.2012.1035. [DOI] [PubMed] [Google Scholar]
- 31.Fyrst H, Oskouian B, Bandhuvula P, Gong Y, Byun HS, Bittman R, Lee AR, Saba JD. Natural sphingadienes inhibit Akt-dependent signaling and prevent intestinal tumorigenesis. Cancer Res. 2009;69:9457–9464. doi: 10.1158/0008-5472.CAN-09-2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bielawski J, Pierce JS, Snider J, Rembiesa B, Szulc ZM, Bielawska A. Sphingolipid analysis by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS) Adv Exp Med Biol. 2010;688:46–59. doi: 10.1007/978-1-4419-6741-1_3. [DOI] [PubMed] [Google Scholar]
- 33.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006;39:82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 34.Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem. 2003;75:3019–3030. doi: 10.1021/ac020361s. [DOI] [PubMed] [Google Scholar]
- 35.Sugawara T, Kinoshita M, Ohnishi M, Tsuzuki T, Miyazawa T, Nagata J, Hirata T, Saito M. Efflux of sphingoid bases by P-glycoprotein in human intestinal Caco-2 cells. Biosci Biotechnol Biochem. 2004;68:2541–2546. doi: 10.1271/bbb.68.2541. [DOI] [PubMed] [Google Scholar]
- 36.Duan RD, Bergman T, Xu N, Wu J, Cheng Y, Duan J, Nelander S, Palmberg C, Nilsson A. Identification of human intestinal alkaline sphingomyelinase as a novel ecto-enzyme related to the nucleotide phosphodiesterase family. J Biol Chem. 2003;278:38528–38536. doi: 10.1074/jbc.M305437200. [DOI] [PubMed] [Google Scholar]
- 37.Duan RD, Cheng Y, Hansen G, Hertervig E, Liu JJ, Syk I, Sjostrom H, Nilsson A. Purification, localization, and expression of human intestinal alkaline sphingomyelinase. J Lipid Res. 2003;44:1241–1250. doi: 10.1194/jlr.M300037-JLR200. [DOI] [PubMed] [Google Scholar]
- 38.Kono M, Dreier JL, Ellis JM, Allende ML, Kalkofen DN, Sanders KM, Bielawski J, Bielawska A, Hannun YA, Proia RL. Neutral ceramidase encoded by the Asah2 gene is essential for the intestinal degradation of sphingolipids. J Biol Chem. 2006;281:7324–7331. doi: 10.1074/jbc.M508382200. [DOI] [PubMed] [Google Scholar]
- 39.Sugawara T, Zaima N, Yamamoto A, Sakai S, Noguchi R, Hirata T. Isolation of sphingoid bases of sea cucumber cerebrosides and their cytotoxicity against human colon cancer cells. Biosci Biotechnol Biochem. 2006;70:2906–2912. doi: 10.1271/bbb.60318. [DOI] [PubMed] [Google Scholar]
- 40.Tavakoli Tabazavareh S, Seitz A, Jernigan P, Sehl C, Keitsch S, Lang S, Kahl BC, Edwards M, Grassme H, Gulbins E, Becker KA. Lack of Sphingosine Causes Susceptibility to Pulmonary Staphylococcus Aureus Infections in Cystic Fibrosis. Cell Physiol Biochem. 2016;38:2094–2102. doi: 10.1159/000445567. [DOI] [PubMed] [Google Scholar]
- 41.Saied EM, Banhart S, Burkle SE, Heuer D, Arenz C. A series of ceramide analogs modified at the 1-position with potent activity against the intracellular growth of Chlamydia trachomatis. Future Med Chem. 2015;7:1971–1980. doi: 10.4155/fmc.15.126. [DOI] [PubMed] [Google Scholar]
- 42.Cukkemane N, Bikker FJ, Nazmi K, Brand HS, Sotres J, Lindh L, Arnebrant T, Veerman EC. Anti-adherence and bactericidal activity of sphingolipids against Streptococcus mutans. Eur J Oral Sci. 2015;123:221–227. doi: 10.1111/eos.12200. [DOI] [PubMed] [Google Scholar]
- 43.Rice TC, Seitz AP, Edwards MJ, Gulbins E, Caldwell CC. Frontline Science: Sphingosine rescues burn-injured mice from pulmonary Pseudomonas aeruginosa infection. J Leukoc Biol. 2016;100:1233–1237. doi: 10.1189/jlb.3HI0416-197R. [DOI] [PMC free article] [PubMed] [Google Scholar]