

Abstract
Phosphoinositide 3-kinase (PI3K) is deregulated in a wide variety of human tumors and triggers activation of protein kinase B (PKB/Akt) and mammalian target of rapamycin (mTOR). Here we describe the preclinical characterization of compound 1 (PQR309, bimiralisib), a potent 4,6-dimorpholino-1,3,5-triazine-based pan-class I PI3K inhibitor, which targets mTOR kinase in a balanced fashion at higher concentrations. No off-target interactions were detected for 1 in a wide panel of protein kinase, enzyme, and receptor ligand assays. Moreover, 1 did not bind tubulin, which was observed for the structurally related 4 (BKM120, buparlisib). Compound 1 is orally available, crosses the blood−brain barrier, and displayed favorable pharmacokinetic parameters in mice, rats, and dogs. Compound 1 demonstrated efficiency in inhibiting proliferation in tumor cell lines and a rat xenograft model. This, together with the compound’s safety profile, identifies 1 as a clinical candidate with a broad application range in oncology, including treatment of brain tumors or CNS metastasis. Compound 1 is currently in phase II clinical trials for advanced solid tumors and refractory lymphoma.
Introduction
Phosphoinositide 3-kinases (PI3Ks) are lipid kinases that play a central role in the control of cancer cell growth, proliferation, and metastasis.1–3 The PI3K family is divided into three classes according to sequence homology and substrate specificity: so-called class I PI3Ks are activated downstream of cell surface receptors, including receptor protein tyrosine kinases (RTKs), immunoglobulin receptors, and G-protein-coupled receptors (GPCRs). Class IA PI3Ks consist of the catalytic subunits p110α, p110β, and p110δ. These interact with dedicated phosphorylated tyrosine motifs on growth factor receptors or their substrates via two src-homology 2 (SH2) domains present in their tightly associated p85 regulatory subunits. The class IB PI3Kγ operates downstream of GPCRs and is composed of a catalytic subunit p110γ and an adapter subunit, which is either p84 or p101.4
Class I PI3Ks phosphorylate phosphatidylinositol (4,5)-bisphosphate [PtdIns(4,5)P2] to yield phosphatidylinositol (3,4,5)-trisphosphate [PtdIns(3,4,5)P3]. PtdIns(3,4,5)P3, produced at the inner leaflet of the plasma membrane, provides a docking site for effector proteins with pleckstrin-homology (PH) domains. The serine/threonine kinases phosphoinositide-dependent kinase 1 (PDK-1) and protein kinase B (PKB/Akt) contain a PH domain and are thus activated by PI3K and relay the activation of the mammalian target of rapamycin complex 1 (mTORC1). In tumor cells, hyperactivation of the PI3K/mTOR pathway can occur on multiple levels, including mutation or overexpression of cell surface receptors such as HER2,5 oncogenes, the protein tyrosine phosphatase non-receptor 12 (PTPN12),6 or the presence of activating hotspot mutations in PIK3CA (encoding the catalytic PI3Kα subunit p110α).7 Other key regulators of the PI3K pathway include the lipid phosphatase PTEN, hydrolyzing PtdIns(3,4,5)P3 back to PtdIns(4,5)P2,2,8 and the inositol polyphosphate 4-phosphatase type II (INPP4B)9 degrading PtdIns(3,4)P2. Loss of either lipid phosphatase activities or gain of function mutations at the level of PI3K, and aberrations up- and downstream in the pathway can thus lead to the constitutive activation of PI3K, PKB/Akt, and mTOR.
Besides promotion of tumor cell growth and angiogenesis, an overactivated PI3K/mTOR pathway has been associated with resistance to other cancer treatments, e.g., receptor tyrosine kinase inhibitors. PI3K inactivating agents are currently in various clinical trials as monotherapy or in combination with other drugs.2,3,10
In our search for the development of a dual pan-PI3K/mTOR inhibitor, we focused on dimorpholino substituted compounds with a triazine core, as their symmetry supports rapid access to a compound library with various aryl substituents. Furthermore, our goal was to develop brain-penetrable compounds to allow for the treatment of brain metastasis and brain tumors. Here we describe a SAR study around the aryl moiety at the C2-position of bismorpholino-triazines, which led to the identification of 1 (PQR309, bimiralisib),11 a clinical candidate and brain penetrable pan-PI3K/mTOR inhibitor.
Results and Discussion
Compound 1 evolved as the lead compound of a series of dimorpholinotriazine based compounds and is currently tested in phase II clinical trials (clinicaltrials.gov). Initially inspired by 2-(2-difluoromethylbenzimidazol-1-yl)-4,6-dimorpholino-1,3,5-triazine (ZSTK474),12 a pan PI3K inhibitor with a triazine core, development of 1 aimed to (i) maximize compound solubility and bioavailability, (ii) achieve blood–brain barrier penetration, (iii) avoid microtubule interactions as observed for 4 (BKM120, buparlisib),11,13 and (iv) introduce moderate mTOR inhibition. The rationale for PI3K/mTOR dual activity was based on the observation that persistent mTOR signaling was reported to confer resistance to PI3K inhibition,14 while selective targeting of the mTOR complex 1 (TORC1) initiates positive feedback leading to PI3K hyperactivation.15 To avoid this, we selected for compounds with a higher potency for PI3K than mTOR kinase. Moreover, drugs targeting both PI3K and mTOR have been shown early on to be more effective in tumors with multiple pathway activation such as melanoma16,17 and to potentially support drug combinations.18
A number of clinically advanced PI3K inhibitors with a pyrimidine, triazine, or fused pyrimidine core contain at least one morpholine moiety. This morpholine was demonstrated to be a crucial feature for PI3K binding, as the morpholine oxygen atom forms a hydrogen bond interaction with the hinge region backbone NH of the amino acid residue Val851 in PI3Kα (Val882 in PI3Kγ, and Val2240 in mTOR). Structurally related compounds, which display distinct PI3K and mTOR kinase inhibitory profiles (Table 1), are shown in Figure 1: 1, 4,19 and 6 (GDC-0980, apitolisib/RG7422)20 target PI3K and mTOR kinase with some preference for PI3Kα, while 3 (PIK-587, gedatolisib/PF-05212384)21 is a potent dual PI3K/mTOR inhibitor and 5 (GDC-0941, pictilisib)22 targets PI3K.
Table 1. Affinity of Triazine and Pyrimidine Core-Containing Molecules to PI3K and PI3K-Related Kinases.
| binding constant Kd [nM]a |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| compd | PI3Kα | PI3Kβ | PI3Kγ | PI3Kδ | mTOR | PI3KC2β | VPS34 | PI4KCβ | DNAPK | mTOR/PI3Kα |
| 1 | 1.5 | 11 | 25 | 25 | 12 | 820 | 230 | 40000 | 1600 | 8.0 |
| 2 | 1.2 | 3.5 | 11 | 3.5 | 61 | 540 | 130 | 27000 | >30000 | 51 |
| 3 | 1.0 | 3.5 | 24 | 76 | 0.94 | 0.46 | 5100 | >30000 | >30000 | 0.94 |
| 4 | 3.5 | 36 | 130 | 260 | 19 | 2100 | 200 | 40000 | 1800 | 5.4 |
| 5 | 0.76 | 2.1 | 7.7 | 3.3 | 48 | 40 | nd | 40000 | 1300 | 63 |
| 6 | 0.62 | 3.8 | 2.6 | 3.3 | 3.3 | 580 | 250 | >30000 | 9800 | 5.3 |
| IC50, kinase assay [nM]b |
||||||||||
| compd | PI3Kα | PI3Kβ | PI3Kγ | PI3Kδ | mTOR | α542K/545K/1047Rc | VPS34 | DNAPK | mTOR/PI3Kα | |
| 1 | 33 | 661 | 708 | 451 | 89 | 63/136/36 | 8486 | 8567 | 2.6 | |
| 4 | 45 | 607 | 778 | 110 | 94 | 111/216/87 | 2581 | 6917 | 13 | |
Dissociation constants (Kd) were determined using ScanMax technology (DiscoveRx) 26with 11-point 3-fold serial dilutions of the indicated compounds. Kd is the mean value from experiments performed in duplicate and calculated from standard dose–response curves using the Hill equation depicted in Experimental Section (n ≥ 2). A TreeSpot representation of a full kinase panel and kinase residual binding values are shown in Supporting Information Figure S1 and Table S2 for all above compounds except compound 2.
Prokinase activity assays of the indicated enzymes and PI3Kα mutants.
Inhibition of PI3Kα E542K, E545K, and H2047R mutant activities are indicated. For these values, the wild type PI3Kα reference IC50 values were 122 nM for 1 and 142 nM for 4.
Figure 1.

Structure of triazine core PI3K inhibitors 1, reference compound 2, reported clinical candidate 3, and pyrimidine or fused pyrimidine core containing molecules 4, 5, and 6.
The above compounds are substituted with various aryl moieties (e.g., indazole, pyrimidine, pyridine, benzoimidazole, or aniline), which are situated in the PI3K’s ATP-binding site toward the affinity binding pocket. The primary amine of 411,19 and 620 interacts via hydrogen bonding with the carboxyl groups of the amino acid residues Asp841 and Asp836 in PI3Kγ. For 3 it has been proposed that the urea-NH groups form hydrogen bonds with Asp810 and the urea oxygen engages the wortmannin-reactive Lys80223,24 in PI3Kα.25 Finally, 5 exploits its indazole N-NH atoms to form hydrogen bonds with Tyr867 and Asp841 in PI3Kγ. As illustrated by the exchange of the aryl moiety from 4-(trifluoromethyl)pyridin-2-amine in 1 to 4-(trifluoromethyl)pyrimidin-2-amine in compound 2, the aryl substitution can be used to fine-tune PI3K vs mTOR activity.
Chemistry
To achieve a balanced PI3K and mTOR inhibition and to adjust physicochemical properties, a library of 4,6-dimorpholino-1,3,5-triazines substituted with various C-2-aryl moieties was prepared from 4,4′-(6-chloro-1,3,5-triazine-2,4-diyl)dimorpholine (7, Scheme 1). The chlorine was displaced by aniline or heteroaryl moieties using Suzuki cross-coupling reactions with boronic acid pinacol esters. The desired products were obtained in moderate to good yield (11–86%) depending on the used boronic acid pinacol ester derivative. Moreover, compound 1 was transformed by acetylation and diazotization followed by bromination and Sandmeyer reaction in order to obtain derivatives bearing different substituents at the C-2 pyridine (17–21). All final compounds were analyzed for purity by analytical HPLC (>95%).
Scheme 1. Synthesis of C-2-Aryl-Substituted 4,6-Dimorpholino-1,3,5-triazine Analogsa.

a. aReagents and conditions: (i) ArBpin, XPhosPdG2 (cat.), K3PO4, dioxane/H2O, 95 °C, 2–16 h, then HCl (aq), 60 °C, 2–8 h; (ii) ArBpin, Pd(dppf)Cl2 (cat.), Na2CO3, 1,2-dimethoxyethane/H2O, 90 °C, 16 h; (iii) ArBpin, XPhosPdG2 (cat.), K3PO4, dioxane/H2O, 95 °C, 2 h; (iv) AcCl, K2CO3, CH2Cl2, 0 °C → rt, 3.5 h; (v) HCl (conc), NaNO2, 0 °C → rt, 1 h, then CuCl, rt, 1 h; (vi) isopentyl nitrite, CH2Br2, rt, 5 min, then (CH3)3SiBr, rt, 1 h; (vii) CH3ONH2·HCl, NaHCO3, DMSO, 100 °C, 6 h. bCompound 1 was prepared according to the procedure recently reported.11 ArBpin: boronic acid pinacol ester (the amino group was protected in certain cases).
We have recently reported an optimized synthetic route to compound 1.11 This strategy allowed for rapid access to large quantities of 1 starting from cyanuric chloride (81% yield) for preclinical and clinical development.
Determination of Cellular Potency and PI3K vs mTOR Kinase Activities
In the following studies, the role of the aryl moiety was investigated systematically for enzymatic activity and selectivity for class IA PI3K versus mTOR (Table 2). A first series of compounds including variations of nitrogen atoms number and position was prepared to study the effect of the polarity of the aryl group (9–12). Interestingly, 10 and 11 substituted with 2-aminopyridine and 2-aminopyrimidine, respectively, were both found to be active in cellular and enzymatic assays. The calculated PSA (polar surface area) value for the pyridine-substituted 10 (PSA = 103) was slightly lower as compared to pyrimidine 11 (PSA = 115), predicting a better brain penetration for 10. Therefore, pyridines were substituted with lipophilic substituents such as methyl and trifluoromethyl at pyridine C3 and C4 positions to generate compounds 13–15. Introduction of a C4-trifluoromethyl group significantly increased cellular potency and enzymatic targeting: compared with 13, 1 showed excellent cellular activities, good potency in vitro (Ki for PI3Kα of 17 nM), targeted mutated forms of the catalytic PI3Kα subunit, and displayed the desired selectivity profile targeting PI3Kα>mTOR (Ki ratio of mTOR/p110α of ~3.6 [~8.0 from KINOMEscan; see Table 1]). On the contrary, introduction of a lipophilic substituent at the C3-position (methyl in 14 and trifluoromethyl in 15) diminished binding to PI3Kα as well as to mTOR.
Table 2. Direction of PI3K/mTOR Selectivity with Aromatic Substituents for Dimorpholinotriazine.
| Cmpd |  |
Cellular IC50 [nM]a,b | in vitro Ki [nM]c | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Growth | Growth | mTOR/PI3Kα | clogPd | PSAd | ||||||
| pPKB/Akt | pS6 | PI3Kα | mTOR | |||||||
| A2058 | SKOV3 | |||||||||
| 9 | 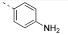 |
1744 | 1282 | 12630 | 8504 | 1513 | 396 | 0.26 | 2.45 | 89.6 |
| 10 | 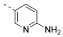 |
367 | 415 | 3819 | 742 | 123 | 110 | 0.89 | 2.23 | 103 |
| 11 |  |
330 | 415 | 2161 | 270 | 46 | 106 | 2.34 | 1.39 | 115 |
| 12 | 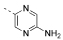 |
1283 | 817 | 9083 | 4428 | 911 | 185 | 0.20 | 1.31 | 115 |
| 13 | 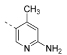 |
401 | 939 | 9113 | 1504 | 61 | 609 | 10.00 | 2.69 | 102 |
| 1 | 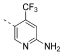 |
139 | 205 | 2333 | 237 | 17 | 62 | 3.60 | 3.11 | 102 |
| 14 | 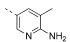 |
1620 | 1105 | 14210 | 6221 | 1205 | 241 | 0.20 | 2.69 | 102 |
| 15 |  |
2370 | 1638 | >20000 | 9213 | 4.3e6 | 284 | 6.5e-5 | 3.11 | 102 |
| 16 | 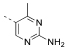 |
169 | 831 | 2934 | 342 | 13 | 224 | 17.60 | 1.56 | 115 |
| 2 | 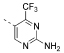 |
85 | 312 | 1636 | 73 | 8.1 | 203 | 25.10 | 2.67 | 115 |
| 20 | 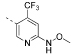 |
3303 | 5588 | 19200 | 4545 | 689 | 2539 | 3.70 | 3.09 | 97.8 |
| 17 | 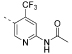 |
208 | 729 | 14480 | 11500 | 9.7 | 1213 | 125.00 | 3.22 | 105 |
| 18 | 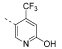 |
1.3e5 | 1.7e7 | >20000 | >20000 | 2299 | n.d. | – | 3.61 | 96.7 |
| 19 |  |
51971 | 1.1e6 | 4347 | 14050 | 32833 | 1.1e5 | 3.22 | 4.08 | 76.5 |
| 21 | 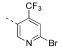 |
1.5e5 | n.d. | 3033 | 3654 | n.d. | 6.6e5 | – | 4.38 | 76.5 |
Cellular phosphorylation of PKB/Akt on Ser473 and ribosomal S6 on Ser235/236 were analyzed in inhibitor treated A2058 melanoma cells using in-cell Western detection. Each experiment was performed in triplicate or as a multiple of n = 3.
IC50 values of A2058 or SKOV3 cell proliferation were determined from 10-point 2-fold serial dilutions (n = 3).
Compounds were tested in a time-resolved FRET (TR) assay (LanthaScreen), and inhibitor Ki values were calculated for PI3Kα and mTOR as described in Experimental Section. The mTOR/PI3Kα column depicts the ratio of compound-specific mTOR Ki over the Ki for PI3Kα. Each experiment was performed at least twice or as a multiple of n = 2.
Marvin/JChem 16.10.17 was used for calculation of log P (partition coefficient) and PSA (polar surface area) values.
While compound 11, bearing an unsubstituted 2-amino-pyrimidine, turned out to be a balanced inhibitor for PI3Kα and for mTOR in vitro, (Ki(mTOR)/Ki(PI3Kα) ≈ 2.3), a substitution at the pyrimidine C4-position with methyl (16) or trifluoromethyl (2) enhanced cellular and in vitro activity toward PI3Kα and diminished affinity for mTOR, raising the mTOR/PI3Kα Ki ratio to ~16–25. Modifications at the C2-pyridine position of the most potent compound 1 were also investigated (17–21). These changes alter both the steric and the electronic properties of the amine group and are likely to affect hydrogen bond interactions with the aspartate carboxy groups in the affinity binding pocket (Asp841 and Asp836 in PI3Kγ). Hydroxylamine and amide moieties replacing primary amine functionality in 1 impaired binding to mTOR (Ki > 1 μM), and replacing the primary amine moiety by a hydroxylamine, amide, hydroxy, or halide decreased cellular potency considerably. Compound 17 retained its affinity for PI3Kα in spite of an amide extension, likely due to a hydrogen bond of the amide carbonyl with Lys802 (Lys833 in PI3Kγ: in analogy to compound 26 in ref 27 [see also PDB code 3IBE]).
The SAR study depicted above led to the identification of two potent compounds 1 and 2, both substituted with a trifluoromethyl group on their aryl moiety. Both of them showed excellent potency for PI3Kα, cellular activity profiles, and a calculated log P value of ~3. Compound 1 was investigated in more detail, as it displayed a mTOR/PI3Kα Ki selectivity ratio in the range of 3–8, while 2 had reduced impact on mTOR (Tables 1 and 2). An additional point in favor of compound 1 was a lower calculated PSA value of 102 as compared to 115 for 2, predicting a better brain penetration for 1.
Binding Mode of Compound 1 to PI3K and mTOR Kinase
Binding of compound 1 to the PI3Kγ catalytic subunit p110γ ATP-binding site was confirmed by a 2.7 Å complex crystal structure and identified key interactions of 1 with residues of p110γ. As reported for other morpholino-substituted PI3K inhibitors including compound 4,11,19 one morpholine of 1 forms a hydrogen bridge with the backbone amide of Val882, a well-known and crucial interaction.28 The compound 1/PI3Kγ complex crystal structure (PDB code 5OQ4; Table S1 in Supporting Information) also showed that the amino group of the 2-amino-4-(trifluoromethyl)pyridine moiety forms H-bonds with Asp836, Asp841, and Asp964 (Figure 2A). Given the high homology of class I PI3K catalytic pockets and the nanomolar in vitro dissociation constants of 1 for all class I PI3Ks (Table 1), an identical binding mode for all PI3K isoforms can be assumed. Besides the high affinity for class I PI3K isoforms (highest affinity for PI3Kα), compound 1’s affinity for mTOR kinase is comparable to PI3Kβ, PI3Kγ, and PI3Kδ (Table 1).
Figure 2.

Interactions of 1 with the ATP-binding site of PI3Kγ catalytic subunit p110γ and mTOR kinase. (A) Crystal structure of 1 in PI3Kγ with a resolution of 2.7 Å (PDB code 5OQ4; Table S1). Compound 1 interacts with the hinge valine V882 in PI3Kγ, and its heteroaryl amine function forms hydrogen bonds with the carboxyl groups of aspartates D836, D841, and D964. (B) Overlay of the compound 1/PI3Kγ complex coordinates with the compound 4/PI3Kγ data from PDB accession code 3SD5.19 The pyrimidine core nitrogen atom positions are corrected for the orientation as proposed in ref 11 (* denotes the position of C–H in compound 4); the two molecules are oriented exactly as in (A). (C) The coordinates of a mTOR kinase/PI103 complex resolved to 3.6 Å (PDB code 4JT6; docked, yellow) and mTOR at a resolution of 3.2 Å (PDB code 4JSN; relaxed, turquoise) were used to dock 1 into the ATP-binding site of mTOR kinase. The yellow structure represents the results of a manual predocking process (docked) where PI103 was replaced by 1 with subsequent energy minimization. The turquoise structure represents the best fit of dynamic docking procedures to 4JSN (relaxed starting conditions). Dotted lines indicate proposed H-bond interactions.
The current compound 1/PI3Kγ X-ray crystal structure combined with our previous structural studies on 4 and an asymmetrically substituted triazine (PIKiN3, PDB code 5JHB)11 underlines the importance of Asp836, Asp841, and Asp964 and associated structured water molecules for the binding of this compound class. Changes in the basicity of the amino-heteroaryl (2-amino-4-(trifluoromethyl)pyridine in 1 are therefore expected to affect the affinity for PI3K, as it was the case for 2-aminopyrimidine 2. The situation in the mTOR catalytic pocket is less well-defined: (i) modeling compound 1/mTOR interactions starting with a predocked inhibitor generate an interaction pattern similar to PI3K, where carboxyl groups of Asp2357, Glu2190, and Asp2195 are at a distance of 3.5–4 Å from the heteroaryl amino group and could execute hydrogen bonding; (ii) starting with less constraints, the inhibitor is slightly displaced by docking procedures, and the sole residue in hydrogen bonding distance remains Glu2190. Although less conventional, the latter structure offers some explanation for the differential effects of heteroaryl modifications for PI3K and mTOR affinity shown in Table 2.
On- and Off-Target Activities
In line with its activities against PI3K and mTOR, compound 1 induced a G1 cell cycle arrest in A2058 melanoma cells (mutated B-Raf, loss of PTEN) and SKOV3 cells (PI3Kα mutated at H1047R), which have both constitutively activated PI3K/mTOR signaling pathways. In contrast, compound 4 triggered a mitotic arrest and cells accumulated in G2/M or subG1 (Figure 3), which is in agreement with the reported binding of 4 to the colchicine-binding site of tubulin.11 In spite of compound 4’s off-target effect, the compound attenuated PI3K and mTOR signaling as determined by protein kinase B (PKB/Akt) phosphorylation on Ser473 and Thr308 and by S6 kinase (S6K, Thr389) and S6 phosphorylation on Ser235 and Ser236. Here, 4 closely matched the action of 1, 5, 6, 3, and 2, while rapamycin by its specific action on TORC1 triggered an upregulation of PI3K activity, which is in agreement with earlier observations.29
Figure 3.

Action of indicated compounds on cell cycle and PI3K and mTOR signaling. Cell cycle distribution of A2058 (A) and SKOV3 (B) cells after 24 h exposure to 5 μM (A) or 2 μM (B) of indicated drugs (1, green; 4, red) or DMSO. After drug exposure cells were collected, fixed, permeabilized and the DNA was stained using propidium iodide. Cell cycle profiles were acquired by fluorescence activated cell sorting. Left panels: Examples of cell cycle histograms. Right panels: Quantification of cells in cell cycle phases G1, S, G2/M, and sub-G1 (as % of total, n = 9, mean ± SD, one way ANOVA test with Dunnett’s multiple comparison test for each cell cycle phase related to DMSO; ns, not significant, (**) p > 0.0021, (****) p > 0.0001). (C, D) A2058 or SKOV3 cells were exposed to 1 μM drug (rapamycin 100 nM) for 1 h, subsequently lysed, and the denatured lysates subjected to SDS–PAGE, Western blotting, and immune detection. (C) Detection of total and phosphorylated (pT308 and pS473) PKB/Akt, and α-tubulin (left, A2058; right, SKOV3). Top: Quantification of phospho-PKB/Akt levels related to DMSO control (blue, pT308; red, pS473; n = 3, mean ± SEM). (D) Detection of total and phosphorylated (pS235/236) ribosomal protein S6 and of total and phosphorylated (S6K T389) S6 kinase (left, A2058; right, SKOV3). Top: Quantification of phospho-protein levels, related to DMSO control (blue, phospho S6; red, phospho S6K; n = 3, mean ± SEM).
To validate the specificity of 1, the compound was tested in a KINOMEScan panel for interactions with a wide range of protein (>400) and lipid kinases in parallel with 4, 5, 6, and 3. At 10 μM concentration 1, 4, 5, and 3 did show negligible interference with protein kinase activities (Figure S1, Table S2), while the dual PI3K/mTOR kinase inhibitor 6 displayed multiple hits. Compound 1 reached excellent selectivity scores of S(35) = 0.025, S(10) = 0.018, as calculated according to ref 30.
The impact of compound 1 on cell proliferation was analyzed in four independent human tumor cell line collections of different tissue origin (NCI60;31 NTRC, The Netherlands; Horizon Discoveries, U.K.; Clovis, U.K.; 135 different cell lines in total). Combined across the four cell panels the mean half-maximal growth inhibition (GI50) was reached at ~0.7 μM (median: ~0.5 μM) for 1 as depicted in Figure 4.
Figure 4.

Cell proliferation in response to 1 represented as a waterfall blot. Concentrations of half-maximal growth inhibition (GI50 for 1 were obtained from dose–response growth curves derived from 4 different tumor cell line panels (a total of 135 cell lines; if a cell line was contained in several panels, its GI50 was averaged). Individual GI50 of a cell line was related to the mean GI50 of all cells lines, and cell lines were sorted by lowest to highest sensitivity for 1 from left to right. Individual cell lines and values for the four cell panels are given in Table S3.
We have previously compared IC50-independent parameters such as Hill slopes of proliferation inhibition curves and dose-dependent induction of nuclear condensation and histone H3 phosphorylation in a 44 cell line panel treated with either 1, 4, 5, or 6, which showed a coclustering of the PI3K inhibitors 1, 5, and 6 and grouped 4 with inhibitors of mitosis such as colchicine, nocodazole, and MTD147, a microtubule-binder derived from 4,11 corroborating that 1 is free of the microtubule-binding off-target action of 4.
To evaluate other off-target effects, compound 1 was tested at 10 μM in a CEREP BioPrint panel, where no or only very weak competition with radioligands for cell surface and nuclear receptors, membrane channels, transporters (Table S4) and enzyme activities of kinases, proteases, and phosphodiesterases (Table S5) were detected. Altogether, 1 qualifies as a specific pan-class I PI3K inhibitor without predicted off-target effects.
Pharmacological Parameters of Compound 1
Before translation of the in vitro and cellular activities to in vivo tumor models, pharmacological properties and metabolic stability of 1 were assessed. Compound 1 showed little clearance when exposed to rat, dog, and human liver microsomes, with a quicker turnover of 1 in mouse liver microsomes, where 40% of the compound was eliminated within 30 min (Table 3). Stability in microsome assays was matched with clearance and half-live measurements in human, cynomolgus monkey, rat, dog, and mouse hepatocyte cultures, where 1 showed a low clearance in all species except for mouse. In the presence of human hepatocytes, the half-life of compound 1 was 9.4 h and ~45 min in mouse hepatocytes (Table 4).
Table 3. Stability in Liver Microsomes of Compound 1.
| % remaining in liver microsomesa |
||||
|---|---|---|---|---|
| compd | human | rat | dog | mouse |
| 1 | 84.6 ± 2.2 | 99.9 ± 0.5 | 92.4 ± 1.6 | 60.7 ± 4.6 |
| 7-ECb | 7.45 ± 0.8 | 28.5 ± 3.1 | 1.8 ± 1.0 | 1.5 ± 0.03 |
| propranololb | 54.8 ± 6.7 | 2.9 ± 0.1 | 30.1 ± 2.6 | 6.8 ± 0.9 |
Percentage of compound remaining after 30 min (mean ± SD, n = 2).
Assay reference compounds. 7-EC: 7-ethoxycoumarin.
Table 4. Stability of Compound 1 in Hepatocyte Culturesa.
| compd | parameter | human | monkey | rat | dog | mouse |
|---|---|---|---|---|---|---|
| 1 | CLintb | 2.5 ± 0.4 | 3.5 ± 0.1 | 1.9 ± 0.1 | 2.4 ± 0.1 | 30.3 ± 0.1 |
| t1/2 [min] | 562.6 ± 95.5 | 396.4 ± 16.0 | 731.7 ± 54.5 | 577.6 ± 0.1 | 45.7 ± 0.2 | |
| 7-ECc | CLintb | 37.8 ± 0.8 | 61.1 ± 3.5 | 16.0 ± 0.8 | 151.4 ± 2.8 | 180.3 ± 0.1 |
| t1/2 [min] | 36.7 ± 0.8 | 22.7 ± 1.3 | 86.8 ± 4.6 | 9.2 ± 0.2 | 7.7 ± 0.1 | |
| 7-HCc | CLintb | 98.2 ± 5.1 | 92.5 ± 3.5 | 192.8 ± 14.4 | 178.3 ± 0.7 | 188.1 ± 8.6 |
| t1/2 [min] | 14.1 ± 0.7 | 15.0 ± 0.6 | 7.2 ± 0.5 | 7.8 ± 0.1 | 7.4 ± 0.3 |
Results are expressed as mean ± SD, n = 2.
CLint [(μL/min)/106 cells].
Assay reference compounds: 7-EC, 7-ethoxycoumarin; 7-HC, 7-hydroxycoumarin.
Subsequently, in vivo pharmacological parameters were determined in female CD-1 mice, female Sprague-Dawley rats, and male Beagle dogs to assess optimal dosing schedules for efficacy studies. Compound 1 was delivered by either a single intravenous (5 mg/kg) bolus or a single oral application (10 mg/kg). Drug levels in extracts of plasma, brain, and liver were determined by LC–MS/MS (Figure 5; PK parameters are summarized in Table 5). In female mice, plasma concentrations of 1 depended on the drug administration route, resulting in half-lives of approximately 13–36 min for po administration vs 9–10 min for iv administration. The fact that oral administration yielded similar concentrations of 1 in brain and plasma samples illustrates that compound 1 readily passes the blood–brain barrier. In mice, both po and iv application routes showed a rapid drop below 200 ng/mL (~0.5 μM, Figure 5A) of 1 within <1 h (iv) to <2 h (po) after administration, which reflects the time point when the drug reaches the median GI50 determined in tumor cell lines.
Figure 5.

PK/PD assessment of 1 in mice, rats, and dogs: time course of 1 abundance in vivo. Compound 1 was administered in all species as single dose either per os (po, 10 mg/kg) or via intravenous injection (iv, 5 mg/kg). At indicated time-points 1 was extracted from tissue and its abundance determined using HPLC/MS–MS. (A) Levels of 1 in CD1-mice tissue. Right graph: Zoomed view on early time points, indicated in left graph by dashed square (n = 3, mean ± SD). (B) Concentrations of 1 in tissues from Sprague-Dawly rats. Right graph: Zoomed view on early time points, indicated in left graph by dashed square (n = 3, mean ± SD). (C) Plasma levels of 1 in male Beagle dogs. Panels on the right: zoomed in to dotted line boxes to visualize early time points (n = 3, mean ± SD).
Table 5. PK Analysis of 1 in Plasma of Mice, Rats, and Dogsa.
| parameter | female CD-1 mouse | female Sprague-Dawley rats | male Beagle dog | |||
|---|---|---|---|---|---|---|
| route | iv | po | iv | po | iv | po |
| dose [mg/kg] | 5 | 10 | 5 | 10 | 5 | 10 |
| Cmax [ng/mL] | 3084 ± 379 | 1514 ± 334 | 3047 ± 557 | 4028 ± 1179 | 8760 | 583 |
| Tmax [min] | 5 | 15 | 5 | 15 | 5 | 120−240 |
| t1/2 [h] | 0.15−0.33 | 0.22−0.60 | 5.6−8.2 | 5.6 | 7.6 | 8.6 |
| AUC [h·ng/mL] | 1583 AUC0−8h | 2151 AUC0−8h | 13974 AUC0.25−12h | 15313 AUC0.25−12h | 12500 AUC0.25−24h | 6395 AUC0.017−24h |
| Cl [mL h−1 kg−1] | 3180 | 266 | 411 | |||
| BA [%] | 68 | 55 | 23 | |||
iv, intravenous (5 mg/kg); po, per os (10 mg/kg); Cmax, maximal concentration; Tmax, time of maximal concentration in hours; t1/2, half-life elimination in hours; AUC, area under curve; Cl, clearance; BA, bioavailability. n = 3, mean ± SD for each time point, species, and route.
In female rats a single oral dose (10 mg/kg) achieved similar drug levels as a single intravenous injection (5 mg/kg) with regard to Cmax (see Table 5). The half-life of 5–8 h and an AUC0.25–12 of around 14 000 h·ng/mL contributed to an excellent oral bioavailability of 1 (>50%). Twenty-four hours after po administration, plasma levels of 1 were still >2 μM (800–1000 ng/mL). Moreover, after 1–2 h exposure to 1, drug levels in rat brain samples were comparable to plasma levels (Figure 5B), confirming rapid access of 1 to the brain.
Male Beagle dogs, exposed to 1 at 10 mg/kg po, showed maximal drug plasma concentrations Cmax of 583 ng/mL (approximately 1.5 μM) after 60–90 min and a half-life of >7 h, which results in drug levels of approximately 0.38 μM (150 ng/mL) after 24 h. Intravenous delivery of 5 mg/kg of 1 resulted in high immediate concentrations (>7000 ng/mL after 1 min), and plasma levels decreased within 8 h to similar concentrations as observed for oral dosing (Figure 4C). The oral bioavailability in male Beagle dogs was estimated to be 23%.
In vitro and in vivo toxicokinetic parameters were assessed and are summarized in Table S6. In rodents and Beagle dogs, compound 1 showed moderate but reversible target organ toxicity, which is expected for this drug class and mechanism of action. Altogether the PK studies in the three models show rapid absorption of 1 and good oral bioavailability. Except for mice with a rapid clearance of 1, the late phase concentrations of 1 in rats and dogs are in the expected range of predicted efficacy, independent of the administration route, and are already reached after a single dose. The collected parameters are compatible with oral dosing in humans once daily (QD).
Altogether, compound 1 displays a superior solubility (Table S7) as compared with compound 2, is orally bioavailable and brain penetrable, and does not display a microtubule-binding off-target effect like compound 4.11
Efficacy Models
Due to the short half-live of compound 1 in mice, a PC3 xenograft model in nude rats was used to assess the antiproliferative effects of 1 in vivo. Rats were injected with 2 × 107 human PC3 prostate cancer cells into one flank and randomized after 16 days. From day 17 the control group received vehicle once daily. Compound 1 was orally administered at 5 mg/kg, 10 mg/kg (both daily, QD), or 15 mg/kg [5 consecutive days, 2 days off drug (QD × 5, 2 days off)] for 28 days to match the timelines of regulatory toxicology studies. A vinorelbine-treated group (2.5 mg/kg iv weekly) was included as a “standard of care” control, with best treated-to-control ratios (T/C) of 17%.
Treatment with 1 led to significant tumor size reductions: tumor growth was inhibited dose-dependently (best T/C of 31–12%, Figure 6A). Compound 1 was best tolerated at 5 mg/kg without significant body weight changes (Figure 6B). At 10 mg/kg, 1 caused a reduction of body weight, which accumulated to a reduction of 15% after 28 days of treatment. Similarly, 15 mg/kg of 1 led to body weight loss after 5 days of treatment, which was reversible during the recovery period. After 28 days of drug exposure (day 44 of the experiment), animals with body weight loss fully recovered within a treatment-free period (days 45–50) without overt signs of tumor cell proliferation. In a subsequent treatment period tumor growth remained inhibited and body weight loss was only observed in the 15 mg/kg group.
Figure 6.

Efficacy model for antitumor activity of 1 in PC3 xenografts in nude rats. (A) Tumor volume: Indicated amounts of 1 (po, daily) or vinorelbine (iv, weekly) were given from day 17 to day 44 and from day 51 to day 58 after tumor inoculation (see Experimental Section; n = 4–8, mean ± SEM). (B) Body weight changes in nude rats (treated as in (A), n = 4–8, mean ± SEM). (C) Time course of 1 abundance in nude rats. Compound 1 was administered as single dose per os (po, 15 mg/kg). At indicated time points 1 was extracted from tissue and its abundance determined using HPLC/MS–MS (n = 3, mean ± SD). (D) Time course of blood glucose and insulin levels tumor bearing rats after a single dose of 1 (15 mg/kg po; n = 3, mean ± SEM; except for time points with values above detection limit). (E) Plasma insulin and (F) plasma glucose concentrations on day 57 of the efficacy model, 4 h after the last administration of 1 at indicated doses (n = 6–7, mean ± SEM; for insulin levels below detection level, values were set constant to 0.04 [lowest measured value]; for glucose levels above quantification limit, levels were set 20 [upper quantification limit of kit]). (G) Time course of phosphorylation of Ser473 in PKB/Akt in PC3 tumor xenograft lysates after a single dose of 1 (15 mg/kg po or 10 mg/kg iv). Lysates were subjected to SDS–PAGE and Western blotting. Phosphorylated Ser473 levels were corrected for total PKB/Akt levels and compared to vehicle-treated xenografts (n = 3, mean ± SEM).
As shown in Figure 6C, Cmax of compound 1 is reached 15 min after a po single dose in PC3-tumor bearing nude rats and triggered a rapid and dose-dependent rise in plasma insulin and glucose levels (Figure 6D–F), which are reliable markers for the on-target action of drugs, as reported for other PI3K inhibitors targeting the PI3Kα isoform.32
To further corroborate on-target activity of compound 1 in vivo, phosphorylation of pPKB/AKT was measured in tumor-derived cell lysates. A single dose of 1 (15 mg/kg po or 10 mg/kg iv) rapidly reduced pPKB/AKT levels as compared to untreated control mice up to 12 h (Figure 6G), which matches high levels of 1 in the xenograft tumors at early time points (Figure 6C). After 24 h, PI3K signaling fully recovered, correlating with a decrease of compound 1’s concentration in tissue and tumors. Pharmacokinetics in plasma, brain, and tumor samples were very similar, suggesting that 1 distributes rapidly and targets PI3K and mTOR in tumor and tissues. Proportionally elevated levels of 1 were detected in the liver, without signs of liver toxicity. The fast reversibility of PI3K inhibition might be of advantage in patients, where mechanism-based adverse effects are expected to resolve with falling drug levels. Moreover, it has been observed that tumor cells reduce cell size in response to PI3K inhibitors, while normal hepatocytes do not show mass reduction.17 This, and the observation that interruption of drug administration (see 15 mg/kg schedule above) leads to body weight recovery but not tumor regrowth, suggests that PI3K inhibition triggers latency in tumor growth that allows for the development of intermittent scheduling protocols. To validate this treatment scheme, PC3 tumor-bearing nude rats were treated every second, third, and fourth day with 15 mg/kg po, which yielded a comparable efficacy but substantially alleviated the loss of body weight (Figure S2).
Conclusion
The preclinical results presented here document that compound 1 is a highly selective pan-PI3K inhibitor with a balanced targeting of mTOR kinase. Compound 1’s potent antiproliferative action in vitro and antitumor activity in vivo, paired with mild and reversible, mostly mechanism-based toxicities, have validated the molecule as a clinical candidate. Compound 1 passed phase I studies and is now in phase II studies in relapsed and refractory lymphoma and advanced solid tumors (clinicaltrials.gov).
Experimental Section
General Information
Reagents were purchased at the highest commercial quality from Acros, Sigma-Aldrich, or Fluorochem and used without further purification. Solvents were purchased from Acros Organics in AcroSeal bottles over molecular sieves. Column chromatographic purifications were performed on Merck KGaA silica gel (pore size 60 Å, 230–400 mesh particle size). Cross-coupling reactions were carried out under nitrogen atmosphere in anhydrous solvents, and glassware was oven-dried prior to use. TLC plates were obtained from Merck KGaA (Polygram SIL/UV254, 0.2 mm silica with fluorescence indicator), and UV light (254 nm) was used to visualize the respective compounds. 1H, 19F, and 13C NMR spectra were recorded on a Bruker Avance 400 spectrometer. NMR spectra were obtained in deuterated solvents such as CDCl3 or (CD3)2SO. The chemical shifts (δ values) are reported in ppm and corrected to the signal of the deuterated solvents (7.26 ppm (1H NMR) and 77.16 ppm (13C NMR) for CDCl3 and 2.50 ppm (1H NMR) and 39.52 ppm (13C NMR) for (CD3)2SO). 19F NMR spectra were calibrated relative to CFCl3 (δ = 0 ppm) as external standard. When peak multiplicities are reported, the following abbreviations are used: s (singlet), d (doublet), dd (doublet of doublets), m (multiplet), br (broadened). Coupling constants, when given, are reported in hertz (Hz). High resolution mass spectra (HRMS) were recorded on a Thermo Fisher Scientific LTQ Orbitrap XL (ESI-MS) spectrometer. MALDI-ToF mass spectra were obtained on a Voyager-De Pro measured in m/z. The chromatographic purities of the final compounds were determined by high performance liquid chromatography (HPLC) analyses on an Ultimate 3000SD system from ThermoFisher with LPG-3400SD pump system, ACC-3000 autosampler and column oven, and DAD-3000 diode array detector. An Acclaim-120 C18 reversed-phase column from ThermoFisher was used as stationary. Gradient elution (5:95 for 0.2 min, 5:95 → 100:0 over 10 min, 100:0 for 3 min) of the mobile phase consisting of CH3CN/MeOH:-H2O(10:90) was used at a flow rate of 0.5 mL/min at 40 °C. The purity of all final compounds was >95%.
General Procedure 1
4,4′-(6-Chloro-1,3,5-triazine-2,4-diyl)-dimorpholine (7, 1.0 equiv), the respective boronic acid pinacol ester (1.0–1.5 equiv), potassium phosphate tribasic (3.0 equiv), and chloro(2-dicyclohexylphosphino-2′,4′,6′-triisopropyl-1,1′-biphenyl)[2-(2′-amino-1,1′-biphenyl)]palladium(II) (XPhos Pd G2, 0.05–0.10 equiv) were charged in a flask. Under nitrogen atmosphere, 1,4-dioxane (approximately 1 mL/0.2 mmol) and deionized H2O (approximately 1 mL/0.4 mmol) were added, and the resulting mixture was placed into an oil bath preheated to 95 °C and stirred at this temperature for 2–16 h. After completion of the reaction, the mixture was allowed to cool to room temperature before addition of a 3 M aqueous HCl solution and then heated at 60 °C for 2–8 h. Then the mixture was allowed to cool to room temperature, the pH was adjusted to 8–9 by addition of a 2 M aqueous NaOH solution, and the aqueous layer was extracted with ethyl acetate (3×). The combined organic layers were dried over anhydrous Na2SO4, filtered, and the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel.
General Procedure 2
To a solution of the respective boronic acid pinacol ester (4.0 equiv), [1,1′-bis(diphenylphosphino)ferrocene]-dichloropalladium(II) (0.05 equiv) and 4,4′-(6-chloro-1,3,5-triazine-2,4-diyl)dimorpholine (7, 1.0 equiv) in 1,2-dimethoxyethane (approximately 1 mL/0.2 mmol) under nitrogen atmosphere, a 2 M aqueous Na2CO3 solution (3.0 equiv) was added. The resulting reaction mixture was heated at 90 °C for 16 h. Then the mixture was allowed to cool to room temperature, poured onto an aqueous saturated NH4Cl solution, and the aqueous layer was extracted with ethyl acetate (3×). The combined organic layers were washed with deionized H2O, dried over anhydrous Na2SO4, filtered, and the solvent was evaporated to dryness under reduced pressure. The crude product was purified by column chromatography on silica gel.
General Procedure 3
N,N-Dimethylformamide dimethyl acetal (1.5 equiv) and the respective amine (1.0 equiv) were dissolved in tetrahydrofuran (approximately 1 mL/0.3 mmol), and the resulting reaction mixture was stirred at 70 °C overnight. The solvent was evaporated under reduced pressure and the residue was triturated with acetonitrile (3×x) before it was dried under vacuum.
5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)-pyridin-2-amine (1)
1 was prepared according to the reported procedure in ref 11.
5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)-pyrimidin-2-amine (2)
2 was prepared according to general procedure 1 from 4,4′-(6-chloro-1,3,5-triazine-2,4-diyl)dimorpholine (7, 300 mg, 1.05 mmol, 1.0 equiv) and 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-4-(trifluoromethyl)pyrimidin-2-amine (455 mg, 1.58 mmol, 1.5 equiv). Purification by column chromatography on silica gel (cyclohexane/ethyl acetate 2:3 → 3:2) gave product 2 as a colorless solid (225 mg, 546 μmol, 52%). 1H NMR (400 MHz, CDCl3): δ 8.94 (s, 1 H), 5.65 (br s, 2 H), 3.39–3.78 (m, 8 H), 3.76–3.69 (m, 8 H). 19F{1H} NMR (376 MHz, (CD3)2SO): δ –63.8 (s, 3 F). 13C{1H} NMR (101 MHz, CDCl3): δ 168.5 (s, 1 C), 164.7 (s, 2 C), 163.1 (s, 1 C), 162.7 (s, 1 C), 154.6 (q, 2JC,F = 35 Hz, 1 C), 121.1 (s, 1 C), 120.8 (q, 1JC,F = 276 Hz, 1 C), 66.9 (s, 4 C), 43.7 (s, 4 C). ESI-HRMS (m/z): [M + H]+ calcd for C16H20F3N8O2, 413.1656; found, 413.1662. HPLC: tR = 7.39 min (>99.9% purity).
4,4′-(6-Chloro-1,3,5-triazine-2,4-diyl)dimorpholine (7)
7 was prepared according to ref 11.
4,4′-(6-Iodo-1,3,5-triazine-2,4-diyldimorpholine (8)
To an aqueous hydriodic acid solution (57 wt %, 2.00 mL, 15.2 mmol, excess) diluted with deionized H2O (2.00 mL), 4,4′-(6-chloro-1,3,5-triazine-2,4-diyl)dimorpholine (7, 500 mg, 1.75 mmol, 1.0 equiv) was added at 0 °C. The reaction mixture was allowed to warm to room temperature overnight and then poured onto a 1 M aqueous NaOH solution. The product was extracted with ethyl acetate (3×). The combined organic layers were dried over anhydrous Na2SO4, filtered, and the solvent was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (cyclohexane/ethyl acetate 1:0 → 1:1) to afford product 8 as a colorless solid (409 mg, 1.08 mmol, 61%). 1H NMR (400 MHz, CDCl3): δ 3.87–3.63 (m, 16 H). 13C{1H} NMR (101 MHz, CDCl3): δ 162.1 (s, 2 C), 140.6 (s, 1 C), 66.8 (s, 4 C), 44.0 (s, 4 C). MALDI-MS: m/z = 378.1 ([M + H]+).
4-(4,6-Dimorpholino-1,3,5-triazin-2-yl)aniline (9)
9 was prepared according to general procedure 1 from 4,4′-(6-chloro-1,3,5-triazine-2,4-diyl)dimorpholine (7, 150 mg, 525 μmol, 1.0 equiv) and tert-butyl (4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)-carbamate (22, 250 mg, 783 μmol, 1.5 equiv). Purification by column chromatography on silica gel (cyclohexane/ethyl acetate 1:0 → 3:2) gave product 9 as a colorless solid (80.9 mg, 236 μmol, 45%). 1H NMR (400 MHz, CDCl3): δ 8.22 (d, 3JH,H = 8.7 Hz, 2 H), 6.69 (d, 3JH,H = 8.7 Hz, 2 H), 3.99–3.81 (m, 10 H), 3.79–3.72 (m, 8 H). 13C{1H} NMR (101 MHz, CDCl3): δ 170.3 (s, 1 C), 165.3 (s, 2 C), 149.7 (s, 1 C), 130.1 (s, 2 C), 127.5 (s, 1 C), 114.2 (s, 2 C), 67.0 (s, 4 C), 43.7 (br s, 4 C). MALDI-MS: m/z = 343.2 ([M + H]+). HPLC: tR = 7.27 min (>99.9% purity). The spectroscopic data are consistent with previous literature reports.34
5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)pyridin-2-amine (10)
10 was prepared according to general procedure 2 from 4,4′-(6-chloro-1,3,5-triazine-2,4-diyl)dimorpholine (7, 200 mg, 702 μmol, 1.0 equiv) and 2-aminopyridine-5-boronic acid pinacol ester (616 mg, 2.80 mmol, 4.0 equiv). Purification by column chromatography on silica gel (dichloromethane/methanol 9:1) gave compound 10 as a colorless solid (166 mg, 483 μmol, 69%). 1H NMR (400 MHz, CDCl3): δ 9.07 (d, 4JH,H = 2.3 Hz, 1 H), 8.37 (dd, 3JH,H = 8.7 Hz, 4JH,H = 2.3 Hz, 1 H), 6.50 (d, 3JH,H = 8.6 Hz, 1 H), 4.71 (br s, 2 H), 3.88 (br s, 8 H), 3.79–3.71 (m, 8 H). 13C{1H} NMR (101 MHz, CDCl3): δ 169.2 (s, 1 C), 165.1 (s, 2 C), 160.4 (s, 1 C), 150.0 (s, 1 C), 137.9 (s, 1 C), 123.6 (s, 1 C), 107.5 (s, 1 C), 67.0 (s, 4 C), 43.7 (s, 4 C). ESI-HRMS (m/z): [M + H]+ calcd for C16H22N7O2, 344.1829; found, 344.1832. HPLC: tR = 8.59 min (96.2% purity).
5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)pyrimidin-2-amine (11)
11 was prepared according to general procedure 2 from 4,4′-(6-chloro-1,3,5-triazine-2,4-diyl)dimorpholine (7, 50.0 mg, 170 μmol, 1.0 equiv) and 2-aminopyrimidine-5-boronic acid pinacol ester (155 mg, 700 μmol, 4.0 equiv). Purification by column chromatography on silica gel (ethyl acetate/methanol 1:0 → 20:1) gave compound 11 as a colorless solid (10.0 mg, 29.0 μmol, 17%). 1H NMR (400 MHz, (CD3)2SO): δ 9.05 (s, 2 H), 7.26 (br s, 2 H), 3.94–3.62 (m, 8 H), 3.64 (br s, 8 H). 13C{1H} NMR (101 MHz, (CD3)2SO): δ 167.2 (s, 1 C), 164.8 (s, 1 C), 164.2 (s, 2 C), 158.6 (s, 2 C), 118.5 (s, 1 C), 66.0 (s, 4 C), 43.2 (br s, 4 C). ESI-HRMS (m/z): [M + H]+ calcd for C15H21N8O2, 345.1782; found, 345.1784. HPLC: tR = 6.06 min (95.4% purity).
5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)pyrazin-2-amine (12)
4,4′-(6-Iodo-1,3,5-triazine-2,4-diyl)dimorpholine (8, 200 mg, 530 μmol, 1.0 equiv), 5-aminopyrazin-2-ylboronic acid pinacol ester (350 mg, 1.58 mmol, 3.0 equiv), potassium phosphate tribasic (339 mg, 1.60 mmol, 3.0 equiv), and chloro(2-dicyclohexylphosphino-2′,4′,6′-triisopropyl-1,1′-biphenyl)[2-(2′-amino-1,1′-biphenyl)]palladium(II) (41.7 mg, 53.0 mmol, 0.10 equiv) were charged in a flask. Under nitrogen atmosphere, 1,4-dioxane (5 mL) and deionized H2O (1.5 mL) were added, and the resulting mixture was placed into an oil bath preheated and allowed to stir for 2 h at 95 °C. After completion of the reaction, the mixture was allowed to cool to room temperature. Dichloromethane and a 2 M aqueous NaOH solution were added. The aqueous layer was separated and extracted with dichloromethane (3×). The combined organic layers were then dried over anhydrous Na2SO4, filtered, and the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel (dichloromethane/methanol 1:0 → 9:1) to obtain compound 12 as a colorless solid (20.1 mg, 58.4 μmol, 11%). 1H NMR (400 MHz, CDCl3): δ 9.08 (br s, 1 H), 8.10 (br s, 1 H), 4.86 (br s, 2 H), 4.08–3.81 (m, 8 H), 3.81–3.71 (m, 8 H). 13C{1H} NMR (101 MHz, CDCl3): δ 168.8 (s, 1 C), 165.2 (s, 2 C), 155.2 (s, 1 C), 144.5 (s, 1 C), 140.1 (s, 1 C), 131.6 (s, 1 C), 67.0 (s, 4 C), 43.8 (br s, 4 C). ESI-345.1780. HPLC: tR = 5.37 min (95.2% purity).
5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-methylpyridin-2-amine (13)
13 was prepared according to general procedure 2 from 4,4′-(6-chloro-1,3,5-triazine-2,4-diyl)dimorpholine (7, 40.0 mg, 140 μmol, 1.0 equiv) and 2-amino-4-methylpyridine-5-boronic acid pinacol ester (132 mg, 560 μmol, 4.0 equiv). Purification by column chromatography on silica gel (ethyl acetate/methanol 1:0 → 10:1) gave product 13 as a colorless solid (13.1 mg, 36.7 μmol, 26%). 1H NMR (400 MHz, CDCl3): δ 8.78 (s, 1 H), 6.33 (s, 1 H), 4.54 (br s, 2 H), 3.90–3.80 (m, 8 H), 3.77–3.72 (m, 8 H), 2.60 (s, 3 H). 13C{1H} NMR (101 MHz, CDCl3): δ 171.4 (s, 1 C), 165.0 (s, 2 C), 159.3 (s, 1 C), 151.4 (s, 1 C), 149.4 (s, 1 C), 124.1 (s, 1 C), 109.8 (s, 1 C), 66.8 (s, 4 C), 43.7 (br s, 4 C), 22.3 (s, 1 C). ESI-HRMS (m/z): [M + H]+ calcd for C17H24N7O2, 358.1986; found, 358.1983. HPLC: tR = 6.57 min (98.5% purity).
5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-3-methylpyridin-2-amine (14)
14 was prepared according to general procedure 1 from 4,4′-(6-chloro-1,3,5-triazine-2,4-diyl)dimorpholine (7, 150 mg, 525 μmol, 1.0 equiv) and boronic acid pinacol ester 23 (180 mg, 622 μmol, 1.2 equiv). Purification by column chromatography on silica gel (cyclohexane/ethyl acetate 1:0 → 4:1) gave product 14 as a colorless solid (116 mg, 32.5 μmol, 62%). 1H NMR (400 MHz, CDCl3): δ 8.89 (d, 4JH,H = 2.2 Hz, 1 H), 8.22 (br s, 1 H), 5.25 (br s, 2 H), 4.02–3.68 (m, 16 H), 2.19 (s, 3 H). 13C{1H} NMR (101 MHz, CDCl3): δ 168.9 (s, 1 C), 165.0 (s, 2 C), 158.9 (s, 1 C), 145.2 (s, 1 C), 138.1 (s, 1 C), 123.5 (s, 1 C), 116.3 (s, 1 C), 67.0 (s, 4 C), 43.7 (br s, 4 C), 17.0 (s, 1 C). ESI-HRMS (m/z): [M + H]+ calcd for C17H24N7O2, 358.1986; found, 358.1981. HPLC: tR = 6.91 min (99.3% purity).
5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-3-(trifluoromethyl)-pyridin-2-amine (15)
15 was prepared according to general procedure 1 from 4,4′-(6-chloro-1,3,5-triazine-2,4-diyl)dimorpholine (7, 83.3 mg, 292 μmol, 1.0 equiv) and boronic acid pinacol ester 24 (100 mg, 292 μmol, 1.0 equiv). Purification by column chromatography on silica gel (cyclohexane/ethyl acetate 1:0 → 1:1) gave product 15 as a colorless solid (89.5 mg, 218 μmol, 75%). 1H NMR (400 MHz, CDCl3): δ 9.19 (s, 1 H), 8.63 (d, 4JH,F = 2.2 Hz, 1 H), 5.24 (br s, 2 H), 4.01–3.80 (m, 8 H), 3.80–3.71 (m, 8 H). 19F{1H} NMR (376 MHz, (CD3)2SO): δ –63.1 (s, 3 F). 13C{1H} NMR (101 MHz, (CD3)2SO): δ 167.3 (s, 1 C), 164.2 (s, 2 C), 157.3 (s, 1 C), 153.1 (s, 1 C), 134.5 (br s, 1 C), 124.1 (q, 1JC,F = 271 Hz, 1 C), 119.9 (s, 1 C), 105.1 (q, 2JC,F = 31 Hz, 1 C), 66.0 (s, 4 C), 43.2 (br s, 4 C). ESI-HRMS (m/z): [M + H]+ calcd for C17H21F3N7O2, 412.1703; found, 412.1703. HPLC: tR = 8.40 min (>99.9% purity).
5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-methylpyrimidin-2-amine (16)
16 was prepared according to general procedure 2 from 4,4′-(6-chloro-1,3,5-triazine-2,4-diyl)dimorpholine (7, 40.0 mg, 140 μmol, 1.0 equiv) and 4-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyrimidin-2-amine (132 mg, 560 μmol, 4.0 equiv). Purification by column chromatography on silica gel (ethyl acetate 100%) gave product 16 as a colorless solid (17.6 mg, 49.2 μmol, 34%). 1H NMR (400 MHz, (CD3)2SO): δ 8.90 (s, 1 H), 7.02 (br s, 2 H), 3.81–3.69 (m, 8 H), 3.66–3.59 (m, 8 H), 2.63 (s, 3 H). 13C{1H} NMR (101 MHz, CDCl3): δ 170.1 (s, 1 C), 168.9 (s, 1 C), 164.6 (s, 2 C), 162.6 (s, 1 C), 160.9 (s, 1 C), 121.3 (s, 1 C), 66.9 (s, 4 C), 43.7 (br s, 4 C), 25.2 (s, 1 C). ESI-HRMS (m/z): [M + H]+ calcd for C16H23N8O2, 359.1938; found, 359.1939. HPLC: tR = 6.22 min (97.3% purity).
N - (5 - (4,6-Dimorpholino-1,3, 5-triazin - 2-yl)-4-(trifluoromethyl)pyridin-2-yl)acetamide (17)
5-(4,6-Dimorpholi-no-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-amine (1, 200 mg, 480 μmol, 1.0 equiv) and K2CO3 (134 mg, 972 mmol, 2.0 equiv) were mixed in dichloromethane (4 mL) and stirred at room temperature for 5 min. Then, the reaction mixture was cooled to 0 °C and acetyl chloride (41.4 μL, 583 mmol, 1.2 equiv) was added dropwise. The resulting mixture was stirred for 3.5 h and allowed to warm up to room temperature. Then, deionized H2O was added, the layers were separated, and the aqueous layer was extracted with dichloromethane (3×). The combined organic layers were dried over anhydrous filtered, and the solvent was evaporated under reduced pressure. The crude product was puri ed by column chromatography on silica gel (cyclohexane/ethyl acetate 1:1 → 1:3) to afford compound 17 (120 mg, 265 mmol, 60%) as a colorless solid. 1H NMR (400 MHz, CDCl3): δ 8.81 (s, 1 H), 8.61 (s, 1 H), 8.34 (s, 1 H), 3.85 (br s, 8 H), 3.78–3.67 (m, 8 H), 2.26 (s, 3 H). 19F{1H} NMR (376 MHz, CDCl3): δ –59.5 (s, 3 F). 13C{1H} NMR (101 MHz, CDCl3): δ 169.3 (s, 1 C), 168.7 (s, 1 C), 164.5 (s, 2 C), 152.6 (s, 1 C), 151.0 (s, 1 C), 138.8 (q, 2JC,F = 33 Hz, 1 C), 128.4 (s, 1 C), 122.5 (q, 1JC,F = 275 Hz, 1 C), 110.5 (q, 3JC,F = 5.1 Hz, 1 C), 66.7 (s, 4 C), 43.5 (br s, 4 C), 24.7 (s, 1 C). ESI-HRMS (m/z): [M + H]+ calcd for C19H23F3N7O3, 454.1809; found, 454.1797. HPLC: tR = 7.94 min (>99.9% purity).
5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)-pyridin-2-ol (18) and 4,4′-(6-(6-Chloro-4-(trifluoromethyl)-pyridin-3-yl)-1,3,5-triazine-2,4-diyl)dimorpholine (19)
To a conc HCl solution (36%, 5.12 mL) at 0 °C, 5-(4,6-dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-amine (1, 500 mg, 1.21 mmol, 1.0 equiv) was added dropwise followed by NaNO2 (1.70 g, 24.6 mmol, 20 equiv). The resulting mixture was then allowed to warm to room temperature over 1 h, and CuCl (4.10 g, 41.4 mmol, 34 equiv) was added. After 1 h, the reaction mixture was quenched with a 2 M aqueous NaOH solution and extracted with ethyl acetate (3×). The combined organic layers were dried over anhydrous Na2SO4, filtered, and the solvent was evaporated under reduced pressure. The products were separated by column chromatography on silica gel (cyclohexane/ethyl acetate 1:0 → 1:1). Compound 19 (247 mg, 573 μmol, 47%) and compound 18 (166 mg, 403 μmol, 33%) were obtained in two separate fractions and isolated as colorless solids.
Compound 19
1H NMR (400 MHz, CDCl3): δ 8.91 (s, 1 H), 7.66 (s, 1 H), 3.89–3.78 (m, 8 H), 3.78–3.68 (m, 8 H). 19F{1H} NMR (376 MHz, CDCl3): δ –59.7 (s, 3 F). 13C{1H} NMR (101 MHz, CDCl3): δ 169.0 (s, 1 C), 164.6 (s, 2 C), 153.2 (s, 1 C), 153.0 (s, 1 C), 154.1 (q, 2JC,F = 34 Hz, 1 C), 131.5 (s, 1 C), 122.1 (q, 1JC,F = 275 Hz, 1 C), 121.4 (q, 3JC,F = 5.4 Hz, 1 C), 66.9 (s, 4 C), 43.8 (br s, 4 C). ESI-HRMS (m/z): [M + H]+ calcd for C17H19F3ClN6O2, 431.1205; found, 431.1199. HPLC: tR = 9.91 min (>99.9% purity).
Compound 18
1H NMR (400 MHz, (CD3)2SO): δ 12.6 (br s, 1 H), 8.15 (s, 1 H), 6.80 (s, 1 H), 3.79–3.67 (m, 8 H), 3.66–3.55 (m, 8 H). 19F{1H} NMR (376 MHz, (CD3)2SO): δ –59.4 (s, 3 F). 13C{1H} NMR (101 MHz, (CD3)2SO): δ 168.0 (s, 1 C), 164.0 (s, 2 C), 161.2 (s, 1 C), 141.4 (br s, 1 C), 138.9 (q, 2JC,F = 32 Hz, 1 C), 122.3 (q, 1JC,F = 275 Hz, 1 C), 119.2 (br s, 1 C), 113.3 (br s, 1 C), 65.9 (s, 4 C), 43.2 (s, 4 C). ESI-HRMS (m/z): [M + H]+ calcd for C17H20F3N6O3, 413.1543; found, 413.1535. HPLC: tR = 6.79 min (99.6% purity).
N-(5-(4,6-Dimorpholino-1,3,5-triazin-2-yl)-4-(trifluoromethyl)pyridin-2-yl)-O-methylhydroxylamine (20)
4,4′-(6-(6-Chloro-4-(trifluoromethyl)pyridin-3-yl)-1,3,5-triazine-2,4-diyl)dimorpholine (19, 200 mg, 464 μmol, 1.0 equiv), methoxyamine hydrochloride (77.5 mg, 928 μmol, 2.0 equiv), and NaHCO3 (79.0 mg, 940 μmol, 2.0 equiv) were mixed in dimethyl sulfoxide (3.0 mL). The resulting reaction mixture was stirred at 100 °C for 6 h. The solvent was partially evaporated under reduced pressure. Then, deionized H2O and dichloromethane were added. The aqueous layer was separated and extracted with dichloromethane (3×). The combined organic layers were dried over anhydrous Na2SO4, filtered, and the solvent was evaporated to dryness under reduced pressure. The crude product was purified by column chromatography on silica gel (cyclohexane/ethyl acetate 1:1 → 0:1) to afford compound 20 (30.0 mg, 34.0 mmol, 15%) as a colorless solid. 1H NMR (400 MHz, CDCl3): δ 8.77 (s, 1 H), 7.77 (br s, 1 H), 7.23 (s, 1 H), 3.90–3.79 (m, 11 H), 3.77–3.70 (m, 8 H). 19F{1H} NMR (376 MHz, CDCl3): δ –59.6 (s, 3 F). 13C{1H} NMR (101 MHz, CDCl3): δ 169.6 (s, 1 C), 164.8 (s, 2 C), 156.0 (s, 1 C), 151.8 (s, 1 C), 138.6 (q, 2JC,F = 274 Hz, 1 C), 127.5 (s, 1 C), 122.6 (q, 1JC,F = 275 Hz, 1 C), 110.1 (q, 3JC,F = 6.2 Hz, 1 C), 66.9 (s, 4 C), 63.1 (s, 1 C), 43.8 (br s, 4 C). ESI-HRMS (m/z): [M + H]+ calcd for C18H23F3N7O3, 442.1809; found, 442.1799. HPLC: tR = 8.33 min (98.9% purity).
4,4′-(6-(6-Bromo-4-(trifluoromethyl)pyridin-3-yl)-1,3,5-triazine-2,4-diyl)dimorpholine (21)
Compound 1 (1.00 g, 2.43 mmol, 1.0 equiv) was dissolved in dibromomethane (10 mL), and isopentyl nitrite (392 μL, 2.92 mmol, 1.2 equiv) was added. After 5 min, a solution of bromotrimethylsilane (401 μL, 3.04 mmol, 1.3 equiv) in dibromomethane (1.5 mL) was added and the resulting reaction mixture was allowed to stir at room temperature for 1 h. After this time, an aqueous saturated NaHCO3 solution was added, the layers were separated, and the aqueous layer was extracted with dichloro-methane (3×). The combined organic layers were dried over anhydrous Na2SO4, filtered, and the solvent was evaporated under reduced pressure. Purification by column chromatography on silica gel (cyclohexane/ethyl acetate 1:0 → 1:1) gave compound 21 (909 mg, 1.91 mmol, 79% yield) as colorless solid. 1H NMR (400 MHz, CDCl3): δ 8.88 (s, 1 H), 7.81 (s, 1 H), 3.88–3.78 (m, 8 H), 3.78–3.68 (m, 8 H). 19F{1H} NMR (376 MHz, CDCl3): δ –59.6 (s, 3 F). 13C{1H} NMR (101 MHz, CDCl3): δ 169.0 (s, 1 C), 164.6 (s, 2 C), 153.2 (s, 1 C), 143.7 (s, 1 C), 138.5 (q, 2JC,F = 34 Hz, 1 C), 131.8–131.7 (m, 1 C), 125.1 (q, 3JC,F = 5.3 Hz, 1 C), 122.0 (q, 1JC,F = 275 Hz, 1 C), 66.8 (s, 4 C), 43.7 (br s, 4 C). ESI-HRMS (m/z): [M + H]+ calcd for C17H19BrF3N6O2, 475.0699; found, 475.0710. HPLC: tR = 10.11 min (97.8% purity).
tert-Butyl (4-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)-phenyl)carbamate (22)
22 was prepared according to the literature.33
N,N-Dimethyl-N′-(3-methyl-5-(4,4,5,5-tetramethyl-1,3,2-di-oxaborolan-2-yl)pyridin-2-yl)formimidamide (23)
23 was prepared according to general procedure 3 from 3-methyl-5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)pyridin-2-amine (200 mg, 854 μmol, 1.0 equiv). The desired product 23 was obtained in quantitative yield as a brownish solid and was used in the next step without further purification. 1H NMR (400 MHz, (CD3)2SO): δ 8.50 (s, 1 H), 8.25 (s, 1 H), 7.61 (s, 1 H), 3.10 (s, 3 H), 3.02 (s, 3 H), 2.19 (s, 3 H), 1.27 (s, 12 H).
N,N-Dimethyl-N′-(5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-(trifluoromethyl)pyridin-2-yl)formimidamide (24)
24 was prepared according to general procedure 3 from 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-(trifluoromethyl)pyridin-2-amine (300 mg, 1.04 mmol, 1.0 equiv). The desired product 24 was obtained in quantitative yield as a yellowish solid and was used in the next step without further purification. 1H NMR (400 MHz, (CD3)2SO): δ 8.70 (s, 1 H), 8.54 (d, J = 1.8 Hz, 1 H), 7.96 (d, J = 1.9 Hz, 1 H), 3.16 (s, 3 H), 3.06 (s, 3 H), 1.29 (s, 12 H). 19F{1H} NMR (376 MHz, (CD3)2SO): δ –61.6 (s, 3 F).
Protein Production and Purification
The ΔABDp110γ was expressed in Sf9 cells using a baculovirus expression system as has been described previously.24,35 Briefly, human ΔABDp110γ was expressed with a C-terminal His6 tag and purified by immobilized metal-affinity chromatography, heparin chromatography, and gel filtration. The protein in gel filtration buffer (20 mM Tris-HCl, 0.5 mM (NH4)2SO4, 5 mM DTT, 1% ethylene glycol, and 0.02% CHAPS) was concentrated to 8 mg/mL and then frozen in liquid nitrogen and stored at –80 °C.
Crystallization of PI3Kγ. Soaking with Compound 1
Drops containing 1 μL of ΔABDp110γ protein at 4 mg/mL and 1 μL of crystallization buffer (16% PEG4K, 250 mM (NH4)2SO4, 0.1 M Tris-HCl, pH 7.5, at rt) were hair seeded with crushed ΔABDp110γ crystals and incubated at 17 °C. The crystals reached their maximum size (0.2 mm × 0.1 mm × 0.1 mm) in about 10 days. The inhibitor 1 was prepared in DMSO and diluted in cryoprotectant containing 25% PEG 4000, 15% glycerol, 250 mM (NH4)2SO4, and 100 mM Tris, pH 7.5, to a final concentration of 1 mM. The inhibitor in cryoprotectant solution was added to the drop containing the crystals in a stepwise manner. The crystals were soaked in the final 1 mM inhibitor solution for 4 h. Crystals were then transferred to fresh inhibitor in cryoprotectant solution for 30 s and flash frozen in liquid nitrogen.
Data Collection and Structure Determination
Diffraction data sets were collected at the ESRF beamline ID23-1 on an ADSC Quantum Q315r detector, using 1.007 20 Å wavelength radiation. Images were processed using Mosflm36 or XDS37 and scaled with SCALA.38 The crystal structure of the ΔABDp110γ/compound 1 complex was solved by molecular replacement using Phaser,39 with the previously published human ΔABDp110γ as the search model (PDB entry 1E8Y). Coot was used to manually build inhibitors in the unaccounted Fo – Fc difference electron density within the active site of the map.40 Initial models of the inhibitors were generated from SMILES strings using the ELBOW module of PHENIX.41 Refmac refinement42 was iterated with manual rebuilding using Coot until the structure converged. In the final model for the ΔABDp110γ/compound 1 complex, 89.2% of residues were in the core regions of the Ramachandran plot, with one residue in a disallowed region.
Cellular PI3K and mTOR Signaling
Downstream signals of PI3K and mTOR were determined in in-cell Western assays detecting phosphorylation of Ser473 of PKB/Akt (pPKB/Akt) and Ser235/236 on the ribosomal protein S6 (pS6), respectively. Briefly, 2 × 104 A2058 cells/well in 96-well plates (Cell Carrier, PerkinElmer) were grown for 24 h, before inhibitors or DMSO was added (1 h; 37 °C, 5% CO2). Cells were subsequently fixed (4% PFA in PBS for 30 min at rt) and then blocked (1% BSA/0.1% Triton X-100/5% goat serum in PBS for 30 min, rt). pPKB/Akt was detected with a rabbit polyclonal antibody (Cell Signaling Technology, no. 4058), and pS6 with a rabbit monoclonal antibody (Cell Signaling Technology, no. 4856). Tubulin staining (mouse anti-α-tubulin, Sigma no. T9026) was assessed as internal standard. As readout, IRDye680-conjugated goat anti-mouse and IRDye800-conjugated goat anti-rabbit antibodies (LICOR no. 926-68070 and no. 926-32211) were used, and fluorescence was measured on an Odyssey CLx infrared imaging scanner (LICOR). Percentage of remaining phospho-substrate signals were calculated in relation to cellular tubulin. Further details and calculations are explained in ref 11.
Determination of Inhibitor Dissociation Constants
Dissociation constants of compounds (Ki) for p110α and mTOR were determined by LanthaScreen technology (Life Technologies) as described in detail in ref 11. Briefly, the AlexaFluor647-labeled kinase tracer 314 (no. PV6087) was used for p110α with a determined Kd of 2.2 nM at 20 nM, and for mTOR with a Kd of 19 nM used at a final concentration of 10 nM. While recombinant p110α was N-terminally (His)6-tagged and combined with a biotinylated anti-(His)6-tag antibody (2 nM, no. PV6089) and LanthaScreen Eu-streptavidin (2 nM, no. PV5899), truncated mTOR (amino acids 1360–2549, no. PR8683B) fused to the C-terminus of GST was detected with a LanthaScreen Eu-labeled anti-GST antibody (2 nM, no. PV5594). The p110α assay buffer was composed of 50 mM HEPES, pH 7.5, 10 mM MgCl2, 1 mM EGTA, and 0.01% (v/v) Brij-35, and the mTOR assay buffer contained 50 mM HEPES, 5 mM MgCl2, 1 mM EGTA, 0.01% Pluronic F-127. Further details and calculations are described in ref 11.
Cell Cycle Analysis
A2058 cells ((1–2) × 105 cells/mL) or SKOV3 cells ((0.25–0.5) × 105 cells/mL) were seeded in DMEM supplemented with 10% heat-inactivated FCS, 1% l-glutamine, and 1% penicillin–streptomycin (2 mL/well of six-well plates). The day after, inhibitors (5 μM for A2058, 2 μM for SKOV3) were added for 24 h. Subsequently nonadherent cells were collected by centrifugation, adherent cells were detached, fixed, and permeabilized (combined with the previously nonadherent cell) in PBS supplemented with 4% paraformaldehyde/1% bovine serum albumin/0.1% TritonX-100 for 30 min at 4 °C and then washed with 1% bovine serum albumin/0.1% TritonX-100 in PBS followed by DNA-staining with 50 μg/mL propidium iodide in 0.1% TritonX-100/0.1% sodium citrate solution (pH 7.4) containing 10 μg/mL DNase-free RNase (>1 h, rt, in the dark). Cell cycle profiles were acquired by fluorescence activated cell sorting (FACSCanto II, Becton Dickinson) and analyzed with FlowJo (Treestar) software.
Western Blotting
Cells were lysed in NP40-based lysis buffer (20 mM Tris-HCl, pH 8, 138 mM NaCl, 2.7 mM KCl, 5% glycerol, 1% NP-40) supplemented with protease and phosphatase inhibitors, cleared by centrifugation, and denatured by adding 5× sample buffer and boiling at 96 °C for 6 min. Equal amount of proteins were subjected to SDS–PAGE and transferred to Immobilon FL membranes (Millipore). Primary antibodies to pSer473-PKB/Akt (no. 4058L), pThr308-PKB/Akt (no. 4056L), PKB/AKT (no. 2929S), pT389-S6K1 (no. 9206S), pS235/236 ribosomal protein S6 (no. 4856S), S6K1 (no. 9202S), and ribosomal protein S6 (no. 2317S) were from Cell Signaling Technology, and primary antibody to α-tubulin (no. T9026) was from Sigma. HRP-conjugated secondary antibodies were visualized using enhanced chemiluminescence (Millipore) on a Fusion FX (Vilber Lourmat) imaging system. Levels of phosphorylated proteins were quantified using ImageJ and normalized to their respective nonphosphorylated proteins.
Kinome Profiling
The inhibitory capacity and selectivity of compound were determined using the ScanMax platform provided by DiscoverX.26 In short, binding of immobilized ligand to DNA-tagged kinases was competed with 10 μM compound. The amount of kinase bound to the immobilized ligand was measured by quantitative PCR of the respective DNA tags and is given as percentage of control. Binding constants of compounds for kinases of interest were determined by competing the immobilized ligand kinase interactions with an 11-point 3-fold serial dilution of compound starting from 30 μM and subsequent quantitative PCR of DNA tags. Binding constants were calculated by a standard dose−response curve using the Hill equation: response = background + (signal − background)/(1 + 10[log Kd−log dose](Hill slope)), with Hill slope set to −1.
Cell Proliferation Assay Panels
NCI60
Detailed procedures on compound and cell handling are available from https://dtp.cancer.gov/discovery_development/nci-60/methodology.htm. Briefly, human tumor cell lines were seeded into 96-well microtiter plates and exposed to five (1/2 log serial) drug dilutions plus control, followed by 48 h (except for two controls of each cell line which were fixed with TCA (cell population at t = 0 h [Tz]). The assay was terminated by fixation with TCA (10% final). Cell density was determined using a sulforhodamine B staining protocol and the absorbance measured at 515 nm. Using seven absorbance measurements [Tz; control growth (C); growth with drugs added (Ti)], the percentage growth was calculated at each of the drug concentrations levels. Percentage growth inhibition is calculated as
for concentrations for which Ti ≥ Tz, and
for concentrations for which Ti < Tz.
The NTRC Oncolines 44 cell lines were exposed for 72 h to 9-point 3-fold serial dilutions of 1 as described in ref 11. The concentration of 50% growth inhibition is associated with the signal ((luminescenceuntreated,t=72h – luminescencet=0)/2) + luminescencet=0. The data set integrated here was used for IC50 calculations in ref 11.
IC50 values of A2058 or SKOV3 cell proliferation given in Table 2 were determined and calculated as described in ref 11 for the indicated compounds.
PC3 Xenograft Model in Nude Rats
Compound Preparation and Dosing
Compound 1 was dissolved in DMSO to 40 mg/mL. This solution was diluted with 20% HPBCD [(2-hydroxypropyl)-β-cyclodextrin] in water for injection (Lavoisier, batch no. 2F274 5) in order to reach a final concentration of 1 of 4 mg/mL. Successive dilutions in vehicle (DMSO/20% HPBCD solution, 10/90, v/v) were performed to reach 1, 2, and 3 mg/mL final concentrations. Vinorelbine was diluted in 0.9% NaCl solution to 0.5 mg/mL. Treatment doses for 1 were 5, 10, and 15 mg/kg, and it was 2.5 mg/kg for vinorelbine. Rats were given compound 1 by oral gavage (po) and given vinorelbine intravenously (iv, as bolus) into the tail vain. The po administration volume and the iv injection volume were 5 mL/kg.
Animals and Animal Handling
Healthy male nude NIH rats (Crl:NIH-Foxn1 rnu, Charles River, Germany) bread in a specific pathogen-free (SPF) animal care unit were fed sterile, irradiated food granules (V1246-703, Soest, Germany). Animal procedures were reviewed by the Animal Care and Use Committee of Pharmacy and Medicine University of Dijon or Oncodesign’s ethical committee.
Cell Culture
PC-3 prostatic adenocarcinoma tumor cells (ATCC, CRL-1435) were grown as monolayer in complete RPMI1640 medium, supplemented with 10% FBS (37 °C, 5% CO2). Tumor cells were detached with trypsin-Versene (Lonza; 5 min in in Hanks’ medium without Ca2+ or Mg2+) and then resuspended in complete RPMI1640.
Tumor Model
2 ×107 PC-3 cells were injected subcutaneously at day 0 (D0) in 200 μL of RPMI1640 into the right flank of male nude rats, 24 h after a whole-body irradiation with a γ-source (5 Gy, 60Co). Tumor-bearing rats were randomized on day 16 (mean volume of 330 ± 70 mm3 according to their individual tumor volume into five groups of each eight animals using Vivo manager software (Bio-systemes, Couternon, France). Analysis of variance was performed to test for homogeneity between groups.
Treatment Schedule
Daily administration on D17–D44 and from D51 to D57: group 1, vehicle; group 2, compound 1 at 5 mg/kg; group 3, 1 at 10 mg/kg. Group 4: 1 at 15 mg/kg from D17 to D21, from D24 to D28, from D34 to D38, from D41 to D4, and from D51 to D56. Group 5: one iv injection of vinorelbine at 2.5 mg/kg on D17, D24, D31, and D38. Final termination of rats was performed on D87.
Body weight was measured at least twice a week. Length and width of tumors were measured and recorded twice a week with calipers, and the tumor volume was estimated by the formula
Glucose and Insulin Levels Determination
Glucose was measured using kit CBA086 from Calbiochem and insulin was determined using kit EZRMI-13K from EMD Millipore, according to the manufacturers’ instructions.
PK/PD Studies in Nude Rat PC3 Xenograft Model
Pharmacokinetic Study of Compound 1 in PC-3 Tumor Bearing Rats
Induction of PC-3 subcutaneous tumors is described above. Animals were randomized on D21 when the tumors reached 575 ± 267 mm3 (D20). Twenty-seven animals were randomized according to their individual tumor volume into 3 groups (one group of 3 rats and two groups of 12 rats) using Vivo manager software (Biosystemes, Couternon, France).
Treatment Q1D × 1. Group 1
Three rats single iv injection of vehicle. Group 2: 12 and 3 satellite rats single iv injection of 1 (10.3 mg/kg). Group 3: 12 and 3 satellite rats single po administration of compound 1 (16.55 mg/kg). On selected time points blood, brain, liver, and tumor from 3 rats from each group were collected to determine levels of 1, glucose, and insulin or pPKB/Akt was analyzed in tissues.
Solubility Assays
Stock solutions of compounds were prepared in DMSO at 20 mM. From these, 200 μM solutions were made up by a direct dilution of the stock solution with a phosphate buffer (PBS, pH 7.4) or citrate buffer (pH 4.0). Reference solutions were made up in 100% MeOH. Solutions in buffer (200 μL) were incubated and agitated at room temperature for 1 h and then centrifuged at 13 000 rpm for 10 min. A volume of 100 μL of the supernatants was diluted with 100 μL of MeOH/PBS/citrate 8:1:1 and analyzed by LC/MS.
Structure Modeling of Compound 1/mTOR Kinase Complexes
The coordinates of mTOR kinase 4JT6 (3.6 Å) and 4JSN (3.2 Å) were used as starting points to dock 1 into the ATP-binding site of mTOR kinase. Docking of the ligand 1 to mTOR was performed using SwissDock (swissdock.ch), and energy minimization was performed using YASARA’s default settings. Alternatively, the PI103 ligand in 4JT6 was manually replaced by 1 docking one of its morpholino groups to Val2240 before energy minimizations were performed. The docking procedures were validated by redocking 1 into the apoprotein of the 1/PI3Kγ complex (PDB code 5OQ4).
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.7b00930.
Molecular formula strings and some data (CSV)
X-ray crystallography parameters and refinement values for 1 (Table S1); a TREEspot representation (Figure S1) and its raw data (Table S2); the antiproliferative action of compound 1 in four cell line panels (Table S3); extended Tables S4 and S5 with in vitro pharmacology data; the safety profile of 1 (Table S6); comparative solubility of 1 (Table S7); efficacy of compound 1 intermittent dosing in tumor bearing rats (Figure S2); 1H NMR spectra; 19F{1H} NMR spectra; 13C{1} NMR spectra; ESI-HRMS spectra; MALDI-MS spectra; HPLC chromatograms; and appendix of chemical formula of compounds synthesized here (PDF)
Acknowledgments
We thank A. Pfaltz, R.A. Ettlin, W. Dieterle, J.-B. Langlois, S. Mukherjee, and M. Lang for advice and discussions; S. Bünger and A. Dall Asen for technical assistance; Master students of the Department of Chemistry for early synthetic efforts; M. Neuburger and D. Häussinger for chemical structure determination. We are grateful to G. Zaman and the NTRC team for help and expertise with high content screening assays. This work was supported by the Swiss Commission for Technology and Innovation (CTI) by PFLS-LS Grants 14032.1, 15811.2, and 17241.1; the Stiftung für Krebsbekampfung Grant 341; Swiss National Science Foundation Grants 310030_153211 and 316030_133860 (to M.P.W.) and Grant 310030B_138659 and in part by European Union’s Horizon 2020 research and innovation program under the Marie Sklodowska-Curie Grant Agreement 675392 and by the MRC to R.L.W. (Grant MC_U105184308).
Abbreviations Used
- DNA-PK
DNA-dependent protein kinase
- ESI
electrospray ionization
- GPCR
G-protein-coupled receptor
- mTOR
mammalian target of rapamycin
- PI3K
phosphoinositide 3-kinase
- PKB/Akt
protein kinase B/Akt
- PSA
polar surface area
- PtdIns(4,5)P2
phosphatidylinositol (4,5)-bisphosphate
- PtdIns(3,4,5)P3
phosphatidylinositol (3,4,5)-trisphosphate
- S6
ribosomal protein S6
- S6K
S6 kinase
- SH2
src-homology 2
- TORC1
mTOR complex 1
- VPS34
vacuolar protein sorting 34, the class III PI3K
Footnotes
Notes
The authors declare the following competing financial interest(s): F.B., N.C., V.C., J.M., P.He., P.Hi., and D.F. are current or past employees of PIQUR Therapeutics AG, Basel; and N.C., V.C., J.M., P.He., P.Hi., D.F., B.G., and M.P.W. are shareholders of PIQUR Therapeutics AG.
Author Contributions
F.B., N.C., and V.C. contributed equally to this work.
References
- (1).Wymann MP, Schneiter R. Lipid signalling in disease. Nat Rev Mol Cell Biol. 2008;9:162–176. doi: 10.1038/nrm2335. [DOI] [PubMed] [Google Scholar]
- (2).Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15:7–24. doi: 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- (4).Wymann M. PI3Ks-Drug targets in inflammation and cancer. Subcell Biochem. 2012;58:111–181. doi: 10.1007/978-94-007-3012-0_5. [DOI] [PubMed] [Google Scholar]
- (5).Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2012;9:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- (6).Sun T, Aceto N, Meerbrey KL, Kessler JD, Zhou C, Migliaccio I, Nguyen DX, Pavlova NN, Botero M, Huang J, Bernardi RJ, et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell. 2011;144:703–718. doi: 10.1016/j.cell.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- (8).Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11:289–301. doi: 10.1038/nrc3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gewinner C, Wang ZC, Richardson A, Teruya-Feldstein J, Etemadmoghadam D, Bowtell D, Barretina J, Lin WM, Rameh L, Salmena L, Pandolfi PP, et al. Evidence that inositol polyphosphate 4-phosphatase type II is a tumor suppressor that inhibits PI3K signaling. Cancer Cell. 2009;16:115–125. doi: 10.1016/j.ccr.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).(a) Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discovery. 2009;8:627–644. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) LoRusso PM. Inhibition of the PI3K/AKT/mTOR pathway in solid tumors. J Clin Oncol. 2016;34:3803–3815. doi: 10.1200/JCO.2014.59.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bohnacker T, Prota AE, Beaufils F, Burke JE, Melone A, Inglis AJ, Rageot D, Sele AM, Cmiljanovic V, Cmiljanovic N, Bargsten K, et al. Deconvolution of Buparlisib’s mechanism of action defines specific PI3K and tubulin inhibitors for therapeutic intervention. Nat Commun. 2017;8:14683. doi: 10.1038/ncomms14683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Yaguchi S, Fukui Y, Koshimizu I, Yoshimi H, Matsuno T, Gouda H, Hirono S, Yamazaki K, Yamori T. Antitumor activity of ZSTK474, a new phosphatidylinositol 3-kinase inhibitor. J Natl Cancer Inst. 2006;98:545–556. doi: 10.1093/jnci/djj133. [DOI] [PubMed] [Google Scholar]; (b) Rewcastle GW, Gamage SA, Flanagan JU, Frederick R, Denny WA, Baguley BC, Kestell P, Singh R, Kendall JD, Marshall ES, Lill CL, Lee WJ, et al. Synthesis and biological evaluation of novel analogues of the pan class I phosphatidylinositol 3-kinase (PI3K) inhibitor 2-(difluoromethyl)-1-[4,6-di(4-morpholinyl)-1,3,5-triazin-2-yl]-1H-benzimidazole (ZSTK474) J Med Chem. 2011;54:7105–7126. doi: 10.1021/jm200688y. [DOI] [PubMed] [Google Scholar]
- (13).Brachmann SM, Kleylein-Sohn J, Gaulis S, Kauffmann A, Blommers MJ, Kazic-Legueux M, Laborde L, Hattenberger M, Stauffer F, Vaxelaire J, Romanet V, et al. Characterization of the mechanism of action of the pan class I PI3K inhibitor NVP-BKM120 across a broad range of concentrations. Mol Cancer Ther. 2012;11:1747–1757. doi: 10.1158/1535-7163.MCT-11-1021. [DOI] [PubMed] [Google Scholar]
- (14).Elkabets M, Vora S, Juric D, Morse N, Mino-Kenudson M, Muranen T, Tao J, Campos AB, Rodon J, Ibrahim YH, Serra V, et al. mTORC1 inhibition is required for sensitivity to PI3K p110alpha inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med. 2013;5:196ra99. doi: 10.1126/scitranslmed.3005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Marone R, Cmiljanovic V, Giese B, Wymann MP. Targetingphosphoinositide3-kinase:movingtowardstherapy. Biochim Biophys Acta Proteins Proteomics. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- (17).Marone R, Erhart D, Mertz AC, Bohnacker T, Schnell C, Cmiljanovic V, Stauffer F, Garcia-Echeverria C, Giese B, Maira SM, Wymann MP. Targeting melanoma with dual phosphoinositide 3-kinase/mammalian target of rapamycin inhibitors. Mol Cancer Res. 2009;7:601–613. doi: 10.1158/1541-7786.MCR-08-0366. [DOI] [PubMed] [Google Scholar]
- (18).Corcoran RB, Rothenberg SM, Hata AN, Faber AC, Piris A, Nazarian RM, Brown RD, Godfrey JT, Winokur D, Walsh J, Mino-Kenudson M, et al. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. ACS Med Chem Lett. 2013;5:196ra98. doi: 10.1126/scitranslmed.3005753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Burger MT, Pecchi S, Wagman A, Ni ZJ, Knapp M, Hendrickson T, Atallah G, Pfister K, Zhang Y, Bartulis S, Frazier K, et al. Identification of NVP-BKM120 as a potent, selective, orally bioavailable class I PI3 kinase inhibitor for treating cancer. ACS Med Chem Lett. 2011;2:774–779. doi: 10.1021/ml200156t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sutherlin DP, Bao L, Berry M, Castanedo G, Chuckowree I, Dotson J, Folks A, Friedman L, Goldsmith R, Gunzner J, Heffron T, et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J Med Chem. 2011;54:7579–7587. doi: 10.1021/jm2009327. [DOI] [PubMed] [Google Scholar]
- (21).Dehnhardt CM, Venkatesan AM, Chen Z, Delos-Santos E, Ayral-Kaloustian S, Brooijmans N, Yu K, Hollander I, Feldberg L, Lucas J, Mallon R. Identification of 2-oxatriazines as highly potent pan-PI3K/mTOR dual inhibitors. Bioorg Med Chem Lett. 2011;21:4773–4778. doi: 10.1016/j.bmcl.2011.06.063. [DOI] [PubMed] [Google Scholar]
- (22).Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, Chuckowree IS, Clarke PA, Depledge P, Eccles SA, Friedman LS, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522–5532. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- (23).Arcaro A, Wymann MP. Wortmannin is a potent phosphatidylinositol 3-kinase inhibitor: the role of phosphatidylinositol 3,4,5-trisphosphate in neutrophil responses. Biochem J. 1993;296:297–301. doi: 10.1042/bj2960297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wymann MP, Bulgarelli-Leva G, Zvelebil MJ, Pirola L, Vanhaesebroeck B, Waterfield MD, Panayotou G. Wortmannin inactivates phosphoinositide 3-kinase by covalent modification of Lys-802, a residue involved in the phosphate transfer reaction. Mol Cell Biol. 1996;16:1722–1733. doi: 10.1128/mcb.16.4.1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Venkatesan AM, Dehnhardt CM, Delos Santos E, Chen Z, Dos Santos O, Ayral-Kaloustian S, Khafizova G, Brooijmans N, Mallon R, Hollander I, Feldberg L, et al. Bis(morpholino-1,3,5-triazine) derivatives: potent adenosine 5′-triphosphate competitive phosphatidylinositol-3-kinase/mammalian target of rapamycin inhibitors: discovery of compound 26 (PKI-587), a highly efficacious dual inhibitor. J Med Chem. 2010;53:2636–2645. doi: 10.1021/jm901830p. [DOI] [PubMed] [Google Scholar]
- (26).Fabian MA, Biggs WH, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, Ford JM, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- (27).Zask A, Verheijen JC, Curran K, Kaplan J, Richard DJ, Nowak P, Malwitz DJ, Brooijmans N, Bard J, Svenson K, Lucas J, et al. ATP-competitive inhibitors of the mammalian target of rapamycin: design and synthesis of highly potent and selective pyrazolopyrimidines. J Med Chem. 2009;52:5013–5016. doi: 10.1021/jm900851f. [DOI] [PubMed] [Google Scholar]
- (28).Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell. 2000;6:909–919. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- (29).O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–1508. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127–132. doi: 10.1038/nbt1358. [DOI] [PubMed] [Google Scholar]
- (31).Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- (32).Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159–169. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- (33).Sugino M, Hatanaka K, Araki Y, Hisaki I, Miyata M, Tohnai N. Amphiphilic inclusion spaces for various guests and regulation of fluorescence intensity of 1,8-bis(4-aminophenyl)-anthracene crystals. Chem—Eur J. 2014;20:3069–3076. doi: 10.1002/chem.201304541. [DOI] [PubMed] [Google Scholar]
- (34).Zhao X, Tan Q, Zhang Z, Zhao Y. 1,3,5-Triazine inhibitors of histone deacetylases: synthesis and biological activity. Med Chem Res. 2014;23:5188–5196. [Google Scholar]
- (35).Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, Balla T, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Leslie AGW. Joint CCP4/ESF-EACBM Newsletter on Protein Crystallography. 26 Daresbury Laboratory; Warrington, U.K.: 1992. Mosflm—a programme for processing X-ray diff;raction data. [Google Scholar]
- (37).Kabsch W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- (38).The CCP4 suite: programs for protein crystallography. Acta Crystallogr Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- (39).McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr Sect D: Biol Crystallogr. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (41).Moriarty NW, Grosse-Kunstleve RW, Adams PD. Electronic ligand builder and optimization workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr Sect D: Biol Crystallogr. 2009;65:1074–1080. doi: 10.1107/S0907444909029436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.