

Abstract
Insufficient lysosomal removal of autophagic cargoes in cardiomyocytes has been suggested as a main cause for the impairment of the autophagic-lysosomal pathway (ALP) in many forms of heart disease including cardiac proteinopathy and may play an important pathogenic role; however, the molecular basis and the correcting strategy for the cardiac ALP insufficiency require further investigation. The present study was sought to determine whether myocardial expression and activity of TFEB, the recently identified ALP master regulator, are impaired in a cardiac proteinopathy mouse model and to determine the effect of genetic manipulation of TFEB expression on autophagy and proteotoxicity in a cardiomyocyte model of proteinopathy. We found that increased myocardial TFEB mRNA levels and a TFEB protein isoform switch were associated with marked decreases in the mRNA levels of representative TFEB target genes and increased mTORC1 activation, in mice with cardiac transgenic expression of a missense (R120G) mutant αB-crystallin (CryABR120G), a well-established model of cardiac proteinopathy. Using neonatal rat ventricular cardiomyocyte cultures, we demonstrated that downregulation of TFEB decreased autophagic flux in cardiomyocytes both at baseline and during CryABR120G overexpression and increased CryABR120G protein aggregates. Conversely, forced TFEB overexpression increased autophagic flux and remarkably attenuated the CryABR120G overexpression- induced accumulation of ubiquitinated proteins, caspase 3 cleavage, LDH leakage, and decreases in cell viability. Moreover, these protective effects of TFEB were dramatically diminished by inhibiting autophagy. We conclude that myocardial TFEB signaling is impaired in cardiac proteinopathy and forced TFEB overexpression protects against proteotoxicity in cardiomyocytes through improving ALP activity.
Keywords: TFEB, mTOR, the autophagic- lysosomal pathway, proteotoxicity, misfolded proteins, cardiomyocytes
1. INTRODUCTION
Protein quality control (PQC) acts to keep the level of misfolded proteins low and to minimize the toxicity of misfolded proteins in the cell, indispensable to protein homeostasis and cell survival and functions. As indicated by increases in the abundance of total ubiquitinated proteins and pre-amyloid oligomers in the vast majority of human hearts with end-stage heart failure, increased proteotoxicity and inadequate PQC may contribute to the genesis of and the progression from a large subset of heart diseases to heart failure [1, 2]. Hence, it is conceivable that improving cardiac PQC has the potential to become a new therapeutic strategy for heart failure, a leading cause of disability and mortality in humans.
PQC is accomplished by intricate collaboration between molecular chaperones and targeted protein degradation; the latter is carried out primarily by the ubiquitin-proteasome system (UPS) and the autophagic- lysosomal pathway (ALP) [3]. It is generally accepted that all abnormal cellular proteins are degraded by the UPS but the proteasome can only degrade protein molecules one at a time. By contrast, the ALP degrades cellular content in a bulky fashion and thereby plays an important role in the quality control of not only proteins but also organelles such as mitochondria. Terminally misfolded proteins, when escaped from or overwhelmed the surveillance of chaperones and the UPS, undergo aggregation via hydrophobic interaction and form aberrant aggregates which are inaccessible by the proteasome and can only be degraded by macroautophagy [3, 4]. Hence, the ALP plays a critical role in PQC.
The lysosomal degradation of cell’s own components, or autophagy, takes three different forms: macroautophagy, chaperone-mediated autophagy (CMA), and microautophagy, among which macroautophagy is most extensively studied [3]. In microautophagy, invagination of lysosomal membrane captures directly a small amount of soluble cytoplasmic content into the lumen of the lysosome. By CMA, the lysosome selectively uptakes specific individual protein molecules one at a time with the help of heat shock proteins (e.g., HSC70) and through a translocating complex formed by lysosome associated membrane protein 2A (LAMP2A). Proteins that are targeted for CMA degradation harbor the KFERQ motif [5]. Neither microautophagy nor CMA is well studied in the heart. Macroautophagy (hereafter referred to as autophagy for simplicity) segregates a portion of cytoplasm through formation of a double membraned vacuole known as an autophagosome for fusion with and degradation by lysosomes. It is increasingly evidenced that the ALP capacity of removing its cargos becomes inadequate in cardiomyocytes under many diseased conditions including cardiac proteinopathy [6]; however, the mechanism underlying the cardiac ALP insufficiency remains largely unknown and, as result, measures tested to improve cardiac ALP are often non-specific, flaunted with unwanted off-target effects [3]. Thus, a better understanding of ALP insufficiency and search for specific strategies to coordinately enhance both the formation and the removal of autophagosomes, i.e., the entire ALP, are urgently needed.
Recent advances in cell biology identify the transcription factor EB (TFEB) as a master regulator of lysosome biogenesis. TFEB is a basic helix- loop-helix- leucine-zipper (bHLH-Zip) transcription factor, belonging to the microphthalmia family (MiT family) [7]. Similar to other bHLH-Zip transcription factors, TFEB recognizes and binds the palindromic E box (i.e., CACGTG). A common 10-base E-box like palindromic sequence, referred to as the coordinated lysosomal expression and regulation (CLEAR), has been identified in lysosomal genes via promoter analysis. The network of genes harboring the CLEAR motif is referred to as the CLEAR network. By directly binding to the CLEAR element, TFEB can activate all genes of the CLEAR network [8, 9], increase lysosome numbers and lysosomal enzyme levels, and thereby promote lysosomal catabolic function [9]. More recently, it has been further shown that TFEB also regulates the expression of many autophagy-related genes to orchestrate autophagosome formation and lysosomal degradation [10]. Overexpression of TFEB was shown to enhance the clearance of misfolded or aggregation-prone proteins in neurons [9, 11–16], hepatocytes and lung epithelia [17, 18]; however, this has not been demonstrated in cardiomyocytes expressing a human disease-linked bona fide misfolded protein. The present study was sought to fill this gap.
Here we report that myocardial TFEB signaling is inhibited in mice with advanced cardiac proteinopathy induced by cardiomyocyte-restricted expression of a missense (R120G) mutant αB-crystallin (CryABR120G), a well-established animal model of cardiac proteotoxicity. Moreover, we demonstrate for the first time in cardiomyocytes that TFEB is required for sustaining ALP activity and forced TFEB expression is sufficient to facilitate ALP activity and thereby protects against misfolded protein-induced proteotoxicity. Our findings suggest that enhancing TFEB should be explored as a therapeutic strategy to ameliorate cardiac proteotoxic stress that is implicated in a large subset of heart disease during their progression to heart failure.
2. METHODS
2.1 Animals
The protocol for the care and use of animals in this study was approved by University of South Dakota Institutional Animal Care and Use Committee. The creation and baseline characterization of the inbred FVB/N mice with transgenic (tg) overexpression of wild type CryAB (CryABWT) or CryABR120G driven by the murine Myh6-promoter were previously described [19–22]. Mixed sex tg and non-tg (Ntg) littermate mice at 6 months of age were used. In-house bred Sprague-Dawley rats at postnatal day 2 were used for isolating ventricular cardiomyocytes for cell cultures.
2.2 Reagents
Phenylmethylsulfonyl Fluoride (PMSF), and 5-Bromo-2'-Deoxyuridine (BrdU) were purchased from Sigma-Aldrich (St. Louis, MO). 3-Methyladenine (3-MA) was purchased from ThermoFisher Scientific (Waltham, MA). Bafilomycin A1 (BFA) was from LC laboratory (Woburn, MA). DNase I and complete protease inhibitor cocktail was from Roche Applied Science (Indianapolis, IN).
2.3 Neonatal rat ventricular myocytes (NRVMs) culture and adenoviral infection
Primary NRVMs were isolated from the ventricles of 2-day old Sprague-Dawley rats and plated on 6-cm plates at a density of 2.0×106 cells in 10% FBS in DMEM as previously described [23]. The plated cells were then cultured in a 5% CO2 incubator at 37°C for at least 24 hours before the medium was changed to meet the needs of the follow-up experiments. Forty-eight hours after plating, cells were infected with recombinant replication-deficient adenoviruses harboring desired transgenes for 4 hours in DMEM media. Adenoviral constructs expressing β-galactosidase (Ad-β-gal) or human influenza hemagglutinin (HA) epitope tagged CryABR120G (Ad-CryABR120G-HA) were previously described [21]. Ad-TFEB was custom made to harbor the expression cassette of human TFEB cDNA (Addgene plasmid # 38119) [24]. Post-infection cells were maintained in 2% FBS, 1% penicillin/streptomycin in DMEM until harvested or fixed.
2.4 Total protein extraction and western blot analysis
Proteins were extracted from ventricular myocardium or cultured NRVMs with 1× sampling buffer (41 mM Tris-HCl, 1.2% SDS, 8% glycerol). The determination of protein concentration used bicinchoninic acid (BCA) reagents (Pierce biotechnology, Rockford, IL). Equal amounts of protein samples were subject to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to PVDF membrane using a Trans-blot apparatus (Bio-Rad, Hercules, CA). The membranes were blocked with 5% nonfat dry milk in phosphate buffered saline (PBS) containing 0.1% Tween-20 (PBS-T) for 1 hour at room temperature before being incubated with the primary antibodies overnight at 4°C. The following primary antibodies were used: anti-ubiquitin (#3933, Cell Signaling; 1:1000), anti-GAPDH (G8795, Sigma-Aldrich; 1:1000), anti-LC3 (#2775, Cell Signaling; 1:1000), anti-HA (#3724, Cell Signaling; 1:1000), anti-mTOR (#2983, Cell Signaling; 1:1000), anti-Phospho-mTOR (Ser2481) (#2974, Cell Signaling; 1:1000), anti-p70 S6 kinase (#2708, Cell Signaling; 1:1000), anti-Phospho-p70 S6 kinase (Thr389) (#9234, Cell Signaling; 1:1000), anti-4E-BP1 (#9644, Cell Signaling; 1:1000), anti-Phospho-4E-BP1 (Thr37/46) (#2855, Cell Signaling; 1:1000), and anti-TFEB (A303-673A, BETHYL; 1:10000). The corresponding horseradish peroxidase-conjugated goat anti-mouse, goat anti-rabbit, or goat anti-guinea secondary antibodies (Santa Cruz Biotechnology) were used respectively for chemiluminescence-based western blot analyses. The bound secondary antibody signals were as detected using either enhanced chemiluminescence (ECL-Plus) reagents (GE Healthcare, Piscataway, NJ) or, for weak signals, ECL Advance Western Blotting Kit (GE Healthcare) and digitalized with a VersaDoc3000 or ChemiDoc MP imaging system (Bio-Rad). The digital signal was quantified with the Quantity One or Image Lab™ software (Bio-Rad). For some of the western blot analyses, the in-lane total protein content derived from the stain-free protein imaging technology was used for loading normalization [25].
2.5 Preparation of soluble and insoluble fractions of myocardial proteins
Frozen ventricular tissues were homogenized in cold phosphate-buffered saline (PBS) at pH 7.4 containing 2% Triton-X100, 2.5mM EDTA, 0.5mM PMSF, and a cocktail of complete protease inhibitors and then incubated on ice for 30 min, vortexed 30 seconds every 5 minutes. The tissue homogenates were centrifuged at 12,000×g for 30 min and the supernatant was collected as the soluble fraction. The pellet was resuspended in 1× loading buffer (50mM Tris-HCl at pH 6.8 containing 2% SDS, 10% glycerol, and a complete protease inhibitor cocktail), sonicated on ice, and boiled for 10 minutes before centrifugation at 12,000×g for 20 minutes at 4°C; the resultant supernatant was obtained as the insoluble fraction of myocardial proteins.
2.6 Immunofluorescence labeling and confocal microscopy
NRVMs cultured in 8-well chamber slides were fixed with 4% of paraformaldehyde for 20 minutes at room temperature and washed thrice for 5 min with PBS, and blocked with 0.5% BSA for 1 hour. The specimens were then incubated with primary antibodies overnight at 4°C. The primary antibody dilutions were as follows: anti-TFEB (A303-673A, BETHYL; 1:200), and anti-HA (C29F4, cell signaling; 1:1000). Subsequently, appropriate Alexa Fluor®-488, -568 or -647 conjugated secondary antibodies (Invitrogen) were applied to detect the bound primary antibodies. Alexa Fluor®568-conjugated phalloidin (Invitrogen) was used to identify cardiomyocytes. DAPI (Sigma-Aldrich) was used to stain nuclei. The fluorescence labeling was visualized and imaged using a confocal microscope (Olympus Fluoview 500, Center Valley, PA).
2.7 siRNA transfection
The small interference RNA (siRNA) specific for rat TFEB (siTFEB: 5’-ACAGUCCCAUGGC CAUGCUACACAU-3’) and the siRNA targeting luciferase serving as a control siRNA (siLuc: 5’-AACGTACGCGGAATACTTCGA-3’) were purchased from Invitrogen (Cat. #: RSS337387). For the transient knockdown of target genes, 2×106 cells were plated in 60-mm dishes, and transfection of cultured NRVMs with siRNA was generally started at 48–72 hours after myocytes were plated. Lipofectamine 2000 transfection reagent (Invitrogen) was used for siRNA transfection following the manufacturer’s protocol. In knock down experiments, the same amounts of luciferase siRNA and TFEB siRNA were applied to the control and experimental groups, respectively. Four hours after the transfection, the siRNA-containing medium was replaced with the fresh medium containing 10% FBS. Two consecutive rounds of transfection of TFEB siRNA at a dose of 200 pmol for 2×106 cells with an interval of 72 hours were performed. Three days after the second TFEB siRNA transfection, the follow-up assessments were performed.
2.8 LDH assa
Lactate dehydrogenase (LDH) activity in the collected medium was measured using a Cytotoxicity Detection Kit (Roche, Indianapolis, IN) following the manufacturer’s protocols. Briefly, both 100 μl of culture medium and 100 μl of reaction reagent were added to each well of a 96-well plate, and the mixture was incubated for 30 minutes at 25°C. Absorbance of the samples at 490 nm was read on a Perkin Elmer VICTOR X3 multimode plate reader. LDH release of an experimental group was presented as a percentage of the mean LDH activity of the corresponding control group [19].
2.9 MTT assay
The MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay, an index of cell viability and cell growth, was performed as described [19]. Briefly, 1.0×106 NRVMs were plated in the 6-well plate. After the experimental treatments were performed, MTT dye (Sigma, 500 μg/mL) was added to each well of the plate, and the plates were incubated at 37°C for 4 hours. At the end of the incubation, the dye solution was completely removed, 400 μl solvent solution (1 volume of 1N HCl in 9 volume of anhydrous isopropanol) was added to each well, and the absorbance was determined at 570 nm in a Perkin Elmer VICTOR X3 multimode plate reader. Cell viability of the experimental group was determined as a percentage of the reading of the control group.
2.10 Reverse transcription-polymerase chain reaction (RT-PCR) analysis
Total RNA was isolated from cultured cardiomyocytes or ventricular myocardium tissue using the TriReagent (Molecular Research Center, Inc., Cincinnati, CA). The concentration and quality of the isolated RNA was determined using Agilent RNA 6000 Nano assay (Agilent technologies, Inc., Germany). For reverse transcription reaction, 1μg of total RNA was used as the template to generate cDNA using the SuperScript III First-Strand Synthesis kit (Invitrogen) according to the manufacturer’s instructions. For PCR amplification, 1 μl of the first-strand solution resulting from the reverse transcription reaction and specific primers towards the gene of interest were used. The transcript levels of TFEB, CryAB, and representative TFEB target genes were assessed with PCR at the minimum number of cycles that can detect the PCR products. Sequences of the specific primers are presented in Supplementary Table 1. GAPDH was probed as a house-keeping gene control and its level was utilized to normalize the PCR product levels of other genes for comparison of the relative expression levels among different groups.
2.11 The LC3-II flux assay
The cultured cells were treated with bafilomycin-A1 (BFA1, LC Laboratories; 6nM) or vehicle control (DMSO) for 24 hours before they were harvested for extraction of total proteins with the 1× sampling buffer. Western blot analyses for LC3-II levels were performed as previously described [26]. The LC3-II flux presented here refers to the net amount of LC3-II accumulated by the BFA-mediated lysosomal inhibition. Mathematically, it is calculated by subtracting the GAPDH normalized LC3-II level of a BFA-treated sample with the mean value of the GAPDH normalized LC3-II levels of the DMSO treated samples of the same group.
2.12 Statistical analysis
All continuous variables are presented as mean±SEM unless indicated otherwise. Differences between two groups were evaluated for statistical significance with a 2-tailed unpaired t-test. When the difference among ≥3 groups was evaluated, 1-way ANOVA or, when appropriate, 2-way ANOVA followed by the Tukey test for pairwise comparisons was performed. A p value < 0.05 is considered statistically significant.
3. RESULTS
3.1 Myocardial TFEB signaling is suppressed in advanced cardiac proteinopathy
Prior studies have demonstrated that ALP insufficiency plays an important role in cardiac pathogenesis, as exemplified by cardiac proteinopathy [6]. Since TFEB has emerged as a central regulator of the ALP, we wish to determine whether myocardial TFEB signaling is altered in cardiac proteinopathy; hence, we examined myocardial expression of TFEB and its target genes in a well-established mouse model of cardiac proteinopathy induced by stable cardiomyocyte-restricted expression of CryABR120G which generates a bona fide misfolded protein and is linked to human desmin-related cardiomyopathy [22, 27], the most studied cardiac proteinopathy. We tested Ntg, CryABWT tg, and CryABR120G tg mice at 6 months of age when advanced cardiomyopathy has been detected in this CryABR120G stable tg line by prior characterization and was verified in the present study (Supplementary Tables 2 and 3). Our western blot analyses for mouse myocardial TFEB proteins revealed two main species falling in between the molecular weight makers 75 and 50 kDa, consistent with a prior report using the same antibodies [28]. Their apparent molecular weights are approximately ~60 and 53 kDa, respectively (Figure 1A) which match pretty well the calculated molecular weight of TFEB isoform a (TFEBa, 59295Da; NCBI Reference Sequence: NP_035679.3) and isoform b (TFEBb, 52483Da; NCBI Reference Sequence: NP_001155195.1), respectively. Two similar TFEB protein species were also detected in cultured NRVMs where both TFEB proteins were markedly decreased by TFEB-specific siRNA mediated knockdown (Supplementary Figure 1), indicating that both bands belong to TFEB. Importantly, myocardial TFEBa was significantly decreased whereas TFEBb was markedly increased in the CryABR120G tg mice compared with Ntg littermates and CryABWT tg mice (Figure 1). Myocardial TFEB proteins were not discernibly altered by a comparable tg overexpression of wild type CryAB (Figure 1A~1C), indicating that the changes in myocardial TFEB proteins are specific to the CryABR120G-based proteinopathy.
Figure 1. Changes in myocardial TFEB protein expression in CryABR120G tg mice.

(A~C) Western blot analyses for TFEB in total myocardial proteins. Ventricular myocardium from 6-month-old CryABR120G tg, CryABWT tg, and their non-tg (NTG) littermate mice was used for extraction of total proteins. Representative images (A) and pooled densitometry data (B, C) are shown. GAPDH was probed for loading controls. (D~F) Changes in myocardial soluble and insoluble TFEB proteins in CryABR120G tg and Ntg littermate mice at 6 months. Triton X-100 soluble and insoluble fractions of ventricular myocardial proteins were subject to western blot analyses for TFEB. Representative images (D) and poled densitometry data (E, F) are shown. A longer exposure (long exp.) is included (middle image of D) to better show the bands in the soluble fraction. In-lane total protein contents derived from the stain-free protein imaging technology (bottom image of D) were used for loading normalization. *p<0.05, **p<0.01, ***p<0.005 vs. NTG; n=3 mice/group.
To decipher the bioavailability of the increased TFEB proteins in the CryABR120G tg hearts, we also assessed the distribution of TFEB proteins in the 2% Triton X-100 soluble and insoluble fractions of myocardium. Interestingly, TFEBa is present primarily in the soluble fraction while TFEBb is mostly present in the insoluble fraction in the Ntg hearts (Figure 1D). TFEBa is not detected in the insoluble fraction. Compared with the Ntg group, soluble TFEBb was increased while TFEBa was decreased in CryABR120G tg hearts; an increase of TFEBb was also observed in the insoluble fraction of CryABR120G tg mice (Figure 1D~1F).
Using a pair of primers capable of detecting all three variants of murine TFEB transcripts, our RT-PCR analyses showed that myocardial TFEB mRNA levels were moderately but statistically significantly increased in the CryABR120G tg mice compared with NTG littermates (Figure 2B, 2C). However, this increased TFEB mRNA and TFEBb protein expression were not translated into an increased TFEB activity because the steady state mRNA levels of all examined TFEB target genes including Mcoln1, M6pr, Vps18 and Uvrag were significantly and uniformly down-regulated in the CryABR120G tg mouse hearts (Figure 2B, 2C), indicating that TFEB activation is suppressed in the heart with advanced cardiac proteinopathy. Phosphorylation of TFEB by mTORC1 (mechanistic target of rapamycin complex 1) is a mechanism that sequesters TFEB in the cytoplasm and thereby suppresses TFEB activation [4]. To explore the underlying cause of suppressed TFEB signaling in the proteinopathic hearts, we assessed myocardial mTORC1 activity. The total and the Ser-2481phosphorylated mTOR proteins were significantly increased in CryABR120G tg mice at 6 months of age compared with Ntg littermates (Figure 2D, 2E; Supplementary Figure 2), indicative of increased mTOR activation in the tg hearts. The p70 S6 kinase and 4E-BP1 are two bona fide targets of mTORC1. Both Thr389-phosphorylated p70 S6 kinase and Thr37/46-phosphorylated 4E-BP1 were significantly increased (Figure 2D, 2E), confirming increased activation of mTORC1 in the CryABR120G tg hearts. Thus, changes in both the activity of a major upstream suppressive kinase mTORC1 and the expression of downstream target genes consistently demonstrate that myocardial TFEB signaling is inhibited in mice with advanced cardiac proteinopathy.
Figure 2. Changes of myocardial TFEB and mTORC1 activities in mice with advanced cardiac proteinopathy.

(A) A schematic illustration of the TFEB signaling pathway. (B, C) Semi-quantitative RT-PCR analyses for myocardial mRNA levels of TFEB and its representative target genes in CryABR120G tg and NTG mice. (D, E) Western blot analyses for representative proteins of the mTORC1 signaling pathway. The soluble protein extracts of ventricular myocardial samples from 6-month-old CryABR120G tg (R120G) and Ntg littermate mice were subject to SDS-PAGE and western blot analyses for total mTOR and phosphorylated mTOR (p-mTOR), total p70 S6 kinase (p70 S6K) and phosphorylated p70 S6K (p-p70 S6K), total 4E-BP1 and phosphorylated 4E-BP1 (p-4E-BP1). Shown are the representative images (D) and pooled densitometry data (E). The in-lane total protein content derived from the stain-free protein imaging technology (see Supplementary Figure 2 for the images) was used for loading normalization. **p<0.005 vs. NTG.
These in vivo findings prompted us to test the effects of genetic manipulation of the Tfeb gene in cardiomyocytes on ALP activity and the cardiac proteotoxicity induced by CryABR120G using a primary cardiomyocyte culture system. Similar to what observed in mouse myocardium, western blot analyses for TFEB in cultured NRVMs also detected two major bands in between molecular weight makers 75kDa and 50kDa (Supplementary Figures 1A, 3A), with the upper band present primarily in the soluble fraction and the lower band present predominantly in the insoluble fraction (Supplementary Figure 3C, 3D). Compared with infection of Ad-β-gal, adenovirus- mediated overexpression of CryABR120G did not significantly alter the level of TFEB proteins in the cardiomyocytes at either 24 or 48 hours after adenoviral gene delivery (Supplementary Figure 3), implicating that the altered myocardial expression of TFEB proteins in mice with CryABR120G-based advanced proteinopathy (Figure 1) likely results from long term effects of misfolded protein expression and that an increased inhibitory regulation from, for example, increased activation of mTORC1 (Figure 2) likely plays a critical role in suppressing TFEB signaling in the proteinopathic hearts.
3.2 Downregulation of TFEB decreases autophagic flux in cardiomyocytes
To test directly the impact of suppressed TFEB signaling on cardiac autophagic activity, we examined changes in the LC3-II flux in cultured NRVMs subject to siRNA-mediated TFEB downregulation. The efficacy of transfection of TFEB-specific siRNA (siTFEB) to reduce TFEB protein expression was confirmed by western blot analyses for TFEB. Both the upper and lower bands of TFEB in cultured NRVMs were significantly decreased by siTFEB transfection (Supplementary Figure 1). The protein level of LC3-II in a cell reflects generally the abundance of autophagosomes in the cell and the rate of LC3-II degradation (i.e., LC3-II flux) serves as a very good indicator of the rate of autophagosome degradation by lysosomes (i.e., autophagic flux) [29]. We found that the LC3-II flux was significantly less in the NRVMs transfected with siTFEB than in those transfected with the control siRNA (siLuc) both at baseline and during CryABR120G overexpression (Figure 3), indicating that autophagic flux is reduced in cardiomyocytes with TFEB downregulation. Immunofluorescence confocal microscopy revealed that the same Ad-HA-CryABR120G infection led to more abundant aberrant protein aggregates in siTFEB transfected NRVMs than in siLuc transfected NRVMs (Supplementary Figure 4), indicative of impairment of autophagic removal of aggregated misfolded proteins by suppressing TFEB signaling.
Figure 3. TFEB knockdown decreases the autophagic flux in cultured neonatal rat ventricular myocytes (NRVMs).

(A~C) Effect of siRNA-mediated TFEB knockdown on the LC3-II flux in NRVMs at baseline. At 48 hours after transfection of siRNA specific for TFEB (siTFEB) or siRNA against luciferase (siLuc), the cells were treated with a lysosomal inhibitor bafilomycin A1 (BFA, 6 nM) or vehicle control (DMSO) and harvested 24 hours later for extraction of total proteins. The extracted proteins were used for western blot analyses for LC3. GAPDH was probed as a loading control. Shown are the representative images (A), a summary of LC3-II densitometry data (B), and the LC3-II flux (C) derived from the data presented in B. The LC3-II flux is the net amount of LC3-II accumulated by BFA treatment and calculated as described in Methods. (D~F) Effect of siRNA-mediated TFEB knockdown on the LC3-II flux in NRVMs overexpressing CryABR120G. NRVMs were infected with Ad-HA-CryABR120G at 48 hours after siTFEB or siLuc. Twenty-four hours later, the cells were treated with BFA or DMSO for 24 hours before they were harvested and subject to the same analyses as described for panels A~C. *p<0.05, **p<0.01 vs. the siLuc+DMSO group (B) or the R120G+siLuc+DMSO group (E).
3.3 TFEB overexpression protects against CryABR120G cytotoxicity in cardiomyocytes
Forced expression of TFEB has been shown to enhance the removal of neurodegenerative disease linked aggregation-prone proteins in neurons but it remains untested whether TFEB overexpression promotes degradation of misfolded proteins in cardiomyocytes. Therefore, we sought to determine the effect of TFEB overexpression on the expression, distribution, and cytotoxicity of CryABR120G in cultured NRVMs. To deliver the TFEB gene into cultured NRVMs, we created a replication-deficient recombinant adenovirus harboring the cDNA expression cassette encoding the human TFEB isoform 1 (Ad-TFEB) which is homologous to both mouse and rat TFEBa. Infection of the Ad-TFEB resulted in an overexpression of a TFEB protein whose apparent molecular weight is close to the upper band of endogenous TFEB (Supplementary Figure 5). More importantly, TFEB overexpression did not discernibly alter the steady state mRNA levels of CryABR120G (Supplementary Figure 6) but resulted in striking decreases in CryABR120G-positive aggregates (Figure 4), in CryABR120G protein levels, and in total ubiquitin conjugates in cardiomyocytes overexpressing CryABR120G (Figure 5), demonstrating an essential role for TFEB in enhancing the removal of aberrant protein aggregates derived from CryABR120G, a bona fide misfolded protein known to cause human proteinopathy.
Figure 4. TFEB overexpression reduces CryABR120G aggregates in NRVMs.

Infection of Ad-TFEB or Ad-β-gal was administered 24 hours after NRVMs were plated; 24 hours later, Ad-HA-CryABR120G infection was applied to the indicated groups; and further 48 hours later, the cells were fixed in 4% paraformaldehyde and used for indirect immunofluorescence labeling for the HA epitope (green). The nuclei were stained with DAPI (blue) and cardiomyocytes were counterstained with Alexa Fluor 568 conjugated phalloidin (red). Representative fluorescence confocal micrographs are shown. Scale bar=40 μm.
Figure 5. TFEB overexpression attenuates the accumulation of CryABR120G proteins and ubiquitin conjugates (Ub-proteins) in cardiomyocytes overexpressing CryABR120G.

NRVMs culture and adenoviral gene delivery were performed as described in Figure 4. The cells were harvested 48 hours after Ad-HA-CryABR120G infection (R120G) and the total cellular proteins were used for western blot analyses of the indicated proteins. GAPDH was probed as loading control. (A) Representative western blot images. (B) A summary of the densitometry data of the HA-tagged CryABR120G protein. (C) A summary of the densitometry data of Ub-proteins. *p<0.05, **p<0.01 vs. the β-gal group; n=3 biological repeats/group.
In agreement with prior reports, overexpression of CryABR120G caused cardiomyocyte injury and cell death as reflected by increased caspase 3 cleavage (Figure 6A, 6B), LDH leakage (Figure 6C), reduced cell viability revealed by the MTT assay (Figure 6D), and increased LDH/MTT ratio (Figure 6E). The rationale for inclusion of the LDH/MTT ratio is to eliminate the impact of a potential variation of total cardiomyocyte number on the amount of LDH leakage. All the indices of cell injury and cell death were blocked or markedly attenuated by TFEB overexpression (Figure 6).
Figure 6. Effect of TFEB overexpression on CryABR120G-induced cytotoxicity.

NRVMs were cultured and treated as described in Figure 4. (A, B) Western blot analyses for the indicated proteins. Shown are the representative images (A) and a summary of cleaved caspase 3 densitometry data (B). (C~E) The culture media and the NRVMs of a dish were simultaneously collected for assessing LDH activity in the media (C) and MTT assays of the cells (D), respectively. The LDH/MTT ratio (E) is calculated to minimize the impact of variation resulting from potential difference in the total cell number in a dish on LDH leakage. NS, not significant, **p<0.01 vs. the β-gal group; n=6 biological repeats/group. Casp 3, caspase 3; C-Casp 3, cleaved caspase 3.
3.4 TFEB overexpression increases autophagic flux in cardiomyocytes
To verify whether TFEB signaling pathway is activated by TFEB overexpression in cultured cardiomyocytes, we assessed the transcript levels of representative TFEB target genes, including genes for lysosomal biogenesis (Ctsb, Ctsd, Lamp1, M6pr, and Mcoln1; Figure 7A, 7C) as well as genes involved in autophagy (Uvrag, Vps18, Becn1, Rab7a, and Sqstm1; Figure 7B, 7D). All the examined genes showed a significant upregulation (Figure 7). Moreover, the LC3-II flux which is the rate of lysosomal degradation of LC3-II and a well-established indicator of autophagic flux, was assess by detecting the differential of LC3-II protein levels between the cells with lysosomal inhibition by BFA and those without. Overexpression of CryABR120G significantly reduced LC3-II flux in NRVMs (Supplementary Figure 7). TFEB overexpression significantly increased LC3-II flux in cultured NRVMs both at baseline (Figure 8A, 8B) and during CryABR120G overexpression (Figure 8C, 8D).
Figure 7. RT-PCR analysis of TFEB target genes induced by forced expression of TFEB.

Total RNAs extracted from NRVMs at 24 hours after infection of Ad-TFEB or Ad-β-gal were subject to semi-quantitative RT-PCR. Representative PCR images (A, B) and summaries of densitometry data (C, D) are shown. (A, C) Changes in the indicated lysosomal genes. (B, D) Changes in the indicated autophagic genes. **p<0.01 vs. the Ad-β-gal group, n=3 biological repeats/group.
Figure 8. TFEB overexpression increases the autophagic flux in NRVMs.
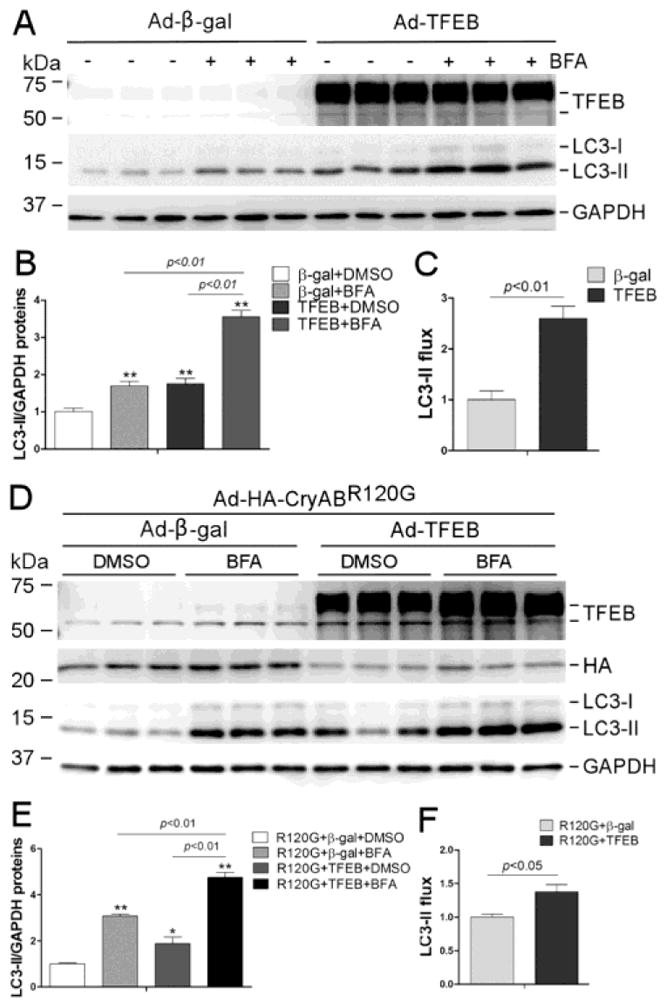
(A~C) Effect of TFEB overexpression on the LC3-II flux at baseline. At 48 hours after infection of Ad-TFEB or Ad-β-gal, the cells were treated with BFA (6 nM) or DMSO for 24 hours before harvest. Shown are the images of western blot analyses for the indicated proteins (A), pooled LC3-II densitometry data (B), and the LC3-II flux (C). (D~F) Effect of forced TFEB overexpression on the LC3-II flux in NRVMs overexpressing CryABR120G. NRVMs were infected with Ad-HA-CryABR120G (R120G) at 24 hours after Ad-TFEB or Ad-β-gal infection. Twenty-four hours later, the cells were treated with BFA or DMSO for 24 hours before they were harvested and subject to the same analyses as described for panels A~C. *p<0.05, **p<0.01 vs. the β-gal+DMSO group (B) or the R120G+β-gal+DMSO group (E); n=3 biological repeats/group.
3.5 Protection of TFEB overexpression against proteotoxicity requires autophagy
3-Methyladenine (3-MA) is known to suppress autophagy at the autophagosome formation stage through inhibition of type III phosphatidylinositol 3-kinases (PI3Ks) and has been extensively used in autophagy studies [30]. Here we have verified that 3-MA is also capable of reducing LC3-II flux in cardiomyocytes with TFEB overexpression (Supplementary Figure 8). As depicted by Figure 9, the protective effects of TFEB overexpression against CryABR120G-based proteotoxicity (Figure 6) including the reduction of the steady state CryABR120G protein levels, caspase 3 cleavage, and LDH leakage and the improvement of cell viability as revealed by the MTT assay, were either significantly attenuated or completely obliterated by co-treating the cells with 3-MA. These findings demonstrate that protection against proteotoxicity by TFEB overexpression is at least in part through enhancing autophagy.
Figure 9. Protection of TFEB overexpression against proteotoxicity is autophagy-dependent.

NRVMs were cultured and treated as described in Figure 6 with the exception of adding a 3-MA (2.5mM) treatment cohort to determine the impact of autophagy inhibition on TFEB’s effects. (A~D) Representative images (A) of western blot analyses for the indicated proteins and the summaries of the detected changes in LC3-II (B), HA-CryABR120G (C), and cleaved-caspase 3 (D). (E~G) The culture media and the NRVMs were simultaneously collected for LDH and MTT assays, respectively. The LDH/MTT ratio is calculated to minimize the impact of the variation resulting from potential difference in the total cell number of the dish on LDH activity in the media. NS, not significant; *p<0.05, **p<0.01 vs. the β-gal group; n=6 biological repeats/group. Casp 3, caspase 3; C-casp 3, cleaved caspase 3.
4. DISCUSSION
In the present study we have discovered that TFEB signaling is suppressed in mouse hearts with advanced cardiac proteinopathy and we have demonstrated for the first time in cardiomyocytes that TFEB is required for sustaining ALP activity in cardiomyocytes and forced TFEB expression is sufficient to facilitate ALP activity and thereby protect against misfolded protein-induced cardiac proteotoxicity, suggesting that promoting TFEB signaling should be an effective strategy to treat heart disease with increased proteotoxic stress. These findings are highly significant because increased cardiac proteotoxicity is implicated in the pathogenesis of a large subset of human heart failure [31].
4.1 Decreased TFEB activity contributes to ALP insufficiency in cardiac proteinopathy
The abundance and functional status of lysosomes not only determine the efficiency and completeness of cargo degradation in both autophagy and heterophagy but also dictate the fusion between lysosomes and autophagosomes. Thus, lysosomal biogenesis and acidification as well as the replenishment of autophagic machinery are crucial to maintaining ALP sufficiency in the cell. In fact, the lysosome surface is recognized as a major signaling hub that integrates and relays external cues and internal signals to regulate the ALP and metabolism in the cell [32, 33]. Exciting recent advance has identified TFEB as not only a master regulator of lysosomal biogenesis but also a key regulator of autophagy [34]. TFEB phosphorylation status determines its subcellular localization and activity. mTORC1 is a major upstream kinase that phosphorylates TFEB at multiple critical serine residues, which enables the chaperone 14-3-3 to bind TFEB and thereby sequesters TFEB in the cytoplasm where TFEB can co-localize with mTOR at the surface of lysosomes [4]. When mTORC1 is inactivated, TFEB phosphorylation by mTORC1 stops and concomitantly phosphatase calcineurin is activated to dephosphorylate TFEB, in which calcineurin activation is triggered by the release of lysosomal Ca2+ via the Ca2+ channel mucolipin 1 (MCOLN1). The dephosphorylated TFEB can then enter the nucleus and activate the transcription of an entire network of genes that are essential to and orchestrate lysosomal biogenesis and autophagy [4]. Here we discovered that the myocardial mRNA levels of all examined representative TFEB target genes involved respectively in autophagy (Vps18 and Uvrag) and lysosomal biogenesis (M6pr and Mcoln1) were significantly decreased in the CryABR120G mice (Figure 2B, 2C). These results unveil for the first time that TFEB transactivation can be markedly suppressed in the heart with elevated proteotoxic stress, which is supported by previously reported downregulation of many other ALP genes in the heart of the same tg mouse model at a similar disease stage [6]. Since simple changes in neither myocardial TFEB mRNA levels nor TFEBb proteins (Figures 1, 2B and 2C) can explain the decreased TFEB activity, we explored for a posttranslational mechanism that could prevent TFEB from activation. Indeed, we detected that the activity of mTORC1, a key suppressive force of TFEB activation, was clearly increased in the proteinopathic hearts as reflected by increased phosphorylation in the bona fide targets (p70 S6 kinase and 4E-BP1) of mTORC1 as well as by the increases in the total and activated forms of mTOR (Figure 2D, 2E). Given that TFEB is a master regulator for the ALP and that ALP insufficiency as evident by accumulation protein aggregates and decreased autophagic flux has been described for, and plays an major pathogenic role in, this mouse model of cardiac proteinopathy [6, 34], our findings suggest that impaired TFEB signaling may contribute to the ALP impairment and the disease progression in cardiac proteinopathy. This postulate is indeed corroborated by our in vitro siRNA-mediated TFEB knockdown experiments which revealed that downregulation of TFEB is sufficient to decrease autophagic flux in cultured NRVMs both at baseline and during CryABR120G overexpression (Figures 3) and exacerbate the abundance of aberrant protein aggregates resulting from CryABR120G overexpression (Supplementary Figure 4).
Our findings show that TFEB protein isoforms a and b distribute differently in mouse myocardium (Figure 1D): TFEBa is present primarily in the soluble fraction while TFEBb is present in both the soluble and insoluble fractions but predominantly in the latter. Similar phenomena also were observed in cultured NRVMs (Supplementary Figure 3C, 3D). Moreover, we have discovered an isoform switch from TFEBa to TFEBb in proteinopathic myocardium (Figure 1). The physiological significance for the differential distribution of TFEB isoforms and for the proteinopathy- induced isoform switch are presently unclear and will be interesting and important future investigation. Nevertheless, we propose that the remarkable segregation of the TFEBb in the insoluble fraction (Figure 1D~1F) may hinder its activation, thereby contributing to the discrepancy between the increased TFEBb and decreased TFEB activity in the proteinopathic hearts. This is because TFEBs in the insoluble fraction are unlikely available for direct activation. Meanwhile, the association of decreased TFEB activity with the TFEB isoform switch from a to b in the proteinopathic hearts also raises a possibility that TFEBa may be more apt to be activated than TFEBb. Further studies are warranted to test these propositions.
4.2 TFEB gene delivery protects against cardiac proteotoxicity via ALP enhancement
Our findings from the CryABR120G tg hearts also prompted us to hypothesize that measures to correct the impaired TFEB signaling protect against cardiac proteinopathy. Hence, we sought to test the effects of forced expression of TFEB on cardiac proteotoxicity in a cell model of cardiac proteinopathy. As reported before [19, 35], expression of CryABR120G in NRVMs caused aberrant protein aggregation (Figure 4), accumulation of ubiquitinated proteins (Figure 5), activation of the apoptotic pathway as reflected by increases in cleaved caspase 3, and cardiomyocyte injury as evidenced by elevated LDH leakage from the cell to the culture media and concomitant decreased cell viability revealed by the MTT assays (Figure 6). All these pathological changes were significantly attenuated by forced expression of TFEB, which demonstrates for the first time in cardiomyocytes that increasing TFEB expression is sufficient to reduce the cytotoxicity of a human disease linked misfolded protein. Importantly, the protection by TFEB was associated with a remarkable decrease of CryABR120G total protein (Figure 5) but not transcript levels (Supplementary Figure 6), suggesting that TFEB promotes the degradation of CryABR120G proteins, consistent with TFEB acting as a master regulator of the ALP. Indeed, our further experimentation confirms that forced expression of TFEB significantly upregulated all examined representative target genes of TFEB in cultured NRVMs (Figure 7). The criteria used to choose these genes are two-folded; first, they are known TFEB target genes in other cell types as previously reported and second, they are well known to play an important role in either lysosomal biogenesis or autophagy. Among the representative lysosomal genes, cathepsin D (Ctsd) and Ctsb are major lysosomal proteases [36], Lamp1 and Mcoln1 are crucial lysosomal membrane proteins with the latter being the calcium channel even involved in TFEB activation [37, 38]; and M6pr (mannose-6-phosphate receptor) is essential for the targeting of lysosomal enzymes from the Golgi complex and cell membrane to the lysosome compartment during lysosomal biogenesis [39]. In terms of the representative autophagic genes, Becn1 (Beclin1) not only promotes the initiation of autophagosome formation but also, when complexing with UVRAG, regulates autophagosome maturation [4]; Vps18 belongs to the Class C VPS genes which mediate vesicle trafficking steps in the endosomal/lysosomal pathway [40]; Rab7a is a small G-protein required for autophagosome maturation while Sqstm1 (AKA p62) plays important role in targeting ubiquitinated cargos to autophagosomes [19, 41, 42]. In agreement with the notion that TFEB activation coordinately enhances both formation and removal of autophagosomes, we observed that forced expression of TFEB significantly increased LC3-II flux, a widely used indicator of autophagic flux, in NRVMs both at baseline and during CryABR120G overexpression (Figure 8).
This increase of autophagic flux by TFEB certainly plays an indispensable role in its protection against proteotoxicity because inhibition of autophagic activation with 3-MA, a class III PI3K inhibitor commonly used for autophagy inhibition, significantly attenuated all the protective effects of forced TFEB overexpression in the cultured NRVMs (Figure 9). These findings demonstrate that protection of forced TFEB expression against cytotoxicity of a bona fide misfolded protein in cardiomyocytes depends at least in part on enhancement of autophagy. Notably, the observed effect of TFEB manipulation on CryABR120G is not due to a property intrinsic to wild type CryAB protein. This is because a prior report showed that manipulating autophagy did not affect the turnover of either endogenous or overexpressed wild type CryAB proteins in cultured cardiomyocytes [43], indicating that the mutation is required to render CryABR120G sensitive to ALP degradation. More specifically, the misfolding nature of the mutation and resultant aggregation-prone feature of CryABR120G are responsible because improved clearance of other misfolded or aggregation-prone proteins by enhancing TFEB signaling have also been observed in other cell types, such as neurons [9, 11–16], hepatocytes, and lung epithelia [17, 18]. Hence, the protection of TFEB against proteotoxicity is common to both cardiomyocytes and non-cardiac cells.
CryABR120G-based cardiomyopathy or even bona fide cardiac proteinopathy is by no means a common form of heart disease; however, cardiac PQC inadequacy and associated increases in misfolded proteins and proteotoxic stress in cardiomyocytes are associated with a large subset of human end-stage heart failure [1, 3]. Therefore, the conclusions drawn from the present study have implication in a much broader spectrum of cardiac disorders than cardiac proteinopathy. To this end, experimental evidence is rapidly emerging that inadequate lysosomal removal of autophagosomes as reflected by the co-existence of elevated LC3-II with decreased LC3-II flux, is a shared phenomenon during the progression to heart failure from common forms of etiology such as ischemic heart disease [44], pressure overloaded cardiac hypertrophy [45], and diabetic cardiomyopathies [46]; hence, optimizing the ALP or the coordinate activation of both the formation and the removal of autophagosomes in the heart has been suggested as potential therapeutic strategy to treat these life-threatening conditions. With the exception of ATG7 overexpression and, to some extent, of Beclin1 overexpression [6, 47], means used to increase or optimize ALP flux, such as mTORC1 inhibitors (rapamycin or its homologs), AMPK activators (e.g., metformin), and even starvation and exercise, can hardly be ALP-specific because they all can elicit many other actions favorable or unfavorable to cardiomyocytes [4]. Given the importance of lysosomal degradation in intracellular quality control and the pivotal roles of TFEB in promoting lysosomal biogenesis and activity and in orchestrating the ALP, enhancing cardiac TFEB activity represents an attractive strategy for therapeutic intervention of many cardiac disorders that are associated with lysosomal or ALP insufficiency and with the accumulation of aberrant aggregates. This is directly supported by the present study and also has been implicated by several reports from the cardiac field. For instance, cardiac glucolipotoxicity in obesity and diabetes has been linked to diminished TFEB and autophagic flux in cardiomyocytes [48]. Disrupting the ALP through impairing lysosome acidification and repressing TFEB signaling was recently reported to contribute to doxorubicin cardiotoxicity [49, 50]. TFEB-mediated transactivation of ALP machinery was proposed to mediate the protection by intermittent fasting against myocardial ischemia-reperfusion injury [28]. The activation of monoamine oxidase-A (MAO-A) can generate robust reactive oxygen species and thereby damage mitochondria. Cardiac tg overexpression of MAO-A led to cardiac autophagosome clearance impairment and heart failure in mice [51]. In cultured cardiomyocytes, forced expression of MAO-A blocked the nuclear translocation of TFEB and suppressed autophagic flux, resulting in cell necrosis. Most importantly, all these pathologies induced by MAO-A activation were significantly attenuated by TFEB overexpression [51], indicating that TFEB activation can protect against oxidative stress in cardiomyocytes both in vitro and in vivo. Cardiac progenitor cells from failing human hearts, compared with those from healthy donors, display impaired autophagy in cultures. This impairment was recently found to associate with depressed TFEB signaling [52]. Elderly patients are generally more vulnerable to sepsis than younger patients. By comparing young and aged mice, a recent report showed that more pronounced endotoxin-induced cardiac injury was observed in aged mice and this was associated with more severe impairment of TFEB-mediated autophagy in the aged mice, compared with the young mice [53].
4.3 Conclusions
The present study has identified impaired TFEB signaling in a bona fide mouse model of advance cardiac proteinopathy and demonstrated in cultured cardiomyocytes that reduction of TFEB decreases autophagic flux and forced TFEB expression is sufficient to increase autophagic flux and thereby ameliorates proteotoxicity resulting from overexpression of a bona fide misfolded protein linked to human proteinopathy. Future studies to define mechanisms underlying the suppression of TFEB signaling in cardiac proteinopathy and to test the in vivo effect of cardiac overexpression of TFEB are warranted.
Supplementary Material
Highlights.
Myocardial TFEB signaling is suppressed in CryABR120G-based proteinopathy
TFEB knockdown suppresses autophagic flux in cultured cardiomyocytes
TFEB overexpression elevates autophagic flux in cultured cardiomyocytes
TFEB facilitates autophagic removal of a bona fide misfolded protein
Protection of TFEB against cardiac proteotoxicity depends on autophagy
Acknowledgments
5. FUNDING SOURCES:
This work was supported in part by the National Institutes of Health (HL072166, HL085629 and HL131667) and by a National Natural Science Foundation of China (81570278-H0203).
Footnotes
6. DISCLOSURES
None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sanbe A, Osinska H, Saffitz JE, Glabe CG, Kayed R, Maloyan A, et al. Desmin-related cardiomyopathy in transgenic mice: a cardiac amyloidosis. Proc Natl Acad Sci U S A. 2004;101:10132–6. doi: 10.1073/pnas.0401900101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weekes J, Morrison K, Mullen A, Wait R, Barton P, Dunn MJ. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. 2003;3:208–16. doi: 10.1002/pmic.200390029. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Robbins J. Proteasomal and lysosomal protein degradation and heart disease. J Mol Cell Cardiol. 2014;71:16–24. doi: 10.1016/j.yjmcc.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Cui T. Autophagy modulation: a potential therapeutic approach in cardiac hypertrophy. Am J Physiol Heart Circ Physiol. 2017;313:H304–H19. doi: 10.1152/ajpheart.00145.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res. 2014;24:42–57. doi: 10.1038/cr.2013.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhuiyan MS, Pattison JS, Osinska H, James J, Gulick J, McLendon PM, et al. Enhanced autophagy ameliorates cardiac proteinopathy. J Clin Invest. 2013;123:5284–97. doi: 10.1172/JCI70877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steingrimsson E, Copeland NG, Jenkins NA. Melanocytes and the microphthalmia transcription factor network. Annu Rev Genet. 2004;38:365–411. doi: 10.1146/annurev.genet.38.072902.092717. [DOI] [PubMed] [Google Scholar]
- 8.Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, et al. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet. 2011;20:3852–66. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 9.Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, et al. A gene network regulating lysosomal biogenesis and function. Science. 2009;325:473–7. doi: 10.1126/science.1174447. [DOI] [PubMed] [Google Scholar]
- 10.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, et al. TFEB links autophagy to lysosomal biogenesis. Science. 2011;332:1429–33. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, et al. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci. 2010;30:12535–44. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A. 2013;110:E1817–26. doi: 10.1073/pnas.1305623110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RA, et al. PGC-1alpha rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, et al. Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J Neurosci. 2014;34:9607–20. doi: 10.1523/JNEUROSCI.3788-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, et al. Neuronal-Targeted TFEB Accelerates Lysosomal Degradation of APP, Reducing Abeta Generation and Amyloid Plaque Pathogenesis. J Neurosci. 2015;35:12137–51. doi: 10.1523/JNEUROSCI.0705-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y, et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med. 2014;6:1142–60. doi: 10.15252/emmm.201303671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pastore N, Blomenkamp K, Annunziata F, Piccolo P, Mithbaokar P, Maria Sepe R, et al. Gene transfer of master autophagy regulator TFEB results in clearance of toxic protein and correction of hepatic disease in alpha-1-anti-trypsin deficiency. EMBO Mol Med. 2013;5:397–412. doi: 10.1002/emmm.201202046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hidvegi T, Stolz DB, Alcorn JF, Yousem SA, Wang J, Leme AS, et al. Enhancing Autophagy with Drugs or Lung-directed Gene Therapy Reverses the Pathological Effects of Respiratory Epithelial Cell Proteinopathy. J Biol Chem. 2015;290:29742–57. doi: 10.1074/jbc.M115.691253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res. 2011;109:296–308. doi: 10.1161/CIRCRESAHA.111.244707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li J, Horak KM, Su H, Sanbe A, Robbins J, Wang X. Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest. 2011;121:3689–700. doi: 10.1172/JCI45709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Q, Liu JB, Horak KM, Zheng H, Kumarapeli AR, Li J, et al. Intrasarcoplasmic amyloidosis impairs proteolytic function of proteasomes in cardiomyocytes by compromising substrate uptake. Circ Res. 2005;97:1018–26. doi: 10.1161/01.RES.0000189262.92896.0b. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, Osinska H, Klevitsky R, Gerdes AM, Nieman M, Lorenz J, et al. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 23.Dong X, Liu J, Zheng H, Glasford JW, Huang W, Chen QH, et al. In situ dynamically monitoring the proteolytic function of the ubiquitin-proteasome system in cultured cardiac myocytes. Am J Physiol Heart Circ Physiol. 2004;287:H1417–25. doi: 10.1152/ajpheart.01233.2003. [DOI] [PubMed] [Google Scholar]
- 24.Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012;5:ra42. doi: 10.1126/scisignal.2002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gilda JE, Gomes AV. Stain-Free total protein staining is a superior loading control to beta-actin for Western blots. Anal Biochem. 2013;440:186–8. doi: 10.1016/j.ab.2013.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su H, Li J, Osinska H, Li F, Robbins J, Liu J, et al. The COP9 signalosome is required for autophagy, proteasome-mediated proteolysis, and cardiomyocyte survival in adult mice. Circ Heart Fail. 2013;6:1049–57. doi: 10.1161/CIRCHEARTFAILURE.113.000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vicart P, Caron A, Guicheney P, Li Z, Prevost MC, Faure A, et al. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–5. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- 28.Godar RJ, Ma X, Liu H, Murphy JT, Weinheimer CJ, Kovacs A, et al. Repetitive stimulation of autophagy- lysosome machinery by intermittent fasting preconditions the myocardium to ischemia-reperfusion injury. Autophagy. 2015;11:1537–60. doi: 10.1080/15548627.2015.1063768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gottlieb RA, Andres AM, Sin J, Taylor DP. Untangling autophagy measurements: all fluxed up. Circ Res. 2015;116:504–14. doi: 10.1161/CIRCRESAHA.116.303787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seglen PO, Gordon PB. 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc Natl Acad Sci U S A. 1982;79:1889–92. doi: 10.1073/pnas.79.6.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Pattison JS, Su H. Posttranslational modification and quality control. Circ Res. 2013;112:367–81. doi: 10.1161/CIRCRESAHA.112.268706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shen HM, Mizushima N. At the end of the autophagic road: an emerging understanding of lysosomal functions in autophagy. Trends Biochem Sci. 2014;39:61–71. doi: 10.1016/j.tibs.2013.12.001. [DOI] [PubMed] [Google Scholar]
- 33.Mony VK, Benjamin S, O'Rourke EJ. A lysosome-centered view of nutrient homeostasis. Autophagy. 2016;12:619–31. doi: 10.1080/15548627.2016.1147671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129:2475–81. doi: 10.1242/jcs.146365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ranek MJ, Terpstra EJ, Li J, Kass DA, Wang X. Protein kinase g positively regulates proteasome-mediated degradation of misfolded proteins. Circulation. 2013;128:365–76. doi: 10.1161/CIRCULATIONAHA.113.001971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu P, Yuan X, Li F, Zhang J, Zhu W, Wei M, et al. Myocardial upregulation of cathepsin D by ischemic heart disease promotes autophagic flux and protects against cardiac remodeling and heart failure. Circ Heart Fail. 2017;10 doi: 10.1161/CIRCHEARTFAILURE.117.004044. pii: e004044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288–99. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sancak Y, Bar-Peled L, Zoncu R, Markhard AL, Nada S, Sabatini DM. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell. 2010;141:290–303. doi: 10.1016/j.cell.2010.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matovcik LM, Goodhouse J, Farquhar MG. The recycling itinerary of the 46 kDa mannose 6-phosphate receptor--Golgi to late endosomes--coincides with that of the 215 kDa M6PR. Eur J Cell Biol. 1990;53:203–11. [PubMed] [Google Scholar]
- 40.Kim BY, Kramer H, Yamamoto A, Kominami E, Kohsaka S, Akazawa C. Molecular characterization of mammalian homologues of class C Vps proteins that interact with syntaxin-7. J Biol Chem. 2001;276:29393–402. doi: 10.1074/jbc.M101778200. [DOI] [PubMed] [Google Scholar]
- 41.Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–97. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 42.Su H, Li F, Ranek MJ, Wei N, Wang X. COP9 signalosome regulates autophagosome maturation. Circulation. 2011;124:2117–28. doi: 10.1161/CIRCULATIONAHA.111.048934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pattison JS, Osinska H, Robbins J. Atg7 induces basal autophagy and rescues autophagic deficiency in CryABR120G cardiomyocytes. Circ Res. 2011;109:151–60. doi: 10.1161/CIRCRESAHA.110.237339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schiattarella GG, Hill JA. Therapeutic targeting of autophagy in cardiovascular disease. J Mol Cell Cardiol. 2016;95:86–93. doi: 10.1016/j.yjmcc.2015.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin Q, Qu C, Niu T, Zang H, Qi L, Lyu L, et al. Nrf2-Mediated Cardiac Maladaptive Remodeling and Dysfunction in a Setting of Autophagy Insufficiency. Hypertension. 2016;67:107–17. doi: 10.1161/HYPERTENSIONAHA.115.06062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sciarretta S, Boppana VS, Umapathi M, Frati G, Sadoshima J. Boosting autophagy in the diabetic heart: a translational perspective. Cardiovasc Diagn Ther. 2015;5:394–402. doi: 10.3978/j.issn.2223-3652.2015.07.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117:1782–93. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trivedi PC, Bartlett JJ, Perez LJ, Brunt KR, Legare JF, Hassan A, et al. Glucolipotoxicity diminishes cardiomyocyte TFEB and inhibits lysosomal autophagy during obesity and diabetes. Biochim Biophys Acta. 2016;1861:1893–910. doi: 10.1016/j.bbalip.2016.09.004. [DOI] [PubMed] [Google Scholar]
- 49.Bartlett JJ, Trivedi PC, Yeung P, Kienesberger PC, Pulinilkunnil T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem J. 2016;473:3769–89. doi: 10.1042/BCJ20160385. [DOI] [PubMed] [Google Scholar]
- 50.Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, et al. Doxorubicin Blocks Cardiomyocyte Autophagic Flux by Inhibiting Lysosome Acidification. Circulation. 2016;133:1668–87. doi: 10.1161/CIRCULATIONAHA.115.017443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santin Y, Sicard P, Vigneron F, Guilbeau-Frugier C, Dutaur M, Lairez O, et al. Oxidative Stress by Monoamine Oxidase-A Impairs Transcription Factor EB Activation and Autophagosome Clearance, Leading to Cardiomyocyte Necrosis and Heart Failure. Antioxid Redox Signal. 2016;25:10–27. doi: 10.1089/ars.2015.6522. [DOI] [PubMed] [Google Scholar]
- 52.Gianfranceschi G, Caragnano A, Piazza S, Manini I, Ciani Y, Verardo R, et al. Critical role of lysosomes in the dysfunction of human Cardiac Stem Cells obtained from failing hearts. Int J Cardiol. 2016;216:140–50. doi: 10.1016/j.ijcard.2016.04.155. [DOI] [PubMed] [Google Scholar]
- 53.Li F, Lang F, Zhang H, Xu L, Wang Y, Hao E. Role of TFEB Mediated Autophagy, Oxidative Stress, Inflammation, and Cell Death in Endotoxin Induced Myocardial Toxicity of Young and Aged Mice. Oxid Med Cell Longev. 2016;2016:5380319. doi: 10.1155/2016/5380319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.