

Abstract
Transcriptional regulatory networks are at the core of establishing cell type specific gene expression programs. In mammalian systems, such regulatory networks are determined by multiple levels of regulation, including by transcription factors, chromatin environment, and three-dimensional organization of the genome. Recent efforts to measure diverse regulatory genomic datasets across multiple cell types and tissues offer unprecedented opportunities to examine the context-specificity and dynamics of regulatory networks at a greater resolution and scale than before. In parallel, numerous computational approaches to analyze these data have emerged that serve as important tools for understanding mammalian cell type specific regulation. In this article, we review recent computational approaches to predict the expression and sequence-based regulators of a gene’s expression level and examine long-range gene regulation. We highlight promising approaches, insights gained, and open challenges that need to be overcome to build a comprehensive picture of cell type specific transcriptional regulatory networks.
Keywords: gene regulation, regulatory networks, cell lineage, transcription factor binding, chromatin state, three-dimensional genome organization
Graphical abstract
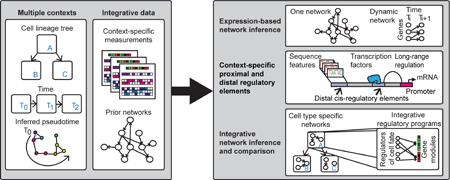
Introduction
Cell type specific gene expression patterns are outputs of transcriptional regulatory networks connecting regulatory proteins such as transcription factors (TFs) and signaling proteins to target genes. These networks are defined by two components (Figure 1A): structure, specifying the regulators for a gene, and, parameters, specifying how the regulator activities drive a gene’s expression level. These networks control the spatial and temporal gene expression patterns, which are important for establishing cell type identity and function (Figure 1C). Hence, the ability to infer regulatory networks of different cell types is critical for understanding gene regulation and its role in dynamic processes such as cell fate specification. Inference of genome-scale mammalian regulatory networks is challenging because multiple factors influence which regulators can regulate the expression of a gene, including TF sequence affinity, chromatin state and three-dimensional genome organization. Advances in single cell genomics, coupled with large-scale measurements of transcriptomes, epigenomes, chromatin accessibility, TF binding, and chromosome conformation, are providing new opportunities to understand mammalian gene regulation. Here we review recent computational approaches to answer three major questions in the analysis of mammalian developmental regulatory networks (Table S1): (a) what TFs regulate the expression level of a gene? (b) what is the full complement of sequence elements that regulate a gene? (c) how does the regulatory network change between cell types?
Figure 1.

Regulatory network concepts. (A) Regulatory networks have two components: structure and parameters. The structure provides the wiring diagram of the network, specifying the regulators of each gene. The parameters (ψ) model the target gene expression level as a function of the combined activities of its regulators. Cis-regulatory elements are regulatory DNA sequence elements and include genomic regions such as transcription factor binding sites. Distal cis-elements have been called enhancers, and are brought into proximity of a gene through three-dimensional looping. Trans-regulatory elements are regulatory proteins that interact directly or indirectly with the gene’s cis-regulatory elements. (B) The context-specific regulatory environment of a cell is measured using various omics technologies. Transcriptomic and proteomic assays measure activity levels of trans-regulators and target genes. Different sequencing technologies, e.g., ChIP-seq, ATAC-seq, DNAse I-seq, are used to measure binding profiles of specific transcription factors, histone modifications, and DNA accessibility. C Cell type specific networks for two cell types A and B. The regulatory networks in each cell type can differ at the structure or at the parameter level. Cell type A can transition to cell type B by appropriate activation of B’s network.
Expression-based regulatory network inference
Expression-based network inference (Figure 2A) is a popular approach to infer genome-scale regulatory networks [1–10] and seeks to infer both the network structure and parameters (Figure 1A). Such methods can be especially useful to predict targets of less studied regulators or regulators with unknown sequence specificity. This class of approaches leverages large number of genome-wide measurements for a given cell type where each measurement profiles a perturbed version of the cell type. Several successful reconstructions have been undertaken for a single cell type, including Th17 cells [11, 12], germ cell tumor [13], glioblastoma multiforme (GBM) [14]. New expression-based network inference methods are either integrating auxiliary non-expression datasets, or inferring regulatory networks from single cell transcriptome data.
Figure 2.

Inference of cell type specific regulatory networks can be addressed using tools for three main types of problems. Shown are the inputs, outputs and the popular types of computational approaches used for addressing these problems. (A) Expression-based network inference uses bulk or single-cell transcriptome profiles to infer dependencies between regulators and targets. Methods can infer a single static regulatory network or infer a dynamic network that describes the change in network structure over time or between cell types. (B) Identification of regulatory elements and predicting their targets via long-range interactions. Methods can either segment the genome based on measured combinations of different regulatory signals (e.g., chromatin marks, TF binding) or predict binding sites of different TFs. Long-range gene regulation is studied using different Chromosome Conformation Capture (3C) technologies, which measure the 3D proximity of two genomic regions. 3C technologies differ in the resolution (genomic region size) and the number of regions they can interrogate. These data can be analyzed to find topological units of organization like TADs, or be used to build classifiers for predicting interactions for new loci. (C) Examining cell type specific regulatory networks that integrate expression and sequence-based regulators of target gene expression. Methods use clustering or dimensionality reduction to study relationships among cell types and loci, predictive models of expression to identify regulatory features explaining expression variation, and probabilistic graphical models. Methods based on Dynamic Bayesian networks and Hidden Markov models provide principled frameworks for modeling networks at each time point and their dynamics.
Inferring constrained networks from bulk transcriptomic data
One direction of research, motivated by the poor agreement of expression-inferred networks and physical regulatory networks (e.g., derived from ChIP-chip/seq experiments) [8], integrates context-agnostic information, such as sequence-specific motifs, to constrain the structure of the inferred network (Table S1). Such constraints are imposed using a penalized linear regression framework [15], or with graph structure priors [16, 17]. In the penalized regression framework (e.g., Inferelator [15]), the penalty of a regulatory edge is reduced if there is previous knowledge supporting this edge. In the graph structure prior approach (e.g, MERLIN-P [16]), the prior probability of an edge with regulatory evidence is higher than other edges. Extending linear regression to a non-linear setting, iRafNet [18] incorporated priors by extending GENIE3 [7], an established state-of-the-art purely expression-based network inference approach, which learns an ensemble of trees to provide a ranking on candidate regulatory edges. All these approaches have shown that adding additional constraints in the network generally improves the agreement with ChIP-chip/seq networks. In addition to requiring sufficient number of samples, these methods assume that the regulator’s expression level is predictive of its target’s expression level. Some approaches are relaxing this assumption by modeling the TF activity as a hidden variable [19–22] however, the only context-specific information here are mRNA levels.
Inferring networks from single cell transcriptomic data
While expression-based network inference from bulk expression measurements is useful, population averages from bulk data can obscure cell-to-cell variability of regulatory networks [23]. Therefore, a recent direction of expression-based network inference has been to infer dynamic regulatory networks from single cell transcriptomic data (Figure 2B, Table S1). Inference of dynamic regulatory networks requires an ordering among the samples, which may be available or can be inferred computationally using cellular trajectory finding algorithms (reviewed in [23, 24]). Once an order is established, a dynamic network is inferred by learning a Boolean network [25, 26], or more complex models, such as dynamic Bayesian networks [27] and Gaussian Processes [28]. Ocone et al. [28] operated on each branch of the inferred trajectory separately. For each branch, a coarse skeleton regulatory network is inferred using GENIE3 [7], followed by detailed regulatory program learning using Gaussian Processes. While many of these approaches use single cell expression data alone, some approaches have integrated other sources of data, e.g., TF ChIP-seq [29] or TF knockdown assays [29, 30] to establish a network structure [29].
So far, single cell network inference has been applied to a relatively small number of manually chosen genes (≤100) in up to 4000 cells [25–29, 31]. Recent single cell RNA-seq measurements of thousands of cells could be used to infer genome-scale networks [32, 33], currently possible only from bulk transcriptomes. However, several technical challenges first need to be addressed including normalization and missing values due to dropout of genes [24, 34, 35].
Identification of regulatory sequence elements and their genomic targets
The regulatory network structure of a cell type depends on the regulatory sequence elements active in the cell type, as well as the three-dimensional proximity of such elements to genes. Accordingly, in addition to mRNA levels, several studies have measured genome-wide binding profiles of transcription factors [36], histone modifications [36–48], and chromatin accessibility [49–51] to identify sequence elements, as well as high-throughput chromosome conformation capture (3C) assays to examine the three-dimensional organization of the genome [51–54]. In parallel, several new computational tools have emerged that use these data to both identify regulatory sequence elements and predict gene targets of these elements.
Identifying regulatory sequence elements
One approach to identify regulatory sequence elements is genome segmentation using one-dimensional regulatory genomic signals, e.g., genome-wide chromatin marks, chromatin accessibility and TF binding (Table S1, [55–62]). These methods assign a state annotation to genomic regions based on combinations of these regulatory signals. A state is a concise description of a region’s regulatory status and can often be mapped to known regulatory elements, including enhancers. Among the earliest approaches for genome segmentation were ChromHMM [55] (a Hidden Markov Model) and Segway [56] (a Dynamic Bayesian Network). More recently, EpiCSeg [59], extended ChromHMM to model count data as negative binomial distributions rather than discretized values. jMOSAiCS [58] also uses count values and considers all possible combinatorial enrichment patterns of different regulatory signals. These methods differ based on the statistical model of the count data, and how they model data from multiple cell types, e.g., either by concatenating the data from multiple cell types [55], or by jointly modeling cell-type [61] or time point specific [57] chromatin mark data. While most of these approaches use one-dimensional regulatory genomic signals, one exception is Graph-based Regularization (GBR) [62], which extended Segway to use high-throughput 3C data as a prior graph to encourage regions that are nearby in 3D space to share the same annotation.
While genome annotation methods identify general sequence elements, another set of methods identify binding events of transcription factors from TF ChIP-seq, DNase I-seq [63, 64] or ATAC-seq [65]. Computational footprinting methods using DNase I-seq data (Table S1, [66]) are specifically geared towards finding footprints of TFs defined as “protected regions” of DNA where a TF might bind. Footprints may be identified using only accessibility data [67–69], or by integrating information of known sequence-specific motifs (see methods reviewed by Gusmao et al.[66]). An alternative is to perform de novo motif discovery on accessible regions using machine learning frameworks that identify sequence features that are predictive of accessibility or ChIP-seq data. In particular, SeqGL [70] and gkm-SVM [71, 72] use a binary classification framework to discriminate peak from non-peak or flanking regions using k-mer features, while the Synergistic Chromatin Model (SCM) [73] performs L1-regularized Poisson regression to predict quantitative accessibility signal. These approaches can identify sequence specific motifs based on the selected k-mers. SeqGL aims to improve the interpretability of the resulting motifs by clustering similar k-mers to account for redundancy and using a Group Lasso penalty to select groups of k-mers. SCM’s regression approach bypasses the need to call peaks and models synergistic relationships among nearby k-mers to directly predict the accessibility of a region. More recently, several deep learning methods [74, 75] aim to predict chromatin features including accessibility [76–78] (Table S1). A recent approach is Basset [78], which uses convolutional neural networks to learn context-specific sequence predictors of DNA accessibility. An advantage of deep learning approaches comes from using convolutional filters to automatically learn informative sequence features from the data, in contrast to manual feature engineering.
Identifying long-range regulatory interactions between enhancers and target genes
Any given cell type can have thousands of active regulatory elements, e.g., enhancers [48, 79], many of which regulate a gene’s expression through long-range interactions by being in three-dimensional proximity to their targets [80–83]. Therefore, a key problem in understanding mammalian regulation is to identify the target genes of enhancers (Figure 2B).
Several approaches have been developed to predict long-range interactions either from statistical analysis of Hi-C data [84], inferring statistical correlation among pairs of genomic loci [49], or by integrating 3C datasets with one-dimensional regulatory signals (Table S1). A correlation-based approach, first proposed by Thurman et al. [49], relied on the correlation of open chromatin profiles of pairs of genomic loci across hundreds of cell types. More recently, the EpiTensor method [85] used tensor decomposition of one-dimensional signals for multiple marks from multiple cell types. This approach uses a tensor to represent three dimensions: cell type, region and signal. PRESTIGE incorporates CTCF domain information with H3k4me1 and RNA-seq expression from multiple cell types to compute a pairwise information theoretic score that is predictive of these interactions [86]. A similar approach developed by Marbach et al. [87] relied on CAGE-seq data from multiple cell types (measured by the FANTOM consortium). Both enhancers and promoters are defined by CAGE-seq expression in a tissue, although CAGE might not identify all enhancers due to its restriction to 5′ capped transcripts from transcription start sites. Enhancers are matched to nearest promoters and filtered based on joint expression in the tissue type. By applying this approach to hundreds of cell types, the authors provided a comprehensive collection of interactions of CAGE-detected enhancers to promoters.
A complementary set of methods use supervised learning, e.g., IM-PET [88], RIPPLE [89], TargetFinder [90], CITD [91]. These methods differ based on the 3C technology used for training, which can be ChIA-PET (IM-PET), 5C (RIPPLE), or Hi-C (TargetFinder, RIPPLE, CITD), input regulatory signals, and whether the methods use data from multiple cell types. TargetFinder and RIPPLE were both trained in a per-cell type manner, while IM-PET combined data from multiple cell types. Beyond chromatin features, IM-PET used sequence conservation as well, while TargetFinder used features associated with regions between the enhancer and promoter.
Both classes of methods are useful. While correlation based methods do not need 3C data and instead rely on the statistical dependencies inferred from one-dimensional signals, the supervised methods combine both 3C data and one-dimensional signals to more directly predict these interactions and identify informative datasets that can predict these interactions.
Analysis of Hi-C data from different cell types has shown that the genome is organized into higher order organizational units, such as A/B compartments, topologically associated domains (TADs) and sub-TADs [92]. TADs are ∼1 megabase in size and remain largely stable across cell types, while A/B compartments partition entire chromosomes into active (A) and inactive regions (B) [92]. Compartments can be identified from a spectral or clustering analysis of the Hi-C interaction matrix [93–95]. TADs can be identified by quantifying the tendency of a region to interact more with its upstream or downstream neighborhood, termed the Directionality Index (DI, [96]), and modeling DI with a Hidden Markov model. Newer TAD finding methods attempt to identify more fine-grained domains (e.g., sub-TADs) that can reveal a hierarchical organization among the domains that could vary across cell types. Both Armatus [97] and TADtree [98] use dynamic programming to identify TADs as well other types of domains. Armatus finds non-overlapping domains by examining the interaction matrix at different resolutions and selecting those that persist across multiple resolutions. In contrast, TADtree identifies a nested hierarchy of TADs, where TADs and sub-TADs can overlap. Computational methods have also been developed to predict TAD boundaries from one-dimensional regulatory signals by training a classifier (e.g., Bayesian Additive Regression Trees (BART) [99]) with examples of TAD boundaries.
The three-dimensional organization of the genome can change substantially during development [51–54, 92, 100]. Relatively few computational approaches have been developed to examine dynamics of Hi-C data across different cell types. One approach constructed meta-TADs by hierarchical clustering of TADs in three cell types from neuronal development [54], and found that while TAD boundaries were conserved across cell types, there were changes at the meta-TAD levels that corresponded with mRNA level changes. Another approach, Arboretum-Hi-C [101], used multi-task graph clustering to identify topological units in multiple cell types and species and concluded that chromosome organization at a coarse megabase scale is largely conserved between cell types and species.
Integrative approaches to examine cell-type specific regulatory networks
With increasing availability of genome-scale measurements for multiple cell types, approaches to identify and compare cell type specific regulatory networks are needed to provide insights into the dynamics of cell type specific behavior. A straightforward way to combine information across multiple cell types is clustering and dimensionality reduction [36, 39, 41, 42, 44, 48, 50, 102]. Such approaches can be used to group cell types based on their genome-wide profiles or group genomic loci based on their signal across multiple cell types. Approaches to examine cell type specific networks and their dynamics are in their infancy; below we summarize two main themes (Figure 2C).
Predictive models of gene expression
Predictive models of gene expression have been powerful to identify regulatory elements driving gene expression programs [103–106] (Figure 2C, Table S1). These approaches model expression level or change in expression as a function of features capturing the chromatin state, TF occupancy, and sequence composition in proximal and distal regions [40, 42, 107]. These approaches differ in whether they predict change in expression level (e.g., [42, 107]) or differential expression status for a gene [40], as well as how they handle distal elements. For example, Gonzalez et al. [107] initially assign distal regions to the nearest gene followed by reassignment based on prediction error, whereas Hagey et al. [42] assign a distal region to all genes within a 500kb window. In all cases, a linear or logistic regression model learns feature weights, which can identify important cis-regulatory elements for cell type specific expression. When profiling multiple cell types using a single active mark (e.g., H3K27ac), one can use the MARGE framework [108] to identify a set of cis-regulatory elements associated with differentially expressed genes. However, these approaches do not infer regulatory connections for individual genes.
Dynamic Bayesian Networks and extensions
Probabilistic graphical models, specifically, Dynamic Bayesian Networks (DBNs) and Hidden Markov Models (HMMs), can model dynamics of networks [27, 109–112] (Figure 2C, Table S1). Early promising work in this area has extended DBNs to capture time-point specific regulatory networks. In particular, Gong et al. [109] used Time-varying Dynamic Bayesian networks (TVDBN) [113] to model regulatory relationships between ∼70 TFs and target genes at four steps of mouse cardiac cell differentiation (ESC, mesoderm, cardiac progenitors and cardiomyocytes). The TVDBN model allows the parameters to change smoothly over time, which overcomes the stationarity assumption of DBNs. Gong et al. integrated 17 TF ChIP-seq data from either ES or the cardiac cell type, stage-specific RNA-seq and histone marks at all four developmental stages in mouse. In addition to modeling the dynamics in the cardiac differentiation network over time, this approach predicted the relevant cis-regulatory regions up to 20kb from a TSS, and identified several known enhancer regions. TVDBNs learn a rich model of regulation for each gene’s expression level as a function of a gene’s cis-regulatory elements, but requires careful regularization that makes each gene’s regulatory program similar between time points to avoid overfitting.
Conclusions
Here we reviewed recent computational approaches to examine mammalian gene regulation with a focus on cell lineages. Prior-constrained, expression-based network inference methods leverage a large number of genome-wide measurements for the same cell type to predict the regulators of a given gene. However, these predictions are a first approximation and need to be experimentally validated with ChIP-seq binding experiments, when possible, and by targeted perturbation experiments of regulators and transcriptome profiling [114, 115]. Genome annotation and TF binding prediction methods use histone modification, accessibility, and TF ChIP-seq profiles to predict the sequence elements that are active in a cell type. However, to construct a regulatory network, we need to infer regulatory connections among sequence elements, target genes and TFs, many of which have yet unknown sequence specificities. Hence, methods to measure sequence specificity of TFs will be key to build a complete picture of a regulatory network. A current open problem is to examine the dynamics and context-specificity of these networks. Early work in this area using predictive models of gene expression and dynamic probabilistic graphical models are promising. However, inferring detailed regulatory mechanisms for multiple cell types in complex lineages will require systematic measurements of transcriptomic, proteomic and epigenomic signals and computational methods to integrate these measurements. We envision that integrative iterative approaches combining experimental measurements and computational modeling will be essential to construct cell type specific networks and identify the regulatory sub-circuits that establish and change cell fates.
Supplementary Material
Highlights.
Transcription factors and chromatin state together determine regulatory networks.
Cell type specific network inference must integrate mRNA and epigenomic datasets.
Expression-based network inference is useful to map genome-wide regulatory networks.
Regulatory sequence element identification helps determine network structure.
Predictive models of expression and graphical models can examine network dynamics.
Acknowledgments
SR is supported by a Sloan Foundation grant and by an NIH grant (1R01GM117339). DC acknowledges support of the NLM training grant 5T15LM007359. This publication was made possible in part by US Environmental Protection Agency grant 83573701.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
* of special interest
** of outstanding interest
- 1.De Smet R, Marchal K. Advantages and limitations of current network inference methods. Nat Rev Microbiol. 2010;8(10):717–29. doi: 10.1038/nrmicro2419. URL: http://dx.doi.org/10.1038/nrmicro2419. [DOI] [PubMed] [Google Scholar]
- 2.Friedman N. Inferring cellular networks using probabilistic graphical models. Science. 2004;303(5659):799–805. doi: 10.1126/science.1094068. [DOI] [PubMed] [Google Scholar]
- 3.Bonneau R, Reiss DJ, Shannon P, Facciotti M, Hood L, Baliga NS, et al. The Inferelator: an algorithm for learning parsimonious regulatory networks from systems-biology data sets de novo. Genome Biology. 2006;7(5):R36. doi: 10.1186/gb-2006-7-5-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Faith JJ, Hayete B, Thaden JT, Mogno I, Wierzbowski J, Cottarel G, et al. Large-scale mapping and validation of Escherichia coli transcriptional regulation from a compendium of expression profiles. PLoS Biol. 2007;5(1):e8. doi: 10.1371/journal.pbio.0050008. URL: http://dx.doi.org/10.1371/journal.pbio.0050008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, Dalla Favera R, et al. ARACNE: an algorithm for the reconstruction of gene regulatory networks in a mammalian cellular context. BMC Bioinformatics. 2006;7(Suppl 1):S7. doi: 10.1186/1471-2105-7-S1-S7. URL: http://dx.doi.org/10.1186/1471-2105-7-S1-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haury AC, Mordelet F, Vera-Licona P, Vert JP. Tigress: Trustful inference of gene regulation using stability selection. BMC Systems Biology. 2012;6:145. doi: 10.1186/1752-0509-6-145. URL: http://dx.doi.org/10.1186/1752-0509-6-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huynh-Thu VA, Irrthum A, Wehenkel L, Geurts P. Inferring regulatory networks from expression data using tree-based methods. PLoS One. 2010;5(9) doi: 10.1371/journal.pone.0012776. URL: http://dx.doi.org/10.1371/journal.pone.0012776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marbach D, Costello JC, Küffner R, Vega NM, Prill RJ, Camacho DM, et al. Wisdom of crowds for robust gene network inference. Nature Methods. 2012;9(8):796–804. doi: 10.1038/nmeth.2016. URL: http://dx.doi.org/10.1038/nmeth.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pe’er D, Tanay A, Regev A. Minreg: A scalable algorithm for learning parsimonious regulatory networks in yeast and mammals. Journal of Machine Learning Research. 2006;7:167–89. URL: http://dl.acm.org/citation.cfm?id=1248547.1248554. [Google Scholar]
- 10.Joshi A, De Smet R, Marchal K, Van de Peer Y, Michoel T. Module networks revisited: computational assessment and prioritization of model predictions. Bioinformatics. 2009;25(4):490–6. doi: 10.1093/bioinformatics/btn658. URL: http://dx.doi.org/10.1093/bioinformatics/btn658. [DOI] [PubMed] [Google Scholar]
- 11.Ciofani M, Madar A, Galan C, Sellars M, Mace K, Pauli F, et al. A validated regulatory network for Th17 cell specification. Cell. 2012;151(2):289–303. doi: 10.1016/j.cell.2012.09.016. URL: http://dx.doi.org/10.1016/j.cell.2012.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yosef N, Shalek AK, Gaublomme JT, Jin H, Lee Y, Awasthi A, et al. Dynamic regulatory network controlling Th17 cell differentiation. Nature. 2013;496(7446):461–8. doi: 10.1038/nature11981. URL: http://dx.doi.org/10.1038/nature11981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kushwaha R, Jagadish N, Kustagi M, Tomishima MJ, Mendiratta G, Bansal M, et al. Interrogation of a context-specific transcription factor network identifies novel regulators of pluripotency. Stem Cells. 2015;33(2):367–77. doi: 10.1002/stem.1870. URL: http://dx.doi.org/10.1002/stem.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plaisier CL, O’Brien S, Bernard B, Reynolds S, Simon Z, Toledo CM, et al. Causal mechanistic regulatory network for glioblastoma deciphered using systems genetics network analysis. Cell Systems. 2016;3(2):172–86. doi: 10.1016/j.cels.2016.06.006. URL: http://dx.doi.org/10.1016/j.cels.2016.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greenfield A, Hafemeister C, Bonneau R. Robust data-driven incorporation of prior knowledge into the inference of dynamic regulatory networks. Bioinformatics. 2013;29(8):1060–7. doi: 10.1093/bioinformatics/btt099. URL: http://dx.doi.org/10.1093/bioinformatics/btt099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Siahpirani AF, Roy S. A prior-based integrative framework for functional transcriptional regulatory network inference. Nucleic Acids Research. 2016 doi: 10.1093/nar/gkw963. URL: http://dx.doi.org/10.1093/nar/gkw963. [DOI] [PMC free article] [PubMed]
- 17.Hill SM, Lu Y, Molina J, Heiser LM, Spellman PT, Speed TP, et al. Bayesian inference of signaling network topology in a cancer cell line. Bioinformatics (Oxford, England) 2012;28:2804–10. doi: 10.1093/bioinformatics/bts514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petralia F, Wang P, Yang J, Tu Z. Integrative random forest for gene regulatory network inference. Bioinformatics. 2015;31(12):i197–205. doi: 10.1093/bioinformatics/btv268. URL: http://dx.doi.org/10.1093/bioinformatics/btv268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liao JC, Boscolo R, Yang YL, Tran LM, Sabatti C, Roychowdhury VP. Network component analysis: reconstruction of regulatory signals in biological systems. Proc Natl Acad Sci U S A. 2003;100(26):15522–7. doi: 10.1073/pnas.2136632100. URL: http://dx.doi.org/10.1073/pnas.2136632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boulesteix AL, Strimmer K. Predicting transcription factor activities from combined analysis of microarray and ChIP data: a partial least squares approach. Theoretical Biology and Medical Modelling. 2005;2:23. doi: 10.1186/1742-4682-2-23. URL: http://dx.doi.org/10.1186/1742-4682-2-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21*.Arrieta-Ortiz ML, Hafemeister C, Bate AR, Chu T, Greenfield A, Shuster B, et al. An experimentally supported model of the bacillus subtilis global transcriptional regulatory network. Molecular Systems Biology. 2015;11(11):839. doi: 10.15252/msb.20156236. The authors propose a strategy for expression-based network inference wherein the expression value of a regulator is replaced by the estimated activity of the regulator using network component analysis. This is demonstrated to improve reconstruction accuracy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gitter A, Huang F, Valluvan R, Fraenkel E, Anandkumar A. Unsupervised learning of transcriptional regulatory networks via latent tree graphical models. 2016 URL: http://arxiv.org/abs/1609.06335.arXiv1609.06335.
- 23.Trapnell C. Defining cell types and states with single-cell genomics. Genome Research. 2015;25(10):1491–8. doi: 10.1101/gr.190595.115. URL: http://dx.doi.org/10.1101/gr.190595.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bacher R, Kendziorski C. Design and computational analysis of single-cell RNA-sequencing experiments. Genome Biology. 2016;17:63. doi: 10.1186/s13059-016-0927-y. URL: http://dx.doi.org/10.1186/s13059-016-0927-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25**.Moignard V, Woodhouse S, Haghverdi L, Lilly AJ, Tanaka Y, Wilkinson AC, et al. Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nat Biotechnol. 2015;33(3):269–76. doi: 10.1038/nbt.3154. URL: http://dx.doi.org/10.1038/nbt.3154. This work proposes a new method for Boolean network inference, single cell network synthesis, from a state transition graph over binarized single cell qRT-PCR expression profiles. Predictions from in silico perturbation experiments on the network were validated to identify novel regulatory interactions in blood development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen H, Guo J, Mishra SK, Robson P, Niranjan M, Zheng J. Single-cell transcriptional analysis to uncover regulatory circuits driving cell fate decisions in early mouse development. Bioinformatics. 2015;31(7):1060–6. doi: 10.1093/bioinformatics/btu777. URL: http://dx.doi.org/10.1093/bioinformatics/btu777. [DOI] [PubMed] [Google Scholar]
- 27.Schütte J, Wang H, Antoniou S, Jarratt A, Wilson NK, Riepsaame J, et al. An experimentally validated network of nine haematopoietic transcription factors reveals mechanisms of cell state stability. Elife. 2016;5:e11469. doi: 10.7554/eLife.11469. URL: http://dx.doi.org/10.7554/eLife.11469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ocone A, Haghverdi L, Mueller NS, Theis FJ. Reconstructing gene regulatory dynamics from high-dimensional single-cell snapshot data. Bioinformatics. 2015;31(12):i89–96. doi: 10.1093/bioinformatics/btv257. URL: http://dx.doi.org/10.1093/bioinformatics/btv257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu H, Ang YS, Sevilla A, Lemischka IR, Ma’ayan A. Construction and validation of a regulatory network for pluripotency and self-renewal of mouse embryonic stem cells. PLOS Computational Biology. 2014;10(8):e1003777. doi: 10.1371/journal.pcbi.1003777. URL: http://dx.doi.org/10.1371/journal.pcbi.1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papatsenko D, Darr H, Kulakovskiy IV, Waghray A, Makeev VJ, MacArthur BD, et al. Single-cell analyses of ESCs reveal alternative pluripotent cell states and molecular mechanisms that control self-renewal. Stem Cell Reports. 2015;5(2):207–20. doi: 10.1016/j.stemcr.2015.07.004. URL: http://dx.doi.org/10.1016/j.stemcr.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunn SJ, Martello G, Yordanov B, Emmott S, Smith AG. Defining an essential transcription factor program for naïve pluripotency. Science. 2014;344(6188):1156–60. doi: 10.1126/science.1248882. URL: http://dx.doi.org/10.1126/science.1248882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu LF, Leng N, Zhang J, Hou Z, Mamott D, Vereide DT, et al. Single-cell rna-seq reveals novel regulators of human embryonic stem cell differentiation to definitive endoderm. Genome Biology. 2016;17(1):173. doi: 10.1186/s13059-016-1033-x. URL: http://dx.doi.org/10.1186/s13059-016-1033-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016;539489:309–13. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stegle O, Teichmann SA, Marioni JC. Computational and analytical challenges in single-cell transcriptomics. Nat Rev Genet. 2015;16(3):133–45. doi: 10.1038/nrg3833. URL: http://dx.doi.org/10.1038/nrg3833. [DOI] [PubMed] [Google Scholar]
- 35.Wagner A, Regev A, Yosef N. Revealing the vectors of cellular identity with single-cell genomics. Nature Biotechnology. 2016;34(11):1145–60. doi: 10.1038/nbt.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsankov AM, Gu H, Akopian V, Ziller MJ, Donaghey J, Amit I, et al. Transcription factor binding dynamics during human ES cell differentiation. Nature. 2015;518(7539):344–9. doi: 10.1038/nature14233. URL: http://dx.doi.org/10.1038/nature14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wamstad JA, Alexander JM, Truty RM, Shrikumar A, Li F, Eilertson KE, et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell. 2012;151(1):206–20. doi: 10.1016/j.cell.2012.07.035. URL: http://dx.doi.org/10.1016/j.cell.2012.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, et al. Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell. 2013;153(5):1134–48. doi: 10.1016/j.cell.2013.04.022. URL: http://dx.doi.org/10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin Q, Chauvistré H, Costa IG, Gusmao EG, Mitzka S, Hänzelmann S, et al. Epigenetic program and transcription factor circuitry of dendritic cell development. Nucleic Acids Research. 2015;43(20):9680–93. doi: 10.1093/nar/gkv1056. URL: http://dx.doi.org/10.1093/nar/gkv1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mateo JL, van den Berg DLC, Haeussler M, Drechsel D, Gaber ZB, Castro DS, et al. Characterization of the neural stem cell gene regulatory network identifies OLIG2 as a multifunctional regulator of self-renewal. Genome Research. 2015;25(1):41–56. doi: 10.1101/gr.173435.114. URL: http://dx.doi.org/10.1101/gr.173435.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ziller MJ, Edri R, Yaffe Y, Donaghey J, Pop R, Mallard W, et al. Dissecting neural differentiation regulatory networks through epigenetic footprinting. Nature. 2015;518(7539):355–9. doi: 10.1038/nature13990. URL: http://dx.doi.org/10.1038/nature13990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hagey DW, Zaouter C, Combeau G, Lendahl MA, Andersson O, Huss M, et al. Distinct transcription factor complexes act on a permissive chromatin landscape to establish regionalized gene expression in CNS stem cells. Genome Research. 2016;26(7):908–17. doi: 10.1101/gr.203513.115. URL: http://dx.doi.org/10.1101/gr.203513.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cavazza A, Miccio A, Romano O, Petiti L, Malagoli Tagliazucchi G, Peano C, et al. Dynamic transcriptional and epigenetic regulation of human epidermal keratinocyte differentiation. Stem Cell Reports. 2016;6(4):618–32. doi: 10.1016/j.stemcr.2016.03.003. URL: http://dx.doi.org/10.1016/j.stemcr.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nature Genetics. 2016 doi: 10.1038/ng.3646. URL: http://dx.doi.org/10.1038/ng.3646. [DOI] [PMC free article] [PubMed]
- 45.van der Veeken J, Gonzalez AJ, Cho H, Arvey A, Hemmers S, Leslie CS, et al. Memory of inflammation in regulatory T cells. Cell. 2016;166(4):977–90. doi: 10.1016/j.cell.2016.07.006. URL: http://dx.doi.org/10.1016/j.cell.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang XL, Wu J, Wang J, Shen T, Li H, Lu J, et al. Integrative epigenomic analysis reveals unique epigenetic signatures involved in unipotency of mouse female germline stem cells. Genome Biology. 2016;17(1):162. doi: 10.1186/s13059-016-1023-z. URL: http://dx.doi.org/10.1186/s13059-016-1023-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, et al. Immunogenetics. Chromatin state dynamics during blood formation. Science. 2014;345(6199):943–9. doi: 10.1126/science.1256271. URL: http://dx.doi.org/10.1126/science.1256271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roadmap Epigenomics Consortium. Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–30. doi: 10.1038/nature14248. URL: http://dx.doi.org/10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489(7414):75–82. doi: 10.1038/nature11232. URL: http://dx.doi.org/10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stergachis AB, Neph S, Reynolds A, Humbert R, Miller B, Paige SL, et al. Developmental fate and cellular maturity encoded in human regulatory DNA landscapes. Cell. 2013;154(4):888–903. doi: 10.1016/j.cell.2013.07.020. URL: http://dx.doi.org/10.1016/j.cell.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518(7539):331336. doi: 10.1038/nature14222. URL: http://dx.doi.org/10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, et al. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290–4. doi: 10.1038/nature12644. URL: http://dx.doi.org/10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schoenfelder S, Furlan-Magaril M, Mifsud B, Tavares-Cadete F, Sugar R, Javierre BM, et al. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Research. 2015;25(4):582–97. doi: 10.1101/gr.185272.114. URL: http://dx.doi.org/10.1101/gr.185272.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fraser J, Ferrai C, Chiariello AM, Schueler M, Rito T, Laudanno G, et al. Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Molecular Systems Biology. 2015;11(12):852852. doi: 10.15252/msb.20156492. URL: http://dx.doi.org/10.15252/msb.20156492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nature Methods. 2012;9(3):215–6. doi: 10.1038/nmeth.1906. URL: http://dx.doi.org/10.1038/nmeth.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoffman MM, Buske OJ, Wang J, Weng Z, Bilmes JA, Noble WS. Unsupervised pattern discovery in human chromatin structure through genomic segmentation. Nature Methods. 2012;9(5):473–6. doi: 10.1038/nmeth.1937. URL: http://dx.doi.org/10.1038/nmeth.1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu P, Xiao S, Xin X, Song CX, Huang W, McDee D, et al. Spatiotemporal clustering of the epigenome reveals rules of dynamic gene regulation. Genome Research. 2013;23(2):352–64. doi: 10.1101/gr.144949.112. URL: http://dx.doi.org/10.1101/gr.144949.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zeng X, Sanalkumar R, Bresnick EH, Li H, Chang Q, Keles S. jMOSAiCS: joint analysis of multiple chip-seq datasets. Genome Biology. 2013;14(4):R38. doi: 10.1186/gb-2013-14-4-r38. URL: http://dx.doi.org/10.1186/gb-2013-14-4-r38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mammana A, Chung HR. Chromatin segmentation based on a probabilistic model for read counts explains a large portion of the epigenome. Genome Biology. 2015;16:151. doi: 10.1186/s13059-015-0708-z. URL: http://dx.doi.org/10.1186/s13059-015-0708-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sohn KA, Ho JWK, Djordjevic D, Jeong HH, Park PJ, Kim JH. hihmm: Bayesian non-parametric joint inference of chromatin state maps. Bioinformatics. 2015;31(13):2066–74. doi: 10.1093/bioinformatics/btv117. URL: http://dx.doi.org/10.1093/bioinformatics/btv117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, An L, Yue F, Hardison RC. Jointly characterizing epigenetic dynamics across multiple human cell types. Nucleic Acids Research. 2016;44(14):6721–31. doi: 10.1093/nar/gkw278. URL: http://dx.doi.org/10.1093/nar/gkw278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Libbrecht MW, Ay F, Hoffman MM, Gilbert DM, Bilmes JA, Noble WS. Joint annotation of chromatin state and chromatin conformation reveals relationships among domain types and identifies domains of cell-type-specific expression. Genome Research. 2015;25(4):544–57. doi: 10.1101/gr.184341.114. URL: http://dx.doi.org/10.1101/gr.184341.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song L, Crawford GE. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harbor Protocols. 2010;2010(2) doi: 10.1101/pdb.prot5384. pdb.prot5384. URL: http://dx.doi.org/10.1101/pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.John S, Sabo PJ, Canfield TK, Lee K, Vong S, Weaver M, et al. Genome-scale mapping of DNase i hypersensitivity. Curr Protoc Mol Biol. 2013 doi: 10.1002/0471142727.mb2127s103. Chapter 27:Unit 21.27. URL: http://dx.doi.org/10.1002/0471142727.mb2127s103. [DOI] [PMC free article] [PubMed]
- 65.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nature Methods. 2013;10(12):1213–8. doi: 10.1038/nmeth.2688. URL: http://dx.doi.org/10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66*.Gusmao EG, Allhoff M, Zenke M, Costa IG. Analysis of computational footprinting methods for dnase sequencing experiments. Nature Methods. 2016;13(4):303–9. doi: 10.1038/nmeth.3772. URL: http://dx.doi.org/10.1038/nmeth.3772. The authors empirically compared multiple computational footprinting methods to establish the cell type specific binding targets of transcription factors from motifs and DNase-seq profiles. Experiments lead the authors to recommend three leading methods (HINT, DNase2TF, and PIQ) and also provide datasets for benchmarking future methods. [DOI] [PubMed] [Google Scholar]
- 67.Boyle AP, Song L, Lee BK, London D, Keefe D, Birney E, et al. High-resolution genome-wide in vivo footprinting of diverse transcription factors in human cells. Genome research. 2011;21(3):456–64. doi: 10.1101/gr.112656.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Piper J, Elze MC, Cauchy P, Cockerill PN, Bonifer C, Ott S. Wellington: a novel method for the accurate identification of digital genomic footprints from dnase-seq data. Nucleic acids research. 2013 doi: 10.1093/nar/gkt850. gkt850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sung MH, Guertin MJ, Baek S, Hager GL. DNase footprint signatures are dictated by factor dynamics and DNA sequence. Molecular Cell. 2014;56(2):275–85. doi: 10.1016/j.molcel.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70**.Setty M, Leslie CS. SeqGL identifies context-dependent binding signals in genome-wide regulatory element maps. PLOS Computational Biology. 2015;11(5):e1004271. doi: 10.1371/journal.pcbi.1004271. URL: http://dx.doi.org/10.1371/journal.pcbi.1004271. SeqGL is a de novo motif discovery method that uses sequence k-mers with Group-LASSO with classification and is applicable to DNase I and ATAC-seq accessibility profiles. It identifies important sequence features that capture the sequence affinity of a TF and has been used to has been to examine the cis-regulatory environment controlling hematopoetic stem cell transition into differentiated cell types [107]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ghandi M, Lee D, Mohammad-Noori M, Beer MA. Enhanced regulatory sequence prediction using gapped k-mer features. PLOS Computational Biology. 2014;10(7):e1003711. doi: 10.1371/journal.pcbi.1003711. URL: http://dx.doi.org/10.1371/journal.pcbi.1003711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee D, Gorkin DU, Baker M, Strober BJ, Asoni AL, McCallion AS, et al. A method to predict the impact of regulatory variants from DNA sequence. Nature Genetics. 2015;47(8):955–61. doi: 10.1038/ng.3331. URL: http://dx.doi.org/10.1038/ng.3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hashimoto T, Sherwood RI, Kang DD, Rajagopal N, Barkal AA, Zeng H, et al. A synergistic DNA logic predicts genome-wide chromatin accessibility. Genome Research. 2016;26(10):1430–40. doi: 10.1101/gr.199778.115. URL: http://dx.doi.org/10.1101/gr.199778.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015;521(7553):436–44. doi: 10.1038/nature14539. URL: http://dx.doi.org/10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- 75.Angermueller C, Pärnamaa T, Parts L, Stegle O. Deep learning for computational biology. Molecular Systems Biology. 2016;12(7):878. doi: 10.15252/msb.20156651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhou J, Troyanskaya OG. Predicting effects of noncoding variants with deep learning-based sequence model. Nature Methods. 2015;12(10):931–4. doi: 10.1038/nmeth.3547. URL: http://dx.doi.org/10.1038/nmeth.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alipanahi B, Delong A, Weirauch MT, Frey BJ. Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat Biotechnol. 2015;33(8):831–8. doi: 10.1038/nbt.3300. URL: http://dx.doi.org/10.1038/nbt.3300. [DOI] [PubMed] [Google Scholar]
- 78*.Kelley DR, Snoek J, Rinn JL. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Research. 2016;26(7):990–9. doi: 10.1101/gr.200535.115. URL: http://dx.doi.org/10.1101/gr.200535.115. Basset is deep learning based method that uses convolutional neural networks to learn context-specific sequence predictors of DNA accessibility. Compared to k-mer features that require feature pre-processing, convolutional networks automatically learn informative features, including position-specific features. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Calo E, Wysocka J. Modification of enhancer chromatin: What, how, and why? Molecular Cell. 2013;49(5):825–37. doi: 10.1016/j.molcel.2013.01.038. URL: http://www.sciencedirect.com/science/article/pii/S1097276513001020. doi: http://dx.doi.org/10.1016/j.molcel.2013.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rubtsov MA, Polikanov YS, Bondarenko VA, Wang YH, Studitsky VM. Chromatin structure can strongly facilitate enhancer action over a distance. Proceedings of the National Academy of Sciences. 2006;103(47):17690–5. doi: 10.1073/pnas.0603819103. URL: http://dx.doi.org/10.1073/pnas.0603819103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miele A, Dekker J. Long-range chromosomal interactions and gene regulation. Molecular BioSystems. 2008;4(11):1046. doi: 10.1039/b803580f. URL: http://dx.doi.org/10.1039/b803580f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.de Laat W, Duboule D. Topology of mammalian developmental enhancers and their regulatory landscapes. Nature. 2013;502(7472):499–506. doi: 10.1038/nature12753. URL: http://dx.doi.org/10.1038/nature12753. [DOI] [PubMed] [Google Scholar]
- 83.Dekker J. Capturing chromosome conformation. Science. 2002;295(5558):1306–11. doi: 10.1126/science.1067799. URL: http://dx.doi.org/10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 84.Ay F, Noble WS. Analysis methods for studying the 3D architecture of the genome. Genome Biology. 2015;16(1) doi: 10.1186/s13059-015-0745-7. URL: http://dx.doi.org/10.1186/s13059-015-0745-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85*.Zhu Y, Chen Z, Zhang K, Wang M, Medovoy D, Whitaker JW, et al. Constructing 3D interaction maps from 1D epigenomes. Nature Communications. 2016;7:10812+. doi: 10.1038/ncomms10812. URL: http://dx.doi.org/10.1038/ncomms10812. EpiTensor performs tensor factorization of 1D epigenomic features and gene expression in multiple cell types to identify significant association between pairs of genomic loci. It is able to identify long-range interactions more accurately than correlation-based methods or simple proximity to the nearest gene. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Corradin O, Saiakhova A, Akhtar-Zaidi B, Myeroff L, Willis J, Cowper-Sal lari R, et al. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome research. 2014;24(1):1–13. doi: 10.1101/gr.164079.113. URL: http://dx.doi.org/10.1101/gr.164079.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marbach D, Lamparter D, Quon G, Kellis M, Kutalik Z, Bergmann S. Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nature Methods. 2016;13(4):366–70. doi: 10.1038/nmeth.3799. URL: http://dx.doi.org/10.1038/nmeth.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.He B, Chen C, Teng L, Tan K. Global view of enhancer-promoter interactome in human cell. Proceedings of the National Academy of Sciences. 2014;111(21):E2191–9. doi: 10.1073/pnas.1320308111. URL: http://dx.doi.org/10.1073/pnas.1320308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Roy S, Siahpirani AF, Chasman D, Knaack S, Ay F, Stewart R, et al. A predictive modeling approach for cell line-specific long-range regulatory interactions. Nucleic Acids Research. 2015;43(18):8694–712. doi: 10.1093/nar/gkv865. URL: http://dx.doi.org/10.1093/nar/gkv865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90**.Whalen S, Truty RM, Pollard KS. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nature Genetics. 2016;48(5):488–96. doi: 10.1038/ng.3539. URL: http://view.ncbi.nlm.nih.gov/pubmed/27064255. TargetFinder is a boosted decision tree classifier to predict enhancer-promoter interactions using epigenomic features. Compared to previous approaches, TargetFinder incorporates epigenomic signals from the “window” region in between the enhancer and promoter of interest. These window features are observed to be particularly important to the classifier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Chen Y, Wang Y, Xuan Z, Chen M, Zhang MQ. De novo deciphering three-dimensional chromatin interaction and topological domains by wavelet transformation of epigenetic profiles. Nucleic Acids Research. 2016;44(11):e106. doi: 10.1093/nar/gkw225. URL: http://dx.doi.org/10.1093/nar/gkw225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bouwman BA, de Laat W. Getting the genome in shape: the formation of loops, domains and compartments. Genome Biology. 2015;16(1) doi: 10.1186/s13059-015-0730-1. URL: http://dx.doi.org/10.1186/s13059-015-0730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nature Methods. 2012;9(10):999–1003. doi: 10.1038/nmeth.2148. URL: http://dx.doi.org/10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell. 2014;159(7):1665–80. doi: 10.1016/j.cell.2014.11.021. URL: http://dx.doi.org/10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Fortin JP, Hansen KD. Reconstructing A/B compartments as revealed by Hi-C using long-range correlations in epigenetic data. Genome Biology. 2015;16:180. doi: 10.1186/s13059-015-0741-y. URL: http://dx.doi.org/10.1186/s13059-015-0741-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376380. doi: 10.1038/nature11082. URL: http://dx.doi.org/10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Filippova D, Patro R, Duggal G, Kingsford C. Identification of alternative topological domains in chromatin. Algorithms for Molecular Biology. 2014;9(1):14. doi: 10.1186/1748-7188-9-14. URL: http://dx.doi.org/10.1186/1748-7188-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98*.Weinreb C, Raphael BJ. Identification of hierarchical chromatin domains. Bioinformatics. 2015;32(11):16011609. doi: 10.1093/bioinformatics/btv485. URL: http://dx.doi.org/10.1093/bioinformatics/btv485. TADTree finds a nested hierarchy of TADs by modeling the relative enrichment of observed interaction counts between bins at a specific distance within a TAD over expected counts at the same distance and uses dynamic programming to efficiently score an entire hierarchy of TADs and sub-TADs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Huang J, Marco E, Pinello L, Yuan GC. Predicting chromatin organization using histone marks. Genome Biology. 2015;16:162. doi: 10.1186/s13059-015-0740-z. URL: http://dx.doi.org/10.1186/s13059-015-0740-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wilson NK, Schoenfelder S, Hannah R, Sánchez Castillo M, Schütte J, Ladopoulos V, et al. Integrated genome-scale analysis of the transcriptional regulatory landscape in a blood stem/progenitor cell model. Blood. 2016;127(13):e12–23. doi: 10.1182/blood-2015-10-677393. URL: http://dx.doi.org/10.1182/blood-2015-10-677393. [DOI] [PubMed] [Google Scholar]
- 101.Fotuhi Siahpirani A, Ay F, Roy S. A multi-task graph-clustering approach for chromosome conformation capture data sets identifies conserved modules of chromosomal interactions. Genome Biology. 2016;17(1) doi: 10.1186/s13059-016-0962-8. URL: http://dx.doi.org/10.1186/s13059-016-0962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Research. 2011;21(10):17571767. doi: 10.1101/gr.121541.111. URL: http://dx.doi.org/10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kundaje A, Lianoglou S, Li X, Quigley D, Arias M, Wiggins CH, et al. Learning regulatory programs that accurately predict differential expression with MEDUSA. Annals of the New York Academy of Science. 2007;1115:178–202. doi: 10.1196/annals.1407.020. URL: http://dx.doi.org/10.1196/annals.1407.020. [DOI] [PubMed] [Google Scholar]
- 104.Karlić R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. Proc Natl Acad Sci U S A. 2010;107(7):2926–31. doi: 10.1073/pnas.0909344107. URL: http://dx.doi.org/10.1073/pnas.0909344107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Dong X, Greven MC, Kundaje A, Djebali S, Brown JB, Cheng C, et al. Modeling gene expression using chromatin features in various cellular contexts. Genome Biology. 2012;13(9):R53. doi: 10.1186/gb-2012-13-9-r53. URL: http://dx.doi.org/10.1186/gb-2012-13-9-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.do Rego TG, Roider HG, de Carvalho FAT, Costa IG. Inferring epigenetic and transcriptional regulation during blood cell development with a mixture of sparse linear models. Bioinformatics. 2012;28(18):2297–303. doi: 10.1093/bioinformatics/bts362. URL: http://dx.doi.org/10.1093/bioinformatics/bts362. [DOI] [PubMed] [Google Scholar]
- 107.González AJ, Setty M, Leslie CS. Early enhancer establishment and regulatory locus complexity shape transcriptional programs in hematopoietic differentiation. Nature Genetics. 2015;47(11):1249–59. doi: 10.1038/ng.3402. URL: http://dx.doi.org/10.1038/ng.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang S, Zang C, Xiao T, Fan J, Mei S, Qin Q, et al. Modeling cis-regulation with a compendium of genome-wide histone H3K27ac profiles. Genome Research. 2016 doi: 10.1101/gr.201574.115. URL: http://dx.doi.org/10.1101/gr.201574.115. [DOI] [PMC free article] [PubMed]
- 109*.Gong W, Koyano-Nakagawa N, Li T, Garry DJ. Inferring dynamic gene regulatory networks in cardiac differentiation through the integration of multi-dimensional data. BMC Bioinformatics. 2015;16:74. doi: 10.1186/s12859-015-0460-0. URL: http://dx.doi.org/10.1186/s12859-015-0460-0. To study cardiac development, the authors developed a Time-varying Dynamic Bayesian Networks approach to infer the regulatory network dynamics over three cell type transitions between four sequential cell types. This approach incorporates multiple time steps and exploits chromatin features, which are important for studying developmental lineages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mendoza-Parra MA, Malysheva V, Mohamed Saleem MA, Lieb M, Godel A, Gronemeyer H. Reconstructed cell fate-regulatory programs in stem cells reveal hierarchies and key factors of neurogenesis. Genome Research. 2016 doi: 10.1101/gr.208926.116. URL: http://dx.doi.org/10.1101/gr.208926.116. [DOI] [PMC free article] [PubMed]
- 111.Parikh AP, Wu W, Curtis RE, Xing EP. Treegl: reverse engineering tree-evolving gene networks underlying developing biological lineages. Bioinformatics. 2011;27(13):i196–204. doi: 10.1093/bioinformatics/btr239. URL: http://dx.doi.org/10.1093/bioinformatics/btr239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jojic V, Shay T, Sylvia K, Zuk O, Sun X, Kang J, et al. Identification of transcriptional regulators in the mouse immune system. Nat Immunol. 2013;14(6):633–43. doi: 10.1038/ni.2587. URL: http://dx.doi.org/10.1038/ni.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Song L, Kolar M, Xing EP. Time-varying dynamic bayesian networks. In: Bengio Y, Schuurmans D, Lafferty JD, Williams CKI, Culotta A, editors. Advances in Neural Information Processing Systems 22. Curran Associates, Inc.; 2009. pp. 1732–40. URL: http://papers.nips.cc/paper/3716-time-varying-dynamic-bayesian-networks.pdf. [Google Scholar]
- 114.Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell. 2016;167(7):1853–66. doi: 10.1016/j.cell.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, et al. Dissecting immune circuits by linking CRISPR-pooled screens with Single-Cell RNA-seq. Cell. 2016;167(7):1883–1896.e15. doi: 10.1016/j.cell.2016.11.039. URL: http://dx.doi.org/10.1016/j.cell.2016.11.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.