

Abstract
Changes in gamma oscillations (20-50 Hz) have been observed in several neurological disorders. However, the relationship between gamma and cellular pathologies is unclear. Here, we show reduced behaviorally-driven gamma before the onset of plaque formation or cognitive decline in a mouse model of Alzheimer's disease (AD). Optogenetically driving FS-PV-interneurons at gamma (40 Hz), but not other frequencies, reduced levels of amyloid-β (A β)1-40 and A β1-42 isoforms. Gene expression profiling revealed induction of genes associated with morphological transformation of microglia and histological analysis confirmed increased microglia co-localization with A β. Subsequently, we designed a non-invasive 40 Hz light-flickering paradigm that reduced A β1-40 and A β1-42 levels in visual cortex of pre-depositing mice and mitigated plaque load in aged, depositing mice. Our findings uncover a previously unappreciated function of gamma rhythms in recruiting both neuronal and glial responses to attenuate AD-associated pathology.
Activation of local circuits of excitatory and fast-spiking inhibitory neurons that resonate at 20-50 Hz gives rise to oscillations in the local field potential (LFP), called gamma oscillations.1–3 Although studies have demonstrated disrupted gamma in various neurological diseases, the interplay between pathology and this emergent circuit property has yet to be determined.4,5 In general, molecular and cellular pathology is thought to alter synaptic activity. However, in at least one disorder, Alzheimer's disease (AD), changes in synaptic activity can also feedback to alter molecular pathology. Studies have shown that increases in synaptic activity in vivo increase levels of amyloid-β (A β),6 a 36-43 amino acid protein, whose aggregation is thought to initiate neurotoxic events, including neuroinflammation, synaptic and neuronal loss, and Tau-associated pathology.7 We aimed to determine how gamma might affect molecular pathology in a mouse model of AD. Understanding how gamma may affect disease pathogenesis has important implications for elucidating both the basic pathology of and possible therapeutic interventions for neurological diseases with altered gamma.
Gamma is decreased during hippocampal sharp wave ripples in 5XFAD mice early in disease
Altered gamma has been observed in multiple brain regions in several neurological and psychiatric disorders, including a reduction in spontaneous gamma synchronization in AD patients and reduced gamma power in multiple AD mouse models.4,5,8,9 However, it is unclear if gamma is altered in other mouse models of AD, if it occurs early in disease progression, and if gamma affects disease pathology. Accordingly, we recorded neural activity from behaving 5XFAD mice, a well-established model of Alzheimer's disease.10 In 3-month-old mice, which have elevated levels of A β but no major plaque accumulation in the hippocampus or manifestation of learning and memory deficits,10 we recorded neural activity from hippocampal subregion CA1, where gamma has been particularly well characterized (e.g.11–14), using a virtual environment (Ext. Data Fig. 1a). In CA1, gamma is present during distinct periods of activity: running, when theta oscillations (4-12 Hz) occur (Ext. Data Fig. 1b, left), and quiescent behavior, when sharp-wave ripples (SWR) occur15,16 (Ext. Data Fig. 1b, right). We found no clear differences in slow gamma power (20 to 50 Hz) between 5XFAD mice and wild-type (WT) littermates during theta (Ext. Data Fig. 1c, d).
We next examined gamma during SWRs, high frequency oscillations of 150-250 Hz lasting around 50-100 ms (Ext. Data Fig. 1b, e).14 Prior work has shown that slow gamma is elevated during SWRs and increased gamma synchrony across CA3 and CA1 during SWR correlates with more coordinated firing between neurons.16 Similarly, we found increased gamma power during SWRs (Fig. 1da yellow arrow indicates elevated gamma, Ext. Data Fig. 1e). The instantaneous frequencies of these slower oscillations (10-50 Hz, Methods) were a unimodal distribution centered around 40 Hz (Fig. 1b, Ext. Data Fig. 1f). Comparing gamma during SWRs in WT and 5XFAD littermates, we found that gamma power was significantly lower in 5XFAD than in WT mice (Methods, Fig. 1c, Ext. Data Fig. 1g, j; examples in Fig. 1c, top). Spiking was phase modulated by gamma in both groups, although the depth of modulation was significantly smaller in 5XFAD than in WT animals (Fig. 1d, Ext. Data Fig. 1h, k). Furthermore, there were fewer SWRs per time in non-theta periods in 5XFAD mice compared to WT (Ext. Data Fig. 1i), reducing periods when gamma power is elevated (Fig. 1a,c, Ext. Data Fig. 1e). These results reveal deficits in gamma modulation of CA1 spiking in a mouse model of AD prior to the development of major amyloid plaque accumulation or evidence of cognitive impairment. This deficit in gamma converges with evidence of gamma deficits in different mouse models of AD, and reports that gamma is altered in humans with AD.5,8,9 Indeed, molecular deficits in Nav1.1 in humans have been linked with gamma deficits in hAPP mice.7
Figure 1. 5XFAD mice have reduced power in gamma during hippocampal SWRs.

a) Average SWR-triggered spectrograms for one mouse (left) showing gamma (yellow arrow) during SWRs (red arrow);right: frequencies below 80 Hz enlarged (n=370 SWRs). b) Histogram of instantaneous gamma frequencies during SWRs for mouse in d. a) Above: Z-scored gamma power around SWR peak for one WT and one 5XFAD mouse (mean ± SEM). Below: Cumulative distribution of gamma power during SWRs (ranksum test, n=2166 and 3085 SWRs in 6 5XFAD and WT mice, respectively). c) Above: Fraction of spikes during SWRs as a function of gamma phase (mean ± SEM). Below: Depth of gamma spiking modulation during SWRs. (ranksum test, bootstrap resampling, n=2500 5XFAD and 3000 WT phase distributions). d) Above: Local field potential trace before and during 40 Hz optogenetic stimulation.Below: Mean and standard deviation of power spectral density (n=4 5XFAD and 3 WT mice). e) Relative A β1-40 levels in CA1 of 5XFAD/PV-Cre mice in each stimulation condition normalized to EYFP controls (n=8 EYFP, n=7 40 Hz, n=4 8 Hz n=6 random mice). f) As in i for A β1-42(n=4 EYFP, n=4 40 Hz, n=3 8 Hz n=3 random mice). g) Relative A β1-40 levels in CA1 of 5XFAD/αCamKII-Cre mice in each stimulation condition normalized to EYFP controls (n=6 40 Hz, n=3 8 Hz n=3 random mice). h) As in k for A β1-42(n=3 mice per group). n.s. not significant, * p<0.05, ** p<0.01, *** p<0.001 by one-way ANOVA; circles indicate n, mean+SEM in bar graphs.
Gamma stimulation reduced A β production in hippocampal CA1
These gamma deficits during SWRs early in disease progression in this mouse model of AD prompt the question of whether gamma could affect molecular and cellular AD pathophysiology. To test this, we induced gamma optogenetically in 5XFAD/PV-Cre mice (Methods, Fig. 1e, left, Ext. Data Fig. 2a, b, c). We chose to drive FS-PV-interneurons at 40 Hz because we found deficits in gamma during SWRs, and instantaneous gamma frequencies during SWRs were centered around 40 Hz. Delivering 1 ms 473 nm light pulses at 40 Hz resulted in increased power at 40 Hz in LFPs in CA1, while random stimulation did not (Fig. 1e, Ext. Data Fig. 1l). Both resulted in similar firing rates (Ext. Data Fig. 1m, n, o).
A β accumulation is thought to initiate multiple neurotoxic events typical for AD pathology. Therefore, we examined whether gamma stimulation affected overall A β peptide levels in the hippocampus of 5XFAD mice. We found that 1 hr of FS-PV-interneuron stimulation reduced A β1-40 by 53.22% and A β1-42 by 44.62% in the 40 Hz group which expresses ChR2 compared to the EYFP control group, as measured in CA1 by A β enzyme-linked immunosorbent assay (ELISA) (Fig. 1f, g, raw concentration (pg/ml) in Ext. Data Table 1). We performed a comprehensive set of control experiments to determine whether the effect was specific to frequency, cell type, and/or rhythmicity. Neither stimulation of CamKII-positive excitatory neurons at 40 Hz, nor FS-PV-interneurons at 8 Hz or random intervals significantly reduced A β levels (Fig. 1f-i, Methods). Immunohistochemical analysis using two β-amyloid-specific antibodies (Cell Signaling Technology; D54D2, BioLegend; 12F417) in CA1 confirmed these results: A β labeling intensity was significantly reduced by 39.5% following 40 Hz stimulation compared to EYFP controls (Fig. 2e, f, D54D2 antibody, Ext. Data Fig. 2e, f, 12F4 antibody).
Figure 2. Driving 40 Hz oscillations optogenetically in hippocampus reduces A β in 5XFAD mice.
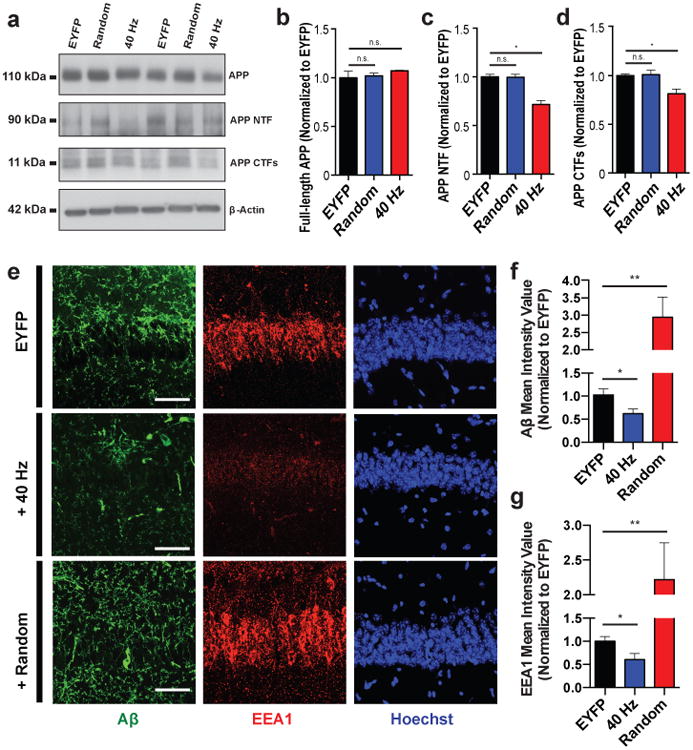
a) Representative western blot showing levels of APP (CT695), APP NTFs (A8967), APP CTFs (CT695), and β-Actin (A5316, loading control) in CA1 of 5XFAD/PV-Cre mice expressing only EYFP or ChR2 with 40 Hz, or random stimulation conditions. 1 mouse per lane, 2 biological replicates. b) Relative immunoreactivity of full-length APP normalized to actin (for b-d, n=6 mice per group). c) Relative immunoreactivity of APP NTF normalized to actin. d) Relative immunoreactivity of APP CTFs normalized to actin. e) Immunohistochemistry with anti-A β (D54D2, green) and anti-EEA1 (610457, red) antibodies in CA1 of 5XFAD/PV-Cre mice (scale bar = 50 μm). f) Relative immunoreactivity of A β normalized to EYFP controls(for f, g, n=3 mice per group). g) Relative immunoreactivity of EEA1 normalized to EYFP controls. n.s. not significant, * p<0.05, ** p<0.01, by one-way ANOVA; mean + SEM in bar graphs.
Brain amyloid concentration depends on A β production from amyloid precursor protein (APP) and A β clearance rates. To elucidate how 40 Hz stimulation reduced A β production, we examined its effects on APP cleavage by measuring levels of the cleavage intermediates of APP, C-terminal fragments (CTFs) and N-terminal fragments (NTFs), in the hippocampus of the 5XFAD/PV-Cre mice. Following 40 Hz stimulation, we found significantly reduced APP CTFs and NTFs compared to EYFP and random controls (Fig. 2a, b, c, d, Ext. Data Fig. 2d).
Prior work has shown that APP is transported and processed in recycling endosomes18, and enlarged early endosomes have been observed in brain tissue from AD patients19. Therefore, we characterized endosomes in CA1 following stimulation using two markers, EEA1 (early endosomal antigen 1) and Rab5 (Ras-related protein encoded by the RAB5A gene). Altogether, the intensity of endosomal labeling of CA1 neurons significantly decreased in both EEA1 (39.7%) and Rab5 (40.1%) following 40Hz stimulation compared to EYFP controls (Fig. 2e, g, Ext. Data Fig. 2g, h). These results suggest that in addition to observed changes in APP cleavage products, 40 Hz stimulation also alters general endosomal processing.
Gamma stimulation induced morphological transformation of microglia
To further explore the cellular and molecular effects of stimulation in an unbiased manner, we performed genome-wide RNA sequencing (RNA-seq) of CA1 tissue following 1 hr of 40 Hz or no stimulation (EYFP) of the 5XFAD/PV-Cre mice (Fig. 3a, Ext. Data Fig. 3a, b, c). Interestingly, 35% of all up-regulated genes had their highest expression in microglia (Fig. 3b). This RNA-seq analysis strongly suggests that 40 Hz stimulation causes an alteration in the state of microglia, which is significant given the accumulating evidence that microglia play a role in AD pathology.20 Transcriptomic changes following 40 Hz stimulation were positively correlated with changes due to increased neural activity (by NMDA and bicuculline), and negatively correlated with changes due to silencing activity (by tetrodotoxin) (Ext. Data Fig. 3d). The immediate early genes Nr4a1, Arc, and Npas4, which are up-regulated by neuronal activity, were elevated as shown by both RNA-seq and RT-qPCR (Ext. Data Fig. 3e).
Figure 3. Driving 40 Hz oscillations optogenetically in hippocampus causes a distinct morphological transformation of microglia in 5XFAD mice.
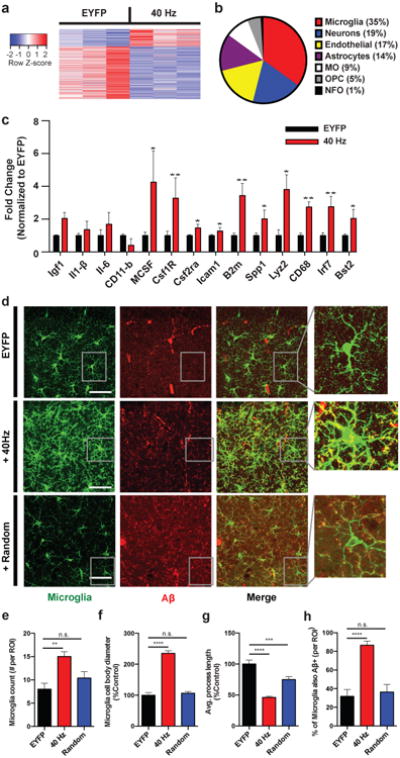
a) Heat map of differentially expressed genes determined by whole-transcriptome RNA-Seq of CA1 from 5XFAD/PV-Cre mice expressing only EYFP or ChR2 with 40 Hz stimulation. Normalized z-score values (high: red, low: blue) were calculated for each differentially expressed gene (row). b) Cell-type-specific expression patterns of up-regulated genes following 40 Hz stimulation (MO: myelinating oligodendrocyte, OPC: oligodendrocyte progenitor cell, NFO: newly formed oligodendrocyte). c) RT-qPCR of specific up-regulated genes:relative RNA levels (fold change) in CA1 of 5XFAD/PV-Cre expressing only EYFP or ChR2 with 40 Hz stimulation, normalized to EYFP controls (Student's t-test; n=6 mice per group). d) Immunohistochemistry with anti-Iba1 (019-19741, green) to identify microglia and anti-A β(12F4, red) antibodies in CA1 of 5XFAD/PV-Cre mice expressing only EYFP or ChR2 with 40 Hz, and random stimulation (40× objective; scale bar = 50 μm). e) Number of Iba1-positive microglia (for f-I, one-way ANOVA; n=4 mice per group). f) Diameter of Iba1-positive microglia cell bodies. g) Average length of Iba1-positive microglia primary processes. h) Percent of Iba1-positive microglia cell bodies that are also A β-positive. n.s. not significant, * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001; mean + SEM in bar graphs.
These transcriptomic results also suggest an engulfing state of microglia. The up-regulated genes were positively correlated with genomic changes induced by macrophage colony-stimulating factor (MCSF) and granulocyte macrophage colony-stimulating factor (GMCSF), known to promote microglial A β uptake (Ext. Data Fig. 3d).21 RT-qPCR confirmed that up-regulated genes included microglial engulfment associated genes Cd68, B2m, Bst2, Icam1, and Lyz2 (Fig. 3c). Microglia-enriched transcriptional regulator Irf7, cell adhesion and migration regulator Spp1, and microglia proliferation markers Csf1r and Csf2ra were also up-regulated (Fig. 3c). Importantly, RT-qPCR showed that the expression levels of pro-inflammatory genes Il6, Il1b (Il1-β), Itgam (CD11-b) and an anti-inflammatory gene Igf1 were not changed (Fig. 3c).
Given that 40 Hz stimulation up-regulated both phagocytosis- and migration/cell adhesion-related genes, we examined morphological features of microglia activation. We used an antibody against microglial marker Iba1 to label microglia in CA1 sections from 5XFAD/PV-Cre mice after 1 hr of 40 Hz or random stimulation, or in mice expressing only EYFP (EYFP, Fig. 3d). We observed almost twice as many microglia in the 40 Hz group compared to EYFP and random control groups (Fig. 3d, e). Furthermore, microglia cell body diameter increased by 135.3% following 40 Hz stimulation compared to EYFP controls and by 138.7% compared to random stimulation (Fig. 3d, f). The length of microglia primary processes were reduced by 54.0% in the 40 Hz stimulation condition compared to EYFP controls and by 38.5%compared to random stimulation (Fig. 3d, g). Iba1 levels did not affect these findings as gene expression analysis showed that Iba1 expression did not differ between conditions (Ext. Data Fig. 3a, b). The increase in cell body size and decrease in process length observed after 40 Hz stimulation are consistent with a shift towards a phagocytic state of microglia.22To evaluate microglia A β uptake specifically, we measured co-localization of A β within microglia by co-immunostaining with an A β antibody (12F4, Methods). The percent of microglia co-localized with A β in the cell body increased to 85.6% following 40 Hz stimulation from 31.7% (EYFP control, Fig. 3d,h). 3D renderings of microglia further demonstrate the presence of Aβ within microglia (Ext. Data Vid. 1, 2, 3). We did not find evidence of neuronal loss by measuring the CA1 cellular layer thickness (Ext. Data Fig. 3f, g). Together, these results suggest that gamma stimulation triggers microglia to increase A β uptake.
Visual stimulation by light flicker drives gamma non-invasively in primary visual cortex
Many studies have shown that visual stimulation can drive oscillations in the gamma range.2,23 In particular, flickering lights at a specific frequency can induce that frequency in primary visual cortex (VC).24 To determine if light flickering could entrain 40 Hz oscillations and subsequently alter A β, we exposed 5XFAD mice to 40 Hz flickering for 1 hr, analogous to optogenetic stimulation that reduced A β described above (Fig. 4a, Ext. Data Vid. 4). In VC, we found that light flickering at 40 Hz increased power in the LFP at 40 Hz, while random interval flickering (random flicker) and dark exposure did not (Fig. 4a, Ext. Data Fig. 4a). All induced similar firing rates (Ext. Data Fig. 4b, c). Spiking increased and decreased concomitantly as the light flickered on and off, resulting in spiking entrained to 40 Hz during 40 Hz flicker but not during random flicker (Fig. 4b). Recordings from saline above the brain exhibited no change in 40 Hz power during 40 Hz flicker, showing that this effect was not due to photoelectric effects or electrical noise (Ext. Data Fig. 4d, e).
Figure 4. Driving 40 Hz oscillations in VC via light flicker reduces A β and amyloid plaques in 5XFAD mice.
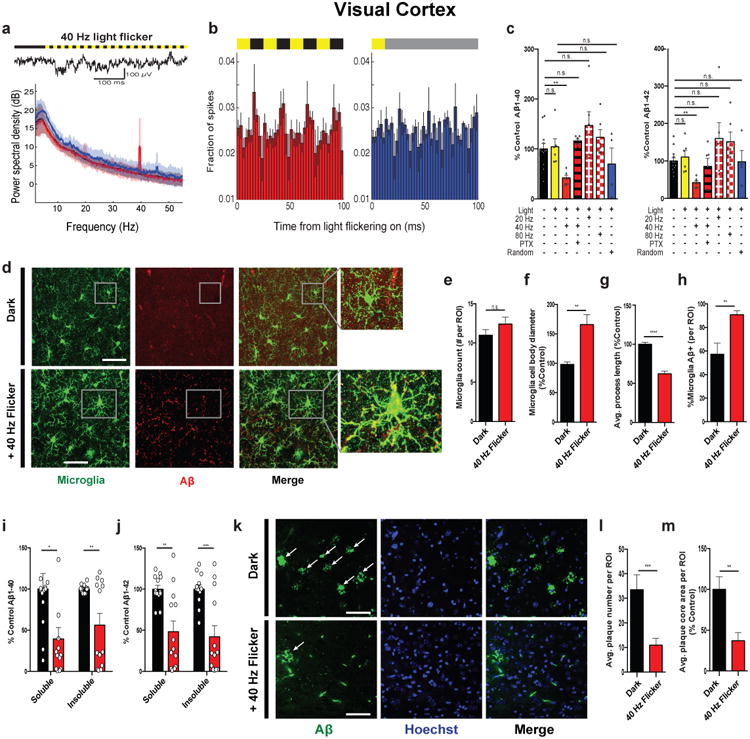
a) Local field potential trace in VC before and during 40 Hz light flicker (above). Power spectral density mean and standard deviation (below, n=4 5XFAD mice, 5 recording sessions). b) Fraction of spikes in VC over 4 cycles of 40 Hz flicker (left) or the equivalent time for random flicker (right, n=4 5XFAD mice from 5 recording sessions, mean ± SEM across animals). For random stimulation, spiking was aligned to light turning on, grey indicates additional light-on flickers occurring randomly (Methods). c) Relative A β1-40 (left) and A β1-42 (right) levels normalized to dark, in VC of 5XFAD mice exposed to dark, light, 40 Hz, 20 Hz, 80 Hz, 40 Hz with picrotoxin (PTX), and random conditions (n=12 dark; n=6 light, 40 Hz, 20 Hz, 80 Hz flicker and PTX; n=4 random mice; one-way ANOVA). d) Immunohistochemistry with anti-Iba1 (019-19741, green) and anti-A β (12F4, red)antibodies in VC of 5XFAD mice exposed to dark or 40 Hz flicker. Right: 120X zoom; arrows indicate +Iba1/+Aβ signal in cell body(scale bar=50 μm). e) Number of Iba1-positive microglia(for e-h Student's t-test unpaired, n=4 mice per group) f) Diameter of Iba1-positive microglia cell bodies. g) Average length of Iba1-positive microglia primary processes. h) Percent of Iba1-positive microglia cell bodies that are also A β-positive. i) Relative A β1-40 levels in VC of 6-month-old 5XFAD mice after 7 days of 1 hr/day dark or 40 Hz flicker (Student's t-test unpaired; n=13 mice per group). j) As in i forA β1-42. k) Immunohistochemistry with anti-A β(D5452, green) antibody in 6-month-old VC of 5XFAD mice after 7 days of 1 hr/day dark or 40 Hz flicker showing plaques (white arrows; scale bar=50 μm). i) Number of A β-positive plaques; (for l,m Student's t-test unpaired, n=8 mice per group). m) Area of A β-positive plaques. n.s. not significant, * p<0.05, ** p<0.01, *** p<0.001; circles indicate n, mean + SEM in bar graphs.
Visual stimulation by light flicker decreases A β levels in primary visual cortex
Given that 40 Hz light flicker entrains 40 Hz oscillations in VC, we aimed to determine whether 40 Hz flicker could reduce A β levels. 3-month-old 5XFAD mice were placed in a dark box and exposed to either light flicker at different frequencies (20, 40, or 80 Hz), random flicker, constant light on (light), or dark for 1 hr. One hour after 1 hr of 40 Hz flicker, we observed that A β1-40 levels in VC were reduced by 57.96% and Aβ1-42levels by 57.97% compared to dark controls (as measured by A β ELISA, Fig. 4c). The effect was specific to 40 Hz flicker as neither constant light nor 20 Hz, 80 Hz, or random flicker significantly reduced A β levels compared to dark and light controls (Fig. 4c). We also found no change in A β levels in somatosensory barrel cortex and hippocampus following 40 Hz flicker (Ext. Data Fig. 5a-h). When we pretreated 5XFAD mice with a low dose GABA-A antagonist (picrotoxin, 0.18 mg/kg25), the effects of 40 Hz flicker on A β levels were completely abrogated, indicating that GABAergic neurotransmission is necessary for this effect (Fig. 4c). To demonstrate that these effects extend beyond the 5XFAD mouse, we examined the effect of 40 Hz flicker in APP/PS1 mice, another well-validated AD model,26 and found significantly reduced Aβ1-40, by 20.80%, and a non-significant trend of reduced Aβ1-42 by 37.68%(Ext. Data Fig. 6a). Furthermore, in 9-month-old WT mice, we found a 58.2% reduction in endogenous mouse A β1-40 following 1 hr of 40 Hz flicker (Ext. Data Fig. 6b). The reduction of endogenous mouse A β1-40 in WT animals reveals that these effects are not restricted to transgenic APP expression or mutant APP; rather, they extend to A β produced from endogenous APP driven by its endogenous promoter.
Next, we investigated whether 40 Hz flicker alters microglia activity in VC in a similar manner that 40 Hz optogenetic stimulation altered CA1 microglia. While microglia number was unchanged (Fig. 4d, e), microglia cell body diameter increased by 165.8% following 40 Hz flicker compared to dark controls (Fig. 4d, f). Microglia primary process lengths were reduced by 37.7% after 40 Hz flicker compared to dark controls (Fig. 4d, g). Consistent with this morphology, which indicates enhanced engulfment activity,22 Aβ/Iba1 co-localization in the cell body was increased to 90.8%after 40 Hz flicker from 57.3% in the dark condition, indicating more A β-bearing microglia (Fig. 4d, h, p<0.01). To better resolve the morphological change in microglia, we used CLARITY27 to create 3D renderings of microglia from 100 μm sections of VC, (Ext. Data Vid. 5, 6). To demonstrate that microglia indeed engulf A β in the 5XFAD mouse, we purified microglia from 5XFAD and WT animals using fluorescence-activated cell sorting (FACS, Methods) and analyzed A β levels via ELISA. We found that microglia-specific levels of A β are significantly higher in 5XFAD animals than in WT controls (Ext. Data Fig. 7a, b). Synaptophysin levels did not change between dark and 40 Hz flicker conditions, indicating that microglia activation likely did not affect synapse number (Ext. Data Fig. 7c, d, n.s. = not significant, n=4 mice). Thus, 40 Hz oscillations induced non-invasively via sensory entrainment reduced A β abundance and promoted microglia/A β interactions.
Chronic visual stimulation by light flicker decreases plaque load in VC
We next assessed whether 40 Hz flicker was effective to treat animals that have amyloid plaques. Because the effects of 40 Hz flicker on soluble A β levels were transient (Ext. Data Fig. 8a), we hypothesized that we would need repeated exposure to affect insoluble A β. Thus, we treated 6-month-old 5XFAD mice, which have amyloid plaque pathology in many brain regions including VC,10 for 1 hr daily over 7 days with 40 Hz flicker or dark, and analyzed VC tissue 24 hr later. We found that 7 days of 1 hr 40 Hz flicker reduced both soluble A β1-40 and A β1-42 levels, by 60.5% and 51.7% respectively, (Fig. 4i, j) and insoluble A β1-40 and A β1-42 levels by 43.7% and 57.9% respectively (Fig. 4i, j). Immunohistochemical analysis showed that 40 Hz flicker significantly reduced plaque number in VC by 67.2% compared to dark controls (Fig. 4k, l) and plaque size by 63.7% (Fig. 4k, m). Taken together, these experiments identify a non-invasive treatment with a profound effect on amyloid plaque pathology. We next determined if 40 Hz flicker reduced tau phosphorylation, another AD-related pathology. Using the TauP301S tauopathy mouse model28 we found that 7 days of 1 hr 40 Hz visual flicker treatment reduced phosphorylated tau serine202 and serine404/threonine403/serine400 levels in VC by 41.2% and 42.3%, respectively, and triggered microglia responses similar to those observed in 5XFAD mice (Ext. Data Fig. 9a-k).
Discussion
Gamma oscillations are thought to be important for higher cognitive functions and sensory responses.2,12,23 Here, we demonstrated that entraining oscillations and spiking at 40 Hz, using optogenetics in the hippocampus of 5XFAD mice and using a non-invasive light flicker treatment to affect primary visual cortex in multiple mouse models, resulted in a marked reduction of A β peptides. We also found a concomitant microglia response following 40 Hz entrainment.
The robust reduction of total amyloid levels was likely mediated by both decreased amyloidogenesis and increased amyloid endocytosis by microglia. Thus, it appears that driving 40 Hz gamma oscillations may induce an overall neuroprotective response that recruits both neurons and microglia. The fact that GABA-A antagonist treatment completely abrogated the effects of 40 Hz stimulation on A β levels strongly suggests that GABAergic neurotransmission is critical for these effects.
40 Hz flicker stimulation reduced A β in multiple mouse models, including 5XFAD, APP/PS1, and WT mice. This replication in multiple mouse models shows that these findings are not specific to one animal model and, importantly, extend to situations where A β is produced from APP expressed by its physiological promoter as it is in WT animals. In addition, we found that 40 Hz oscillations reduced phosphorylated tau in a mouse model of tauopathy, TauP301S, showing that the protective effects of gamma stimulation generalize to other pathogenic proteins. In summary, these findings uncover previously unknown cellular and molecular processes mediated by gamma oscillations and establish a functional connection between brain gamma rhythms, microglia function, and AD-related pathology.
These observations indicate that entraining gamma oscillations may provide a broad spectrum of systemic effects in the brain, including in non-neuronal cells, to attenuate Alzheimer's-related pathology. Because this approach is fundamentally different from prior AD therapies,29 further study is needed to determine whether this approach will be therapeutic in human AD.
Methods
Animals
All animal work was approved by the Committee for Animal Care of the Division of Comparative Medicine at the Massachusetts Institute of Technology. Adult (3-month-old) male double transgenic 5XFAD Cre mice were produced by crossing 5XFAD transgenic mice with the transgenic PV or CW2 promoter driven Cre line. Adult (5-month-old) male and female APP/PS1 mice were gifted from the Tonegawa Laboratory. Adult (4-month-old) male TauP301S mice were obtained from the Jackson Laboratory. 9-month-old WT mice (C57Bl/6) were obtained from the Jackson Laboratory. Mice were housed in groups of 3-5 on a standard 12 hr light / 12 hr dark cycle, and all experiments were performed during the light cycle. Food and water were provided ad libitum unless otherwise noted. Littermates were randomly assigned to each condition by the experimenter. Experimenter was blind to animal genotypes during tissue processing and electrophysiological recording and analysis. No animals were excluded from analysis.
AAV vectors
Adeno-associated viral particles of serotype 5 were obtained from the Vector Core Facility at The University of North Carolina at Chapel Hill. The AAV5 virus contained a channelrhodopsin-2 (ChR2) fused to enhanced yellow fluorescent protein (EYFP) in a double-floxed, inverted, open-reading-frame (DIO) driven by the EF1 α promoter (Ext. Data Fig. 2a). An AAV-DIO-EYFP construct was used as a control.
Surgical procedures
3-month-old 5XFAD/PV-Cre or CW2 mice were anesthetized with an intraperitoneal (i.p.) injection of a mixture of ketamine (1.1 mg kg-1) and xylazine (0.16 mg kg-1). A small craniotomy was made 2.0 mm posterior to bregma and 1.8 mm lateral to the midline on the left side. Virus was delivered through a small durotomy by a glass micropipette attached to a Quintessential Stereotaxic Injector (Stoelting). The glass micropipette was lowered to 1.2 mm below the brain surface. A bolus of 1 μ L of virus (AAV-DIO-ChR2-EYFP or AAV-DIO-EYFP; 2 × 1012 viral molecules per ml) was injected into the CA1 region of hippocampus at 0.075 μ L min-1. The pipette remained in place for 5 min following the injection before being retracted from the brain. A unilateral optical fiber implant (300 μ m core diameter; Thor Labs) was lowered to 0.9 mm below the brain surface about the injection site. Two small screws anchored at the anterior and posterior edges of the surgical site were bound with dental glue to secure the implant in place.
For electrophysiological recordings adult (3-month-old) male 5XFAD/PV-Cre and 5XFAD negative littermates (for CA1 recordings), or 5XFAD and their wild type littermates (for VC recordings) mice were anesthetized using isoflurane and placed in a stereotactic frame. The scalp was shaved, ophthalmic ointment (Puralube Vet Ointment, Dechra) was applied to the eyes, and Betadine and 70% ethanol were used to sterilize the surgical area. For CA1 recordings, a craniotomy (in mm, from bregma: -2 A/P, 1.8 M/L) was opened to deliver 1 μ L of virus to CA1 (as described above). The target craniotomy site for LFP recordings was marked on the skull (in mm, from bregma: -3.23 A/P, 0.98 M/L for CA1 and 2.8 A/P, 2.5 M/L for VC), three self-tapping screws (F000CE094, Morris Precision Screws and Parts) were attached to the skull, and a custom stainless steel headplate was affixed using dental cement (C&B Metabond, Parkell). On the day of the first recording session, a dental drill was used to open the LFP craniotomies (300-400 μ m diameter) by first thinning the skull until ∼100 μ m thick, and then using a 30 gauge needle to make a small aperture. The craniotomy was then sealed with a sterile silicone elastomer (Kwik-Sil WPI) until recording that day and in between recording sessions.
Optogenetic stimulation protocol
Two to four weeks following virus injection and implant placement (which provides time for the mice to recover and undergo behavior training for animals used for electrophysiology) and the virus to express in the neurons, CA1 neurons were optogenetically manipulated. A 200 mW 4793 nm DPSS laser was connected to a patch cord with a fiber channel/physical contact connector at each end. During the experiment, 1 mW (measured from the end of the fiber) of optical stimulation was delivered for 1 hr. For molecular and biochemical analyses, each animal received one of three stimulation protocols: 8 Hz, 40 Hz, or random stimulation (light pulses were delivered with a random interval determined by a Poisson process with an average frequency of 40 Hz). EYFP control animals received 40 Hz stimulation. For electrophysiological recordings each animal received all stimulation conditions interleaved during recordings.
Visual stimulation protocol
15 minutes prior to the experiment 5XFAD mice were treated with saline (Control) or picrotoxin (0.18 mg/kg).25 For molecular and biochemical analyses mice were then placed in a dark chamber illuminated by a light emitting diode (LED) bulb and exposed to one of five stimulation conditions: dark, light, 20 Hz, 40 Hz, 80 Hz flicker (12.5 ms light on, 12.5 ms light off, 60 W) for 1h. For electrophysiological recordings each animal received dark, light, 40 Hz flicker, or random (light pulses were delivered with a random interval determined by a Poisson process with an average interval of 40 Hz) stimulation conditions interleaved in 10 s blocks during recordings.
Behavior training and virtual reality environment (VR) for electrophysiology
For CA1 recordings, headfixed animals ran on an 8” spherical treadmill supported by an air cushion through a virtual reality environment, as described in Harvey et al.30 The motion of the spherical treadmill was measured by an optical mouse and fed into virtual reality software31, running in MATLAB (version 2013b, Mathworks). The virtual environment consisted of a linear track with two small enclosures at the ends where the animal could turn (Ext. Data Fig. 1a). Animals were rewarded with sweetened condensed milk (diluted 1:2 in water) at each end of the track for alternating visits to each end of the track.
Animals learned to run on the virtual linear track over approximately one week. The animals were left to recover from the surgery for one week, and habituated to handling for one to two days before behavioral training began. To learn to maneuver on the treadmill and get comfortable in the testing environment, on the first two days of training the animals were placed on the spherical treadmill with the virtual reality system off and were rewarded with undiluted sweetened condensed milk. On the second day of training on the spherical treadmill, animals' food was restricted to motivate them to run. Animals were restricted to no more than 85% of their baseline weight and typically weighed over 88% of their baseline weight. From the third day until the end of training (typically 5-7 days) the animals were placed on the treadmill for increasing amounts of time (30 min to 2 hr) running in the VR linear track. Animals were rewarded with diluted (1:2) sweetened condensed milk at the end of the linear track after traversing the length of the track. Between recording sessions, animals were given refresher training sessions to maintain behavioral performance.
For VC recordings, animals ran on the spherical treadmill while exposed to dark, light, or light flickering conditions (described below in data acquisition). Prior to recordings animals learned to maneuver on the treadmill and get comfortable in the testing environment by being placed on the spherical treadmill (with the virtual reality system off) and receiving reward of undiluted sweetened condensed milk.
Electrophysiology data acquisition
For optogenetic stimulation of CA1 during recording, a 300 μ m core optical fiber was advanced through the craniotomy used to deliver virus to CA1 to a depth of 900 μ m into the brain. Light pulses that were 1 ms and 1 mW (measured from the end of the fiber) were delivered via a 473 nm DPSS (diode pumped solid state) laser (as described above).
To avoid photoelectric artifacts, neural activity was recorded with glass electrodes. LFP electrodes were pulled from borosilicate glass pipettes (Warner) on a filament-based micropipette puller (Flaming-Brown P97, Sutter Instruments), to a fine tip, which was then manually broken back to a diameter of ∼10-20 μ m and then filled with sterile saline. For CA1 recordings the LFP electrode was advanced through the LFP recording craniotomy at an angle 60 degrees posterior to the coronal plane and 45 degrees inferior to the horizontal plane until clear electrophysiological signatures of the hippocampal stratum pyramidale layer were observed (∼600-1000 μ V theta waves while the animal was running, clearly distinguishable sharp-wave ripples during immobility, and multiple spikes greater than 150 μ V, Ext. Data Fig. 1b). For VC recordings the LFP electrode was advanced vertically through the LFP recording craniotomy to a depth of 600-900 μm and multiple spikes greater than 150 μ V were observed.
Data was acquired with a sampling rate of 20 kHz and bandpass filtered 1 Hz-1 kHz. Animals ran on the spherical treadmill or rested for prolonged periods. For optogenetic simulation sessions, data was recorded for 30 minutes before any stimulation began. Then stimulation was delivered at gamma (40 Hz), random (as described under optogenetic stimulation protocol), or theta (8 Hz) frequency for 10 s periods interleaved with 10 s baseline periods (no stimulation). In two animals, stimulation of each type or baseline was delivered for 5 min periods instead of 10 s periods. Each 30 minutes of stimulation recordings were followed by 5-30 minutes of recording with no stimulation. For visual light flicker simulation sessions, LED strip lights surrounding the animal lights were flickered at gamma (40 Hz), random (described above in Visual stimulation protocol), theta (8 Hz), or 20 Hz frequency for 10 s periods, or were on continuously for 10 s periods, interleaved with 10 s periods with lights off. A few recordings were made above the brain surface during light flicker to ensure that the lights did not create electrical or photoelectric noise during recording. Recording sessions were terminated after approximately 3-5 hr. Animals were 3-4 months old at the time of recording.
Analysis of electrophysiology recordings
Spike detection
Spikes were detected by thresholding the 300-6000 Hz bandpassed signal. Threshold was the median of the filtered signal plus five times a robust estimator of the standard deviation of the filtered signal (median/0.675) to avoid contamination of the standard deviation measure by spikes. (Rossant 2015: http://biorxiv.org/content/biorxiv/early/2015/02/16/015198.full.pdf)
Local field potential (LFP)
Recorded traces were downsampled to 2 kHz and then bandpass filtered between 1 to 300 Hz.
Theta and SWR detection
Activity across the hippocampal network changes markedly when animals run or sit quietly and these changes are often referred to as different network states. These network states are clearly distinguishable by the presence or absence of LFP oscillations in different frequency bands12,13. When animals ran, we observed large theta (4-12 Hz) oscillations in CA1 as others have shown (Ext. Data Fig. 1b, left)13,30,32,33. When animals sat quietly, theta oscillations were no longer visible and we recorded sharp wave ripples (SWRs), high frequency oscillations of 150-250 Hz that last around 50-100 ms and are associated with bursts of population activity, as others have observed (Ext. Data Fig. 1b, right)15,16.
SWRs were detected (Fig. 1a, b, c, d, Ext. Data Fig. 1d, e, f, g, h, i) when the envelope amplitude of the filtered trace was greater than four standard deviations above the mean for at least 15 ms. The envelope amplitude was calculated by taking the absolute value of the Hilbert transform of the filtered LFP. We also confirmed our results held when using a higher threshold for SWR detection, 6 standard deviations above the mean, which detects larger SWRs (Ext. Data Fig 1j, k). To detect theta (Ext. Data Fig. 1c,d), the LFP was bandpass filtered for theta (4-12 Hz), delta (1-4 Hz), and beta (12-30 Hz) using an FIR equiripple filter. The ratio of theta to delta and beta (“theta ratio”) was computed as the theta envelope amplitude divided by the sum of the delta and beta envelope amplitudes. Theta periods were classified as such when the theta ratio was greater than one standard deviation above mean for at least two seconds and the ratio reached a peak of at least two standard deviations above mean. Non-theta periods were classified as such when the theta ratio was less than one for at least two seconds. Sharp wave ripples, theta periods, and non-theta periods were visually inspected to ensure that these criteria accurately detected sharp wave ripple, theta periods, and non-theta periods, respectively.
Power spectrum
Spectral analysis was performing using multitaper methods (Chronux toolbox, time-bandwidth product = 3, number of tapers = 5). For examining power spectra without stimulation (Ext. Data Fig. 1c, d), only theta periods were included: theta periods greater than 5 s long were divided into 5 s trials and the average power spectral density was computed for each animal over these trials. For examining power spectra during optogenetic (Fig. 1e, Ext. Data Fig. 1l) and visual stimulation (Fig. 4a, Ext. Data Fig. 4a), data was divided into 10 s trials of each stimulation condition or baseline periods, and the average power spectral density was computed for each animal over these trials.
Gamma during SWRs
Spectrograms were computed using multitaper methods (Chronux toolbox). The spectrogram was computed for each SWR including a window of 400 ms before and after the peak of the SWR. Then a z-scored spectrogram was computed in each frequency band using the mean and standard deviation of the spectrogram computed across the entire recording session to create a normalized measure of power in units of standard deviation (Fig. 1a, Ext. Data Fig. 1e). Instantaneous frequency of gamma during SWRs was computed by bandpass filtering the LFP for 10-50 Hz, taking the Hilbert transform, then taking the reciprocal of the difference in peaks of the transformed signal (Fig. 1b, Ext. Data Fig. 1f). Gamma power before, during, and after SWRs was computed by filtering the LFP for low gamma (20-50 Hz) and taking the amplitude of the envelope of the Hilbert transform to get the mean gamma power in 100 ms bins centered on the SWR peak. This was normalized by the mean and standard deviation of the amplitude of the envelope for the entire recording session to get z-scored gamma power for each bin around each SWR (Fig. 1c, Ext. Data Fig. 1g, j). Phase modulation by gamma during SWRs was computed by bandpass filtering the LFP for gamma (20-50 Hz), taking the Hilbert transform, and determining the phase of the resulting signal for each spike that occurred during SWRs (Ext. Data Fig. 1h). To measure differences in phase modulation between 5XFAD and WT animals, we used resampling with replacement: a subset of 100 spikes from each recording was randomly selected to create a phase modulation distribution and this was repeated 500 times for each recording (Fig. 1d, Ext. Data Fig. 1k). We then measured the depth of modulation for the spike-gamma phase distribution by computing the difference between the peak and trough divided by the sum of the peak and trough for each distribution (Fig. 1d, Ext. Data Fig. 1k).
Differences in firing during stimulation
To plot stimulus-evoked multiunit firing histograms, spikes were binned in 2.5 ms bins for 100 ms after the start of each light on pulse and the fraction of spikes in each bin was computed. Mean and standard error was then computed across all light-on periods. To compute differences in multi-unit firing rate between conditions, firing rates were computed for each 10 s period of stimulation or baseline (total number of spikes divided by duration of period). Differences in firing rate were taken between nearby periods of the relevant type of stimulation (firing rate in gamma stimulation period minus baseline or random periods for optogenetic stimulation, firing rate in gamma stimulation period minus baseline, continuous on, or random periods for light flicker stimulation). Differences from all animals were plotted in histograms (Ext. Data Fig. 1m, 4c) and the median and quartiles of the multiunit firing rates per 40 Hz stimulation, random stimulation, and no stimulation period for each animal were plotted in box plots (Ext. Data Fig. 1o, 4d).
Immunohistochemistry
Mice were perfused with 4% paraformaldehyde under deep anesthesia, and the brains were post-fixed overnight in 4% paraformaldehyde. Brains were sectioned at 40 μ m using a vibratome (Leica). Sections were permeabilized and blocked in PBS containing 0.2% Triton X-100 and 10% normal donkey serum at room temperature for 1 hr. Sections were incubated overnight at 4 °C in primary antibody in PBS with 0.2% Triton X-100 and 10% normal donkey serum. Primary antibodies were anti-EEA1 (BD Transduction Laboratories; 641057), anti-β-amyloid (Cell Signaling Technology; D54D2), anti-Iba1 (Wako Chemicals; 019-19741), anti-parvalbumin (Abcam; ab32895), anti-Rab5 (Enzo Life Sciences; ADI-KAP-GP006-E). To confirm ELISA experiments, the anti-A β antibody D54D2 was used because it allowed for co-labeling with EEA1 and the anti A β antibody 12F4 was used because it does not react with APP allowing us to determine if our labeling was specific to A β. For co-labeling experiments, the anti-A β antibody 12F4 (Biolegend; 805501) was used. Primary antibodies were visualized with Alexa-Fluor 488 and Alex-Fluor 647 secondary antibodies (Molecular Probes), and cell nuclei visualized with Hoechst 33342 (Sigma-Aldrich; 94403). Images were acquired using a confocal microscope (LSM 710; Zeiss) with a 40X objective at identical settings for all conditions. Images were quantified using ImageJ 1.42q by an experimenter blind to treatment groups. For each experimental condition, 2 coronal sections from at least 3 animals were used for quantification. Scale bars are 50 μ m. For CA1 imaging, the analysis was restricted to the pyramidal cell layer, except in the case of Iba1+ cells analysis, where the whole field of view was required to image an adequate number of cells. ImageJ was used to measure the diameter of Iba1+ cell bodies and to trace the processes for length measurement. In addition, the Coloc2 plugin was used to measure co-localization of Iba1 and Aβ. Imarisx64 8.1.2 (Bitplane, Zurich, Switzerland) was used for 3D rendering. For counting the “plaque number,” deposits ≥10 μ m were included.
Clarity
Fixed brains were sliced into 100uM coronal sections on a vibratome (Leica VT100S) in 1XPBS. Sections containing VC were selected, with reference to the Allen Mouse Brain Atlas, and incubated in clearing buffer (pH 8.5-9.0, 200mM sodium dodecylsulfate, 20mM lithium hydroxide monohydrate, 4mM boric acid in ddH2O) for 2 hours, shaking at 55°C. Cleared sections were washed 3 ×10mins in 1XPBST (0.1% Triton-X100/1XPBS) and put into blocking solution (2% bovine serum albumin/1XPBST) overnight, shaking at RT.27 Subsequently, three 1hour washes in 1X PBST were performed, shaking at RT. Sections were then incubated at 4°C for 2 days, shaking, with anti-β-amyloid (Biolegend;805501) and anti-Iba1 (Wako Chemicals; 019-19741) primary antibodies, diluted to 1:100 in 1X PBST. Another set of 3×1 h washes in 1XPBST was conducted before sections were incubated for 9 hours, shaking at RT, in 1:100 1X PBS diluted secondary antibody mixture. Fragmented Donkey Anti-Rabbit Alexa Fluor® 488 (Abcam; ab175694) and Anti-Mouse 568 (Abcam; ab150101) secondary antibodies were used to visualize the primary antibody labeling. Halfway through this incubation period, Hoechst 33258 (Sigma-Aldrich; 94403) was spiked into each sample at a 1:250 final dilution. Sections were then washed overnight in 1×PBS, shaking at RT. Prior to mounting for imaging, slices were incubated in RIMS (Refractive Index Matching Solution: 75g Histodenz, 20mL 0.1M phosphate buffer, 60mL ddH2O) for 1 hour, shaking at RT. Tissue sections were mounted onto microscopy slides with coverslips (VWR VistaVision, VWR International, LLC, Radnor, PA) using Fluromount G Mounting Medium (Electron Microscopy Sciences, Hatfield, PA, USA). Images were acquired on a Zeiss LSM 880 microscope with the accompanying Zen Black 2.1 software (Carl Zeiss Microscopy, Jena, Germany). Section overview and cellular level images used for 3D reconstruction were taken using a Plan-Apochromat 63×/1.4 Oil DIC objective. Imaris×64 8.1.2 (Bitplane, Zurich, Switzerland) was used for 3D rendering and analysis.
Western blot
CA1 whole cell lysates were prepared using tissue from 3-month-old male 5XFAD/PV-Cre mice. Tissue was homogenized in 1 ml RIPA (50 mM Tris HCl pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) buffer with a hand homogenizer (Sigma), incubated on ice for 15 min, and rotated at 4 °C for 30 min. Cell debris was isolated and discarded by centrifugation at 14,000 rpm for 10 minutes. Lysates were quantitated using a nanodrop and 25 μg protein was loaded on a 10% acrylamide gels. Protein was transferred from acrylamide gels to PVDF membranes (Invitrogen) at 100 V for 120 min. Membranes were blocked using bovine serum albumin (5% w/v) diluted in TBS:Tween. Membranes were incubated in primary antibodies overnight at 4 °C and secondary antibodies at room temperature for 90 minutes. Primary antibodies were anti-APP (Invitrogen; PAD CT695), anti-APP (Sigma;A8967), anti-β-actin (Abcam; ab9485). Secondary antibodies were horseradish peroxidase-linked (GE Healthcare). Signal intensities were quantified using ImageJ 1.46a and normalized to values of β-actin. We examined tau protein solubility using sequential protein extraction as described in Yoshiyama et al., 2007.28 We then probed the detergent insoluble tau fraction using an antibody against Tau5 (Thermo Fisher Scientific; AHB0042).
Elisa
CA1 or VC was isolated from male mice, lysed with PBS or 5M Guanidine HCl, and subjected to A β measurement with the use of mouse (for WT experiments) or human (for all other experiments) A β1-40 or Aβ1-42 ELISA kit (Invitrogen) according to the manufacturer's instructions. We lysed the tissue in phosphate-buffered saline (PBS) to extract the PBS soluble A β fraction. The soluble A β fraction likely contained monomeric and oligomeric A β. Tissue was further treated with guanidine HCl to extract the insoluble A β fraction. Aβ1-42 was below detectable levels for both flicker and control groups in WT VC and microglia-specific samples.
Genome-Wide RNA Sequencing
Total RNA was extracted from CA1 isolates using the RNeasy kit (Qiagen). Purified mRNA was used for RNA-seq library preparation using the BIOO NEXTflex™ kit (BIOO# 5138-08) per the manufacturer's instructions. 1 μg of total mRNA was subject to a sequential workflow of poly-A purification, fragmentation, 1st strand and 2nd strand synthesis, DNA end-adenylation, and adapter ligation. The libraries were enriched by 15 cycles of PCR reactions and cleaned with Agencourt AMPure XP magnetic beads (Beckman Coulter). The quality of the libraries was assessed using an Advanced Analytical-fragment Analyzer. The bar-coded libraries were equally mixed for sequencing in a single lane on the Illumina HiSeq 2000 platform at the MIT BioMicro Center. The raw fastq data of 50-bp single-end sequencing reads were aligned to the mouse mm9 reference genome using TopHat2.0. The mapped reads were processed by Cufflinks 2.2 with UCSC mm9 reference gene annotation to estimate transcript abundances, and test for differential expression. An average of 26,518,345 sequencing reads was obtained from 3 stimulated and 3 non-stimulated mice. Relative abundance of transcript was measured by Fragments Per Kilobase of exon per Million fragments mapped (FPKM). Gene differential expression test between treated and untreated groups was performed using Cuffdiff module with an adjusted p-value<0.05 for statistical significance (GEO accession: GSE77471).
To understand the cellular and molecular mechanisms from our RNA-seq data, 14 of publicly available RNA-seq datasets34 were processed for cell-type specific analysis. Additionally, 60 publicly available neuron-, microglia-, and macrophage- specific RNA-seq datasets under different chemical and genetic perturbations35–40 were downloaded and processed using TopHat/Cufflinks pipeline for gene set enrichment (GSEA) statistical analysis. GSEA was used to determine whether a defined gene set from our RNA-seq data is significantly enriched at either direction of a ranked gene list from a particular perturbation study. Genes detected in the public RNA-seq datasets were ranked by log2 values of fold change (case versus control), from positive to negative values. A defined gene set (in our case, up- or down-regulated genes upon gamma treatment) was considered significantly correlated with a perturbation-induced transcriptomic change (either up- or down-regulation), when both nominal p-value and false discovery rate (FDR) q-value were less than 0.05. The sign of the calculated normalized enrichment score (NES) indicates whether the gene set is enriched at the top or the bottom of the ranked list. The heatmap for differentially expressed genes was generated using a custom R script, and z-score values across all libraries for each gene were calculated based on the gene FPKM values. The box plots for cell-type specificity analysis were also generated by R program, based on gene FPKM values.
Quantitative RT-PCR
The CA1 subregion was isolated from hippocampus of 3-month-old male 5XFAD/PV-Cre mice. Tissue was rapidly frozen using liquid nitrogen and stored at -80 °C, and RNA extracted using the RNeasy kit according to the manufacturer's protocol (Qiagen). RNA (3 μ g) was treated with DNase I (4 U, Worthington Biochemical Corporation), purified using RNA Clean and Concentrator-5 Kit (Zymo Research) according to manufacturers' instructions and eluted with 14 μ L DEPC-treated water. For each sample, 1 μ g RNA was reverse transcribed in a 20 μ Lreaction volume containing random hexamer mix and Superscript III reverse transcriptase (50 U, Invitrogen) at 50 °C for 1 hr. First strand cDNAs were diluted 1:10 and 1μ L were used for RT-qPCR amplification in a 20 μ L reaction (SsoFast EvaGreen Supermix, Bio-Rad) containing primers (0.2 μ M). Relative changes in gene expression were assessed using the 2-ΔΔCt method.
Isolation of microglia from VC
The primary visual cortex (V1 region) was rapidly dissected and placed in ice cold Hanks' Balanced Salt Solution (HBSS) (Gibco by Life Technologies, Catalog number 14175-095). The tissue was then enzymatically digested using the Neural Tissue Dissociation Kit (P) (Miltenyi Biotec, Catalog number 130-092-628) according to the manufacturer's protocol, with minor modifications. Specifically, the tissue was enzymatically digested at 37 °C for 15 minutes instead of 35 minutes and the resulting cell suspension was passed through a 40 μ m cell strainer (Falcon Cell Strainers, Sterile, Corning, Product #352340) instead of a MACS SmartStrainer, 70 μ m. The resulting cell suspension was then stained using allophycocyanin (APC)-conjugated CD11b mouse clone M1/70.15.11.5 (Miltenyi Biotec, 130-098-088) and phycoerythrin (PE)-conjugated CD45 antibody (BD Pharmingen, 553081) according to the manufacturer's (Miltenyi Biotec) recommendations. Fluorescence-activated cell sorting (FACS) was then used to purify CD11b and CD45 positive microglial cells. The cells were sorted directly into 1XPBS (Ext. Data Fig. 6a).
Code Availability
Code is publically available upon request from the corresponding author.
Statistics
For electrophysiological data that was not normally distributed, results are presented as medians and quartiles unless otherwise noted. Two-sided Wilcoxon rank sum tests for equal medians were performed to determine if distributions were significantly different, and Wilcoxon signed rank tests were performed to determine if distributions were significantly different from zero as these do not assume data is normally distributed. Variability was similar between the groups that were statistically compared. The Bonferroni method was used to correct for multiple comparisons. No statistical method was used to estimate sample size, but it is consistent with previous publications.
Molecular and biochemical results are presented as mean +SEM. Percentages stated in manuscript are group means. All statistical analysis was performed using Prism GraphPad software. Normality was determined using the D'Agostino & Pearson omnibus normality test. Variability was similar between the groups that were statistically compared. Comparison data for normally distributed data consisting of two groups was analyzed by two-tailed unpaired t tests. Comparison of data for normally distributed data consisting of three or more groups was analyzed by one-way ANOVA followed by Tukey's multiple comparisons test. Comparison data for non-normally distributed data was carried out using Mann Whitney tests. The statistical test, exact P values, and sample size (n) for each experiment is specified in the figure legend. For optogenetic ELISA data, two-sided unpaired Student's t-tests were performed to compare mice from the same litter that received different conditions. No statistical method was used to estimate sample size, but is consistent with previous publications. Molecular and biochemical analysis was performed using a minimum of three biological replicates per condition.
Data availability
Data is publically available upon request from the corresponding author.
Extended Data
Extended Data Figure 1. 5XFAD mice have reduced power in gamma during hippocampal SWRs.

b) Mouse in virtual reality environment.
c) Local field potential recorded in CA1, above, filtered for theta (left) or sharp wave ripples (right), middle, and gamma, below.
d) Mean and standard deviation of the normalized power spectrum during theta. Each animal's power spectral density was normalized to its peak (n=6 mice per group).
e) Normalized power spectral densities during theta periods in 3-month-old 5XFAD (green, n=6 mice) and WT (black/grey, n=6 mice) mice. Each animal's power spectral density was normalized to its peak (in theta).
f) Average SWR-triggered spectrograms for one WT and one 5XFAD mouse shows an increase in the gamma band during SWRs. This increase is lower in the 5XFAD mouse than in the WT mouse (n=370 and 514 SWRs in WT and 5XFAD, respectively; WT mouse shown here is the same as in Fig. 1a).
g) Distributions for each recording (left) and the mean and standard error across sessions (right) of instantaneous gamma frequencies during SWRs in 5XFAD (green) and WT (black) mice show distributions around 40 Hz (n=820, 800, 679, 38, 1875, 57 gamma cycles per session in 6 5XFAD animals and 181, 1075, 919, 1622, 51, 1860, 1903 gamma cycles per session in 6 WT animals).
h) Cumulative distribution of the Z-scored gamma power during the 100 ms around the peak of the SWR for WT (black) and 5XFAD animals (green) for each animal (left) and the mean and standard error (shaded) across animals (right) (n=514, 358, 430, 22, 805, 37 SWRs per session in 6 5XFAD animals and 82, 311, 370, 776, 18, 710, 818 SWRs per session in 6 WT animals)
i) Fraction of spikes in CA1 during SWRs as a function of the phase of gamma in 5XFAD (green) and WT (black) mice for each animal (left) and the mean and standard error across animals (right, n=2475, 1060, 3092, 25, 6521, 123 spikes during SWRs per session in 6 5XFAD mice and 360, 4741, 1564, 2961, 88, 3058, 4270 spikes during SWRs per session in 6 WT mice).
j) SWR rate per non-theta period in 5XFAD (green) and WT (black) mice for each animal (left) and all animals combined (right, ranksum test, p < 10-10, n=117, 210, 151, 55, 100, 1 non-theta periods per session in 6 5XFAD mice and 80, 68, 115, 95, 15, 159, 218 non-theta periods per session in 6 WT mice).
k) The cumulative distribution of gamma power during large SWRs (detection threshold greater than 6 standard deviations above the mean, Methods) shows significantly smaller increases in 5XFAD (green) than WT (black) mice (ranksum test, p<10-5, n=1000 SWRs in 6 5XFAD mice and 1467 SWRs in 6 WT mice).
l) Fraction of spikes as a function of the phase of gamma during large SWRs (detection threshold greater than 6 standard deviations above the mean, Methods), mean ± SEM (left) and histogram of the depth of modulation of spiking (right) as a function of gamma phase in 3-month-old 5XFAD (green, n=6 mice) and WT (black, n=6 mice) mice (ranksum test, bootstrap resampling p < 10-10, n=2500 5XFAD spike-gamma phase distributions and 3000 WT distributions).
m) Power spectral density during 40 Hz stimulation (red, left), random stimulation (blue, center), or no stimulation (black, right) of FS-PV-interneurons in CA1 for each mouse (n=4 5XFAD mice with 169, 130, 240, 73 40 Hz, 143, 129, 150, 72 random, and 278, 380, 52, 215 no stimulation periods per animal and n=3 WT mice with 65, 93, 91 40 Hz, 64, 93, 90 random, and 187, 276, 270 no stimulation periods per animal).
n) Above: Example raw LFP trace (above) and the trace filtered for spikes (300-6000 Hz, below), with spikes indicated with red stars after optogenetic stimulation (blue vertical lines). Below: histogram of spikes per pulse after the onset of the 1 ms laser pulse during 40 Hz stimulation (red), random stimulation (blue), or no stimulation (black, n=345762 40 Hz, 301559 random pulses, and 32350 randomly selected no stimulation times at least 500 ms apart from 552 40 Hz, 543 random, and 1681 no stimulation periods in 4 5XFAD and 3 WT mice).
o) Histogram of the difference in firing rates between 40 Hz stimulation and random stimulation periods shows that both types of stimulation elicit similar amounts of spiking activity (Wilcoxon signed rank test for zero median, p>0.6, n=538 stimulation periods from 4 5XFAD and 3 WT mice, n.s. indicates not significant).
p) Multiunit firing rates per 40 Hz stimulation (red), random stimulation (blue), and no stimulation (black) period for each animal. Box and whisker plots show median (white lines in box) and quartiles (top and bottom of box). In all animals firing rates between 40 Hz and random stimulation were not significantly different, showing that the random stimulation condition serves as a control for spiking activity (ranksum tests for each animal, 3 WT and 4 5XFAD mice, p's>0.09, n=87, 130, 8, 65, 93, 91, 73 40 Hz stimulation periods and 85, 129, 5, 64, 93, 90, 72 random stimulation periods per animal). We also examined whether 40 Hz stimulation caused neuronal hyperactivity relative to no stimulation, because according to a recent report, this could have negative effects on neural circuit function.26 In most animals the firing rates between 40 Hz or random stimulation and no stimulation were not significantly different (ranksum tests for each animal, 2 WT and 2 5XFAD, p's>0.25, n=8, 93, 91, 73 40 Hz stimulation periods and 15, 277, 270, 215 baseline periods per animal) or the firing rates during 40 Hz or random stimulation were lower than during no stimulation (ranksum tests for each animal, 1 WT and 1 5XFAD, p's<10-5, which is significant when corrected for performing multiple comparisons, n=130, 65 40 Hz stimulation periods and 379, 187 baseline periods per animal) indicating that 40 Hz stimulation did not cause neuronal hyperactivity. In one animal there was significantly more activity with 40 Hz or random stimulation than during baseline (ranksum test for 1 5XFAD, mouse, p<10-5, n=87 40 Hz stimulation periods and 251 baseline periods per animal). Therefore in six out of seven animals we see no evidence that the 40 Hz optogenetic stimulation of FS-PV-interneurons causes hyperactivity.
Extended Data Figure 2. ChR2 was expressed in FS-PV-interneurons for optogenetic stimulation.
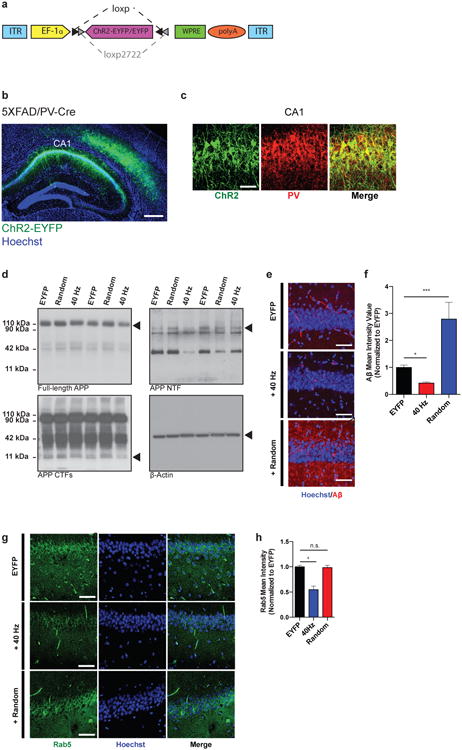
a) AAV-DIO-ChR2-EYFP or AAV-DIO-EYFP drives Cre-dependent expression of ChR2-EYFP or EYFP to produce celltype-specific targeting of ChR2 or EYFP, respectively. In the presence of Cre, ChR2-EYFP or EYFP is inverted into the sense direction and expressed from the EF-1α promoter in PV-positive cells. ITR, inverted terminal repeat; polyA; WPRE, woodchuck hepatitis B virus post-transcriptional element.
b) ChR2-EYFP was strongly expressed in PV-positive interneurons in CA1 of 3-month-old 5XFAD/PV-Cre mice (scale bar = 100 μm).
c) Immunohistochemistry with anti-EYFP and anti-PV antibodies in CA1 of 3-month-old 5XFAD/PV-Cre mice expressing AAV-DIO-ChR2-EYFP shows EYFP expression only in PV-positive cells (scale bar = 50 μm).
d) Representative western blots showing levels of full-length APP (top left, CT695), APP CTFs (bottom left, CT695), APP NTFs (top right, A8967) and β-actin (bottom right, A5316, loading control) in CA1 in EYFP, random, and 40 Hz stimulation conditions, 1 mouse per lane, with 2 biological replicates of each condition.
e) Immunohistochemistry with anti-A β (12F4, red) antibodies in CA1 of 5XFAD/PV-Cre mice expressing only EYFP or ChR2 with 40 Hz, and random stimulation conditions (scale bar = 50 μm).
f) Bar graphs represent the relative immunoreactivity of A β normalized to EYFP (n=4 mice per group; * indicates p<0.05 and *** indicates p<0.001 by one-way ANOVA). Bar graphs show mean + SEM.
g) Immunohistochemistry with anti-Rab5 antibody (ADI-KAP-GP006-E, green) in CA1 of 5XFAD/PV-Cre mice (scale bar = 50 μm).
h) Relative Rab5 intensity levels normalized to EYFP controls (n=3 mice per group).
Extended Data Figure 3. Optogenetically driven 40 Hz oscillations in CA1 cause changes in gene regulation and immediate early gene expression.

a) Table of 130 genes up-regulated by 40 Hz FS-PV-interneuron stimulation determined by whole transcriptome RNA-Seq of CA1 from 3-month-old 5XFAD/PV-Cre mice (p<0.05 by Cufflinks 2.2).52
b) Table of 393 genes down-regulated by 40 Hz FS-PV-interneuron stimulation determined by whole transcriptome RNA-Seq of CA1 from 3-month-old 5XFAD/PV-Cre mice (p<0.05 by Cufflinks 2.2).52
c) Box plot showing fragments per kilobase (FPKM) values of up- and down-regulated genes in EYFP and 40 Hz groups. Box shows median (black lines in box) and quartiles (top and bottom of box), whiskers show minimum and maximum values, and circles show outliers.
d) GSEA statistics tables showing statistical significance of correlation between genes up- or down- regulated by 40 Hz stimulation and publicly available neuron, microglia, and macrophage specific RNA-Seq data under different chemical and genetic perturbations; the perturbation terms were ranked based on the FDR q-values for the up-regulated gene list, from the smallest to the largest (Methods).
e) RT-qPCR verification of specific gene targets in the RNA-Seq data set. Bar graph shows relative RNA levels (fold change) from EYFP (black) and 40 Hz stimulation (red) conditions (* indicates p<0.05, ** indicates p<0.01, and *** indicates p<0.001 by Student's t-test, n=3 mice per group). All bar graphs show mean + SEM.
f) Immunohistochemistry with Hoechst to label all cell nuclei in CA1 of 5XFAD/PV-Cre mice expressing only EYFP or ChR2 with 40 Hz stimulation conditions (scale bar = 50 μm).
g) Bar graph represents the estimated CA1 thickness in 5XFAD/PV-Cre mice expressing only EYFP or ChR2 with 40 Hz stimulation conditions (n=4 mice per group; n.s. indicates not significant, by Student's t-test).
Extended Data Figure 4. 40 Hz light flicker drives 40 Hz oscillations in VC, while random flickering does not.

a) Power spectral densities of local field potentials in VC during 40 Hz light flicker (red, far left), random light flicker (blue, center left), dark (black, center right), or light (green, far right) in VC for each recording session for each mouse (n=5 recordings from 4 5XFAD mice with 47, 51, 64, 49, 16 40 Hz flicker, 47, 50, 64, 50, 16 random flicker, 279, 301, 382, 294, 93 dark and 47, 50, 64, 49, 15 light periods). Light flicker at other frequencies increased power in the flicker frequency, as others have found previously23,24 (data not shown).
b) Histogram of the difference in firing rates between 40 Hz light flicker and random light flicker (n=226 stimulation periods from 5 recording sessions in 4 5XFAD mice).
c) Multiunit firing rates in VC during 40 Hz light flicker (red), random light flicker (blue), dark (black), or light (green) periods. Box plots show median (white lines in box) and quartiles (top and bottom of box). In all animals, firing rates between 40 Hz flicker and random flicker conditions were not significantly different showing that the random stimulation condition serves as a control for spiking activity (ranksum tests for each of 5 recording session from 4 5XFAD mice, p's>0.06, n=47, 51, 64, 49, 16 40 Hz flicker periods and 47, 50, 64, 50, 16 random flicker periods per recording). There were no significant differences in firing rates between 40 Hz flicker and light conditions indicating that 40 Hz light flicker generally did not cause neuronal hyperexcitability (ranksum tests for each of 5 recording session from 4 5XFAD mice, p's > 0.2 for 4 recording sessions, p<0.01 for 1 recording session, which is not significant when corrected for performing multiple comparisons, n=47, 51, 64, 49, 16 40 Hz periods and 47, 50, 64, 49, 16 light periods per recording). In one session, there was more activity in the 40 Hz flicker than in the dark condition.
d) Example traces of LFPs recorded above the brain during light flicker (above, yellow indicates light on, black indicates light off), during three different recording sessions.
e) Power spectral densities of LFPs recorded above the brain during 40 Hz light flicker show no increase in power at 40 Hz. Thus, the effect is not due to photoelectric effects on recording equipment or electrical noise (n= 4, 2,1, 1, 17, 42, 36, 55, 53 40 Hz flicker periods from 4 recording sessions in 3 5XFAD mice undergoing VC recordings and from 5 recording sessions in 2 5XFAD and 3 WT mice undergoing hippocampal recordings). Mean (solid line) and standard deviation (shaded area) across recordings are shown on the left and per animal on the right. Recordings with less than 3 flicker periods (light red) resulted in noisier power spectral densities than recordings with more data (dark red) but none showed evidence of peaks at 40 Hz.
Extended Data Figure 5. 40 Hz light flicker does not affect Aβ levels in hippocampus or barrel cortex.

a) Example local field potential trace in hippocampal CA1 before and during 40 Hz light flicker (above). Mean (solid line) and standard deviation (shaded area) of power spectral density during 40 Hz light flicker (red), random light flicker (blue), or dark (black) in CA1 (n=2 5XFAD and 3 WT mice).
b) Histogram of the fraction of spikes in hippocampus as a function of time for 4 cycles of 40 Hz light flicker (left, red) or the equivalent period of time for random light flicker (right, blue, n=2 5XFAD and 3 WT mice, mean ± SEM across animals). Bar above indicates when light was on (yellow) or off (black). For random stimulation, spiking was aligned to the start of the light turning on, additional periods with light-on occurred at random intervals indicated by grey (Methods).
c) Histogram of the difference in firing rates between 40 Hz light flicker and random light flicker (bottom n=168 stimulation periods from 5 recording sessions in 2 5XFAD and 3 WT mice).
d) Power spectral densities of local field potentials in CA1 during 40 Hz light flicker (red, far left), random light flicker (blue, center left), dark (black, center right), or light (green, far right) for each recording session for each mouse (n=5 recordings from 2 5XFAD and 3 WT mice with 22, 54, 42, 71, 55, 40 Hz flicker, 12, 34, 32, 54, 36 random flicker, 115, 240, 224, 342, 282 dark and 12, 33, 33, 54, 35 light periods).
e) Multiunit firing rates in CA1 during 40 Hz light flicker (red), random light flicker (blue), dark (black), or light (green) periods. Box plots show median (white lines in box) and quartiles (top and bottom of box). In all animals firing rates between 40 Hz flicker and random flicker conditions were not significantly different showing that the random stimulation condition serves as a control for spiking activity (ranksum tests for each of 5 recordings from 2 5XFAD and 3 WT animals, p's>0.2, n=22, 54, 42, 71, 55 40 Hz flicker periods and 12, 34, 32, 54, 36 random flicker periods per recording). There were no significant differences in firing rates between 40 Hz flicker and light conditions indicating that 40 Hz light flicker generally did not cause neuronal hyperexcitability (ranksum tests for each of 5 recordings from 2 5XFAD and 3 WT animals, p's > 0.3, n=22, 54, 42, 71, 55 40 Hz periods and 12, 34, 33, 54, 35 light periods per recording).
f) Bar graphs of relative A β1-40 levels in VC of 5XFAD mice in dark, 40 Hz flicker, and random flicker conditions, normalized to dark (n=4 mice per group; n.s. indicates not significant). Bar graphs represent mean + SEM. Circles superimposed on bars in bar graphs indicate individual data points in each group.
g) Bar graphs of relative A β1-42 levels in VC of 5XFAD mice in dark, 40 Hz flicker, and random flicker conditions, normalized to dark (n=4 mice per group; n.s. indicates not significant). Bar graphs represent mean + SEM. Circles superimposed on bars in bar graphs indicate individual data points in each group.
h) Bar graph of relative A β1-40 and A β1-42 levels in barrel cortex of 5XFAD mice in dark and 40 Hz flicker conditions, normalized to dark (n=3 mice per group; n.s. indicates not significant by Student's t-test).
Extended Data Figure 6. Acute reduction in A β after light flicker in APP/PS1 and WT mice and at various time points.

a) Bar graph of relative A β1-40 and A β1-42 levels of APP/PS1 in VC in dark and 40 Hz flicker conditions, normalized to dark (n=5 mice per group for dark and n=4 mice per group for 40 Hz flicker conditions; n.s. indicates not significant and * indicates p<0.05, by Student's t-test). All bar graphs show mean + SEM throughout this figure. Circles superimposed on bars in bar graphs indicate individual data points in each group.
b) Bar graph of relative mouse A β1-40 and Aβ1-42 levels in VC of 9-month-old WT mice in dark and 40 Hz flicker conditions, normalized to dark (n=11 mice per group for dark and n=9 mice per group for 40 Hz flicker conditions; * indicates p<0.05, by Student's t-test).
Extended Data Figure 7. 40 Hz light flicker does not decrease synaptic density in VC.

a) Schematic depicting isolation of microglia from VC. VC was dissected, then single cells were suspended and labeled with CD11b and CD45 antibodies. Subsequently, cells were sorted via fluorescence-activated cell sorting (FACS) and lysed. A β1-40 levels were analyzed by ELISA.
b) Bar graph of A β1-40 levels in microglia purified using FACS (Methods) from VC of 3-month-old 5XFAD and WT mice (n=8 mice per group for 5XFAD and n=4 mice per group for WT mice; * indicates p<0.05 by Student's t-test). Circles superimposed on bars in bar graphs indicate individual data points in each group.
c) Immunohistochemistry with SVP38 (red) antibodies to detect synaptophysin in VC of3-month-old 5XFAD mice in dark and 40 Hz flicker conditions (Images were taken with 40x objective; scale bar = 50 μm). Right: 100X rendering of dark and 40 Hz flicker conditions.
d) Bar graph of relative SVP38 intensity levels in VC of 5XFAD mice after dark (black) and 40 Hz (red) flicker conditions, normalized to dark (n=4 mice per group; n.s. indicates not significant, by Student's t-test).
Extended Data Figure 8. Aβ levels in VC return to baseline 24 hr after 1 hr of 40 Hz light flicker.

a) Bar graph of relative Aβ1-40 and Aβ1-42 levels in VC of 5XFAD mice 1, 4, 12, and 24 hours after 1 hour of dark or 40 Hz flicker treatment, normalized to dark (n=4 mice per group for 4 and 12 hr wait, n=6 for 1 and 24 hr wait, n=12 for dark; n.s. indicates not significant, * indicates p<0.05 and ** indicates p<0.01, by one-way ANOVA).
Extended Data Figure 9. Driving 40 Hz oscillations in VC via light flicker reduces phosphorylated tau in a tauopathy mouse model.

a) Immunohistochemistry with anti-pTau (S202, green) and anti-MAP2 (red) antibodies in VC of 4-month-old P301S mice after 7 days of 1 hr/day dark or 40 Hz flicker conditions (Images were taken with 40× objective; scale bar = 50 μm). Insets: 100X rendering of representative cell body in dark and 40 Hz flicker conditions. No changes were observed by western blot (Data not shown).
b) Bar graph of relative pTau (S202) intensity levels in P301S mice after 7 days of 1 hr/day dark (black) and 40 Hz flicker (red) conditions (n=8 mice per group; * indicates p<0.05 by Student's t-test). All bar graphs show mean + SEM throughout this figure.
c) Bar graph of relative MAP2 intensity levels in P301S mice after 7 days of 1 hr/day dark (black) and 40 Hz flicker (red) conditions (n=8 mice per group; n.s. indicates not significant by Student's t-test).
d) Immunohistochemistry with anti-pTau (S396, red) antibodies in P301S mice after 7days of 1 hr/day dark and 40 Hz flicker conditions (scale bar = 50 μm).
e) Bar graph of relative pTau (S396) fluorescence intensity levels in P301S mice after 7 days of 1 hr/day dark (black) and 40 Hz flicker (red) conditions (n=8 mice per group; **** indicates p<0.0001 by Student's t-test).
f) Immunohistochemistry with anti-pTau (S404, green) antibodies in P301S mice after 7 days of 1 hr/day dark and 40 Hz flicker conditions (cale bar = 50 μm).
g) Bar graph of relative pTau (S400/T403/S404) fluorescence intensity levels in P301S mice after 7 days of 1 hr/day dark (black) and 40 Hz flicker (red) conditions (n=8 mice per group; ** indicates p<0.01 by Student's t-test). Bar graphs show mean + SEM.
h) Immunohistochemistry with anti-Iba1 (019-19741, green) antibodies in 4-month-old P301S mice after 7 days of 1 hr/day dark and 40 Hz flicker conditions (Images were taken with 40× objective; scale bar = 50 μm.) Insets: 100X rendering of representative microglia in EYFP and 40 Hz stimulation conditions.
i) Bar graph of the number of microglia in P301S mice after 7 days of 1 hr/day dark (black) and 40 Hz flicker (red) conditions (n=8 mice per group; n.s. indicates not significant by Student's t-test).
j) Bar graph of the diameter of microglia cell bodies in P301S mice normalized to dark after 7 days of 1 hr/day dark (black) and 40 Hz flicker (red) conditions (n=8 mice per group; **** indicates p<0.0001 by Student's t-test).
k) Bar graph of the average length of microglia primary processes in P301S mice normalized to control after 7 days of 1 hr/day dark (black) and 40 Hz flicker (red) conditions (n=8 mice per group; **** indicates p<0.0001 by Student's t-test).
Extended Data Table 1. Raw A β1-40 and A β1-42 concentrations.
Table displaying raw A β1-40 and A β1-42 levels with ELISA dilution ratios for each experimental group. Equal tissue masses were compared for each ELISA experiment. For 7-day experiments, values were normalized to within litter controls such that raw values 1-4 in each condition were normalized to the mean of “Dark” values 1-4; raw values 5-9 in each condition were normalized to the mean of “Dark” values 5-9; raw values 10-13 in each condition were normalized to the mean of “Dark” values 10-13.
| Treatment | Dilution Factor | Avgerage Aβ1-40 Concentration (pg/ml) | Average Aβ1-42 Concentration (pg/ml) |
|---|---|---|---|
| Optogenetics | |||
| PV-Cre EYFP | 1:2 | 100.01, 61.598, 65.462, 82.509, 69.023, 70.831, 82.152, 74.314 | 58.777, 54.546, 30.585 |
| PV-Cre 40 Hz | 1:2 | 46.604, 31.041, 26.639, 55.612, 69.326, 17.711, 3.9951 | 27.271, 41.950, 18.790, 18.262 |
| PV-Cre 8 Hz | 1:2 | 101.268, 54.283, 90.190, 151.690 | 50.699, 122.85, 35.507 |
| PV-Cre Random | 1:2 | 235.68, 89.962, 157.37, 323.902, 451.78, 241.63 | 54.029, 137.78, 144.63 |
| αCaMKII-Cre EYFP | 1:2 | 45.813, 59.069, 40.404, 66.810 | 72.052, 36.573, 67.243, 59.295 |
| αCaMKII-Cre 40 Hz | 1:2 | 55.942, 44.270, 57.498, 47.382, 115.08, 75.673 | 70.847, 79.683, 61.429 |
| αCaMKII-Cre 8 Hz | 1:2 | 52.829, 46.604, 57.720 | 95.939, 21.640, 102.987 |
| αCaMKII-Cre Random | 1:2 | 218.00, 191.72, 159.07 | 66.203, 168.867, 176.404 |
| Light flicker | |||
| Dark 1 hr VC | 1:2 | 343.8, 245.3, 210.6, 343.8, 588.4, 394.9, 123.3, 336.3, 328.2, 579.1, 420.0, 339.2 | 449.5, 320.7, 275.2, 449.5, 769.2, 516.2, 449.4, 320.6, 275.2, 449.4, 769.1, 516.1 |
| Light 1 hr VC | 1:2 | 366.9, 632.4, 378.2, 314.1, 266.9, 264.1 | 616.4, 592.3, 802.9, 394.5, 330.7, 337.8 |
| 20 Hz 1 hr VC | 1:2 | 944.4, 313.2, 595.9, 530.0, 456.5, 289.9 | 1624, 302.4, 816.0, 687.2, 676.6, 343.0 |
| 40 Hz 1 hr VC | 1:2 | 146.4, 143.6, 104.9, 99.6, 179.7, 219.8 | 191.4, 187.7, 137.2, 130.2, 234.9, 287.3 |
| 80 Hz 1 hr VC | 1:2 | 332.5, 328.7, 363.5, 390.6, 530.0, 673.3 | 558.3, 418.9, 510.7, 609.5, 1186, 921.9 |
| 40 Hz + PTX 1 hr VC | 1:2 | 367.2, 431.4, 445.2, 392.4, 386.7, 445.2 | 396.6, 540.5, 532.7, 705.0, 104.5, 104.5 |
| Random 1 hr VC | 1:2 | 461.8, 100.2, 9.819, 416.6 | 423.9, 157.9, 389.9, 841.5 |
| Dark 1 hr HPC | 1:2 | 97.949, 107.33, 119.92, 139.33 | 499.30, 355.13, 469.53, 598.03 |
| 40 Hz 1 hr HPC | 1:2 | 88.136, 104.78, 161.52, 197.36 | 364.53, 408.41, 436.62, 873.83 |
| Random 1 hr HPC | 1:2 | 95.816, 136.77, 70.004, 125.47 | 466.39, 500.87, 311.26, 582.355 |
| Dark 7 days soluble | 1:50 | 1216.9, 1181.3, 1173.4, 1199.5, 134.73, 151.34, 113.26, 145.14, 127.91, 127.48, 143.02, 127.48, 141.07 | 5217.2, 8057.9, 9051.3, 6773.7, 244.11, 236.96, 235.38, 240.62, 286.19, 8.382, 11.21, 14.03, 13.56 |
| Dark 7 days insoluble | 1:100 | 1173.2, 1208.2, 1205.3, 1214.6, 994.86, 1059.2, 1176.6, 1065.4, 1002.9, 306.16, 690.70, 3442.7, 152.73 | 8572.7, 9127.1, 6349.3, 10138, 6852.2, 7056.7, 7039.7, 7094.2, 7289.0, 748.21, 1117.1, 1055.5, 504.95 |
| 40 Hz 7 days soluble | 1:50 | 476.71, 283.83, 336.87, 237.22, 7.0175, 4.1480, 4.0580, 1.5205, 91.864, 152.73, 148.84, 141.07, 162.44 | 419.7, 248.1, 242.7, 90.974, 95.626, 56.936, 67.577, 47.586, 200.87, 13.56, 9.794, 15.44, 3.677 |
| 40 Hz 7 days insoluble | 1:100 | 281.97, 270.37, 86.199, 239.71, 23.557, 15.166, 22.714, 1038.9, 1099.8, 1760.8, 1558.8, 187.69, 22.64 | 202.96, 130.71, 195.73, 193.70, 1646.89, 1579.1, 503.44, 1400.0, 7536.62, 955.23, 1208.8, 694.57, 784.91 |
| Dark 1 hr BC | 1:2 | 81.874, 18.343, 86.554 | 391.95, 883.69, 604.97 |
| 40 Hz 1 hr BC | 1:2 | 81.307, 27.986, 30.113 | 300.34, 1152.5, 616.92 |
| 40 Hz 1 hr wait 4 hr | 1:2 | 91.06, 141.8, 111.2, 12.30 | 108.0, 168.1, 157.3, 35.158 |
| 40 Hz 1 hr wait 12 hr | 1:2 | 167.2, 101.6, 89.31, 119.9 | 236.1, 134.6, 124.8, 152.4 |
| 40 Hz 1 hr wait 24 hr | 1:2 | 246.7, 177.6, 281.2, 175.0, 257.3, 204.2 | 231.8, 107.0, 402.7, 184.6, 245.1, 179.7 |
| Dark APP/PS1 | 1:2 | 1050.16, 1085.25, 1522.45, 1153.69, 1750.77 | 19.22, 30.68, 28.08, 14.25, 25.30 |
| 40 Hz APP/PS1 | 1:2 | 512.42, 947.80, 850.45, 793.63 | 18.85, 15.58, 18.92, 11.44 |
| Dark WT | 1:1 | 0.038, 0.813, 2.016, 1.913, 0.313, 4.11, 7.23, 20.2, 40.4, 38.7, 11.9 | N/A |
| 40 Hz WT | 1:1 | 0.139, 0.325, 0.346, 0.390, 8.92, 12.1, 6.34, 12.4, 13.1 | N/A |
Supplementary Material
Extended Data Video 1. 40 Hz optogenetic stimulation causes microglia morphological transformation 3D rendering of Iba1-positive microglia and A β in hippocampal CA1 region of 5XFAD/PV-Cre mice expressing only EYFP. Immunohistochemistry was performed with anti-Iba1 (019-19741, green) and anti-A β (12F4, red) antibodies. Images were taken with 40× objective.
Extended Data Video 2. 40 Hz optogenetic stimulation causes microglia morphological transformation 3D rendering of Iba1-positive microglia and A β in hippocampal CA1 region of 5XFAD/PV-Cre mice after 40 Hz stimulation. Immunohistochemistry was performed with anti-Iba1 (019-19741, green) and anti-A β (12F4, red) antibodies. Images were taken with 40× objective.
Extended Data Video 3. 40 Hz optogenetic stimulation causes microglia morphological transformation 3D rendering of Iba1-positive microglia and A β in hippocampal CA1 region of 5XFAD/PV-Cre mice after random stimulation. Immunohistochemistry was performed with anti-Iba1 (019-19741, green) and anti-A β (12F4, red) antibodies. Images were taken with 40× objective.
Extended Data Video 4. Mouse undergoes light flicker behavioral paradigm Video of a mouse exposed to light flicker. Video shows 20 Hz because the frame rate obscures the on/off dynamics of 40 Hz light flicker.
Extended Data Video 5. 40 Hz light flicker causes microglia morphological transformation 3D rendering of Iba1-positive microglia after dark condition from CLARITY treated 100 μm tissue sections. Immunohistochemistry was performed with anti-Iba1 (019-19741, green) in 5XFAD visual cortex. Images were taken with 63× objective.
Extended Data Video 6. 40 Hz light flicker causes microglia morphological transformation 3D rendering of Iba1-positive microglia after40 Hz flicker from CLARITY treated 100 μm tissue sections. Immunohistochemistry was performed with anti-Iba1 (019-19741, green) in 5XFAD visual cortex. Images were taken with 63× objective.
Acknowledgments
We are grateful to S. Tonegawa and D. Roy for APP/PS1 mice and E. Demmons, W. Raja, E. Wu, and B. Arkhurst and the Boyden laboratory for technical assistance. We thank members of the Tsai and Boyden laboratories, C. Moore, C. Deister, D. Rei, J. Penney, R. Madabhushi, A. Mungenast, A. Bero, and J. Young for discussions and comments on the paper. H.F.I. acknowledges the Cameron Hayden Lord Foundation and Barbara J. Weedon Fellowship; E.S.B. acknowledges the New York Stem Cell Foundation-Robertson Award, NIH 1R01EY023173, and NIH 1DP1NS087724; L.H.T. acknowledges the JPB Foundation, Belfer Neurodegeneration Consortium, Halis Family Foundation, and NIH RO1 AG047661.
Footnotes
Author Contributions: H.F.I, A.C.S., E.N.B, E.S.B., and L.-H.T. designed experiments. H.F.I. and F.G. performed RNA sequencing experiments. H.F.I. and A.C.S. performed electrophysiology. A.C.S. analyzed electrophysiology data. H.F.I. performed and analyzed optogenetics and ELISA experiments. T.Z.G., J.S., and O.K. performed western blots. H.F.I., A.R., F.A., R.R., and R.G.C. performed and analyzed imaging experiments. F.G. analyzed RNA sequencing data. H.F.I., A.J.M., and C.A. performed visual stimulation experiments. H.M. performed FACS experiments. H.F.I, A.C.S., A.R., F.G., E.S.B., and L.-H.T. wrote the manuscript.
RNA-seq data available at https://www.ncbi.nlm.nih.gov/geo/ code: GSE77471. Other data is publically available upon request.
Competing financial interests: LHT and ESB are scientific founders and serve on the scientific advisory board of Cognito Therapeutics and HFI and ACS own shares of Cognito Therapeutics.
References
- 1.Bartos M, Vida I, Jonas P. Synaptic mechanisms of synchronized gamma oscillations in inhibitory interneuron networks. Nat Rev Neurosci. 2007;8:45–56. doi: 10.1038/nrn2044. [DOI] [PubMed] [Google Scholar]
- 2.Fries P, Nikolić D, Singer W. The gamma cycle. Trends Neurosci. 2007;30:309–16. doi: 10.1016/j.tins.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Cardin JA, et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palop JJ, et al. Aberrant Excitatory Neuronal Activity and Compensatory Remodeling of Inhibitory Hippocampal Circuits in Mouse Models of Alzheimer's Disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verret L, et al. Inhibitory Interneuron Deficit Links Altered Network Activity and Cognitive Dysfunction in Alzheimer Model. Cell. 2012;149:708–721. doi: 10.1016/j.cell.2012.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bero AW, et al. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Selkoe DJ, et al. The role of APP processing and trafficking pathways in the formation of amyloid beta-protein. Ann N Y Acad Sci. 1996;777:57–64. doi: 10.1111/j.1749-6632.1996.tb34401.x. [DOI] [PubMed] [Google Scholar]
- 8.Stam CJ, et al. Generalized synchronization of MEG recordings in Alzheimer's disease: evidence for involvement of the gamma band. J Clin Neurophysiol. 2002;19:562–574. doi: 10.1097/00004691-200212000-00010. [DOI] [PubMed] [Google Scholar]
- 9.Gillespie AK, et al. Apolipoprotein E4 causes age-dependent disruption of slow gamma oscillations during hippocampal sharp-wave ripples. Neuron. 2016;90:1–12. doi: 10.1016/j.neuron.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oakley H, et al. Intraneuronal beta-Amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colgin LL, et al. Frequency of gamma oscillations routes flow of information in the hippocampus. Nature. 2009;462:353–357. doi: 10.1038/nature08573. [DOI] [PubMed] [Google Scholar]
- 12.Buzsáki G. Rhythms of the brain. Oxford University Press; 2006. [Google Scholar]
- 13.Buzsáki G, et al. Hippocampal network patterns of activity in the mouse. Neuroscience. 2003;116:201–211. doi: 10.1016/s0306-4522(02)00669-3. [DOI] [PubMed] [Google Scholar]
- 14.Carr MF, Karlsson MP, Frank LM. Transient slow gamma synchrony underlies hippocampal memory replay. Neuron. 2012;75:700–713. doi: 10.1016/j.neuron.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foster DJ, Wilson MA. Reverse replay of behavioural sequences in hippocampal place cells during the awake state. Nature. 2006;440:680–3. doi: 10.1038/nature04587. [DOI] [PubMed] [Google Scholar]
- 16.Carr MF, Jadhav SP, Frank LM. Hippocampal replay in the awake state: a potential substrate for memory consolidation and retrieval. Nat Neurosci. 2011;14:147–153. doi: 10.1038/nn.2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helwig M, et al. The neuroendocrine protein 7B2 suppresses the aggregation of neurodegenerative disease-related proteins. J Biol Chem. 2013;288:1114–24. doi: 10.1074/jbc.M112.417071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Das U, et al. Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron. 2013;79:447–460. doi: 10.1016/j.neuron.2013.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cataldo AM, et al. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–286. doi: 10.1016/s0002-9440(10)64538-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gjoneska E, et al. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature. 2015;518:365–369. doi: 10.1038/nature14252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chitu V, Stanley ER. Colony-stimulating factor-1 in immunity and inflammation. Curr Opin Immunol. 2006;18:39–48. doi: 10.1016/j.coi.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell. 2015;160:1061–1071. doi: 10.1016/j.cell.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gray CM, König P, Engel AK, Singer W. Oscillatory responses in cat visual cortex exhibit inter-columnar synchronization which reflects global stimulus properties. Nature. 1989;338:334–7. doi: 10.1038/338334a0. [DOI] [PubMed] [Google Scholar]
- 24.Eckhorn R, et al. Coherent oscillations: a mechanism of feature linking in the visual cortex? Biol Cybern. 1988;60:121–30. doi: 10.1007/BF00202899. [DOI] [PubMed] [Google Scholar]
- 25.Pericic D, Bujas M. Sex differences in the response to GABA antagonists depend on the route of drug administration. Exp Brain Res. 1997;25:187–90. doi: 10.1007/pl00005681. [DOI] [PubMed] [Google Scholar]
- 26.Jankowsky JL, et al. Mutant presenilins specifically elevate the levels of the 42 residue β-amyloid peptide in vivo: Evidence for augmentation of a 42-specific β-amyloid secretase. Human Mol Genet. 2004;13:159–70. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- 27.Chung K, et al. Structural and molecular interrogation of intact biological systems. Nature. 2013;497:332–7. doi: 10.1038/nature12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yoshiyama Y, et al. Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model. Neuron. 2007;53:337–51. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 29.Canter RG, Penney J, Tsai LH. The road to restoring neural circuits for the treatment of Alzheimer's disease. Nature. 2016 doi: 10.1038/nature20412. in press. [DOI] [PubMed] [Google Scholar]
Extended Data and Methods References
- 30.Harvey CD, Collman F, Dombeck DA, Tank DW. Intracellular dynamics of hippocampal place cells during virtual navigation. Nature. 2009;461:941–6. doi: 10.1038/nature08499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aronov D, Tank DW. Engagement of Neural Circuits Underlying 2D Spatial Navigation in a Rodent Virtual Reality System. Neuron. 2014;84:442–456. doi: 10.1016/j.neuron.2014.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Buzsaki G. Theta oscillations in the hippocampus. Neuron. 2002;33:325–340. doi: 10.1016/s0896-6273(02)00586-x. [DOI] [PubMed] [Google Scholar]
- 33.Ravassard P, et al. Multisensory control of hippocampal spatiotemporal selectivity. Science. 2013;340:1342–1346. doi: 10.1126/science.1232655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. doi: 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crotti A, et al. Mutant Huntingtin promotes autonomous microglia activation via myeloid lineage-determining factors. Nat Neurosci. 2014;17:513–521. doi: 10.1038/nn.3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Erny D, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. 2015;18:965–977. doi: 10.1038/nn.4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chiu IM, et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013;4:385–401. doi: 10.1016/j.celrep.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gosselin D, et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell. 2014;159:1327–1340. doi: 10.1016/j.cell.2014.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang SCC, et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol. 2014;15:846–855. doi: 10.1038/ni.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yu H, et al. Tet3 regulates synaptic transmission and homeostatic plasticity via DNA oxidation and repair. Nat Neurosci. 2015;18:836–43. doi: 10.1038/nn.4008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Extended Data Video 1. 40 Hz optogenetic stimulation causes microglia morphological transformation 3D rendering of Iba1-positive microglia and A β in hippocampal CA1 region of 5XFAD/PV-Cre mice expressing only EYFP. Immunohistochemistry was performed with anti-Iba1 (019-19741, green) and anti-A β (12F4, red) antibodies. Images were taken with 40× objective.
Extended Data Video 2. 40 Hz optogenetic stimulation causes microglia morphological transformation 3D rendering of Iba1-positive microglia and A β in hippocampal CA1 region of 5XFAD/PV-Cre mice after 40 Hz stimulation. Immunohistochemistry was performed with anti-Iba1 (019-19741, green) and anti-A β (12F4, red) antibodies. Images were taken with 40× objective.
Extended Data Video 3. 40 Hz optogenetic stimulation causes microglia morphological transformation 3D rendering of Iba1-positive microglia and A β in hippocampal CA1 region of 5XFAD/PV-Cre mice after random stimulation. Immunohistochemistry was performed with anti-Iba1 (019-19741, green) and anti-A β (12F4, red) antibodies. Images were taken with 40× objective.
Extended Data Video 4. Mouse undergoes light flicker behavioral paradigm Video of a mouse exposed to light flicker. Video shows 20 Hz because the frame rate obscures the on/off dynamics of 40 Hz light flicker.
Extended Data Video 5. 40 Hz light flicker causes microglia morphological transformation 3D rendering of Iba1-positive microglia after dark condition from CLARITY treated 100 μm tissue sections. Immunohistochemistry was performed with anti-Iba1 (019-19741, green) in 5XFAD visual cortex. Images were taken with 63× objective.
Extended Data Video 6. 40 Hz light flicker causes microglia morphological transformation 3D rendering of Iba1-positive microglia after40 Hz flicker from CLARITY treated 100 μm tissue sections. Immunohistochemistry was performed with anti-Iba1 (019-19741, green) in 5XFAD visual cortex. Images were taken with 63× objective.
Data Availability Statement
Data is publically available upon request from the corresponding author.