

Abstract
Purpose
Optic nerve hypoplasia (ONH) is the most common cause of childhood congenital blindness in developed nations, yet the fundamental pathobiology of ONH remains unknown. The objective of this study was to employ a ‘face validated' murine model to determine the timing of onset and the pathologic characteristics of ONH.
Methods
Based on the robust linkage between X-linked CASK haploinsufficiency and clinically diagnosed ONH, we hypothesized that heterozygous deletion of CASK (CASK(+/−)) in rodents will produce an optic nerve pathology closely recapitulating ONH. We quantitatively analyzed the entire subcortical visual system in female CASK(+/−) mice using immunohistochemistry, anterograde axonal tracing, toluidine blue staining, transmission electron microscopy, and serial block-face scanning electron microscopy.
Results
CASK haploinsuffiency in mice phenocopies human ONH with complete penetrance, thus satisfying the ‘face validity'. We demonstrate that the optic nerve in CASK(+/−) mice is not only thin, but is comprised of atrophic retinal axons and displays reactive astrogliosis. Myelination of the optic nerve axons remains unchanged. Moreover, we demonstrate a significant decrease in retinal ganglion cell (RGC) numbers and perturbation in retinothalamic connectivity. Finally, we used this mouse model to define the onset and progression of ONH pathology, demonstrating for the first time that optic nerve defects arise at neonatally in CASK(+/−)mice.
Conclusions
Optic nerve hypoplasia is a complex neuropathology of the subcortical visual system involving RGC loss, axonopathy, and synaptopathy and originates at a developmental stage in mice that corresponds to the late third trimester development in humans.
Keywords: optic nerve hypoplasia, CASK, axonopathy, synaptopathy
Childhood blindness (prevalence ∼0.7/1000) poses a huge socioeconomic challenge due to early affliction.1 Optic nerve hypoplasia (ONH) is the most common form of optic neuropathy and accounts for 15% to 25% of childhood blindness in developed nations.2 Optic nerve hypoplasia is not always associated with vision loss,2 and may be associated with other ophthalmologic manifestations like microphthalmia, aniridia, nystagmus, and strabismus.3 Fundoscopy detects ONH as a small pale optic disc and there may be an increase in the ratio of disc-macula to disc diameter (DM:DD, the ratio of the horizontal distance between the center of the optic disc and the macula to the mean diameter of the optic disc).3 Sometimes, the optic disc also appears to be surrounded by double rings in fundoscopic images.4 The first case of ONH was described in 1884 and at present it is classified into three distinct forms: (1) ONH simplex, (2) septo-optic dysplasia (SOD), and (3) septo-optic-pituitary dysplasia.2
The etiopathogenesis of ONH and its differences from perinatal optic nerve atrophy (ONA) remains unclear.5 Maternal age and primiparity are the most consistent risk factors associated with ONH.6 Although ONH has been observed in a murine model of fetal alcohol syndrome,7 large scale studies have failed to uncover evidence for association of ONH with maternal consumption of alcohol and other recreational drugs.8 Most cases of ONH are sporadic in nature, with only few genes that have been directly implicated. For instance, mutations in HESX1 may be associated with SOD in limited number of cases.9,10 Similarly mutations in transcription factors like PAX6, ATOH7, SOX2, OTX2, and VAX1 have also been clinically associated with ONH.11 Involvement of transcription factors in a developmental disorder is not surprising because they regulate several cell biological pathways crucial for cell survival and differentiation. Mutations in these genes in animal models therefore produce numerous phenotypes including those in eyes, most notably microphthalmia/anophththalmia.9,12,13 Large scale cellular loss in the retinal ganglion cell (RGC) layer, producing optic nerve agenesis or dysgenesis, has also been noted.14 Besides transcription factors, genes involved in metabolism, RNA splicing, chromatin remodeling, and dytroglycan glycosylation have also been reported to be associated with occasional ONH.11 For many of these genes either a good mouse model is not available or the mouse model does not accurately replicate the human conditions.11,15,16
The majority of cases of ONH, however, have no defined genetic association and there is a paucity of genotype–phenotype correlation of patients with ONH.6 Mutations in the X-linked gene CASK encoding a peripheral scaffolding protein have been strongly associated with ONH17 and mutations in CASK therefore represent a potential avenue to generate a novel model of ONH for mechanistic and pathologic evaluation.
CASK was initially described as a candidate gene for X-linked ONA18 and mutations in CASK are associated with ONH.17,19,20 We report new subjects with CASK mutations and ONH, further confirming the association, and we show that heterozygous deletion of CASK causes optic nerve pathology indicating that these phenotypes represent CASK loss of function. Furthermore, our results demonstrate that the optic nerve of CASK(+/−) mice have atrophic axons with increased interaxonal space and astrogliosis. The numbers of retinogeniculate synapses are reduced with specific loss of smaller boutons; furthermore the large boutons exhibit an overall decrease in number of active zones. In retina, we observe a decrease in number of RGCs in CASK(+/−) mice. Finally, we demonstrate that the optic nerve pathology in CASK(+/−) mice occurs very early in postnatal rodent developmental, a stage considered equivalent to the late third trimester and neonatal period of human fetal development.21 Haploinsufficiency of CASK therefore produces a combination of retinopathy, optic nerve pathology, as well as synaptopathy. Thus, our results demonstrate that a face validated mouse model provides new insight into the pathogenesis and timing of ONH.
Methods
Ethical Statement
The Virginia Tech Universities institutional review board approved the collection and use of data from subjects. Informed consent was obtained from the family prior to participation. All animal work was done in accordance to with the guidelines for the animal care of laboratory animals issued by Virginia Tech and adhered to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Identification of a Splice Mutation in CASK
We performed whole-exome sequencing (WES) on genomic DNA of the patient. Using the Illumina HiSeq2000 platform (Illumina, San Diego, CA, USA) and the acquired reads were aligned to the reference human genome (UCSC Genome Browser hg19. Data processing was performed with the Genome Analysis ToolKit (GATK) and variants were annotated using Annovar (http://annovar.openbioinformatics.org/en/latest/, in the public domain) and an in-house developed web interface called Annotate-it.22 In the first stage, we analyzed all genes that were previously associated with microcephaly. The heterozygous splice variant in exon 24 (NM_001126054:exon24:c.2233+1G>A) of CASK, was predicted to lead to exon skipping, and was confirmed by Sanger sequencing. The variant arose de novo and was absent in both biological parents. To further test the effect of the mutation, we performed PCR with primers in exons 25 and 23 on cDNA made from RNA extracted from Epstein-Barr virus–transformed lymphocytes of the patient.23 The RT-PCR on the patient clearly showed two bands and sequencing of the smallest band confirmed exon 24 skipping.
Mouse Breeding and Genotyping
CASK(+/−) female mice, generated in house,24 were crossed with C57BL6 male mice to propagate CASK(+/−) female pups and their CASK(+/+) female littermates. Genotyping was done using a PCR-based method with following primer pair (forward: TTTGGGGACTAGATGGGTGTGGTG, reverse: CTTGGTCGCAGCTTGGGAGTA).
Immunohistochemistry (IHC)
Mice were anesthetized and perfused with PBS and then 4% paraformaldehyde (PFA). Brain, retina, or optic nerve were dissected and postfixed in 4% PFA. Tissues were cryopreserved in 30% sucrose before embedding in Neg50 matrix (Richard-Allan Scientific, San Diego, CA, USA) and cryosectioned using Leica Cryostat (Leica Biosystems, Wetzlar, Germany) to generate 16- to 25-μm thick tissue sections. Sections were permeabilized and blocked with 0.25% Triton X-100 and 5% BSA in PBS. Immunostaining were performed using primary antibodies at an appropriate concentration followed by labeling with fluorophore conjugated secondary antibodies. Primary antibodies include: Calretinin (rabbit polyclonal Ab, Cat. No. AB5054, 1:2000; Millipore, Billerica, MA, USA), RNA-binding protein with multiple splicing (RBPMS; rabbit polyclonal Ab, Cat. No. 1830, 1:500; PhosphoSolution, Aurora, CO, USA), glial fibrillary acidic protein (GFAP; mouse monoclonal Ab, Cat. No. 75240, 1:1000; Neuromab, UC Davis, CA, USA). Secondary antibodies include: Alexa Fluor 488 (goat anti-rabbit, Cat. No. A-11008, 1:1000; Invitrogen) and Dylight 633 (anti-mouse IgG, Cat. No. 35512, 1:1000; Thermo Scientific, Waltham, MA, USA). Sections were mounted with VectaShield containing 4′,6-diamidino-2-phenylindole (DAPI; Vector Laboratories, Burlingame, CA, USA) and were imaged using a Zeiss 700 or 710 laser scanning confocal microscope (Oberkochen, Germany).
Immunoblotting
Optic nerves dissected from young female wild-type and CASK(+/−) mice (postnatal day [P] 40) were boiled in sample buffer and proteins separated by 8% SDS-PAGE. Proteins were then transferred onto nitrocellulose membrane and blocked in 5% skimmed milk for 2 hours and incubated using appropriate primary and secondary antibodies. For quantitative immunoblots, secondary antibodies conjugated with fluorophore were used and images were captured using appropriate filter in Chemidoc (Biorad, Hercules, CA, USA). Antibodies used are CASK (mouse monoclonal Ab, Cat. No. 75000, 1:1000; NeuroMab), neurofilament-associated antigen (NFAA; mouse monoclonal Ab, Cat. No. 3A10, 1:1000l; DSHB, Iowa City, IA, USA), and tubulin (mouse monoclonal Ab, Cat. No. 12G10 anti-alpha-tubulin, 1:1000; DSHB).
Optic Nerve Samples Preparation for Toluidine Blue Staining and Transmission Electronic Microscopy (TEM)
Mice were anesthetized and perfused with PBS and then sodium cacodylate (NaCa) buffer (2% glutaraldehyde/2% PFA [2G/2PF] in 0.1 M sodium cacodylate in PBS, pH 7.4). After perfusion, optic nerves were dissected and immersed in NaCa buffer overnight and then put in 2% OSO4 for 2 hours for postfixation. Dehydration was achieved by sequentially adding 15% to 100% ethanol. Samples were embedded in 100% epoxy resin for 24 hours and then placed in oven at 58°C for 2 days. Sectioning of samples and successive toluidine blue stain or TEM was performed at Virginia-Maryland College of Veterinary Medicine at Virginia Tech.
G-Ratio Distribution Spectra
G-ratio, defined as the ratio of inner axonal diameter to the outer axonal diameter (including myelin sheath),25 of 240 optic nerve axons were measured on TEM images (×4000) using ImageJ software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA). The measurements of inner and outer axonal diameters (d) were calculated from inner and outer axonal areas (a), respectively, to reduce error because the sections were not perfectly round in shape, where 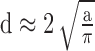 . Probability density distribution of G-ratio values was plotted based on a smooth kernel density estimate by using the function of “SmoothHistogram” offered in the software of Mathematica (Wolfram, Champaign, IL, USA). The smooth kernel density estimate based probability density function is given by a linearly interpolated version of
. Probability density distribution of G-ratio values was plotted based on a smooth kernel density estimate by using the function of “SmoothHistogram” offered in the software of Mathematica (Wolfram, Champaign, IL, USA). The smooth kernel density estimate based probability density function is given by a linearly interpolated version of
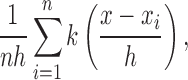 |
where x is the value of G-ratio, n is the sample size, bandwidth h is selected as “Automatic” and kernel smoothing function k(x) was given by “Gaussian” (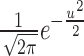 , u ∈ ℝ) in Mathematica. Above functions can be found in any advanced level statistics textbook. The Kolmogorov-Smirnov test was used to determine if the two probability density distributions are from the same population.
, u ∈ ℝ) in Mathematica. Above functions can be found in any advanced level statistics textbook. The Kolmogorov-Smirnov test was used to determine if the two probability density distributions are from the same population.
Serial Block-Face Scanning Electron Microscopy
Control and CASK(+/−) mice were transcardially perfused with PBS and 4% paraformaldehyde/2% glutaradehyde in 0.1 M cacodylate buffer. Brains were removed, 300-μm coronal sections were obtained using vibratome and dorsolateral geniculate nucleus (dLGN) were dissected. Tissues were stained, embedded, sectioned, and imaged as described previously26 by Renovo Neural, Inc. (Cleveland, OH, USA). Images were acquired at a resolution of 6 nm/pixel and image sets included greater than 200-serial sections (with each section representing 65 nm in the z axis). Data sets were analyzed in TrakEM2 and retinal terminals were unequivocally identified by the presence of synaptic vesicles and pale mitochondria.26,27
Intraocular Injections of Anterograde Tracers and Quantification
Intraocular injection of cholera toxin subunit B (CTB) conjugated to AlexaFluor 555 (Invitrogen) was performed as described previously.28 After 2 days, mice were euthanized, perfused with 4% PFA, and brains were dissected and postfixed in 4% PFA overnight. On a vibratome, 80- to 100-μm coronal slices were sectioned and mounted using VectaShield (Vector Laboratories). Confocal Z-stack images from coronal dLGN sections were acquired on a Zeiss LSM 700 confocal microscope. Z-stacks were obtained with a Zeiss ×20 Plan-Apochromat objective and contained 14- to 20-optical sections (in 3-μm steps). Confocal images were obtained from four adult CASK(+/−) mutants and four littermate controls. The size and quantity of synapses in each dLGN (from the center of its rostral-caudal extent), was performed in ImageJ, blind to the genotype of the sample. Five images of each dLGN Z-stacks were randomly selected, images were manually thresholded, and the quantity and surface area of the isolated puncta was measured. Data was exported into Microsoft Excel (Redmond, WA, USA), binned by size, and plotted into a histogram.
Cell Counts and Lamina Quantification
Cell counts were performed on 16-μm cryosectioned 4% PFA-fixed retinal tissue as described previously.29,30 Slides were stained with DAPI (diluted 1:5000 in PBS) for 60 seconds and then mounted with VectaShield (Vector Laboratories). Images of central retina were acquired on a Zeiss LSM 700 confocal microscope or a Zeiss Axio Imager A2 fluorescent microscope. All DAPI and RBPMS-positive cells in the ganglion cell layer were counted manually under ×20 objective. Cell counts were analyzed from at least four animals for each age and genotype. Five images per retina per mouse were analyzed. Retinal lamination was quantified for each genotype in the central retina in DAPI-stained retinas using ImageJ.
Description of Coarse and Fine Axons
Because the axons in CASK(+/−) mice are overall small, it is difficult to apply the same threshold for fine and coarse axons on these mice as with those of wild type. On examining the distribution of the axonal area we found they are distributed in a bimodal manner (2 different slopes; see Fig. 3C). Because the CASK(+/−) axons were smaller we applied a different threshold for each genotype, the value of the threshold was the point where the data points start to lose linearity (see Fig. 3C).
Figure 3.

CASK(+/−) ON display axonal thinning. (A, B) Representative transmission electron micrograph of optic nerves from indicated genotypes. (C) Scatter plot of individual axonal areas from control and mutant ON obtained from three mice per genotype. Arrows indicate the “threshold” where the data points start to lose linearity (correlation coefficient R2 value starts and continue decreasing). Axons with area values that are greater than or equal to the threshold are considered as coarse axons, and axons with volume values that are smaller than the threshold are considered as fine axons. Threshold value for (+/+) is 4 and (+/−) is 2 μm2. (D) Mean and SDs of all axons from (C) are plotted, N = 240. (E, F) Comparisons of fine and coarse axons area in mutant (+/−) and control (+/+) ON (n = 3 mice per genotype). Data are plotted as mean ± SD; N = 240. (G) G-ratio probability density distributions of myelinated optic nerve axons of (+/+) and (+/−), N = 240. (H) Immunoblot of optic nerves from six randomly selected animals of indicated genotype. The antigens are indicated, NFAA is neurofilament associated antigen, which is axonal marker. (I) Quantitation of the blots normalized to ponceau staining. Data are relative to wild-type levels and is plotted as mean ± SEM, n = 3 (all panels, *P < 0.05; **P < 0.01).
Results
Mutations in the CASK Gene are Associated With ONH
Mutations in the CASK gene have been previously associated with ONH, ONA, and glaucoma.17,20 Here we describe a 3-year-old girl with a heterozygous splice site mutation in CASK (NM_001126054:exon24:c.2233+1G>A) leading to skipping of exon 24 that exhibits microcephaly, global developmental delays, and spastic quadriparesis (Figs. 1A–C). Magnetic resonance imaging (MRI) did not reveal any specific midline structural defect as seen in septo-optic dysplasia or any white matter lesion (Figs. 1D, 1E). Although truncated proteins may be generated in the subject from the mutated CASK allele, they are unlikely to be functional in absence of the Hook motif and the guanylate kinase domain, especially because the interdomain interaction with the guanylate kinase domain is critical for proper folding and function of membrane-associated guanylate kinase proteins.31,32 Furthermore, the transcripts with premature stop codon are likely to get degraded due to nonsense-mediated decay. Thus, the pathogenic mutation described here is likely to produce haploinsufficiency. The girl displays ONH and other ophthalmic conditions including hyperopia, nystagmus, and infantile esotropia. Fundoscopy revealed a bilaterally normal anterior segment, however both optic discs were pale, small, and hypoplastic (Fig. 1F). Hypoplastic optic nerves have also been observed in both MRI and fundoscopic examination of a second girl with heterozygous nonsense mutation in the N-terminal calcium/calmodulin-dependent kinase domain of CASK (c.661G>T; p.G221X [het]) (personal communications). This highly premature stop codon would either result in a nonfunctioning small peptide or no CASK protein product due to nonsense-mediated decay. The second girl displays severe visual impairment as well as nystagmus and intermittent exotropia (Mukherjee K, personal communications, 2017). These observations, and a previously published report,17 indicate that heterozygous loss of function mutations of CASK gene associates with ONH.
Figure 1.

Genetic, brain, and retinal alterations associated with heterozygous CASK mutation. (A) Sanger sequences showing the de novo heterozygous splice variant in exon 24 (NM_001126054:exon24:c.2233+1G>A). (B) The schematic of exon skipping. (C) CASK. Reverse-transcription PCR with primers (arrows in panel B) in exons 25 and 23 on cDNA of the patient and subsequent sequencing confirmed exon 24 skipping (a lower band indicated by the arrow compared with control). (D, E) Magnetic resonance images of brain indicate no specific midline structural defect or white matter lesion. (F) A fundoscopic image from this haploinsufficient 3-year-old girl who was diagnosed with ONH. Note the pale, hypoplastic optic disc (as compared with a fundoscopy image from a 3.5-year-old girl without ONH; Supplementary Fig. S1).
Heterozygous Deletion of CASK in Mice Produces Optic Nerve Pathology
To test whether heterozygous mutation of CASK is sufficient to alter the ON and subcortical visual system in mice, we analyzed female mouse mutants with a single allele of CASK deleted (CASK(+/−) mice). Previously, we documented that CASK(+/−) mice exhibit secondary microcephaly, disproportionate cerebellar hypoplasia, as well as thinning and hypoplasia of the optic nerve indicating that it recapitulates the human phenotypes associated with CASK mutation or loss.24 We examined optic nerves following toluidine blue staining of semithin cross-sections—a method that labels myelin and allows the quantification of myelinated retinal axons in the ON. Surprisingly, besides being small (Figs. 2A, 2B), the optic nerve displayed an overall decrease in axonal density (Figs. 2C, 2D). The number of axons in a given area was reduced by approximately 25% and the interaxonal space was expanded (Fig. 2E). Histopathology on a single ONH case suggested that there might be an increase in number of astrocytes in ONH.33 We therefore examined the optic nerve of CASK(+/−) mice for levels of GFAP, a marker of astrocytes. We found a large increase in the GFAP staining in the optic nerve of CASK(+/−) mice compared with sex-matched littermate controls suggesting that haploinsufficiency of CASK gene may be associated with increased astrocytes in optic nerve (Fig. 2F).
Figure 2.

CASK(+/−) ON display reduced axons and increased astrogliosis. (A, B) Representative images of semithin cross section of optic nerves from indicated genotype stained with toluidine blue. Scale bar: 100 μm. (C, D) Higher magnification of toluidine blue–stained semithin optic nerve sections of indicated genotype. Note that CASK(+/−) optic nerve has lesser axonal density and increased interaxonal space (indicated by arrows). (E) Axonal density from optic nerve of four different mice is quantified, data is plotted as mean ± SEM. (*P < 0.05). (F) Glial fibrillary acidic protein immunolabeling of mutant (+/−) and control (+/+) optic nerves. Scale bar: 50 μm. (G) Quantification of pixels of GFAP per unit area of optic nerve has been plotted as mean ± SEM; n = 4 mice.
Axonopathy is Present in Optic Nerves of CASK(+/−) Mice
A reduced axonal density in the CASK(+/−) mice optic nerve may reflect not only a loss of RGC axons, but also a thinning of individual axons of RGCs. Very small atrophic axons may escape counting from toluidine blue–stained semithin sections. We therefore performed TEM on cross sections of ON and quantified axons in CASK(+/−) mutants (Figs. 3A, 3B). Indeed, our data indicate that the cross-sectioned area of individual axons was significantly decreased in CASK(+/−) mice (Figs. 3A–D), with the presence of many very small axons in mutant ON. Because optic nerves contain populations of coarse and fine retinal axons,34,35 we measured the area of coarse and fine axons separately. Our data demonstrate that the area of both the types of axons are decreased in CASK(+/−) mice (Figs. 3E, 3F).
We next turned our attention to myelin content in mutant ON, to assess whether the reduction in ON diameter reflects myelin loss. G-ratio distribution spectra, which assess the ratio of axonal diameter to the diameter of the myelinated axon,25 were applied to compare myelin of wild-type littermate mice with that of the CASK(+/−) mice. The probability density distribution of G-ratios in mutant mice appears shifted to the left, indicating a higher myelin-to-axon ration compared with controls (Fig. 3G). We interpret these results to indicate that while CASK haploinsufficiency influences axon diameter it does not appear to limit the development of compact myelin on these axons.
Because CASK is an X-linked gene and is therefore subject to X-linked random inactivation, two types of RGCs exist in CASK(+/−) mutant mice, those which produce CASK and those which do not. Differences in axonal diameter of CASK+ and CASK− RGCs should produce an increased variability in axonal diameter within the ON in these studies. We therefore measured the mean of deviation (how far on average all values are from the mean), which is a measure of variability of the axonal areas. Surprisingly, the mean deviation for the axonal area in CASK(+/−) optic nerve is smaller than the wild type (Supplementary Fig. S2) indicating a tighter distribution. Overall, the distribution of the area of individual axons was smaller but roughly parallel in CASK(+/−) mice and their sex-matched wild-type littermate indicating random inactivation of deleted CASK allele had no bearing on the development or health of individual axons (Fig. 3C). Indeed, we have previously documented that CASK promotes postnatal brain growth in a non-cell autonomous manner24 (i.e., CASK+ and CASK− cells are equally affected). We therefore tested if there is specific loss of only CASK negative axons in CASK(+/−) mice. Specific loss of axons from cells, which are CASK negative will lead to an artificial skewing of RGCs toward being CASK positive. To determine if such is the case, we blotted optic nerve homogenates for CASK, an axonal marker NFAA, and a general cellular marker tubulin-α (Fig. 3H). Our data suggest that there is a slight reduction in NFAA, which did not reach a statistical significance. Our data thus indicate that the decrease in axonal count from toluidine blue–stained section may indeed be an overestimate due to omission of very small gauge atrophic axons. However, the CASK content within optic nerve was approximately 47%. Secondary selection of CASK positive RGCs would have produced a favorable skewing of X-chromosome inactivation among RGCs.36 Under such circumstances, we would not expect to see a decrease in CASK protein amount (Fig. 3I). Therefore, our data suggest that ONH in CASK(+/−) mice may not be due to specific loss of CASK negative axons.
Retinogeniculate Synaptopathy is Observed in CASK(+/−) Mice
Axonopathy may alter axonal transport and produce defects in synapse formation.37 CASK has also been shown to be both a trafficking molecule38 and a synaptic scaffolding protein. It is also known to interact and phosphorylate presynaptic adhesion molecule neurexin in an activity dependent manner.39,40 Neurexins are synaptogenic,41 therefore, we examined retinogeniculate synapses in CASK(+/−) mice. It is pertinent to note in previous animal model studies involving worms, flies, and mice, deletion of CASK did not produce defects in synapse formation.42–44 We initially examined RGC terminals in thalamus by anterogradely labeling RGCs with intraocular injection of fluorescently conjugated cholera toxin subunit B. We found that there is a significant reduction in small diameter RGC terminals in CASK(+/−) mice compared with wild-type littermate (Fig. 4A). Because of this change in terminal populations, we examined the ultrastructure of RGC terminals using serial block-face scanning electron microscopy. With this technique, retinal terminals can be unequivocally identified based on their ultrastructural morphology.42 Overall synapse volume in CASK(+/−) mice remained largely unchanged (Figs. 4B–D). In order to test if the defect associated with CASK haploinsufficiency is restricted to smaller synapses only or not, we carefully analyzed the ultrastructure of all reconstructed RGC terminals. Our data demonstrate that the number of active zones (release sites) per unit volume of RGC terminals are reduced in CASK(+/−) (Figs. 4E, 4F). Furthermore, smaller terminals have significant increase in volume (Figs. 4G, 4H). Thus, besides the thinning of axons produced by CASK haploinsufficiency we also observed defects in retinogeniculate synapse morphology and ultrastructure.
Figure 4.
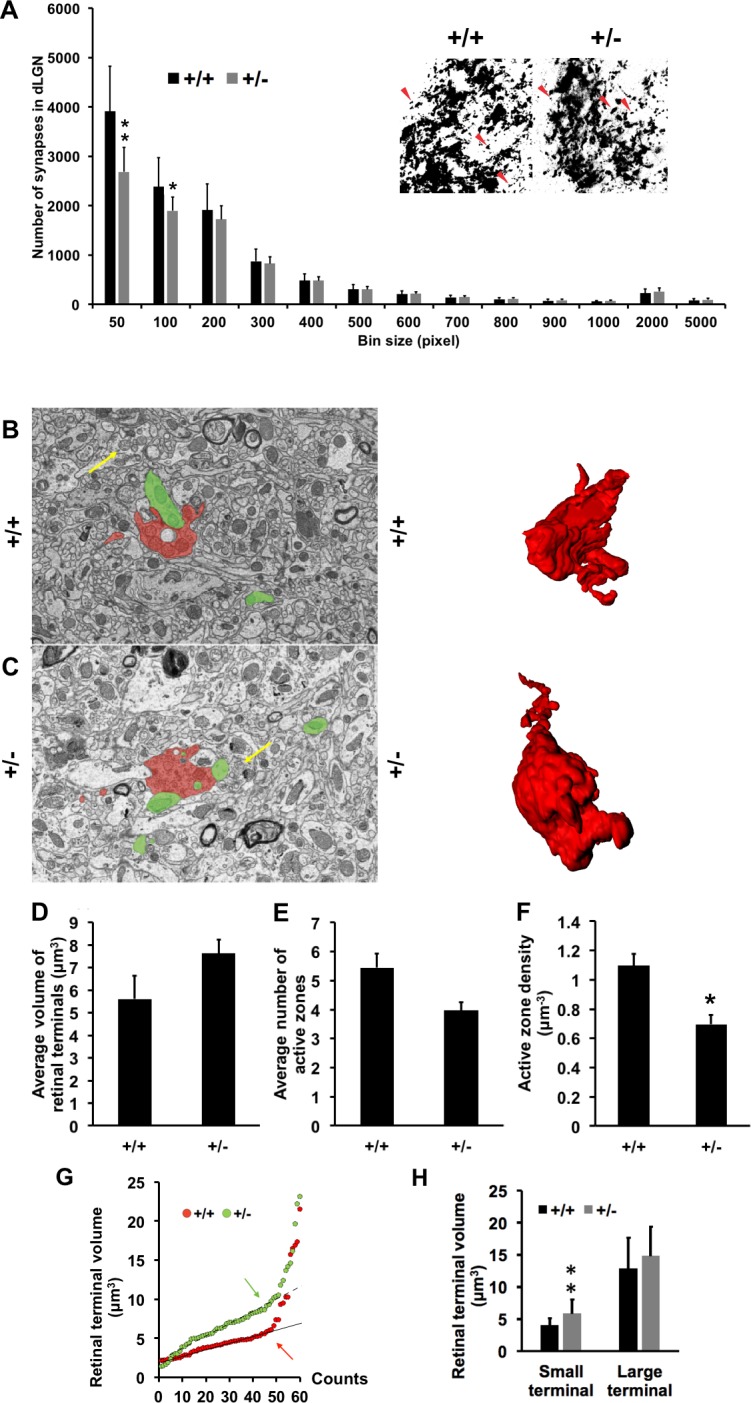
CASK(+/−) mice display a decrease number of retinogeniculate synapses and a reduced number of active zones in the remaining synapses. (A) Quantification of the area of retinal terminals in dLGN by anterograde CTB-labeling. Red arrows represent distinct CTB-labeled retinal terminals of different sizes. Data represents mean ± SEM, n = 4. (B, C) Representative image of Serial Block-Face Scanning Electron Microscopy (SBFSEM) ultramicrograph with a retinogeniculate presynapse (identified by presence of pale mitochondria indicated with yellow arrows) labeled red and a postsynapse labeled green. Examples of reconstructed retinal terminals from a series of SBFSEM micrographs are shown on the right. Volumetric quantitation (D), number of active zones (E), and active zone density (F), of the presynaptic boutons from 60 reconstructed retinogeniculate synapses (SBFSEM analysis) from control and mutant mice. Data represent mean ± SEM, n = 3 mice per genotype. (*P < 0.05; **P < 0.01). (G) Scattered plot of terminal volumes showing two linear regressions for (+/+) and (+/−). Arrows indicate the threshold where the data points start to lose linearity (correlation coefficient R2 value starts and continue decreasing). Terminals with volumes greater than or equal to threshold are classified as large terminals, and terminals with volume values that are smaller than the threshold are considered as small terminals. Threshold value for (+/+) is 7 and (+/−) is 10 μm3. (H) Comparisons of small and large axons between CASK(+/+) and CASK(+/−) by terminal volume (bouton), data are plotted as mean ± SD; n = 3.
RGC Numbers are Reduced in CASK(+/−) Mice
CASK is present in all retinal cells45 and because we detected both a thinner optic nerve and a lowered axonal density in optic nerve of CASK(+/−) mice, we next examined if there was also a loss of retinal integrity or RGCs in CASK(+/−) mice. Immunolabeling mutant and control retinal cross sections with antibodies against Calretinin (Calr) (Fig. 5A), revealed that the general cytoarchitecture and laminated structure of the retina was preserved in the absence of CASK. In an order to be comprehensive in our analysis we also measured the thickness of other layers of the retina to evaluate if these layers are affected by CASK loss. Our data indicate the lamination of retinal layers is not perturbed in CASK(+/−) mice (Supplementary Fig. S3).
Figure 5.

Decreased number of retinal ganglion cells in CASK(+/−) mice. (A) Immunostaining of retina with antibodies against calretinin reveals normal lamination in CASK(+/−) mutants at P26. (B) Representative images of retinas stained with DAPI and RBPMS (a marker of RGCs) reveal a significant loss of cells in the ganglion cell layer in CASK(+/−) mutants compared with controls. Scale bar: 25 μm. Lower panel is a grayscale image for better visualization. (C) Quantition of cells in the ganglion cell layer in indicated staining. (*P < 0.05). Data are relative to wild-type levels and is plotted as mean ± SEM, n = 4.
To assess whether there was a specific loss of retinal ganglion cells in CASK(+/−) mice we counted the total number of cells within the ganglion cell layer of the retina, by labeling nuclei with DAPI and staining for RBPMS, a RNA binding protein selectively expressed by RGCs in the rodent retina(Fig. 5A). Together these data reveal a significant loss of RGCs in CASK(+/−) mutants (Figs. 5A, 5B). Taken together, we find that ONH in CASK(+/−) mice is a complex developmental disorder involving a decrease in the number of RGCs, thinning of individual axons, reactive astrogliosis within the ON, and alterations in retinogeniculate synapses. Thus, mutation of CASK appears to phenocopy many of the features observed in human patients with ONH, and therefore represents an appropriate mouse model to elucidate the mechanisms underlying ONH.
ONH Develops Postnatally in CASK(+/−) Mice
The first mechanistic question regarding ONH pathogenesis that has eluded our understanding is when exactly the disease is initiated. The decrease in RGCs and their axons could occur pre- or postnatally. While it is difficult to parse these differences out in humans, in this mouse model the difference can be studied by observing ON diameter throughout development. We therefore examined toluidine blue–stained semithin section of CASK(+/−) and control optic nerves during the first 3 weeks of postnatal mouse development (Fig. 6A). It is important to point out that the first 2-postnatal weeks of rodent development correspond to developmental milestones that occur in the third trimester of human fetal development.21 Moreover, neonatal mice do not open their eyes until the end of the second postnatal week of development. Surprisingly, the ON of P1 CASK(+/−) mice was of similar size to that of the wild-type controls (although, a slight trend toward being smaller was observed). A more clear and significant difference between the size optic nerves became apparent by the end of the first week of postnatal development and the difference from the wild type reaches the difference observed in adults by P22 (Fig. 6B). Although myelinogenesis starts around P5 in mouse ON it peaks during the third and fourth postnatal week.46 By P22 myelinated axons are easily quantifiable in the ON. To confirm that the decrease in axonal density occurs at an early age and not as a part of later degeneration, we examined the number of axons in P22 optic nerve per unit area. Our data indicate that axonal density is already reduced (at par as noted in adults Fig. 2C) by this stage suggesting that CASK-linked ONH can clearly be classified as a developmental and not a degenerative process. Because, we observed thinning of optic nerve only from P6, we repeated the measurement of RGCs at P6. Our data indicates that at P6 there is no reduction in RGC numbers in CASK(+/−) mice, indicating a potential loss of RGCs early in development (Figs. 6D, 6E). Overall, our data demonstrate that ONH in CASK(+/−) mice occurs early in postnatal development (P6–P22), prior to the full maturation of the subcortical visual system.
Figure 6.

CASK(+/−) mice optic nerve display secondary reduction in size. (A) Representative images of toluidine blue–stained semithin-sections derived from optic nerve of indicated ages and genotypes. Scale bars: 100 μm. (B) Quantitation of optic nerve areas from mice of indicated age and genotype. Data are plotted as mean ± SEM, n = 3. (C) Quantitation of axonal density. Data are plotted as mean ± SD. (*P < 0.05; **P < 0.01). (D) Representative images of retinas from P6 mice stained with DAPI and RNA-RBPMS reveal no change in cell number in the ganglion cell layer of CASK(+/−) mutants compared with controls. Scale bar: 25 μm. (E) Quantification of DAPI- and RBPMS-positive cells in the ganglion cell layer in P6 mutant and control retina.
Discussion
Optic nerve hypoplasia is the most common cause of childhood blindness in developed nations, and its prevalence is growing at an alarming rate.47 A validated genetic animal model of ONH recapitulating the clinical condition with high fidelity will serve not only as a necessary tool to understand the mechanism of disease pathogenesis, but also to test potential therapeutic interventions. Optic nerve hypoplasia may occur with midline defects as observed in SOD, with other brain malformations like atrophic brain or even as isolated condition.6 The coincidence of ONH with other brain malformation is not surprising given that the retina and optic nerves are a part of our central nervous system and subject to the same developmental processes as the brain. Therefore, a clear understanding of ONH may also help unlock the pathophysiology of other neurodevelopmental disorders. In fact, subjects with ONH display a high incidence of autistic phenotype such as stereotypy.48,49 In this regard, mutations in CASK gene as a causal factor for ONH is particularly important because CASK mutations are also known to produce autistic traits.47 CASK directly binds and phosphorylates neuronal adhesion molecule neurexin.39,40 It is pertinent to note that mutations in both neurexins and their transsynaptic interacting partners also are associated with autism.50 Despite known interactions between CASK and neurexin, no synaptic ultrastructural defect has been shown to be associated with CASK mutation in any animal model. Here, we show for the first time clear morphologic defects at retinogeniculate synapse involving changes in size and number of release site. Such changes align with the idea that CASK may be involved in trafficking of molecules and/or acts as a molecular scaffold at the synapse. Although it may also be possible that changes in synapses reflect the atrophy in axons. Mutations in the CASK gene are also associated with postnatal microcephaly.24 It is possible to argue that ONH may simply be a part of microcephaly. However, other monogenic syndromes associated with postnatal microcephaly such as Rett or Angelman syndrome do not present with ONH,51,52 indicating that ONH is an independent manifestation of CASK mutation.
CASK is a membrane associated guanylate kinase protein, which interacts with a large number of other molecules. Specifically, CASK has been considered to be a synaptic scaffolding molecule both at the pre- and postsynaptic compartments.53,54 We have recently made two independent observations, first, CASK stabilizes neuronal adhesion molecule neurexin and links it to the signaling molecule liprin-α.55 Liprin-αs are critical for photoreceptor axonal targeting in drosophila56 as well as for forming active zones.57 Neurexins themselves may play a critical role in intercellular adhesion and signaling.58 A reduction in neurexin signaling and defect in axonal targeting may produce ONH, secondary to defect at the retinogeniculate synapses due to retrograde degeneration.59 Second, CASK may regulate cellular metabolism including mitochondrial respiration.24 Retina and optic nerve are highly susceptible to metabolic defects, and mitochondrial damage and mutations in mitochondrial genes are frequently associated with optic neuropathies.60 In fact, as many as 12% cases of nonsyndromic mitochondrial cytopathies may display ONH.61 Thus, a change in metabolic status of retinal cells may also lead to ONH in CASK(+/−) mice and CASK haploinsufficient girls.
Mutations in CASK have been associated with diagnosis of both ONH and ONA. The difference between these two conditions is often blurred and it has been speculated that better imaging techniques may differentiate these conditions. Many authorities even suggest that both ONH and ONA may be similar pathology with the only difference being in their timing.5 What does our study reveal regarding the pathology of optic nerve? First of all, we present case reports of CASK haploinsufficiency displaying ONH, confirming the genetic associations. Second, using a rodent model, we are able to causally link ONH with CASK haploinsufficiency. Due to a common underlying biology (CASK haploinsufficiency) and similar optic nerve pathology CASK(+/−) mice satisfies the criteria for ‘face validity' of an animal model. Previous rodent models of ONH using alcohol produced thinning of optic nerve only during adulthood (after 9 weeks).62 In contrast, significant decrease in optic nerve area in CASK(+/−) mice was observed as early as P6, which corresponds to late third trimester in human fetal development.21 The time period of optic nerve pathology in CASK(+/−) mice is indeed developmental in nature, and is therefore a superior rodent model of ONH. Several histopathologic hallmarks of atrophy however are present in ONH such as astrogliosis and thinning of axons. A detailed examination of optic nerve, retina, and dLGN in CASK(+/−) mice undermine the idea that simply better imaging tool may be able to distinguish between ONH and ONA in human patients, these optic neuropathies may not be amenable to straightforward binary classification simply by morphology. We suggest that mutations in CASK is typically associated with ONH, the diagnosis of ONA may simply stem from the timing when originally uncovered. In fact, many infants that are diagnosed with ONA may actually have ONH, which was not diagnosed earlier.
Supplementary Material
Acknowledgments
The authors thank the participation and cooperation of families of children with CASK mutation in our studies. They also thank Jeffery Willis and Helen Clark for providing technical help.
Supported by grants from the National Eye Institute (Bethesda, MD, USA) R01EY024712 (KM). The Fox laboratory is supported by RO1AI124677 and RO1EY021222 (MAF).
Disclosure: C. Liang, None; A. Kerr, None; Y. Qiu, None; F. Cristofoli, None; H. Van Esch, None; M.A. Fox, None; K. Mukherjee, None
References
- 1. Rahi JS, Gilbert CE, Foster A, Minassian D. . Measuring the burden of childhood blindness. Br J Ophthalmol. 1999; 83: 387– 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kaur S, Jain S, Sodhi HB, Rastogi A, Kamlesh. Optic nerve hypoplasia. Oman J Ophthalmol. 2013; 6: 77– 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zeki SM, Dudgeon J, Dutton G. . Reappraisal of the ratio of disc to macula/disc diameter in optic nerve hypoplasia. Br J Ophthalmol. 1991; 75: 538– 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frisén L, Holmegaard L. . Spectrum of optic nerve hypoplasia. Br J Ophthalmol. 1978; 62: 7– 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hoyt CS, Good WV. . Do we really understand the difference between optic nerve hypoplasia and atrophy? Eye (Lond). 1992; 6 Pt 2: 201– 204. [DOI] [PubMed] [Google Scholar]
- 6. Garcia-Filion P, Borchert M. . Optic nerve hypoplasia syndrome: a review of the epidemiology and clinical associations. Curr Treat Options Neurol. 2013; 15: 78– 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Parson SH, Dhillon B, Findlater GS, Kaufman MH. . Optic nerve hypoplasia in the fetal alcohol syndrome: a mouse model. J Anat. 1995; 186 Pt 2: 313– 320. [PMC free article] [PubMed] [Google Scholar]
- 8. Garcia-Filion P, Fink C, Geffner ME, Borchert M. . Optic nerve hypoplasia in North America: a re-appraisal of perinatal risk factors. Acta Ophthalmol. 2010; 88: 527– 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dattani MT, Martinez-Barbera JP, Thomas PQ,et al. . Mutations in the homeobox gene HESX1/Hesx1 associated with septo-optic dysplasia in human and mouse. Nat Genet. 1998; 19: 125– 133. [DOI] [PubMed] [Google Scholar]
- 10. Tajima T, Hattorri T, Nakajima T,et al. . Sporadic heterozygous frameshift mutation of HESX1 causing pituitary and optic nerve hypoplasia and combined pituitary hormone deficiency in a Japanese patient. J Clin Endocrinol Metab. 2003; 88: 45– 50. [DOI] [PubMed] [Google Scholar]
- 11. Chen CA, Yin J, Lewis RA, Schaaf CP. . Genetic causes of optic nerve hypoplasia. J Med Genet. 2017; 54: 441– 449. [DOI] [PubMed] [Google Scholar]
- 12. Taranova OV, Magness ST, Fagan BM,et al. . SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 2006; 20: 1187– 1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hill RE, Favor J, Hogan BL,et al. . Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature. 1991; 354: 522– 525. [DOI] [PubMed] [Google Scholar]
- 14. Brown NL, Patel S, Brzezinski J, Glaser T. . Math5 is required for retinal ganglion cell and optic nerve formation. Development. 2001; 128: 2497– 2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Molotkov A, Molotkova N, Duester G. . Retinoic acid guides eye morphogenetic movements via paracrine signaling but is unnecessary for retinal dorsoventral patterning. Development. 2006; 133: 1901– 1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Inloes JM, Hsu KL, Dix MM,et al. . The hereditary spastic paraplegia-related enzyme DDHD2 is a principal brain triglyceride lipase. Proc Natl Acad Sci U S A. 2014; 111: 14924– 14929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moog U, Kutsche K, Kortum F,et al. . Phenotypic spectrum associated with CASK loss-of-function mutations. J Med Genet. 2011; 48: 741– 751. [DOI] [PubMed] [Google Scholar]
- 18. Dimitratos SD, Stathakis DG, Nelson CA, Woods DF, Bryant PJ. . The location of human CASK at Xp11.4 identifies this gene as a candidate for X-linked optic atrophy. Genomics. 1998; 51: 308– 309. [DOI] [PubMed] [Google Scholar]
- 19. Moog U, Bierhals T, Brand K,et al. . Phenotypic and molecular insights into CASK-related disorders in males. Orphanet J Rare Dis. 2015; 10: 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Burglen L, Chantot-Bastaraud S, Garel C,et al. . Spectrum of pontocerebellar hypoplasia in 13 girls and boys with CASK mutations: confirmation of a recognizable phenotype and first description of a male mosaic patient. Orphanet J Rare Dis. 2012; 7: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dobbing J, Sands J. . Comparative aspects of the brain growth spurt. Early Hum Dev. 1979; 3: 79– 83. [DOI] [PubMed] [Google Scholar]
- 22. Sifrim A, Van Houdt JK, Tranchevent LC,et al. . Annotate-it: a Swiss-knife approach to annotation, analysis and interpretation of single nucleotide variation in human disease. Genome Med. 2012; 4: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van Esch H, Bauters M, Ignatius J,et al. . Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am J Hum Genet. 2005; 77: 442– 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Srivastava S, McMillan R, Willis J,et al. . X-linked intellectual disability gene CASK regulates postnatal brain growth in a non-cell autonomous manner. Acta Neuropathol Commun. 2016; 4: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guy J, Ellis EA, Kelley K, Hope GM. . Spectra of G ratio, myelin sheath thickness, and axon and fiber diameter in the guinea pig optic nerve. J Comp Neurol. 1989; 287: 446– 454. [DOI] [PubMed] [Google Scholar]
- 26. Mukherjee K, Clark HR, Chavan V, Benson EK, Kidd GJ, Srivastava S. . Analysis of brain mitochondria using serial block-face scanning electron microscopy. J Vis Exp. 2016; 113: 54214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hammer S, Monavarfeshani A, Lemon T, Su J, Fox MA. . Multiple retinal axons converge onto relay cells in the adult mouse thalamus. Cell Rep. 2015; 12: 1575– 1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Su J, Haner CV, Imbery TE,et al. . Reelin is required for class-specific retinogeniculate targeting. J Neurosci. 2011; 31: 575– 586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fox MA, Sanes JR, Borza DB,et al. . Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell. 2007; 129: 179– 193. [DOI] [PubMed] [Google Scholar]
- 30. Su J, Stenbjorn RS, Gorse K,et al. . Target-derived matricryptins organize cerebellar synapse formation through α3β1 integrins. Cell Rep. 2012; 2: 223– 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tavares GA, Panepucci EH, Brunger AT. . Structural characterization of the intramolecular interaction between the SH3 and guanylate kinase domains of PSD-95. Mol Cell. 2001; 8: 1313– 1325. [DOI] [PubMed] [Google Scholar]
- 32. Li YJ, Wei ZY, Yan Y, Wan QW, Du QS, Zhang MJ. . Structure of Crumbs tail in complex with the PALS1 PDZ-SH3-GK tandem reveals a highly specific assembly mechanism for the apical Crumbs complex. Proc Natl Acad Sci U S A. 2014; 111: 17444– 17449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saadati HG, Hsu HY, Heller KB, Sadun AA. . A histopathologic and morphometric differentiation of nerves in optic nerve hypoplasia and leber hereditary optic neuropathy. Arch Ophthalmol-Chic. 1998; 116: 911– 916. [DOI] [PubMed] [Google Scholar]
- 34. Guillery R, Polley E, Torrealba F. . The arrangement of axons according to fiber diameter in the optic tract of the cat. J Neurosci. 1982; 2: 714– 721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Williams R, Chalupa L. . An analysis of axon caliber within the optic nerve of the cat: evidence of size groupings and regional organization. J Neurosci. 1983; 3: 1554– 1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sharp A, Robinson D, Jacobs P. . Age- and tissue-specific variation of X chromosome inactivation ratios in normal women. Hum Genet. 2000; 107: 343– 349. [DOI] [PubMed] [Google Scholar]
- 37. Sterman AB, Sposito N. . Motoneuron axosomatic synapses are altered in axonopathy. J Neuropathol Exp Neurol. 1984; 43: 201– 209. [DOI] [PubMed] [Google Scholar]
- 38. Wei Z, Zheng S, Spangler SA, Yu C, Hoogenraad CC, Zhang M. . Liprin-mediated large signaling complex organization revealed by the liprin-alpha/CASK and liprin-alpha/liprin-beta complex structures. Mol Cell. 2011; 43: 586– 598. [DOI] [PubMed] [Google Scholar]
- 39. Mukherjee K, Sharma M, Urlaub H,et al. . CASK Functions as a Mg2+-independent neurexin kinase. Cell. 2008; 133: 328– 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mukherjee K, Sharma M, Jahn R, Wahl MC, Sudhof TC. . Evolution of CASK into a Mg2+-sensitive kinase. Sci Signal. 2010; 3:ra33. [DOI] [PMC free article] [PubMed]
- 41. Scheiffele P, Fan JH, Choih J, Fetter R, Serafini T. . Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000; 101: 657– 669. [DOI] [PubMed] [Google Scholar]
- 42. Atasoy D, Schoch S, Ho A,et al. . Deletion of CASK in mice is lethal and impairs synaptic function. Proc Natl Acad Sci U S A. 2007; 104: 2525– 2530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Slawson JB, Kuklin EA, Ejima A, Mukherjee K, Ostrovsky L, Griffith LC. . Central regulation of locomotor behavior of Drosophila melanogaster depends on a CASK isoform containing CaMK-like and L27 domains. Genetics. 2011; 187: 171– 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hoskins R, Hajnal AF, Harp SA, Kim SK. . The C. elegans vulval induction gene lin-2 encodes a member of the MAGUK family of cell junction proteins. Development. 1996; 122: 97– 111. [DOI] [PubMed] [Google Scholar]
- 45. Anjum R, Ayoubian H, Schmitz F. . Differential synaptic distribution of the scaffold proteins Cask and Caskin1 in the bovine retina. Mol Cell Neurosci. 2014; 62: 19– 29. [DOI] [PubMed] [Google Scholar]
- 46. Dangata YY, Kaufman MH. . Myelinogenesis in the optic nerve of (C57BL x CBA) F1 hybrid mice: a morphometric analysis. Eur J Morphol. 1997; 35: 3– 17. [DOI] [PubMed] [Google Scholar]
- 47. Hackett A, Tarpey PS, Licata A,et al. . CASK mutations are frequent in males and cause X-linked nystagmus and variable XLMR phenotypes. Eur J Hum Genet. 2010; 18: 544– 552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ek U, Fernell E, Jacobson L. . Cognitive and behavioural characteristics in blind children with bilateral optic nerve hypoplasia. Acta Paediatr. 2005; 94: 1421– 1426. [DOI] [PubMed] [Google Scholar]
- 49. Parr JR, Dale NJ, Shaffer LM, Salt AT. . Social communication difficulties and autism spectrum disorder in young children with optic nerve hypoplasia and/or septo-optic dysplasia. Dev Med Child Neurol. 2010; 52: 917– 921. [DOI] [PubMed] [Google Scholar]
- 50. Cao X, Tabuchi K. . Functions of synapse adhesion molecules neurexin/neuroligins and neurodevelopmental disorders. Neurosci Res. 2017; 116: 3– 9. [DOI] [PubMed] [Google Scholar]
- 51. Michieletto P, Bonanni P, Pensiero S. . Ophthalmic findings in Angelman syndrome. J AAPOS. 2011; 15: 158– 161. [DOI] [PubMed] [Google Scholar]
- 52. Saunders KJ, Mcculloch DL, Kerr AM. . Visual function in Rett-syndrome. Dev Med Child Neurol. 1995; 37: 496– 504. [DOI] [PubMed] [Google Scholar]
- 53. Hu HT, Umemori H, Hsueh YP. . Postsynaptic SDC2 induces transsynaptic signaling via FGF22 for bidirectional synaptic formation. Sci Rep. 2016; 6: 33592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Samuels BA, Hsueh YP, Shu T,et al. . Cdk5 promotes synaptogenesis by regulating the subcellular distribution of the MAGUK family member CASK. Neuron. 2007; 56: 823– 837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. LaConte LEW, Chavan V, Liang C,et al. . CASK stabilizes neurexin and links it to liprin-alpha in a neuronal activity-dependent manner. Cell Mol Life Sci. 2016; 73: 3599– 3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hofmeyer K, Maurel-Zaffran C, Sink H, Treisman JE. . Liprin-alpha has LAR-independent functions in R7 photoreceptor axon targeting. Proc Natl Acad Sci U S A. 2006; 103: 11595– 11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhen M, Jin YS. . The liprin protein SYD-2 regulates the differentiation of presynaptic termini in C-elegans. Nature. 1999; 401: 371– 375. [DOI] [PubMed] [Google Scholar]
- 58. Graf ER, Zhang XZ, Jin SX, Linhoff MW, Craig AM. . Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell. 2004; 119: 1013– 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mosier MA, Lieberman MF, Green WR, Knox DL. . Hypoplasia of the optic nerve. Arch Ophthalmol. 1978; 96: 1437– 1442. [DOI] [PubMed] [Google Scholar]
- 60. Carelli V, Ross-Cisneros FN, Sadun AA. . Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 2004; 23: 53– 89. [DOI] [PubMed] [Google Scholar]
- 61. Taban M, Cohen BH, David Rothner A, Traboulsi EI. Association of optic nerve hypoplasia with mitochondrial cytopathies. J Child Neurol. 2006; 21: 956– 960. [DOI] [PubMed] [Google Scholar]
- 62. Parson SH, Dhillon B, Findlater GS, Kaufman MH. . Optic-nerve hypoplasia in the fetal alcohol syndrome - a mouse model. J Anat. 1995; 186: 313– 320. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.