

Abstract
The pathogenesis of schizophrenia is considered to be multi-factorial, with likely gene-environment interactions (GEI). Genetic and environmental risk factors are being identified with increasing frequency, yet their very number vastly increases the scope of possible GEI, making it difficult to identify them with certainty. Accumulating evidence suggests a dysregulated complement pathway among the pathogenic processes of schizophrenia. The complement pathway mediates innate and acquired immunity, and its activation drives the removal of damaged cells, autoantigens and environmentally-derived antigens. Abnormalities in complement functions occur in many infectious and auto-immune disorders that have been linked to schizophrenia. Many older reports indicate altered serum complement activity in schizophrenia, though the data are inconclusive. Compellingly, recent genome-wide association studies suggest repeat polymorphisms incorporating the complement 4A (C4A) and 4B (C4B) genes as risk factors for schizophrenia. The C4A/C4B genetic associations have re-ignited interest not only in inflammation-related models for schizophrenia pathogenesis, but also in neurodevelopmental theories, because rodent models indicate a role for complement proteins in synaptic pruning and neurodevelopment. Thus, the complement system could be used as one of the ‘staging posts’ for a variety of focused studies of schizophrenia pathogenesis. They include GEI studies of the C4A/C4B repeat polymorphisms in relation to inflammation-related or infectious processes, animal model studies and tests of hypotheses linked to auto-immune diseases that can co-segregate with schizophrenia. If they can be replicated, such studies would vastly improve our understanding of pathogenic processes in schizophrenia through GEI analyses and open new avenues for therapy.
INTRODUCTION
The multi-factorial polygenic threshold model (MFPT) of schizophrenia posits a large number of genetic risk factors with discrete, cumulative, small effects and environmental factors that can act discretely or interact with the genetic risk factors.1 The MFPT model has been supported by recent genome wide association studies (GWAS).2 In parallel, many environmental risk factors have also been identified, including maternal infection, season of birth (winter excess), urban birth and residence, obstetric complications, maternal malnutrition, substance abuse (particularly cannabis use) and childhood trauma.3–11 Though the MFPT model provides a sound foundation for etiological research in schizophrenia, it challenges simplistic notions of causality. In particular, risk could increase through interactions between genetic and environmental risk factors.12 Initial GEI studies relied on familiality as a proxy for genetic risk.13, 14 As more DNA variant data were generated, the amount of data and the complexity of GEI analyses has increased.15–17 With the availability of GWAS data, the complexity has mushroomed.18–20 Using SNP-based GEI analyses, even if one analyzes the phenotype of schizophrenia as a syndrome, ignoring secondary features, simple ‘two hit’ models involving one genetic and one environmental risk factor can invoke several models of interactions, increasing the number of analyses and the likelihood of false positive results.21–23 For example, Avramopoulos et al19 evaluated multiple infections agents, as well as indices of inflammation in conjunction with genome-wide DNA variant data; interestingly, they found suggestive associations with cytomegalovirus infections, reminiscent of an earlier study by Borglum and colleagues.18, 19 Furthermore, typical case-control designs can be confounded if a correlation exists between the genetic and environmental risk factors, or the risk variables confer risk through more than one pathway.22, 24–28 These complexities explain the difficulties in identifying GEI consistently.29, 30
What can be done in the face of the analytic challenges? One practical solution is a step-wise progression, starting with well-accepted genetic risk factors that are paired with established or plausible environmental risk factors or pathogenic processes. We illustrate this approach with respect to the complement pathway. Recent GWAS analyses implicate complement gene variation in schizophrenia pathogenesis. The complement system is also dysfunctional in many other disorders linked to schizophrenia; thus it provides a nexus for numerous lines of enquiry, including GEI analyses. In the following sections, we initially provide an overview of the complement system and its roles in the immune system, as well as its recently discovered effects on the brain. We next review the putative links between the complement system and schizophrenia: through a possible role in aberrant neurodevelopment, through links to infectious risk factors and through auto-immune disorders that can segregate with schizophrenia. We conclude by suggesting avenues for future research.
The complement system in innate immunity
The complement system encompasses a dynamic, orchestrated array of soluble plasma factors, proteases, cleavage products, cell surface receptors and regulatory protein complexes, all of which serve immune protection of the host.31 This system is best known for its role in halting and destroying invading pathogens by augmenting the effects of antibodies and phagocytes on target antigens and microorganisms.32 The complement system can be activated via three pathways, namely the classical, lectin and alternative pathways, all of which converge on complement C3 (Figure 1). C3 and its activated products covalently bind to cell surface residues to localize the related innate immune inflammatory cascade to specific cellular and tissue sites. The classical pathway is primarily initiated after complement C1q binds to immune complexes composed of immunoglobulin antibodies bound to antigen. Activation of the classical pathway leads to the cleavage and activation products of C4 and C2, which fuse and drive amplification and cleavage of C3. C3 amplification through C4 and C2 cleavage also follows activation of the lectin pathway that occurs when pattern recognition receptors such as mannose-binding lectin (MBL) and the ficolins recognize carbohydrate patterns on damaged cell surfaces or invading microbes. The third pathway, called the alternative pathway, is activated by spontaneous hydrolysis of C3 which prompts a perpetual cycle of amplification that in turn also activates downstream components C5 through C8 and eventually the membrane attack complex. These pathways are intricately controlled, enabling amplification and suppression via complement inhibitors, binding proteins and factors, control genes and cell surface receptors. Thus the complement system maintains a critical role in immune surveillance with important ramifications for protection against infectious agents. Genetically encoded disruption/s could alter responses to environmentally-derived or endogenous antigens perceived as foreign (Figure 1).33–35 As such, susceptibility to infection and autoimmune disorders is increased when there are defects in the complement pathway.36
Figure 1. The complement pathway.

The complement system can be activated along three major pathways. The classical pathway is initiated after C1q interacts with IgM and IgG class antibodies bound to antigen. The lectin pathway is activated by carbohydrate pattern recognition receptors such as mannose-binding lectin (MBL) and the ficolins which are complexed with enzymes known as MBL-associated serine proteases (MASPs). Both the classical and lectin pathways cleave C4 and C2, with subsequent activation of C3. Cleavage of C3 causes C3b to bind to the surface of pathogens and accelerate phagocytic activity. The alternative pathway is activated by spontaneous hydrolysis of C3 and functions as an amplification loop for the cleavage of C3; the generation of C3b involves interactions with the protease factors B and D. In addition to the covalent attachment of C3b to target surfaces, C3b can change substrate specificity of C3 convertases to C5, which leads to assembly of the C5b-C9 membrane attack complex that can lyse targeted cells.
C3*: C3 in its hydrolyzed state.
The complement system AND the brain
The role of complement proteins in synapse formation and elimination has been one of the most fascinating recent discoveries in neuroscience research.37 In the healthy brain, complement proteins are expressed at relatively low levels that vary with stages of maturation.37, 38 The complement proteins C1q, C3 and C4 are detectable on cell bodies, neuronal processes and synapses of discrete neuronal groups. Although neurons express complement proteins, microglia and astrocytes are the major sources of these proteins, suggesting diverse roles.37, 39, 40 In rodent models, the complement system is recruited for removing dysfunctional neuronal cells and dendritic processes.41–43 Through elegant experiments, Stevens and colleagues have suggested that the complement system could also be involved in sculpting neurons even during normal neurodevelopment. They reported that C1q and C3 proteins mediate activity-dependent synaptic elimination in the developing rodent brain, preferentially tagging less active synapses for later elimination by microglia.39, 44, 45 In support, C1q and C3 knockout mice have deficits in synapse elimination.37 As these landmark findings in rodents have invigorated the study of the complement system in neurodevelopment, they merit replications by independent laboratories. In particular, it is important to investigate whether similar processes also occur in other brain regions implicated in the pathogenesis of schizophrenia, e.g., the prefrontal and temporal regions.
Aging, as well as several human brain diseases associated with abnormalities in complement systems, usually stem from infectious or inflammatory pathology. They include Alzheimer’s disease, Down syndrome, multiple sclerosis, amyotrophic lateral sclerosis, Huntington’s disease, Parkinson’s disease and Rett’s Syndrome.38, 46–48 In Alzheimer’s disease, for example, specific protein motifs found in amyloid plaques can trigger the activation of the classical complement pathway.38, 42, 49 A recent translational study in mice and humans documented the accumulation of C1q at synapses in aging animals, suggesting that age-associated cognitive decline may be the result of synapse level vulnerability to extra-CNS and environmental insults. Triggers such as ischemia, trauma and infection could activate the complement cascade and result in inappropriate synapse loss at locations where C1q is over-represented.50 Findings from this study as well as from Hong et al’s study of Alzheimer’s disease are important because of the links they suggest between synaptic C1q and cognitive decline - an indication that disorders like schizophrenia may also be impacted by similar processes.42 Indeed, as elaborated in the following section, Sekar et al suggested a similar synaptic role for C4 in schizophrenia, but with an exquisite genetic twist.40 In summary, components of the complement system could not only help to mold the brain during neurodevelopment39, 51 but also could contribute to dysfunction in the adult brain.
The complement system and the pathogenesis of schizophrenia
Early studies of the complement pathway in schizophrenia utilized the complement hemolytic activity assay to quantify activity of total complement and of specific component proteins in serum samples, based on the percentage of antibody-coated erythrocytes that were lysed in vitro following exposure to the serum (Spivak et al52, and reviewed by Mayilyan et al53). Results from these studies were varied, though altered complement activity was noted in schizophrenia by several investigators.54 Studies that specifically included C4 targets indicated significant elevations of hemolysis by serum from patients with schizophrenia55–58 and similar examinations of C1, C2, and C3 also demonstrated significant schizophrenia-associated complement abnormalities.56, 57 Using immunoassays to quantify serum or plasma concentrations of specific components, Maes and colleagues59 reported higher levels of complement components C3C and C4 and Mayilyan et al58 reported increased lectin-activated complement activation capacity. On the other hand, non-significant case-control differences or even reduced complement activity were also reported.52, 60, 61 The enzymatic nature and varied half-lives of activated complement components likely contributed to difficulties in replicating associations.
A role for complement in schizophrenia could arguably be detected more reliably in the presence of infection or other environmentally-derived antigenic stimuli. For example, complement C1q-bound immune complexes containing food antigens were found at increased rates in individuals with schizophrenia compared with controls.62 Furthermore, elevated levels of complement C1q have been found in the mothers of infants who later developed schizophrenia63, suggesting a role for the complement pathway in early neurodevelopmental events associated with schizophrenia. Notably in this study of unselected mothers, levels of C1q-associated antibodies were significantly correlated with a number of viral antigens, including herpes simplex virus, type 2 (HSV-2) and adenovirus. Thus, the activation of maternal complement by external and intrinsic antigens during a critical period of synaptic pruning may represent an important risk factor for the future development of schizophrenia. In a later section, we discuss more fully the complement system in relation to neurodevelopmental hypotheses of schizophrenia.
Several genetic association studies of complement gene polymorphisms have also been reported. The early studies utilized relatively small samples; unsurprisingly, they yielded inconsistent results.60, 64 Greater clarity has emerged from a recent GWAS. Through a collaborative effort, the Psychiatric Genomics Consortium (PGC) analyzed DNA from 28,799 patients with schizophrenia and 35,986 controls to identify 108 uncorrelated single nucleotide polymorphisms (SNPs) that confer risk for schizophrenia.65 Among the most statistically significant risk variants were those in the human leukocyte antigen (HLA) region; like other schizophrenia-associated SNPs, the risk conferred by individual alleles was modest (odds ratios <2.0). Sekar and colleagues subsequently determined that variation at three uncorrelated loci could explain the observed associations in the HLA region (rs13194504, C4A and rs210133; localized to the distal extended HLA region, the HLA class III and the HLA class II regions, respectively).40 Further, they demonstrated that variation at a polymorphic copy number variant (CNV) spanning the C4A–C4B complement genes accounted for the risk in the HLA Class III region. The primary risk variants represented by the two other SNPs in the HLA region have not been identified yet.
To understand the genetic associations of C4 with schizophrenia, it is necessary to understand the functional impact of the CNV. The CNV cassette (denoted RCCX) comprises STK19 (RP1), C4 (C4A or C4B), CYP21A2, and TNXB.66 A recombination site at CYP21A2 leads to mono-, bi-, and trimodular cassettes with 1–3 functional copies of C4A or C4B, respectively, while retaining just one functional copy of the remaining genes (Figure 2). The C4A and C4B genes, which have over 95% sequence homology, nevertheless encode proteins with different substrate affinities.67 Earlier studies indicated positive correlations between the gene copy number and serum protein concentrations for C4A and C4B.68 To add further complexity, the C4A and the C4B genes can be present in long or short forms, based on the insertion of a human endogenous retroviral (HERV) element. The HERV sequence insertions are associated with increased gene expression, but the mechanism is uncertain. The HERV sequence is present more frequently in the C4A genomic sequences and likely accounts for the observation that C4A expression levels are two to three times greater than expression levels of C4B, even after controlling for relative copy numbers in each genome.69 There is substantial ethnic variation in the distribution of alleles comprising the CNV.70, 71
Figure 2. Copy number variation at the C4A and C4B loci.
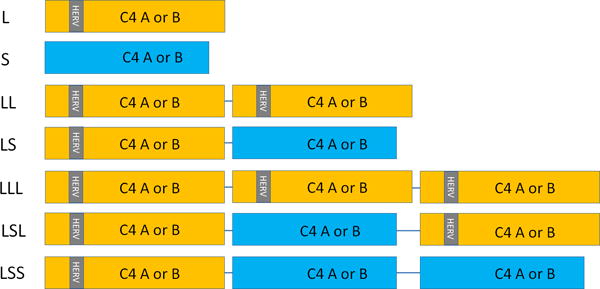
The figure illustrates the range of structural variation at the C4A and C4B loci, indicated as boxes. For clarity, flanking genes (RP1, RP2, CYP21A, CYP21B, TNXA, and TNXB) are not shown; nor are some variants that are less frequent in Caucasian ancestry samples. The gray bar labeled ‘HERV’ indicates a retroviral insertion that produces longer variants (C4A-L or C4B-L, shown in ochre); its absence indicates shorter variants (C4A-S or C4B-S, shown in blue). Each individual can have 0–6 copies of C4A and 0–5 copies of C4B. L: long variant; S: short variant. Additional mutations that can yield non-functional ‘null alleles’ are not shown.
Using an innovative droplet digital PCR (ddPCR) assay, Sekar and colleagues assayed the CNVs in a family-based sample and used this information in conjunction with SNP-based data to impute the number of copies of the C4A and the C4B genes in the PGC dataset. In post-mortem brain samples from the Stanley Medical Research Institute (SMRI) and the Genome Tissue Expression (GTEx) consortia, they showed that the levels of C4A and C4B mRNA increased proportionally with the number of copies of C4A and C4B, respectively. Separately, C4A was also expressed at significantly higher levels in five brain regions in post-mortem samples from patients with schizophrenia, a consistent result was reported with the larger CommonMind Consortium post-mortem dataset.72
In sum, three lines of evidence support the involvement of complement C4A in the pathogenesis of schizophrenia: the early serological studies, the genetic association studies and the post-mortem brain expression analyses. Even though the magnitude of the risk conferred by C4A is similar to, or less than other well established risk factors, the independent lines of evidence lend credence in altering risk for schizophrenia.
Links between the complement system and the neurodevelopmental hypothesis of schizophrenia
Many risk factors for schizophrenia, such as obstetric complications, season of birth effects or nutritional deficiencies could be traced to prenatal maternal influences in utero. The maternal influences, in turn, suggest pathology during the early neurodevelopmental period (spanning prenatal to early postnatal life, when much neuronal proliferation and differentiation occurs). Other lines of evidence implicate the late neurodevelopmental period; i.e., until late adolescence to young adulthood, when synaptic pruning predominantly occurs.
Schizophrenia was proposed as a disorder of faulty programmed synaptic elimination by Feinberg (1982)73 based on convergent evidence from studies of sleep EEG, evoked response potential, brain metabolic rate, dendritic spine variations from new born through age 90 years, and patterns of onset of schizophrenia. These abnormalities were synthesized with evidence that indicated similar temporal patterns for onset of schizophrenia and known age related changes in synaptic density and dendritic spine density. Huttenlocher (1979)74 showed that synaptic density in the middle frontal gyrus increases to a peak in early childhood, and subsequently decreases in late childhood and reaches a plateau in early adolescence, although the pruning continues during the third decade of life before stabilizing at adult level. In some regions that are critical to schizophrenia pathogenesis, e.g. dorsolateral prefrontal cortex, a protracted pruning is observed starting at age 9 years to 22 years. However, different dendritic segments prune dendritic spines with different chronology, e.g. basal and proximal dendrites started to prune at 7–9 years but the distal dendrites do not begin until 17 years of age.75 In primates, substantial reduction in the dendritic spine density occurred in adolescence.76 Overall, the number of synapses decrease in an age-related manner in monkeys and humans77, 78 and these changes could underlie age-related gray matter reductions observed in neuroimaging studies of schizophrenia.79 The factors determining the type or timing of synaptic pruning are uncertain, though much research suggests that immature synapses or those showing lower levels of activity are more likely to be eliminated.80
Thus, the work of Stevens and colleagues37 regarding complement proteins C1q and C3 as mediators of synaptic sculpting in the developing visual system, has important implications for Feinberg’s hypotheses. From a neurodevelopmental perspective, the inappropriate activation of complement or the failure of complement to function correctly in the developing CNS could conceivably disrupt neuronal networks. Faulty complement activity could be generated through environmental factors (such as maternal infection). For example, in a rodent study, adult offspring of dams exposed to prenatal poly(I:C) had significantly elevated expression of prefrontal cortical C1q compared with adult offspring of vehicle treated mothers.81 As discussed earlier, Sekar and colleagues have suggested that risk variants of the C4A CNV could also mediate accelerated synaptic pruning in schizophrenia, consistent with Feinberg’s hypothesis.40 However, there are important caveats: human C4A and C4B sequences do not occur in the mouse genome. Instead, there are two other forms of C4, namely C4 and Slp (sex limited protein).82 Insertion of a retroviral long-terminal repeat in the promoter region of Slp leads to restricted expression of C4 in the mouse.83 Future studies will also need to assay point mutations in C4A that can abrogate function.84, 85 In summary, Sekar’s hypothesis needs to be tested in humans, keeping genomic variations and variations related to brain region and chronological age in mind.
Links between the complement system and the possible role of infection and inflammation in the etiopathogenesis of schizophrenia
Can the C4A genetic associations inform infection as an environmental risk factor for the pathogenesis of schizophrenia or for some aspect of schizophrenia? The answer depends on the strength of evidence linking not only complement system dysfunction and infectious agents, but also the evidence linking infectious agents with schizophrenia.
Complement deficiencies are associated not only with increased levels of bacterial infections,86 but also with viral infections.87–98 Separately, a role for complement in the immune response to Toxoplasma gondii, a protozoan organism was first suggested based on an increased susceptibility of C5-deficient mice to Toxoplasma infection.99 It was subsequently reported that virulent strains of Toxoplasma have a diminished ability to activate the classical complement pathway though interactions with C3.100 In a rodent model, chronic Toxoplasma infection could lead to complement-induced changes in cell connectivity and synaptic pruning,101 as well as the generation of antibodies to the NMDA receptor.102 Thus, complement dysfunction is demonstrable in the pathogenesis of several types of infectious diseases.
With regard to the second question, it is challenging to determine etiological links between infectious agents and schizophrenia due to many technical limitations. Most infections in immune competent individuals result in viral replication for 3–14 days. Thus, evidence for active infection are not expected even in the premorbid period among individuals at high risk for schizophrenia. Hence, most studies of viral infections rely on the immune response to viral proteins, such as circulating antibody molecules or immunoglobulins, but they do not indicate the precise timing of the initial exposure. The difficulty in measuring antibodies within the central nervous system without the performance of cerebrospinal puncture is another substantial barrier. Still, there are several possible mechanisms linking infectious agents such as Toxoplasma gondii infection with schizophrenia.103–106 Conceptually, infectious agents could also elevate risk for certain features of a disorder. For example, several studies have linked the neurotropic herpes simplex virus, type 1 (HSV-1) with cognitive impairment, particularly among patients with schizophrenia107. In sum, proving a link between infections and schizophrenia is challenging; the bulk of evidence suggests several indirect effects.
Several lines of investigation are needed to gain a better understanding of these mechanisms. It would be instructive to evaluate correlations between peripheral and central indices of complement function; e.g., through post-mortem or animal model studies. As it can be difficult to prove causality based on epidemiological studies alone, animal models could be invoked to test causal effects in the association between infection and schizophrenia-relevant brain dysfunctions, as reviewed by Kannan et al.108 The links between infections and complement activation also indicate an intriguing paradox. The genetic association studies of Sekar et al, as well as other studies suggest increased complement activity among patients with schizophrenia. On the other hand, reduced complement system activity facilitates infection and/or increases bacterial/viral loads, and infection or the infectious disease process is a putative risk factor for schizophrenia.109 Thus, it would be of interest to investigate whether individuals with deficiencies in complement system proteins have elevated risk for schizophrenia.
Links between the complement system, auto-immune diseases and schizophrenia
Dysfunction in the complement system can also predispose to well-recognized auto-immune diseases, such as systemic lupus erythematosus (SLE), systemic sclerosis, and rheumatoid arthritis (RA).110 Complete or partial C4 deficiency leads to increased risk of infection and autoimmune diseases, such as SLE.70 It is well-established that reduced concentrations of complement C4 protein or reduced serum complement activity occur with active disease in SLE.111 Though infrequent, absence of complement components C4A and C4B are strongly associated with risk for SLE or lupus-like disease, after controlling for HLA background and ethnicity. A review of 35 studies indicated that heterozygous and homozygous deficiencies of C4A were present in 40–60% of SLE patients from almost all ancestral groups investigated.111 Complement dysfunction has also been linked to other non-infectious diseases, including age-related macular degeneration.112, 113 The prevalence of several auto-immune diseases, including SLE is increased among patients with schizophrenia and their relatives, whereas RA prevalence is reduced among schizophrenia patients and their relatives.114, 115 Systematic studies of complement levels among schizophrenia patients in relation to these auto-immune diseases are, therefore, needed.
The complement system as a PLATFORM for investigating schizophrenia pathogenesis
The application of current knowledge about the complement proteins to schizophrenia research could be fruitful in several directions. Examples include GEI analyses of C4A polymorphisms alongside infection exposure data. Similarly, neurodevelopmental processes in brain imaging studies could be combined with C4A polymorphism data. On a different plane, studies of Toxoplasma gondii infections could be paired with C1q, C3 and C5 levels in the serum. It would also be instructive to investigate whether abnormalities in the complement system, including alterations in levels of complement 4, explain the co-morbidity of schizophrenia and auto-immune diseases.
Most components of the human complement system have a characteristic domain structure; it is likely that the current complexity, exemplified by over 30 proteins, arose partly through multiple gene duplication events.116, 117 In a similar vein, the CNV bearing C4A and the HERV insertion provides a rich source of information about human population history.70, 71 Those data, combined with haplotype analyses may enable future dissection of the origins and geographical variations of schizophrenia.
Such focused analyses, followed by replicative studies could identity pathologic processes for some aspects or sub-groups of schizophrenia, motivating focused therapeutics in the future. More broadly, this scheme could be extended to other genetic risk factors. Indeed, several SNPs identified through schizophrenia GWAS have been linked to immune regulation65 and other studies indicate that genetic factors play an important role in the control of infectious agents and the generation of the immune response to infection.118 The step-wise progression would begin with a single reproducible genetic risk factor - a choice that reflects the difficulty in establishing causal links conclusively for some non-genetic risk factors. Based on its known biological functions, plausible environmental risk factors for schizophrenia could be picked and analyzed next in relation to the selected genetic risk factor. The design and the samples for the joint analyses would be dictated by the biological question. For example, if genetic and environmental risk factors are independent, case only analyses are suitable.28, 119 In other contexts, such as tests of neurodevelopmental hypotheses, premorbid analyses in population-based cohorts may be needed. If plausible GEIs are detected, independent replications would be sought.
CONCLUSIONS
Consistent with the MFPT model of pathogenesis, recent genetic association studies indicated that a portion of the risk for schizophrenia is conferred by copy number variation in the C4A gene; it was also proposed that the pathogenic effects of C4A may be mediated through dysfunction in synaptic pruning. The complement pathway also mediates innate and acquired immunity, suggesting additional plausible mechanisms of pathogenesis and future opportunities for testing novel therapies for schizophrenia – a concept being considered for other diseases.120 We also advocate additional studies of complement function in complement deficient individuals, those with auto-immune disorders and carefully selected animal models studies, as well as post-mortem human samples.
Table 1.
Genetic associations between copy number variants incorporating Complement 4A and Complement 4B genes and selected disorders/diseases.
| Disease / disorder | Genetic Associations | Genotype assays | References |
|---|---|---|---|
| Schizophrenia (SZ) | Increased C4A copy number associated with risk for SZ | Droplet digital PCR | Sekar et al40 |
| Behcet’s disease (BD) | Increased C4A expression and IL-6 levels with 2 or >2 C4A copy number. | qPCR | Hou S et al122 |
| Systemic lupus erythematosus (SLE) | Deficiency - high risk for SLE; 0 or 1 copy of C4A - elevated risk for SLE; 3 or more copies of C4A - protective against SLE | PFGE of PmeI-Digested DNA qPCR qPCR |
Yang et al123 Wu et al124 Yih et al125 |
| Grave’s disease (GD) | <2 copies of C4A associated with risk for vitiligo in patients with GD | qPCR | Liu et al126 |
| Crohn's disease (CD) | CD patients have overall lower C4L and higher C4S copies compared to controls | qPCR | Cleynen et al127 |
| Type1 Diabetes Mellitus | >2 copies of HERV-C4 in patients | qPCR | Mason et al66 |
| Alzheimer’s disorder | Overall increased copies of C4A or C4B in patients | qPCR | Zorzetto et al128 |
PCR – polymerase chain reaction; PFGE - Pulsed-field gel electrophoresis; qPCR – quantitative PCR.
Acknowledgments
This study was funded by grants from the Stanley Medical Research Institute to RHY and grant 07R-1712 to VLN. Additional support was provided by the National Institute of Health (MH63480, MH93246 and D43 TW009114 to VLN; MH101566 to KMP; MH94268 to RHY). We thank Mr. Joel Wood for help with illustrations.
Footnotes
CONFLICT OF INTEREST
The authors declare no competing financial interests in relation to the work described.
References
- 1.Gottesman II, Shields J. A polygenic theory of schizophrenia. Proceedings of the National Academy of Sciences. 1967;58:199–205. doi: 10.1073/pnas.58.1.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Purcell SM, Wray NR, Stone JL, Visscher PM, O’Donovan MC, Sullivan PF, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. doi: 10.1038/nature08185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mednick SA, Machon RA, Huttunen MO, Bonett D. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry. 1988;45(2):189–192. doi: 10.1001/archpsyc.1988.01800260109013. [DOI] [PubMed] [Google Scholar]
- 4.Selten JP, Termorshuizen F. The serological evidence for maternal influenza as risk factor for psychosis in offspring is insufficient: critical review and meta-analysis. Schizophr Res. 2016 doi: 10.1016/j.schres.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 5.Davies G, Welham J, Chant D, Torrey EF, McGrath J. A systematic review and meta-analysis of Northern Hemisphere season of birth studies in schizophrenia. Schizophr Bull. 2003;29(3):587–593. doi: 10.1093/oxfordjournals.schbul.a007030. [DOI] [PubMed] [Google Scholar]
- 6.Pedersen CB. Persons with schizophrenia migrate towards urban areas due to the development of their disorder or its prodromata. Schizophr Res. 2015;168(1–2):204–208. doi: 10.1016/j.schres.2015.08.028. [DOI] [PubMed] [Google Scholar]
- 7.Suvisaari JM, Taxell-Lassas V, Pankakoski M, Haukka JK, Lönnqvist JK, Häkkinen LT. Obstetric complications as risk factors for schizophrenia spectrum psychoses in offspring of mothers with psychotic disorder. Schizophr Bull. 2013;39(5):1056–1066. doi: 10.1093/schbul/sbs109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kirkbride JB, Susser E, Kundakovic M, Kresovich JK, Davey Smith G, Relton CL. Prenatal nutrition, epigenetics and schizophrenia risk: can we test causal effects? Epigenomics. 2012;4(3):303–315. doi: 10.2217/epi.12.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Forti M, Morgan C, Dazzan P, Pariante C, Mondelli V, Marques TR, et al. High-potency cannabis and the risk of psychosis. Br J Psychiatry. 2009;195(6):488–491. doi: 10.1192/bjp.bp.109.064220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perroud N, Courtet P, Vincze I, Jaussent I, Jollant F, Bellivier F, et al. Interaction between BDNF Val66Met and childhood trauma on adult's violent suicide attempt. Genes Brain Behav. 2008;7(3):314–322. doi: 10.1111/j.1601-183X.2007.00354.x. [DOI] [PubMed] [Google Scholar]
- 11.Ruby E, Rothman K, Corcoran C, Goetz RR, Malaspina D. Influence of early trauma on features of schizophrenia. Early Interv Psychiatry. 2015 doi: 10.1111/eip.12239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Os J, Rutten BP, Myin-Germeys I, Delespaul P, Viechtbauer W, van Zelst C, et al. Identifying gene-environment interactions in schizophrenia: contemporary challenges for integrated, large-scale investigations. Schizophr Bull. 2014;40(4):729–736. doi: 10.1093/schbul/sbu069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nimgaonkar VL, Wessely S, et al. Response to Drugs in Schizophrenia: the Influence of Family History, Obstetric Complications and Ventricular Enlargement. Psychological Medicine. 1988;18:583–592. doi: 10.1017/s0033291700008266. [DOI] [PubMed] [Google Scholar]
- 14.Forsyth JK, Ellman LM, Tanskanen A, Mustonen U, Huttunen MO, Suvisaari J, et al. Genetic risk for schizophrenia, obstetric complications, and adolescent school outcome: evidence for gene-environment interaction. Schizophr Bull. 2013;39(5):1067–1076. doi: 10.1093/schbul/sbs098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shirts BH, Wood J, Yolken RH, Nimgaonkar VL. Association study of IL10, IL1ß, and IL1RN and schizophrenia using tag SNPs from a comprehensive database: suggestive association with rs16944 at IL1ß. Schizophrenia Research. 2006;88(1–3):235–244. doi: 10.1016/j.schres.2006.06.037. [DOI] [PubMed] [Google Scholar]
- 16.Kim JJ, Shirts BH, Dayal M, Bacanu S, Wood J, Xie W, et al. Are exposure to cytomegalovirus and genetic variation on chromosome 6p joint risk factors for schizophrenia? Annals of Medicine. 2007;39:145–153. doi: 10.1080/07853890601083808. [DOI] [PubMed] [Google Scholar]
- 17.McGrath JJ, Mortensen PB, Visscher PM, Wray NR. Where GWAS and epidemiology meet: opportunities for the simultaneous study of genetic and environmental risk factors in schizophrenia. Schizophr Bull. 2013;39(5):955–959. doi: 10.1093/schbul/sbt108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borglum AD, Demontis D, Grove J, Pallesen J, Hollegaard MV, Pedersen CB, et al. Genome-wide study of association and interaction with maternal cytomegalovirus infection suggests new schizophrenia loci. Mol Psychiatry. 2014;19(3):325–333. doi: 10.1038/mp.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Avramopoulos D, Pearce BD, McGrath J, Wolyniec P, Wang R, Eckart N, et al. Infection and inflammation in schizophrenia and bipolar disorder: a genome wide study for interactions with genetic variation. PLoS One. 2015;10(3):e0116696. doi: 10.1371/journal.pone.0116696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bani-Fatemi A, Graff A, Zai C, Strauss J, De Luca V. GWAS analysis of suicide attempt in schizophrenia: Main genetic effect and interaction with early life trauma. Neurosci Lett. 2016;622:102–106. doi: 10.1016/j.neulet.2016.04.043. [DOI] [PubMed] [Google Scholar]
- 21.Karl T, Arnold JC. Schizophrenia: a consequence of gene-environment interactions? Frontiers in behavioral neuroscience. 2014;8:435. doi: 10.3389/fnbeh.2014.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Os J, Rutten BP, Poulton R. Gene-environment interactions in schizophrenia: review of epidemiological findings and future directions. Schizophr Bull. 2008;34(6):1066–1082. doi: 10.1093/schbul/sbn117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mittal VA, Ellman LM, Cannon TD. Gene-environment interaction and covariation in schizophrenia: the role of obstetric complications. Schizophr Bull. 2008;34(6):1083–1094. doi: 10.1093/schbul/sbn080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Botto LD, Khoury MJ. Commentary: facing the challenge of gene-environment interaction: the two-by-four table and beyond. Am J Epidemiol. 2001;153(10):1016–1020. doi: 10.1093/aje/153.10.1016. [DOI] [PubMed] [Google Scholar]
- 25.Ottman R. Gene-environment interaction: definitions and study designs. Prev Med. 1996;25(6):764–770. doi: 10.1006/pmed.1996.0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khoury MJ, Flanders WD. Nontraditional epidemiologic approaches in the analysis of gene-environment interaction: case-control studies with no controls. American Journal of Epidemiology. 1996;144(3):207–213. doi: 10.1093/oxfordjournals.aje.a008915. [DOI] [PubMed] [Google Scholar]
- 27.Gatto NM, Campbell UB, Rundle AG, Ahsan H. Further development of the case-only design for assessing gene-environment interaction: evaluation of and adjustment for bias. Int J Epidemiol. 2004;33(5):1014–1024. doi: 10.1093/ije/dyh306. [DOI] [PubMed] [Google Scholar]
- 28.Chatterjee N, Kalaylioglu Z, Carroll RJ. Exploiting gene-environment independence in family-based case-control studies: increased power for detecting associations, interactions and joint effects. Genet Epidemiol. 2005;28(2):138–156. doi: 10.1002/gepi.20049. [DOI] [PubMed] [Google Scholar]
- 29.Modinos G, Iyegbe C, Prata D, Rivera M, Kempton MJ, Valmaggia LR, et al. Molecular genetic gene-environment studies using candidate genes in schizophrenia: a systematic review. Schizophr Res. 2013;150(2–3):356–365. doi: 10.1016/j.schres.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 30.Prasad KM, Talkowski ME, Chowdari KV, McClain L, Yolken RH, Nimgaonkar VL. Candidate genes and their interactions with other genetic/environmental risk factors in the etiology of schizophrenia. Brain Res Bull. 2009;83(3–4):86–92. doi: 10.1016/j.brainresbull.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 - The “Swiss Army Knife” of innate immunity and host defense. Immunol Rev. 2016;274(1):33–58. doi: 10.1111/imr.12500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murphy K, Weaver C. Innate Immunity: the First Lines of Defense page 49, Janeway’s Immunobiology (9th ed) Garland Science [Google Scholar]
- 33.Stoermer KA, Morrison TE. Complement and viral pathogenesis. Virology. 2011;411(2):362–373. doi: 10.1016/j.virol.2010.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carroll MC. The complement system in regulation of adaptive immunity. Nat Immunol. 2004;5(10):981–986. doi: 10.1038/ni1113. [DOI] [PubMed] [Google Scholar]
- 35.Mathern DR, Heeger PS. Molecules Great and Small: The Complement System. Clin J Am Soc Nephrol. 2015;10(9):1636–1650. doi: 10.2215/CJN.06230614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kirschfink M, Mollnes TE. Modern complement analysis. Clin Diagn Lab Immunol. 2003;10(6):982–989. doi: 10.1128/CDLI.10.6.982-989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, et al. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–1178. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- 38.Veerhuis R, Nielsen HM, Tenner AJ. Complement in the brain. Molecular immunology. 2011;48(14):1592–1603. doi: 10.1016/j.molimm.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. doi: 10.1016/j.neuron.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sekar A, Bialas AR, de Rivera H, Davis A, Hammond TR, Kamitaki N, et al. Schizophrenia risk from complex variation of complement component 4. Nature. 2016;530(7589):177–183. doi: 10.1038/nature16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, et al. Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell. 2016;165(4):921–935. doi: 10.1016/j.cell.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–716. doi: 10.1126/science.aad8373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534(7608):538–543. doi: 10.1038/nature18283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bialas AR, Stevens B. TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci. 2013;16(12):1773–1782. doi: 10.1038/nn.3560. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 45.Ransohoff RM, Stevens B. Neuroscience. How many cell types does it take to wire a brain? Science. 2011;333(6048):1391–1392. doi: 10.1126/science.1212112. [DOI] [PubMed] [Google Scholar]
- 46.Watkins LM, Neal JW, Loveless S, Michailidou I, Ramaglia V, Rees MI, et al. Complement is activated in progressive multiple sclerosis cortical grey matter lesions. J Neuroinflammation. 2016;13(1):161. doi: 10.1186/s12974-016-0611-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rus H, Cudrici C, David S, Niculescu F. The complement system in central nervous system diseases. Autoimmunity. 2006;39(5):395–402. [Google Scholar]
- 48.Lin P, Nicholls L, Assareh H, Fang Z, Amos TG, Edwards RJ, et al. Transcriptome analysis of human brain tissue identifies reduced expression of complement complex C1Q Genes in Rett syndrome. BMC Genomics. 2016;17:427. doi: 10.1186/s12864-016-2746-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veerhuis R, Janssen I, De Groot CJ, Van Muiswinkel FL, Hack CE, Eikelenboom P. Cytokines associated with amyloid plaques in Alzheimer's disease brain stimulate human glial and neuronal cell cultures to secrete early complement proteins, but not C1-inhibitor. Exp Neurol. 1999;160(1):289–299. doi: 10.1006/exnr.1999.7199. [DOI] [PubMed] [Google Scholar]
- 50.Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, et al. A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci. 2013;33(33):13460–13474. doi: 10.1523/JNEUROSCI.1333-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Benard M, Raoult E, Vaudry D, Leprince J, Falluel-Morel A, Gonzalez BJ, et al. Role of complement anaphylatoxin receptors (C3aR, C5aR) in the development of the rat cerebellum. Molecular immunology. 2008;45(14):3767–3774. doi: 10.1016/j.molimm.2008.05.027. [DOI] [PubMed] [Google Scholar]
- 52.Spivak B, Radwan M, Brandon J, Baruch Y, Stawski M, Tyano S, et al. Reduced total complement haemolytic activity in schizophrenic patients. Psychol Med. 1993;23(2):315–318. doi: 10.1017/s0033291700028397. [DOI] [PubMed] [Google Scholar]
- 53.Mayilyan KR, Weinberger DR, Sim RB. The complement system in schizophrenia. Drug News Perspect. 2008;21(4):200–210. doi: 10.1358/dnp.2008.21.4.1213349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mayilyan KR, Dodds AW, Boyajyan AS, Soghoyan AF, Sim RB. Complement C4B protein in schizophrenia. World J Biol Psychiatry. 2008;9(3):225–230. doi: 10.1080/15622970701227803. [DOI] [PubMed] [Google Scholar]
- 55.Shcherbakova IV, Neshkova EA, Dotsenko VL, Kozlov LV, Mishin AA, Platonova TP, et al. [Activation of kallikrein-kinin system, degranulating activity of neutrophils and blood-brain barrier in schizophrenia] Zh Nevrol Psikhiatr Im S S Korsakova. 1998;98(6):38–41. [PubMed] [Google Scholar]
- 56.Shcherbakova I, Neshkova E, Dotsenko V, Platonova T, Shcherbakova E, Yarovaya G. The possible role of plasma kallikrein-kinin system and leukocyte elastase in pathogenesis of schizophrenia. Immunopharmacology. 1999;43(2–3):273–279. doi: 10.1016/s0162-3109(99)00099-5. [DOI] [PubMed] [Google Scholar]
- 57.Hakobyan S, Boyajyan A, Sim RB. Classical pathway complement activity in schizophrenia. Neurosci Lett. 2005;374(1):35–37. doi: 10.1016/j.neulet.2004.10.024. [DOI] [PubMed] [Google Scholar]
- 58.Mayilyan KR, Arnold JN, Presanis JS, Soghoyan AF, Sim RB. Increased complement classical and mannan-binding lectin pathway activities in schizophrenia. Neurosci Lett. 2006;404(3):336–341. doi: 10.1016/j.neulet.2006.06.051. [DOI] [PubMed] [Google Scholar]
- 59.Maes M, Delange J, Ranjan R, Meltzer HY, Desnyder R, Cooremans W, et al. Acute phase proteins in schizophrenia, mania and major depression: modulation by psychotropic drugs. Psychiatry Res. 1997;66(1):1–11. doi: 10.1016/s0165-1781(96)02915-0. [DOI] [PubMed] [Google Scholar]
- 60.Fananas L, Moral P, Panadero MA, Bertranpetit J. Complement genetic markers in schizophrenia: C3, BF and C6 polymorphisms. Hum Hered. 1992;42(3):162–167. doi: 10.1159/000154060. [DOI] [PubMed] [Google Scholar]
- 61.Idonije OB, Akinlade KS, Ihenyen O, Arinola OG. Complement factors in newly diagnosed Nigerian schizoprenic patients and those on antipsychotic therapy. Niger J Physiol Sci. 2012;27(1):19–21. [PubMed] [Google Scholar]
- 62.Severance EG, Gressitt KL, Halling M, Stallings CR, Origoni AE, Vaughan C, et al. Complement C1q formation of immune complexes with milk caseins and wheat glutens in schizophrenia. Neurobiol Dis. 2012;48(3):447–453. doi: 10.1016/j.nbd.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Severance EG, Gressitt KL, Buka SL, Cannon TD, Yolken RH. Maternal complement C1q and increased odds for psychosis in adult offspring. Schizophr Res. 2014;159(1):14–19. doi: 10.1016/j.schres.2014.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schroers R, Nothen MM, Rietschel M, Albus M, Maier W, Schwab S, et al. Investigation of complement C4B deficiency in schizophrenia. Hum Hered. 1997;47(5):279–282. doi: 10.1159/000154424. [DOI] [PubMed] [Google Scholar]
- 65.Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mason MJ, Speake C, Gersuk VH, Nguyen QA, O’Brien KK, Odegard JM, et al. Low HERV-K(C4) copy number is associated with type 1 diabetes. Diabetes. 2014;63(5):1789–1795. doi: 10.2337/db13-1382. [DOI] [PubMed] [Google Scholar]
- 67.Blanchong CA, Chung EK, Rupert KL, Yang Y, Yang Z, Zhou B, et al. Genetic, structural and functional diversities of human complement components C4A and C4B and their mouse homologues, Slp and C4. International immunopharmacology. 2001;1(3):365–392. doi: 10.1016/s1567-5769(01)00019-4. [DOI] [PubMed] [Google Scholar]
- 68.Yang Y, Chung EK, Zhou B, Blanchong CA, Yu CY, Fust G, et al. Diversity in intrinsic strengths of the human complement system: serum C4 protein concentrations correlate with C4 gene size and polygenic variations, hemolytic activities, and body mass index. J Immunol. 2003;171(5):2734–2745. doi: 10.4049/jimmunol.171.5.2734. [DOI] [PubMed] [Google Scholar]
- 69.Chung EK, Yang Y, Rennebohm RM, Lokki ML, Higgins GC, Jones KN, et al. Genetic sophistication of human complement components C4A and C4B and RP-C4-CYP21-TNX (RCCX) modules in the major histocompatibility complex. Am J Hum Genet. 2002;71(4):823–837. doi: 10.1086/342777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Blanchong CA, Zhou B, Rupert KL, Chung EK, Jones KN, Sotos JF, et al. Deficiencies of human complement component C4A and C4B and heterozygosity in length variants of RP-C4-CYP21-TNX (RCCX) modules in caucasians. The load of RCCX genetic diversity on major histocompatibility complex-associated disease. J Exp Med. 2000;191(12):2183–2196. doi: 10.1084/jem.191.12.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chung EK, Yang Y, Rupert KL, Jones KN, Rennebohm RM, Blanchong CA, et al. Determining the one, two, three, or four long and short loci of human complement C4 in a major histocompatibility complex haplotype encoding C4A or C4B proteins. Am J Hum Genet. 2002;71(4):810–822. doi: 10.1086/342778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci. 2016;19(11):1442–1453. doi: 10.1038/nn.4399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Feinberg I. Schizophrenia: caused by a fault in programmed synaptic elimination during adolescence? J Psychiatr Res. 1982;17(4):319–334. doi: 10.1016/0022-3956(82)90038-3. [DOI] [PubMed] [Google Scholar]
- 74.Huttenlocher PR. Synaptic density in human frontal cortex - developmental changes and effects of aging. Brain Res. 1979;163(2):195–205. doi: 10.1016/0006-8993(79)90349-4. [DOI] [PubMed] [Google Scholar]
- 75.Petanjek Z, Judas M, Simic G, Rasin MR, Uylings HB, Rakic P, et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc Natl Acad Sci U S A. 2011;108(32):13281–13286. doi: 10.1073/pnas.1105108108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bhatt DH, Zhang S, Gan WB. Dendritic spine dynamics. Annual review of physiology. 2009;71:261–282. doi: 10.1146/annurev.physiol.010908.163140. [DOI] [PubMed] [Google Scholar]
- 77.Huttenlocher PR, Dabholkar AS. Regional differences in synaptogenesis in human cerebral cortex. J Comp Neurol. 1997;387(2):167–178. doi: 10.1002/(sici)1096-9861(19971020)387:2<167::aid-cne1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 78.Bourgeois JP, Goldman-Rakic PS, Rakic P. Synaptogenesis in the prefrontal cortex of rhesus monkeys. Cereb Cortex. 1994;4(1):78–96. doi: 10.1093/cercor/4.1.78. [DOI] [PubMed] [Google Scholar]
- 79.Giedd JN, Jeffries NO, Blumenthal J, Castellanos FX, Vaituzis AC, Fernandez T, et al. Childhood-onset schizophrenia: progressive brain changes during adolescence. Biol Psychiatry. 1999;46(7):892–898. doi: 10.1016/s0006-3223(99)00072-4. [DOI] [PubMed] [Google Scholar]
- 80.Jennings C. Developmental neurobiology. Death of a synapse Nature. 1994;372(6506):498–499. doi: 10.1038/372498a0. [DOI] [PubMed] [Google Scholar]
- 81.Han M, Zhang JC, Hashimoto K. Increased Levels of C1q in the Prefrontal Cortex of Adult Offspring after Maternal Immune Activation: Prevention by 7,8-Dihydroxyflavone. Clin Psychopharmacol Neurosci. 2017;15(1):64–67. doi: 10.9758/cpn.2017.15.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Beurskens FJ, Kuenen JD, Hofhuis F, Fluit AC, Robins DM, Van Dijk H. Sex-limited protein: in vitro and in vivo functions. Clin Exp Immunol. 1999;116(3):395–400. doi: 10.1046/j.1365-2249.1999.00907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stavenhagen JB, Robins DM. An ancient provirus has imposed androgen regulation on the adjacent mouse sex-limited protein gene. Cell. 1988;55(2):247–254. doi: 10.1016/0092-8674(88)90047-5. [DOI] [PubMed] [Google Scholar]
- 84.Fernando MM, Boteva L, Morris DL, Zhou B, Wu YL, Lokki ML, et al. Assessment of complement C4 gene copy number using the paralog ratio test. Hum Mutat. 2010;31(7):866–874. doi: 10.1002/humu.21259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Barba G, Rittner C, Schneider PM. Genetic basis of human complement C4A deficiency. Detection of a point mutation leading to nonexpression. J Clin Invest. 1993;91(4):1681–1686. doi: 10.1172/JCI116377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ram S, Lewis LA, Rice PA. Infections of people with complement deficiencies and patients who have undergone splenectomy. Clin Microbiol Rev. 2010;23(4):740–780. doi: 10.1128/CMR.00048-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen N, Reiss CS. Innate immunity in viral encephalitis: role of C5. Viral Immunol. 2002;15(2):365–372. doi: 10.1089/08828240260066288. [DOI] [PubMed] [Google Scholar]
- 88.Crisci E, Ellegard R, Nystrom S, Rondahl E, Serrander L, Bergstrom T, et al. Complement Opsonization Promotes Herpes Simplex Virus 2 Infection of Human Dendritic Cells. J Virol. 2016;90(10):4939–4950. doi: 10.1128/JVI.00224-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fuchs A, Pinto AK, Schwaeble WJ, Diamond MS. The lectin pathway of complement activation contributes to protection from West Nile virus infection. Virology. 2011;412(1):101–109. doi: 10.1016/j.virol.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kotwal GJ, Fernando N, Zhou J, Valter K. Exploring the potential benefits of vaccinia virus complement control protein in controlling complement activation in pathogenesis of the central nervous system diseases. Molecular immunology. 2014;61(2):204–209. doi: 10.1016/j.molimm.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 91.Rhoades RE, Tabor-Godwin JM, Tsueng G, Feuer R. Enterovirus infections of the central nervous system. Virology. 2011;411(2):288–305. doi: 10.1016/j.virol.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Speth C, Schabetsberger T, Mohsenipour I, Stockl G, Wurzner R, Stoiber H, et al. Mechanism of human immunodeficiency virus-induced complement expression in astrocytes and neurons. J Virol. 2002;76(7):3179–3188. doi: 10.1128/JVI.76.7.3179-3188.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Da Costa XJ, Brockman MA, Alicot E, Ma M, Fischer MB, Zhou X, et al. Humoral response to herpes simplex virus is complement-dependent. Proc Natl Acad Sci U S A. 1999;96(22):12708–12712. doi: 10.1073/pnas.96.22.12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eriksson CE, Studahl M, Bergstrom T. Acute and prolonged complement activation in the central nervous system during herpes simplex encephalitis. J Neuroimmunol. 2016:295–296. 130–138. doi: 10.1016/j.jneuroim.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 95.Mullick J, Kadam A, Sahu A. Herpes and pox viral complement control proteins: 'the mask of self. Trends Immunol. 2003;24(9):500–507. doi: 10.1016/s1471-4906(03)00207-2. [DOI] [PubMed] [Google Scholar]
- 96.Fujita T. Evolution of the lectin-complement pathway and its role in innate immunity. Nat Rev Immunol. 2002;2(5):346–353. doi: 10.1038/nri800. [DOI] [PubMed] [Google Scholar]
- 97.Kittlesen DJ, Chianese-Bullock KA, Yao ZQ, Braciale TJ, Hahn YS. Interaction between complement receptor gC1qR and hepatitis C virus core protein inhibits T-lymphocyte proliferation. J Clin Invest. 2000;106(10):1239–1249. doi: 10.1172/JCI10323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nascimento EJ, Silva AM, Cordeiro MT, Brito CA, Gil LH, Braga-Neto U, et al. Alternative complement pathway deregulation is correlated with dengue severity. PLoS One. 2009;4(8):e6782. doi: 10.1371/journal.pone.0006782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Araujo FG, Rosenberg LT, Remington JS. Experimental Toxoplasma gondii infection in mice: the role of the fifth component of complement. Proceedings of the Society for Experimental Biology and Medicine Society for Experimental Biology and Medicine (New York, NY. 1975;149(3):800–804. doi: 10.3181/00379727-149-38902. [DOI] [PubMed] [Google Scholar]
- 100.Fuhrman SA, Joiner KA. Toxoplasma gondii: mechanism of resistance to complement-mediated killing. J Immunol. 1989;142(3):940–947. [PubMed] [Google Scholar]
- 101.Xiao J, Li Y, Gressitt KL, He H, Kannan G, Schultz TL, et al. Cerebral complement C1q activation in chronic Toxoplasma infection. Brain Behav Immun. 2016;58:52–56. doi: 10.1016/j.bbi.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kannan G, Crawford JA, Yang C, Gressitt KL, Ihenatu C, Krasnova IN, et al. Anti-NMDA receptor autoantibodies and associated neurobehavioral pathology in mice are dependent on age of first exposure to Toxoplasma gondii. Neurobiol Dis. 2016;91:307–314. doi: 10.1016/j.nbd.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Torrey EF, Bartko JJ, Yolken RH. Toxoplasma gondii and other risk factors for schizophrenia: an update. Schizophr Bull. 2012;38(3):642–647. doi: 10.1093/schbul/sbs043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.King DJ, Cooper SJ, Earle JA, Martin SJ, McFerran NV, Rima BK, et al. A survey of serum antibodies to eight common viruses in psychiatric patients. Br J Psychiatry. 1985;147:137–144. doi: 10.1192/bjp.147.2.137. [DOI] [PubMed] [Google Scholar]
- 105.Pedersen MG, Stevens H, Pedersen CB, Nørgaard-Pedersen B, Mortensen PB. Toxoplasma infection and later development of schizophrenia in mothers. Am J Psychiatry. 2011;168(8):814–821. doi: 10.1176/appi.ajp.2011.10091351. [DOI] [PubMed] [Google Scholar]
- 106.Campbell BM, Charych E, Lee AW, Moller T. Kynurenines in CNS disease: regulation by inflammatory cytokines. Front Neurosci. 2014;8:12. doi: 10.3389/fnins.2014.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Prasad KM, Watson AM, Dickerson FB, Yolken RH, Nimgaonkar VL. Exposure to herpes simplex virus type 1 and cognitive impairments in individuals with schizophrenia. Schizophr Bull. 2012;38(6):1137–1148. doi: 10.1093/schbul/sbs046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kannan G, Sawa A, Pletnikov MV. Mouse models of gene-environment interactions in schizophrenia. Neurobiol Dis. 2013;57:5–11. doi: 10.1016/j.nbd.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yolken RH, Torrey EF. Are some cases of psychosis caused by microbial agents? A review of the evidence Mol Psychiatry. 2008;13(5):470–479. doi: 10.1038/mp.2008.5. [DOI] [PubMed] [Google Scholar]
- 110.Ballanti E, Perricone C, Greco E, Ballanti M, Di Muzio G, Chimenti MS, et al. Complement and autoimmunity. Immunologic research. 2013;56(2–3):477–491. doi: 10.1007/s12026-013-8422-y. [DOI] [PubMed] [Google Scholar]
- 111.Yang Y, Chung EK, Zhou B, Lhotta K, Hebert LA, Birmingham DJ, et al. The intricate role of complement component C4 in human systemic lupus erythematosus. Current directions in autoimmunity. 2004;7:98–132. doi: 10.1159/000075689. [DOI] [PubMed] [Google Scholar]
- 112.Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308(5720):385–389. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007;357(6):553–561. doi: 10.1056/NEJMoa072618. [DOI] [PubMed] [Google Scholar]
- 114.Eaton WW, Hayward C, Ram R. Schizophrenia and rheumatoid arthritis: a review. Schizophrenia Research. 1992;6(3):181–192. doi: 10.1016/0920-9964(92)90001-l. [DOI] [PubMed] [Google Scholar]
- 115.Benros ME, Nielsen PR, Nordentoft M, Eaton WW, Dalton SO, Mortensen PB. Autoimmune diseases and severe infections as risk factors for schizophrenia: a 30-year population-based register study. Am J Psychiatry. 2011;168(12):1303–1310. doi: 10.1176/appi.ajp.2011.11030516. [DOI] [PubMed] [Google Scholar]
- 116.Nonaka M, Yoshizaki F. Primitive complement system of invertebrates. Immunol Rev. 2004;198:203–215. doi: 10.1111/j.0105-2896.2004.00118.x. [DOI] [PubMed] [Google Scholar]
- 117.Nonaka M. Evolution of the complement system. Sub-cellular biochemistry. 2014;80:31–43. doi: 10.1007/978-94-017-8881-6_3. [DOI] [PubMed] [Google Scholar]
- 118.Rubicz R, Yolken R, Drigalenko E, Carless MA, Dyer TD, Kent J, Jr, et al. Genome-wide genetic investigation of serological measures of common infections. Eur J Hum Genet. 2015;23(11):1544–1548. doi: 10.1038/ejhg.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pierce BL, Ahsan H. Case-only genome-wide interaction study of disease risk, prognosis and treatment. Genet Epidemiol. 2010;34(1):7–15. doi: 10.1002/gepi.20427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ghebrehiwet B. The complement system: an evolution in progress. F1000Research. 2016;5:2840. doi: 10.12688/f1000research.10065.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Beltrame MH, Catarino SJ, Goeldner I, Boldt AB, de Messias-Reason IJ. The lectin pathway of complement and rheumatic heart disease. Front Pediatr. 2014;2:148. doi: 10.3389/fped.2014.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hou S, Qi J, Liao D, Zhang Q, Fang J, Zhou Y, et al. Copy number variations of complement component C4 are associated with Behcet's disease but not with ankylosing spondylitis associated with acute anterior uveitis. Arthritis Rheum. 2013;65(11):2963–2970. doi: 10.1002/art.38116. [DOI] [PubMed] [Google Scholar]
- 123.Yang Y, Chung EK, Wu YL, Savelli SL, Nagaraja HN, Zhou B, et al. Gene copy-number variation and associated polymorphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in European Americans. Am J Hum Genet. 2007;80(6):1037–1054. doi: 10.1086/518257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wu YL, Yang Y, Chung EK, Zhou B, Kitzmiller KJ, Savelli SL, et al. Phenotypes, genotypes and disease susceptibility associated with gene copy number variations: complement C4 CNVs in European American healthy subjects and those with systemic lupus erythematosus. Cytogenetic and genome research. 2008;123(1–4):131–141. doi: 10.1159/000184700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yih Chen J, Ling Wu Y, Yin Mok M, Jan Wu YJ, Lintner KE, Wang CM, et al. Effects of Complement C4 Gene Copy Number Variations, Size Dichotomy, and C4A Deficiency on Genetic Risk and Clinical Presentation of Systemic Lupus Erythematosus in East Asian Populations. Arthritis Rheumatol. 2016;68(6):1442–1453. doi: 10.1002/art.39589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu YH, Wan L, Chang CT, Liao WL, Chen WC, Tsai Y, et al. Association between copy number variation of complement component C4 and Graves' disease. Journal of biomedical science. 2011;18:71. doi: 10.1186/1423-0127-18-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cleynen I, Konings P, Robberecht C, Laukens D, Amininejad L, Theatre E, et al. Genome-Wide Copy Number Variation Scan Identifies Complement Component C4 as Novel Susceptibility Gene for Crohn's Disease. Inflamm Bowel Dis. 2016;22(3):505–515. doi: 10.1097/MIB.0000000000000623. [DOI] [PubMed] [Google Scholar]
- 128.Zorzetto M, Datturi F, Divizia L, Pistono C, Campo I, De Silvestri A, et al. Complement C4A and C4B Gene Copy Number Study in Alzheimer's Disease Patients. Current Alzheimer research. 2017;14(3):303–308. doi: 10.2174/1567205013666161013091934. [DOI] [PubMed] [Google Scholar]