

Abstract
Sickle retinopathy reflects disease-related vascular injury of the eye, which can potentially result in visual loss from vitreous hemorrhage or retinal detachment. Here we review sickle retinopathy among children with sickle cell disease, describe the epidemiology, pediatric risk factors, pathophysiology, ocular findings and treatment. Newer, more sensitive ophthalmological imaging modalities are available for retinal imaging, including ultra-widefield fluorescein angiography, spectral-domain optical coherence tomography, and optical coherence tomography angiography. Optical coherence tomography angiography provides a non-invasive view of retinal vascular layers that could previously not be imaged and can be quantified for comparative or prospective analyses. Ultra-widefield fluorescein angiography provides a more comprehensive view of the peripheral retina than traditional imaging techniques. Screening for retinopathy by standard fundoscopic imaging modalities detects a prevalence of approximately 10%. In contrast, these more sensitive methods allow for more sensitive examination that includes the retina perimeter where sickle retinopathy is often first detectable. Use of these new imaging modalities may detect a higher prevalence of early sickle pathology among children than has previously been reported. Earlier detection may help in better understanding the pathogenesis of sickle retinopathy and guide future screening and treatment paradigms.
Keywords: Sickle cell, Retinopathy, Fluorescein angiography, Optical coherence tomography
Introduction
Sickle cell disease (SCD) is a severe hematological disorder of chronic hemolysis, vascular injury and tissue ischemia impacting multiple organ systems. While homozygous hemoglobin S disease (HbSS) is associated with more severe clinical manifestations, HbSC disease is associated with more severe and earlier retinal disease.[1-5] This pathology leads to chronic anemia, vascular damage, and interrupted blood flow. As a result of the endothelial damage, perhaps exacerbated by anemia, arteries and arterioles can become damaged, leading to “sickle vasculopathy.”
Ocular manifestations of SCD in the anterior segment include conjunctival comma vessels, hyphema, cataracts, and iris atrophy. Retinal vascular manifestations of SCD are the most important ocular changes by frequency and risk of visual impairment [6] and are classified as either proliferative or non-proliferative based on the presence of neovascularization. Visual loss in SCD patients primarily occurs in patients with proliferative sickle retinopathy (PSR), an outcome that can occur or be initiated in children with SCD. Pediatric screening for early detection of neovascularization can prevent consequences of undetected proliferative retinopathy including vitreous hemorrhage, retinal detachment, and epiretinal membranes.[7] Newer sensitive ophthalmological imaging modalities are increasingly used in non-sickle cell adult populations for early detection of retinal vascular pathology. Application to pediatric populations has not occurred, in large part due to low prevalence of pathology in non-sickle populations. In contrast, use of these techniques may play a role in earlier detection of sickle retinopathy in children, alone or as a precursor to increased risk of pathology in adult patients.
Here we systematically review the available evidence regarding sickle retinopathy, focusing on pediatric populations and how newer ophthalmological techniques are more sensitive than current methodologies currently used for the early detection of retinopathy.
Methods
A comprehensive literature review [8] was performed using PubMed as the information source, restricted to papers published in English. Search terms used were: “Retinopathy and sickle cell,” “Eye and sickle cell,” “Choroid and sickle cell,” “Retina and sickle cell,” “Retinopathy and sickle,” “Fluorescein and sickle cell,” “Optical coherence tomography and sickle cell,” “Optical and sickle cell”, “Retinal and sickle,” “Macular and sickle,” and “Ophthalmological and sickle.” Studies included were published between August 1954 and August 15, 2016. Studies with 3 or more patients (6 eyes) were reviewed. A total of 103 papers were identified and reviewed.
Results
Pediatric Epidemiology
Sickle retinopathy is an age-dependent process, with older people being at substantially higher risk [9]. As found in adult SCD,[10] the prevalence of proliferative sickle retinopathy (PSR) in pediatric patients is several times higher for HbSC than HbSS or HbS-Beta thalassemia.[11, 12] Prevalence of pathology reported has differed among studies in pediatric populations. These discrepancies are in large part due to differing definitions of retinopathy among the different studies, differing examiner ability, differing age ranges of patients studied and, more recently, the use of therapies such as hydroxyurea.[13] Table 1 summarizes the rates of overall retinopathy, as well as proliferative and non-proliferative retinopathy in several pediatric sickle populations. In contrast to pediatric populations, a prevalence study in a population aged 11-63 found the prevalence of proliferative retinopathy to be 45% in HbSC disease, 14% in HbSS disease and 17% in sickle-beta thalassemia.[10]
Table 1. Literature on the epidemiology of pediatric sickle cell retinopathy.
| Study | Number of patients | Age Range | Retina Exams Done | Prevalence | Study Type | Additional Findings |
|---|---|---|---|---|---|---|
| Abiose et al. (1978) [24] | 91 (91 HbSS, 0 HbSC) | 5 to 14 |
|
Any retinopathy - 58% PSR – 1% with neovascularization and 5.5% with peripheral arteriolar occlusion |
Cross-sectional | Vascular tortuosity - 27%; Black sunbursts - 11%; Salmon patch hemorrhage - 4%; Abnormal conjunctival vasculature – 81% |
| Condon et al. (1974) [79] | 54 (0 HbSS, 54 HbSC) | 2 to 15 |
|
Any retinopathy (peripheral retinal vessel disease) - 94% PSR - 11% |
Cross-sectional | Vascular tortuosity - 32%; Black sunbursts - 4%; Arterio-venous fistulae - 17%; Angioid streaks - 0%; Retinoschisis – 2% |
| Downes et al. (2005) [1] | 474 (307 HbSS, 166 HbSC) | 5 to 26 |
|
PSR - 43% of HbSC and 14% of HbSS by ages 24-26 CHANGE TO 18-20 AGE RANGE |
Longitudinal observed over 20 yrs | Spontaneous regression in 32% of PSR-affected eyes |
| El-Ghamrawy et al. (2014) [16] | 40 (26 HbSS, 14 HbS/Beta-thal) | 2 to 28 |
|
Any retinopathy - 47.5% (46.2% of HbSS and 50% of HbS/Beta-thal) PSR - 32.5% NPR - 27.5% |
Cross-sectional | Vascular tortuosity - 25%; Black sunbursts - 5% |
| Eruchalu et al. (2006) [77] | 37 (37 HbSS, 0 HbSC) | 3 to 14 |
|
Any retinopathy - 32.4% PSR - 5.4% NPR - 27% |
Cross-sectional | Retinal infarcts - 14.9%; Vascular tortuosity - 54%; Black sunbursts - 6.8%; Salmon patch hemorrhage - 6.8%; Comma vessels – 70.3% |
| Estepp et al. (2013) [13] | 123 (123 HbSS, 0 HbSC) | 11 to 16 |
|
Any retinopathy - 10.6% PSR- 1.6% NPR - 8.9% |
Retrospective review between 2003-2010 | |
| Gill et al. (2008) [11] | 236 (163 HbSS, 73 HbSC, 27 HbS/Beta-thal) | 1 to 18 |
|
Any retinopathy - 18.7% PSR- 2.7% (8.2% of HbSC, 0.6% of HbSS, and 0% of HbS/Beta-thal) NPR - 18.6% (24.7% of HbSC, 14.1% of HbSS, and 11.1 % of HbS/Beta-thal |
Retrospective longitudinal survival analysis between 1987-2005 | |
| Kimmel et al. (1987) [15] | 135 (Breakdown not available) | 0 to 20 |
|
PSR - 5.2% | Retrospective review between 1976 to 1985 | |
| Oliveira et al. (2014) [12] | 51 (36 HbSS, 15 HbSC) | 4 to 18 |
|
Any retinopathy (ages 10 to 18) - 17.5% (80.0% of HbSS and 22.2% of HbSC ages 10-18) PSR (ages 4 to 18) - 39.2% (30.6% of HbSS and 60.0% of HbSC) NPR (ages 4 to 18) - 39.3% (38.9% of HbSS and 40.0% of HbSC) |
Cross-sectional | Neovascularization - 0%; Vascular tortuosity - 19.6%; Black sunbursts - 9.8%; Salmon patch hemorrhages - 2.0%; Angioid streaks - 0%; Iridescent spots - 7.8% |
| Rosenberg et al. (2011) [17] | 258 (186 HbSS, 55 HbSC, 12 HbS/Beta-thal | 10 to 18 |
|
Any retinopathy - 20.9% (17.2% of HbSS, 32.7% of HbSC, 33.3% of HbS/Beta-thal) PSR - 4.3% (0.54% of HbSS, 16.4% of HbSC, 8.3% of HbS/Beta-thal) NPR - 16.3% |
Retrospective review over 10 year period | |
| Talbot et al. (1982) [25] | 96 (59 HbSS, 37 HbSC) | 5 to 7.5 |
|
PSR - 0.0% | Cohort | Peripheral arteriolar closure- 24% HbSS and 16% HbSC; Arteriolar sheathing- 51% HbSS and 30% HbSC; Arteriovenous anastomoses - 3.1%; Retinal patches - 37% of HbSS and 24% of HbSC |
| Talbot et al. (1983) [19] | 85 (54 HbSS, 31 HbSC) | 6.5 to 8.5 |
|
Cohort | Peripheral arterial closure - 66.6% HbSS and 51.6% HbSC | |
| Talbot et al. (1988) [14] | 389 (219 HbSS, 135 HbSC, 24 HbS/Beta-thal, 11 HbS/Beta-thal0) | 5 to 13 |
|
PSR - 0.3% | Cohort | Peripheral arteriolar closure – 90% of HbSS and HbSC by age 12 |
| Tantawy et al. (2013) [78] | 60 (28 HbSS, 32 HbS/Beta-thal) | 3 to 18 |
|
Any retinopathy – 17% (25% of HbSS and 9.4% of HbS/Beta-thal) PSR - 0% NPR- 17% (25% of HbSS and 9.4% of HbS/Beta-thal) |
Cohort |
NPR- non-proliferative retinopathy; PSR - proliferative sickle retinopathy
Risk Factors Associated with PSR
Risk of PSR increases with age in both genotypes. Prevalence of PSR is much higher in HbSC disease than in HbSS (HR 6.32) or other sickle cell disease types.[1, 2] The youngest case of PSR reported to date in HbSC was an 8 year-old patient and in HbSS in a 13 year-old patient.[14, 15] Several different risk factors have been reported by multiple independent cross-sectional studies of patients with sickle disease.[2, 3, 9, 14, 16-21] Not all of these cohorts reported the same risk factors as significantly associated with sickle retinopathy, possibly due to patient population, sample size, age range, method of eye examination, and concurrent disease therapies.
For HbSS, one or more of these studies have identified older age, male sex, and history of splenic sequestration or splenectomy as risk factors for PSR. One common factor, G6PD deficiency, has been identified as a risk factor, but not at a level of statistical significance (OR 4.20, p=0.054).[17] Overall, more serious disease phenotype, such as previous multiple pain crises or lower hemoglobin or fetal hemoglobin (HbF) levels have been associated with higher prevalence of PSR.[2, 17] To summarize, clinical and hematological factors that have been associated with PSR in HbSS patients are: older age (OR 1.12), longer disease duration (p=0.00), splenectomy (p=0.004), splenic sequestration (OR 4.00), pain crisis (OR 5.00), male sex (OR 2.58-4.20), acute pyelonephritis (OR 2.81), low fetal hemoglobin (p<0.001), and low weight (p<0.05).
For HbSC, risk factors are: splenic sequestration (OR 4.00), pulmonary involvement (OR 3.65), deafness or tinnitus (OR 3.49), no history of osteomyelitis (OR .10 – history of osteomyelitis), pain crisis (OR 5.00), male sex (OR 4.20), high mean cell volume in males (p<0.001), low fetal hemoglobin (p=0.01), low platelet count (p<0.01), and high reticulocyte count (p<0.05).[2-4, 9, 14, 16-25]
Pathophysiology
Prior to the expanded understanding of pathophysiology, development of retinopathy was believed to be initiated by vaso-occlusion at the junction of nonperfused and perfused peripheral retina leading to neovascularization.[26] Contemporary understanding of sickle vasculopathy is of a complex interaction between sickled erythrocytes, vasoactive factors, inflammation, activated endothelium, and other blood cells.[27-30]
Rodent models have been used for uncovering the pathophysiology of PSR. A rat model demonstrated that RBC trapping, not adhesion, is responsible for the retention of RBCs in retinal vasculature in HbSS, as preferential retinal retention of cells with low adherence propensity were observed. In contrast, for HbSC, low retention of HbSC cells was observed, suggesting that extra-erythrocytic factors like adhesive molecule and cytokines may be involved.[31]
The mechanism of angiogenesis in the formation of so-called “sea fans” has been an active area of study in PSR. Both vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) appear to be associated with sea fan formation in PSR. [32] Pigment epithelium-derived factor (PEDF), a known inhibitor of angiogenesis and a neurotrophic factor in the eye, may be elevated in viable vessels of sea fans.[33] Elevation of PEDF production appears to follow increased angiogenesis.[34] In addition, plasma angiopoietins (Ang-1 and Ang-2) and von Willebrand factor are present in higher levels in SCD patients compared to controls.[35]
Endothelial cell adhesion molecules, inflammatory cytokines, and leukocytes also play an important role in the pathophysiology of retinopathy. Circulating intercellular adhesion molecule 1 (sICAM-1) is significantly decreased in HbSC patients with retinopathy. sICAM-1 has immunosuppressive effects, [36] so high levels may be protective against retinopathy by preventing leukocyte extravasation in the retina. Additionally, increased levels of ICAM-1, VCAM-1, and P-selectin immunoreactivities have been demonstrated in SCD patients compared to controls.[37] Also, studies have shown that the number of intraretinal PMNs increases with disease progression which suggests, similar to previous sICAM-1 findings, that leukocyte adhesion might have an important role in the vaso-occlusive phase of sickle cell retinopathy.[37] Finally, certain circulating cytokines have been shown to play a role in the pathophysiology of retinopathy. For example, TNF-alpha stimulates retention of sickled erythrocytes in the retinal vasculature.[38, 39]
Ocular Findings
Retinal pathology is not readily visible except under ophthalmologic examination. Common clinical findings in non-proliferative retinopathy include salmon patch hemorrhages, iridescent spots, and black sunburst lesions, all of which are the sequelae of peripheral arterial occlusions.[27, 28, 40-42] Other findings include angioid streaks, posterior vascular tortuosity, sickle disc sign, retinal depression sign, and retinoschisis.[27, 28] These finding are defined in Table 2.
Table 2. Definitions of major ophthalmological findings in sickle cell retinopathy.
| Finding | Definition [27-29, 69] |
|---|---|
| Salmon Patch Hemorrhage | Oval-shaped, orange, well-defined area of bleeding on the retina surface |
| Black Sunburst Lesion | Circular, black, stellate and spiculate lesion from the migration of retinal pigment epithelium |
| Iridescent Spot | Small retinal cavity with yellow spots containing hemosiderin-laden macrophages resulting from a resorbed salmon patch hemorrhage |
| Disc Sign | Dark red spots on the optic disc due to plugs of deoxygenated, sickled erythrocytes |
| Posterior Vessel Tortuosity | Vessels that make a twisted rather than normal straight path |
| Angioid Streaks | Gray, bilateral, irregular lines emanating from the optic disc representing small breaks in Bruch's membrane (membrane that separates the retina and choroid) |
| Retinal depression sign | Abnormal light reflex from the retina due to ischemia of the macula causing atrophy |
| Retinoschisis | Splitting of the retina's neurosensory layers |
| Sea Fan | Neovascular growth in PSR that resembles the marine species Gorgonia flabellum |
Proliferative retinopathy is defined by the presence of neovascularization, traditionally identified by slit-lamp examination and indirect ophthalmoscopy. PSR begins with retinal vessel occlusion, which occurs most often in the peripheral retina. PSR has classically been defined by five stages developed by Goldberg et al.: 1) only peripheral arteriolar occlusions; 2) arteriolar-venous anastomoses at the retinal border; 3) neovascularization as evidenced by sea-fans; 4) vitreous hemorrhage; 5) retinal detachment.[43] Penman later revised this classification scheme of the peripheral vascular border, and more strictly defined proliferative retinopathy as requiring the presence of neovascularization (Goldberg Stage 3-5).[22]
While findings on clinical exam are important in detecting early signs of retinopathy and prevention of visual loss, newer advances in imaging modalities have made earlier detection and screening for retinopathy possible.
Current Ophthalmic Imaging Techniques
Recent advances in retinal imaging modalities including ultra-widefield fluorescein angiography (UWFA), spectral-domain optical coherence tomography (SD-OCT), and optical coherence tomography angiography (OCT-A) have revealed significant retinal findings in asymptomatic sickle cell patients (Figure 1).[44-51] Some of these silent findings have been shown to be significantly associated with the presence of PSR.[49]
Figure 1.
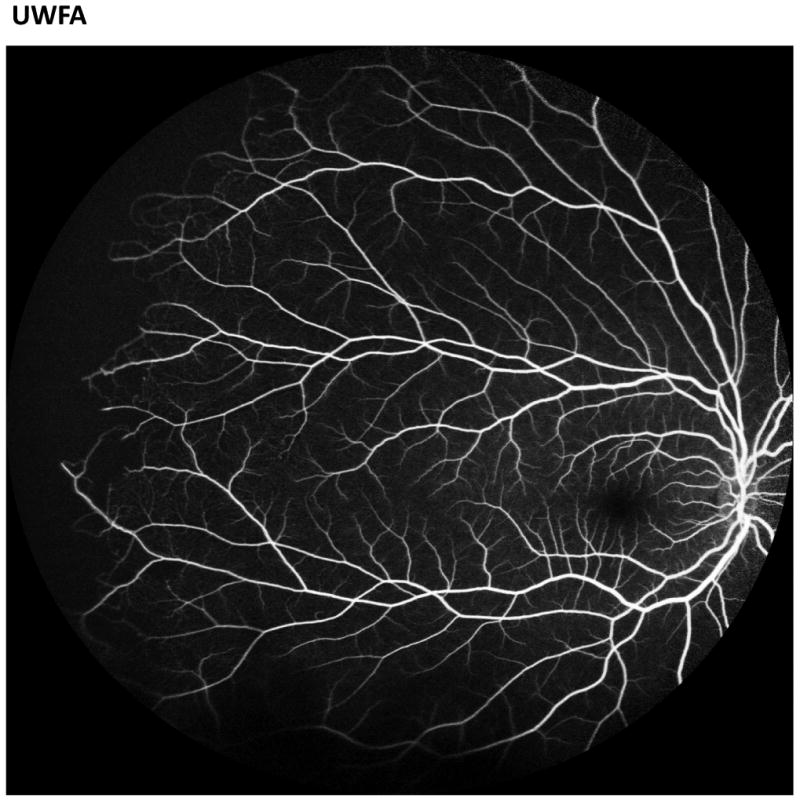
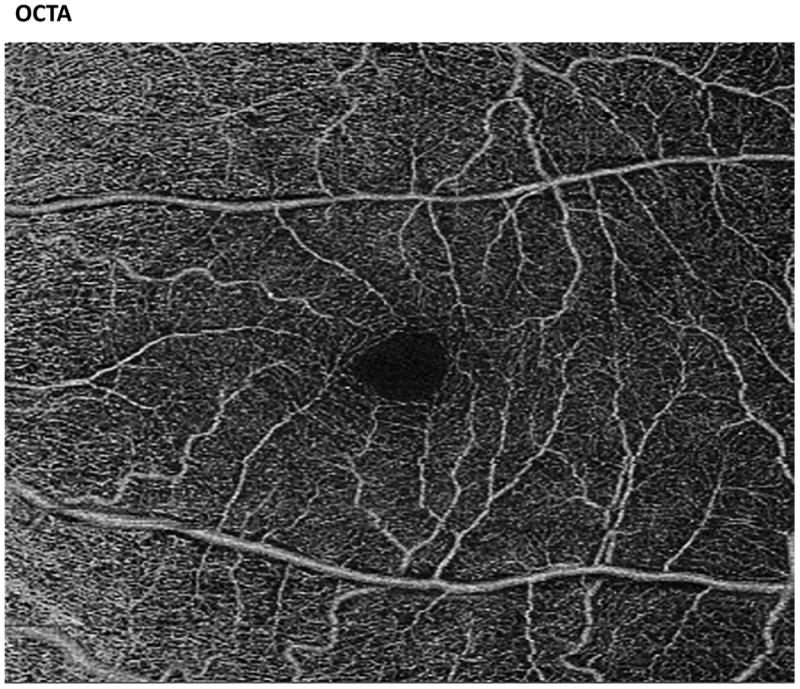
Images of the fundus by ultra-widefield fluorescein angiography (UWFA, left) and optical coherence tomography angiography (OCT-A, right). The UWFA image demonstrates peripheral arteriolar disease in a sickle cell patient. The OCT-A demonstrates a healthy superficial vascular plexus.
Ultra-widefield Fluorescein Angiography
Fluorescein angiography (FA) has served as the gold standard for the detection of proliferative sickle retinopathy for over 40 years. [43] This study allows dynamic visualization of blood flow and observation of areas of dye leakage, pooling, and staining so that vascular abnormalities particularly in the periphery of the retina can be assessed and graded on the Goldberg scale. Traditional FA cameras offered views ranging from 30° to 60° in one exposure. 7-standard fields were used to cover an approximately 75° field of view. Previous techniques to expand the field of view posed technical challenges and required significant patient cooperation.[52]
Ultra-widefield fluorescein angiography (UWFA) captures up to 200° of the retina in a single image and has been shown to capture up to twice as much retinal areas as conventional FA.[53] Since it is more efficient in imaging the retina, this technology reduces the amount of patient cooperation and technical expertise necessary for operation.[52] As mentioned earlier, PSR particularly affects the peripheral vasculature and therefore UWFA has significant utility. In 2011, a retrospective case series of 12 eyes of 6 patients with SCD was performed. In all but one eye, UWFA detected peripheral vascular changes missed on the classical 7-standard fields photograph. Additionally, in 25% of eyes, peripheral vascular changes missed on clinical exam by experienced ophthalmologists were detected by UWFA.[45] Although the data are limited in comparisons of UWFA vs standard FA, UWFA is likely more sensitive in the detection of peripheral proliferative retinopathy.
Spectral Domain Optical Coherence Tomography and Optical Coherence Tomography Angiography
SD-OCT and OCT-A are both non-invasive imaging techniques that utilize reflected light, similar to ultrasound's use of sound, to produce detailed cross-sectional and en-face images of the retina and allow visualization of blood flow in retinal layers. SD-OCT provides near-histological cross-sectional images of the retina and can highlight areas of photoreceptor loss, nerve fiber layer change, and thinning or thickening of the retina in response to ischemic insults and neovascularization. OCT-A is a more recent modality that harnesses the speed and sensitivity of SD-OCT to capture detailed maps of the different layers of retinal and choroidal vasculature. Both have emerged as important modalities for the detection of early signs of PSR.
Recently, multiple groups have reported using SD-OCT imaging to assess the presence of subclinical macular thinning due to macular ischemia. SD-OCT has revealed temporal and central macular thinning (also referred to as macular splaying) in some patients with SCD.[44, 48, 49] Thinning of inner retinal layers in SCD patients with both symptomatic and asymptomatic retinopathy has also been reported in previous case series and histopathological studies.[41, 54-56] A 2015 retrospective study demonstrated that these discrete areas of macular thinning are significantly associated with PSR.[49] This finding was corroborated by a 2016 retrospective, case-controlled study that examined the predictive ability of temporal macular atrophy (thinning) for neovascularization in PSR in 38 patients. Temporal macular atrophy was found to have a positive predictive value of 83% and a negative predictive value of 13% for identifying neovascularization, demonstrating that the presence of temporal macular atrophy suggests the concurrent presence of neovascularization and PSR.[57] Finally, a recent retrospective case series reported that areas of macular thinning on SD-OCT correlated to the degree of peripheral ischemia on UWFA.[58] Taken together, these findings suggest that SD-OCT may be useful for diagnosis and screening of retinopathy because macular thinning observed on SD-OCT is associated with PSR.
The etiology of this macular thinning seen on SD-OCT is not entirely understood. Various studies have suggested that macular thinning may be in part due to ischemia of the deep capillary plexus.[59, 60] Additionally, the functional consequence of macular thinning is not entirely clear. A prospective study in 19 patients demonstrated that SCD patients with focal macular thinning on SD-OCT have significantly decreased retinal sensitivities using microperimetry (a sensitive measurement of macular function) compared to those without thinning or controls suggesting functional consequences of this observed macular thinning.[61] Furthermore, SCD patients with focal macular thinning have significant thinning in the peripapillary retinal nerve fiber layer (characteristic of glaucoma) compared to SCD patients without focal macular thinning. [46] Finally, a recent report found no evidence to support an association between retinal thinning and neurocognitive function.[62]
When compared to traditional FA, OCT-A is probably more sensitive in identifying early regions of ischemia in the macula.[63] This difference was corroborated by a 2016 study that reported that OCT-A demonstrated microvascular abnormalities in the macula in 18 eyes of 9 patients, whereas FA appeared normal in nine of 18 eyes.[50]
Ophthalmological Imaging in Pediatric Populations
SD-OCT has dramatically altered the practice patterns of ophthalmologists over the last decade and is frequently used in patients seen by both adult and pediatric retina specialists for early detection of macular pathology. Both SD-OCT and OCT-A are non-invasive, and individual scans take only seconds to perform. While no studies have reported SD-OCT or OCT-A findings in pediatric sickle populations, the above findings raise the question of whether these technologies may prove useful in early identification of retinopathy. Our own preliminary data suggest that children with sickle cell disease commonly exhibit retinal vascular abnormalities by these sensitive testing modalities, including Goldberg Stage I and II retinopathy identified on UWFA, temporal macular thinning on SD-OCT and vessel dropout on OCT-A. (DP, manuscript in preparation). These abnormalities were not seen in controls within the same age range. Clinical implications of finding abnormalities in retinal vessels by sensitive testing remain unclear and will require longitudinal testing to assess what risk these patients have for serious eye pathology and what, if any, future intervention may be required for vision preservation.
Treatment
In a large prospective cohort of Jamaican children with SCD, proliferative disease developed in 43% of HbSC patients and 14% of HbSS patients over a 20-year period, [1] underscoring the increased prevalence in children with HbSC. However, most patients with PSR do not progress to visual loss. In patients with PSR, neovascular sea fans frequently undergo spontaneous autoinfarction, with the prevalence of this outcome reported as high as 60%.[26, 64-68] This outcome decreases the risk of visual complications, hence treatment has not been standardized. If autoinfarction does not occur, neovascularization can lead to visual loss from vitreous hemorrhage, rhegmatogenous and tractional retinal detachment.[6] In a study of 120 adult patients with HbSS and 222 with HbSC, 10% of untreated eyes developed visual acuity loss over a period of ten years due to PSR.[7]
Treatment is usually undertaken for patients with bilateral proliferative disease, spontaneous hemorrhage, large elevated sea fans, or rapid growth of neovascular tissue.[69] Treatment options have been well summarized and described by previous reviews.[27, 28, 69] The preferred treatment for stage III proliferative lesions is scatter laser photocoagulation. It is effective in reducing the incidence of loss of visual acuity as well as the incidence of vitreous hemorrhage.[64, 70, 71] One study of 21 eyes with PSR found complete regression in 24 of 28 sea fan lesions treated with scatter laser photocoagulation.[72] Others have found complete regression in 30.2% of treated eyes compared to 22.4% of untreated control eyes.[64] Finally, treatment options for the vitreoretinal complications of PSR, which include epiretinal membranes, vitreous hemorrhage and retinal detachment, are pars plana vitrectomy and scleral buckling.[73, 74] However, as previously indicated treatment is not typically required in pediatric populations as advanced stages of PSR and the vision-threatening consequences of retinopathy do not generally occur until later in life.
Prevention by Referral for Ophthalmic Evaluation
Screening for early detection and treatment remain the mainstay of prevention. Based on the risk factors previously discussed, current recommendation by the American Academy of Pediatrics is for retinopathy screening of children by dilated fundoscopic examination with HbSS and HbSC beginning at age 10 years.[75] Other recommendations include biannual or annual examinations as early as age 9. [11, 16, 76, 77]
In addition to the current screening recommendations, patients with more severe systemic disease and a higher number of risk factors for PSR (e.g. splenic sequestration, increased number of pain crises) should be referred to an ophthalmologist. Patients with complaints of sudden or gradual decreased vision, floaters, or flashing lights should also be referred, as these symptoms could reflect the development of macular ischemia, vitreous hemorrhage, retinal tears, or retinal detachment.
A retrospective review of 123 children with SCD demonstrated an association between elevated HbF and lower prevalence of retinopathy. [13] These data suggest that induction of HbF by hydroxyurea, along with other possible drug effects, may prevent pediatric retinopathy and the development of visual loss from proliferative disease.
Future Directions
Given the ubiquity of SD-OCT and the ease of image acquisition in both adult and pediatric populations, it is likely that SD-OCT will be used more frequently in the screening examination of pediatric patients with SCD. OCT-A imaging is a newer technology that is less widely adopted than SD-OCT, but its ability to image the retinal vasculature in a non-invasive fashion makes it a useful research tool for the characterization of microvascular changes in SCD patients.
Future studies will need to explore the connection between temporal macular thinning on SD-OCT, microvascular changes on OCT-A, and proliferative retinopathy as detected on UWFA. If correlations between the imaging modalities are found in the pediatric population as they have been found in adults, abnormal SD-OCT studies may prompt differing monitoring strategies for patients with SCD.
Conclusion
Retinopathy is a serious chronic complication of SCD. By standard dilated fundoscopic examination of pediatric patients, the prevalence of retinal vascular pathology has been estimated to be anywhere between 17% and 94%.[78, 79] Most of the pathology found reflects early vascular changes rather than the more serious injury that can progress to visual loss found in adult patients. Future studies could include the prevalence, progression, clinical outcomes and clinical utility of the newer diagnostic imaging techniques such as SD-OCT, OCT-A, and UWFA and the impact of new early screening and therapeutic approaches on eye health in pediatric SCD.
Future studies using SD-OCT, OCT-A and UWFA may generate more refined data on SCD retinal vascular disease. Understanding the pathogenesis and evolution of retinal vasculopathy could also be targets for these techniques. For example, improved detection of vascular pathology and its sequellae could be compared to effects of SCD vasculopathy in other organs, such as those appreciated through MRI evaluation of pediatric sickle vasculopathy in pediatric central nervous systems.[27, 80]
Acknowledgments
Supported by an Imaging Award from Columbia's Irving Institute for Clinical and Translational Science (RWC; 1UL1TR001873, PI H. Ginsberg) and 2T35HL007616 (DP; R. Liebel, PI).
References
- 1.Downes SM, Hambleton IR, Chuang EL, et al. Incidence and natural history of proliferative sickle cell retinopathy: observations from a cohort study. Ophthalmology. 2005;112:1869–1875. doi: 10.1016/j.ophtha.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 2.Fox PD, Dunn DT, Morris JS, et al. Risk factors for proliferative sickle retinopathy. Br J Ophthalmol. 1990;74:172–176. doi: 10.1136/bjo.74.3.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayes RJ, Condon PI, Serjeant GR. Haematological factors associated with proliferative retinopathy in sickle cell-haemoglobin C disease. Br J Ophthalmol. 1981;65:712–717. doi: 10.1136/bjo.65.10.712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kent D, Arya R, Aclimandos WA, et al. Screening for ophthalmic manifestations of sickle cell disease in the United Kingdom. Eye (Lond) 1994;8(Pt 6):618–622. doi: 10.1038/eye.1994.155. [DOI] [PubMed] [Google Scholar]
- 5.van Meurs JC. Relationship between peripheral vascular closure and proliferative retinopathy in sickle cell disease. Graefes Arch Clin Exp Ophthalmol. 1991;229:543–548. doi: 10.1007/BF00203319. [DOI] [PubMed] [Google Scholar]
- 6.Fadugbagbe AO, Gurgel RQ, Mendonca CQ, et al. Ocular manifestations of sickle cell disease. Ann Trop Paediatr. 2010;30:19–26. doi: 10.1179/146532810X12637745451870. [DOI] [PubMed] [Google Scholar]
- 7.Moriarty BJ, Acheson RW, Condon PI, et al. Patterns of visual loss in untreated sickle cell retinopathy. Eye (Lond) 1988;2(Pt 3):330–335. doi: 10.1038/eye.1988.62. [DOI] [PubMed] [Google Scholar]
- 8.Khan KS, Kunz R, Kleijnen J, et al. Five steps to conducting a systematic review. J R Soc Med. 2003;96:118–121. doi: 10.1258/jrsm.96.3.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leveziel N, Bastuji-Garin S, Lalloum F, et al. Clinical and laboratory factors associated with the severity of proliferative sickle cell retinopathy in patients with sickle cell hemoglobin C (SC) and homozygous sickle cell (SS) disease. Medicine (Baltimore) 2011;90:372–378. doi: 10.1097/MD.0b013e3182364cba. [DOI] [PubMed] [Google Scholar]
- 10.Clarkson JG. The ocular manifestations of sickle-cell disease: a prevalence and natural history study. Trans Am Ophthalmol Soc. 1992;90:481–504. [PMC free article] [PubMed] [Google Scholar]
- 11.Gill HS, Lam WC. A screening strategy for the detection of sickle cell retinopathy in pediatric patients. Can J Ophthalmol. 2008;43:188–191. doi: 10.3129/i08-003. [DOI] [PubMed] [Google Scholar]
- 12.de Almeida Oliveira DC, Carvalho MO, do Nascimento VM, et al. Sickle cell disease retinopathy: characterization among pediatric and teenage patients from northeastern Brazil. Rev Bras Hematol Hemoter. 2014;36:340–344. doi: 10.1016/j.bjhh.2014.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Estepp JH, Smeltzer MP, Wang WC, et al. Protection from sickle cell retinopathy is associated with elevated HbF levels and hydroxycarbamide use in children. Br J Haematol. 2013;161:402–405. doi: 10.1111/bjh.12238. [DOI] [PubMed] [Google Scholar]
- 14.Talbot JF, Bird AC, Maude GH, et al. Sickle cell retinopathy in Jamaican children: further observations from a cohort study. Br J Ophthalmol. 1988;72:727–732. doi: 10.1136/bjo.72.10.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kimmel AS, Magargal LE, Maizel R, et al. Proliferative sickle cell retinopathy under age 20: a review. Ophthalmic Surg. 1987;18:126–128. [PubMed] [Google Scholar]
- 16.El-Ghamrawy MK, El Behairy HF, El Menshawy A, et al. Ocular manifestations in egyptian children and young adults with sickle cell disease. Indian J Hematol Blood Transfus. 2014;30:275–280. doi: 10.1007/s12288-014-0333-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rosenberg JB, Hutcheson KA. Pediatric sickle cell retinopathy: correlation with clinical factors. J AAPOS. 2011;15:49–53. doi: 10.1016/j.jaapos.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 18.Hayes RJ, Condon PI, Serjeant GR. Haematological factors associated with proliferative retinopathy in homozygous sickle cell disease. Br J Ophthalmol. 1981;65:29–35. doi: 10.1136/bjo.65.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Talbot JF, Bird AC, Rabb LM, et al. Sickle cell retinopathy in Jamaican children: a search for prognostic factors. Br J Ophthalmol. 1983;67:782–785. doi: 10.1136/bjo.67.11.782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serjeant BE, Mason KP, Acheson RW, et al. Blood rheology and proliferative retinopathy in homozygous sickle cell disease. Br J Ophthalmol. 1986;70:522–525. doi: 10.1136/bjo.70.7.522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serjeant BE, Mason KP, Condon PI, et al. Blood rheology and proliferative retinopathy in sickle cell-haemoglobin C disease. Br J Ophthalmol. 1984;68:325–328. doi: 10.1136/bjo.68.5.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Penman A, Talbot JF, Chuang EL, et al. New classification of peripheral retinal vascular changes in sickle disease disease. British Journal of Ophthalmolgy. 1994;78:681–689. doi: 10.1136/bjo.78.9.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lemaire C, Lamarre Y, Lemonne N, et al. Severe proliferative retinopathy is associated with blood hyperviscosity in sickle cell hemoglobin-C disease but not in sickle cell anemia. Clin Hemorheol Microcirc. 2013;55:205–212. doi: 10.3233/CH-2012-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abiose A, Lesi FE. Ocular findings in children with homozygous sickle cell anemia in Nigeria. J Pediatr Ophthalmol Strabismus. 1978;15:92–95. doi: 10.3928/0191-3913-19780301-10. [DOI] [PubMed] [Google Scholar]
- 25.Talbot JF, Bird AC, Serjeant GR, et al. Sickle cell retinopathy in young children in Jamaica. Br J Ophthalmol. 1982;66:149–154. doi: 10.1136/bjo.66.3.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goldberg MF. Natural history of untreated proliferative sickle retinopathy. Arch Ophthalmol. 1971;85:428–437. doi: 10.1001/archopht.1971.00990050430006. [DOI] [PubMed] [Google Scholar]
- 27.Kassim AA, DeBaun MR. Sickle cell disease, vasculopathy, and therapeutics. Annu Rev Med. 2013;64:451–466. doi: 10.1146/annurev-med-120611-143127. [DOI] [PubMed] [Google Scholar]
- 28.Ballas SK, Kesen MR, Goldberg MF, et al. Beyond the definitions of the phenotypic complications of sickle cell disease: an update on management. ScientificWorldJournal. 2012;2012:949535. doi: 10.1100/2012/949535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Elagouz M, Jyothi S, Gupta B, et al. Sickle cell disease and the eye: old and new concepts. Surv Ophthalmol. 2010;55:359–377. doi: 10.1016/j.survophthal.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 30.Lutty GA, M DS. Angiogenesis in Sickle Cell Retinopathy. Retinal and Choroidal Angiogenesis. 2008:389–405. [Google Scholar]
- 31.Lutty GA, Phelan A, McLeod DS, et al. A rat model for sickle cell-mediated vaso-occlusion in retina. Microvasc Res. 1996;52:270–280. doi: 10.1006/mvre.1996.0064. [DOI] [PubMed] [Google Scholar]
- 32.Cao J, Mathews MK, McLeod DS, et al. Angiogenic factors in human proliferative sickle cell retinopathy. Br J Ophthalmol. 1999;83:838–846. doi: 10.1136/bjo.83.7.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim SY, Mocanu C, McLeod DS, et al. Expression of pigment epithelium-derived factor (PEDF) and vascular endothelial growth factor (VEGF) in sickle cell retina and choroid. Exp Eye Res. 2003;77:433–445. doi: 10.1016/s0014-4835(03)00174-x. [DOI] [PubMed] [Google Scholar]
- 34.Cruz PR, Lira RP, Pereira Filho SA, et al. Increased circulating PEDF and low sICAM-1 are associated with sickle cell retinopathy. Blood Cells Mol Dis. 2015;54:33–37. doi: 10.1016/j.bcmd.2014.08.003. [DOI] [PubMed] [Google Scholar]
- 35.Mohan JS, Lip PL, Blann AD, et al. The angiopoietin/Tie-2 system in proliferative sickle retinopathy: relation to vascular endothelial growth factor, its soluble receptor Flt-1 and von Willebrand factor, and to the effects of laser treatment. Br J Ophthalmol. 2005;89:815–819. doi: 10.1136/bjo.2004.058164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang HW, Babic AM, Mitchell HA, et al. Elevated soluble ICAM-1 levels induce immune deficiency and increase adiposity in mice. FASEB J. 2005;19:1018–1020. doi: 10.1096/fj.04-3094fje. [DOI] [PubMed] [Google Scholar]
- 37.Kunz Mathews M, McLeod DS, Merges C, et al. Neutrophils and leucocyte adhesion molecules in sickle cell retinopathy. Br J Ophthalmol. 2002;86:684–690. doi: 10.1136/bjo.86.6.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lutty GA, Otsuji T, Taomoto M, et al. Mechanisms for sickle red blood cell retention in choroid. Curr Eye Res. 2002;25:163–171. doi: 10.1076/ceyr.25.3.163.13481. [DOI] [PubMed] [Google Scholar]
- 39.Lutty GA, Taomoto M, Cao J, et al. Inhibition of TNF-alpha-induced sickle RBC retention in retina by a VLA-4 antagonist. Invest Ophthalmol Vis Sci. 2001;42:1349–1355. [PubMed] [Google Scholar]
- 40.Gagliano DA, Goldberg MF. The evolution of salmon-patch hemorrhages in sickle cell retinopathy. Arch Ophthalmol. 1989;107:1814–1815. doi: 10.1001/archopht.1989.01070020896034. [DOI] [PubMed] [Google Scholar]
- 41.Romayanada N, Goldberg MF, Green WR. Histopathology of sickle cell retinopathy. Trans Am Acad Ophthalmol Otolaryngol. 1973;77:OP642–676. [PubMed] [Google Scholar]
- 42.Welch RB, Goldberg MF. Sickle-cell hemoglobin and its relation to fundus abnormality. Arch Ophthalmol. 1966;75:353–362. doi: 10.1001/archopht.1966.00970050355008. [DOI] [PubMed] [Google Scholar]
- 43.Goldberg MF. Classification and pathogenesis of proliferative sickle retinopathy. Am J Ophthalmol. 1971;71:649–665. doi: 10.1016/0002-9394(71)90429-6. [DOI] [PubMed] [Google Scholar]
- 44.Brasileiro F, Martins TT, Campos SB, et al. Macular and peripapillary spectral domain optical coherence tomography changes in sickle cell retinopathy. Retina. 2015;35:257–263. doi: 10.1097/IAE.0000000000000309. [DOI] [PubMed] [Google Scholar]
- 45.Cho M, Kiss S. Detection and monitoring of sickle cell retinopathy using ultra wide-field color photography and fluorescein angiography. Retina. 2011;31:738–747. doi: 10.1097/IAE.0b013e3181f049ec. [DOI] [PubMed] [Google Scholar]
- 46.Chow CC, Shah RJ, Lim JI, et al. Peripapillary retinal nerve fiber layer thickness in sickle-cell hemoglobinopathies using spectral-domain optical coherence tomography. Am J Ophthalmol. 2013;155:456–464 e452. doi: 10.1016/j.ajo.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 47.Grover S, Sambhav K, Chalam KV. Capillary nonperfusion by novel technology of OCT angiography in a patient with sickle cell disease with normal fluorescein angiogram. Eur J Ophthalmol. 2016:0. doi: 10.5301/ejo.5000765. [DOI] [PubMed] [Google Scholar]
- 48.Hoang QV, Chau FY, Shahidi M, et al. Central macular splaying and outer retinal thinning in asymptomatic sickle cell patients by spectral-domain optical coherence tomography. Am J Ophthalmol. 2011;151:990–994 e991. doi: 10.1016/j.ajo.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mathew R, Bafiq R, Ramu J, et al. Spectral domain optical coherence tomography in patients with sickle cell disease. Br J Ophthalmol. 2015;99:967–972. doi: 10.1136/bjophthalmol-2014-305532. [DOI] [PubMed] [Google Scholar]
- 50.Minvielle W, Caillaux V, Cohen SY, et al. Macular Microangiopathy in Sickle Cell Disease Using Optical Coherence Tomography Angiography. Am J Ophthalmol. 2016;164:137–144 e131. doi: 10.1016/j.ajo.2015.12.023. [DOI] [PubMed] [Google Scholar]
- 51.Sanfilippo CJ, Klufas MA, Sarraf D, et al. Optical Coherence Tomography Angiography of Sickle Cell Maculopathy. Retin Cases Brief Rep. 2015;9:360–362. doi: 10.1097/ICB.0000000000000210. [DOI] [PubMed] [Google Scholar]
- 52.Patel M, Kiss S. Ultra-wide-field fluorescein angiography in retinal disease. Curr Opin Ophthalmol. 2014;25:213–220. doi: 10.1097/ICU.0000000000000042. [DOI] [PubMed] [Google Scholar]
- 53.Friberg TR, Gupta A, Yu J, et al. Ultrawide angle fluorescein angiographic imaging: a comparison to conventional digital acquisition systems. Ophthalmic Surg Lasers Imaging. 2008;39:304–311. doi: 10.3928/15428877-20080701-06. [DOI] [PubMed] [Google Scholar]
- 54.Murthy RK, Grover S, Chalam KV. Temporal macular thinning on spectral-domain optical coherence tomography in proliferative sickle cell retinopathy. Arch Ophthalmol. 2011;129:247–249. doi: 10.1001/archophthalmol.2010.357. [DOI] [PubMed] [Google Scholar]
- 55.Shakoor A, Blair NP, Shahidi M. Imaging retinal depression sign in sickle cell anemia using optical coherence tomography and the retinal thickness analyzer. Arch Ophthalmol. 2005;123:1278–1279. doi: 10.1001/archopht.123.9.1278. [DOI] [PubMed] [Google Scholar]
- 56.Witkin AJ, Rogers AH, Ko TH, et al. Optical coherence tomography demonstration of macular infarction in sickle cell retinopathy. Arch Ophthalmol. 2006;124:746–747. doi: 10.1001/archopht.124.5.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hood MP, Diaz RI, Sigler EJ, et al. Temporal Macular Atrophy as a Predictor of Neovascularization in Sickle Cell Retinopathy. Ophthalmic Surg Lasers Imaging Retina. 2016;47:27–34. doi: 10.3928/23258160-20151214-04. [DOI] [PubMed] [Google Scholar]
- 58.Ghasemi Falavarjani K, Scott AW, Wang K, et al. Correlation of Multimodal Imaging in Sickle Cell Retinopathy. Retina. 2016 doi: 10.1097/IAE.0000000000001230. [DOI] [PubMed] [Google Scholar]
- 59.Chen X, Rahimy E, Sergott RC, et al. Spectrum of Retinal Vascular Diseases Associated With Paracentral Acute Middle Maculopathy. Am J Ophthalmol. 2015;160:26–34 e21. doi: 10.1016/j.ajo.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 60.Han IC, Tadarati M, Scott AW. Macular Vascular Abnormalities Identified by Optical Coherence Tomographic Angiography in Patients With Sickle Cell Disease. JAMA Ophthalmol. 2015;133:1337–1340. doi: 10.1001/jamaophthalmol.2015.2824. [DOI] [PubMed] [Google Scholar]
- 61.Chow CC, Genead MA, Anastasakis A, et al. Structural and functional correlation in sickle cell retinopathy using spectral-domain optical coherence tomography and scanning laser ophthalmoscope microperimetry. Am J Ophthalmol. 2011;152:704–711 e702. doi: 10.1016/j.ajo.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oltra EZ, Chow CC, Wubben T, et al. Cross-Sectional Analysis of Neurocognitive Function, Retinopathy, and Retinal Thinning by Spectral-Domain Optical Coherence Tomography in Sickle Cell Patients. Middle East Afr J Ophthalmol. 2016;23:79–83. doi: 10.4103/0974-9233.150632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chalam KV, Sambhav K. Optical Coherence Tomography Angiography in Retinal Diseases. J Ophthalmic Vis Res. 2016;11:84–92. doi: 10.4103/2008-322X.180709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farber MD, Jampol LM, Fox P, et al. A randomized clinical trial of scatter photocoagulation of proliferative sickle cell retinopathy. Arch Ophthalmol. 1991;109:363–367. doi: 10.1001/archopht.1991.01080030065040. [DOI] [PubMed] [Google Scholar]
- 65.Goldberg MF, Jampol LM. Treatment of neovascularization, vitreous hemorrhage, and retinal detachment in sickle cell retinopathy. Trans New Orleans Acad Ophthalmol. 1983;31:53–81. [PubMed] [Google Scholar]
- 66.Nagpal KC, Patrianakos D, Asdourian GK, et al. Spontaneous regression (autoinfarction) of proliferative sickle retinopathy. Am J Ophthalmol. 1975;80:885–892. doi: 10.1016/0002-9394(75)90285-8. [DOI] [PubMed] [Google Scholar]
- 67.Condon PI, Serjeant GR. Behaviour of untreated proliferative sickle retinopathy. Br J Ophthalmol. 1980;64:404–411. doi: 10.1136/bjo.64.6.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McLeod DS, Merges C, Fukushima A, et al. Histopathologic features of neovascularization in sickle cell retinopathy. Am J Ophthalmol. 1997;124:455–472. doi: 10.1016/s0002-9394(14)70862-1. [DOI] [PubMed] [Google Scholar]
- 69.Emerson GG, Lutty GA. Effects of sickle cell disease on the eye: clinical features and treatment. Hematol Oncol Clin North Am. 2005;19:957–973. ix. doi: 10.1016/j.hoc.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 70.Fox PD, Minninger K, Forshaw ML, et al. Laser photocoagulation for proliferative retinopathy in sickle haemoglobin C disease. Eye (Lond) 1993;7(Pt 5):703–706. doi: 10.1038/eye.1993.160. [DOI] [PubMed] [Google Scholar]
- 71.Myint KT, Sahoo S, Thein AW, et al. Laser therapy for retinopathy in sickle cell disease. Cochrane Database Syst Rev. 2015 doi: 10.1002/14651858.CD010790.pub2. CD010790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rednam KR, Jampol LM, Goldberg MF. Scatter retinal photocoagulation for proliferative sickle cell retinopathy. Am J Ophthalmol. 1982;93:594–599. doi: 10.1016/s0002-9394(14)77374-x. [DOI] [PubMed] [Google Scholar]
- 73.Chen RW, Flynn HW, Jr, Lee WH, et al. Vitreoretinal management and surgical outcomes in proliferative sickle retinopathy: a case series. Am J Ophthalmol. 2014;157:870–875 e871. doi: 10.1016/j.ajo.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Williamson TH, Rajput R, Laidlaw DA, et al. Vitreoretinal management of the complications of sickle cell retinopathy by observation or pars plana vitrectomy. Eye (Lond) 2009;23:1314–1320. doi: 10.1038/eye.2008.296. [DOI] [PubMed] [Google Scholar]
- 75.Section on Hematology/Oncology Committee on G and American Academy of P. Health supervision for children with sickle cell disease. Pediatrics. 2002;109:526–535. doi: 10.1542/peds.109.3.526. [DOI] [PubMed] [Google Scholar]
- 76.Babalola OE, Wambebe CO. When should children and young adults with sickle cell disease be referred for eye assessment? Afr J Med Med Sci. 2001;30:261–263. [PubMed] [Google Scholar]
- 77.Eruchalu UV, Pam VA, Akuse RM. Ocular findings in children with severe clinical symptoms of homozygous sickle cell anaemia in Kaduna, Nigeria. West Afr J Med. 2006;25:88–91. doi: 10.4314/wajm.v25i2.28255. [DOI] [PubMed] [Google Scholar]
- 78.Tantawy AA, Andrawes NG, Adly AA, et al. Retinal changes in children and adolescents with sickle cell disease attending a paediatric hospital in Cairo, Egypt: risk factors and relation to ophthalmic and cerebral blood flow. Trans R Soc Trop Med Hyg. 2013;107:205–211. doi: 10.1093/trstmh/trt008. [DOI] [PubMed] [Google Scholar]
- 79.Condon PI, Serjeant GR. Ocular findings in hemoglobin SC disease in Jamaica. Am J Ophthalmol. 1972;74:921–931. doi: 10.1016/0002-9394(72)91213-5. [DOI] [PubMed] [Google Scholar]
- 80.DeBaun MR, Kirkham FJ. Central nervous system complications and management in sickle cell disease. Blood. 2016;127:829–838. doi: 10.1182/blood-2015-09-618579. [DOI] [PubMed] [Google Scholar]