

Abstract
Three-dimensional pluripotent stem cell (PSC) cultures have the ability to undergo differentiation, self-organization, and morphogenesis to yield complex, in vitro tissue models that recapitulate key elements of native tissues. These tissue models offer a system for studying mechanisms of tissue development, investigating disease mechanisms, and performing drug screening. It remains challenging, however, to standardize PSC aggregate differentiation and morphogenesis methods due to heterogeneity stemming from biological and environmental sources. It is also difficult to monitor and assess large numbers of individual samples longitudinally throughout culture using typical batch-based culture methods. To address these challenges, we have developed a microfluidic platform for culture, longitudinal monitoring, and phenotypic analysis of individual stem cell aggregates. This platform uses a hydrodynamic loading principle to capture pre-formed stem cell aggregates in independent traps. We demonstrated that multi-day culture of aggregates in this platform reduces heterogeneity in phenotypic parameters such as size and morphology. Additionally, we showed that culture and analysis steps can be performed sequentially in the same platform, enabling correlation of multiple modes of analysis for individual samples. We anticipate this platform being applied to improve abilities for phenotypic analysis of PSC aggregate tissues and to facilitate research in standardizing culture systems in order to dually increase the yield and reduce the heterogeneity of PSC-derived tissues.
Graphical abstract
We present a microfluidic platform for culture, longitudinal tracking, and imaging-based analysis of individual stem cell aggregates.
Introduction
Pluripotent stem cells (PSCs) have the ability to self-renew and differentiate to the three germ lineages.1–3 In particular, when PSCs are cultured as three-dimensional (3D) cellular aggregates, they mimic many of the biochemical and biophysical interactions that occur during in vivo early embryonic development.4, 5 Efforts to direct stem cell aggregate morphogenesis have demonstrated that complex 3D tissue structures, termed “organoids”, can be generated in vitro,6 and that these tissues recapitulate many key features of native tissue structure and function.7–9 Thus, the generation of PSC-derived tissue models has broad applications in studying mechanisms of tissue development, in disease modeling, and in drug screening.
Within the 3D stem cell culture microenvironment, both exogenous and endogenous biophysical and biochemical cues mediate cellular differentiation and morphogenesis. Maintaining uniformity of aspects of the culture microenvironment is important for standardizing differentiation and morphogenesis of cultures. For example, changes in aggregate size can influence differentiation by affecting diffusive transport of oxygen, nutrients, and morphogens.10 In addition, modulating the frequency of media exchange or perfusion also affects delivery of biochemical cues to cells.11–14 Despite development of various strategies to improve uniformity of cell aggregate cultures, for example by controlling aggregate size,15–17 a significant amount of heterogeneity often remains within cultures. It is unclear how factors such as biological stochasticity, environmental parameters, and experimental procedures each contribute to the heterogeneity, thus motivating the development of technologies that enhance the ability to standardize stem cell aggregate differentiation and morphogenesis by providing improved control of the 3D cellular microenvironment.
A variety of culture systems have been developed for stem cell aggregates. One category of these is batch-based methods, in which aggregates are cultured under static18 or stirred19–21 suspension conditions in petri dishes or larger vessels. Batch-based methods enable high-throughput culture (1,000s per experiment) and have been shown to improve uniformity in aggregate size, such as in stirred suspension cultures.17 However, in batch systems, aggregates can physically interact with one another in a stochastic manner, which can contribute to heterogeneity in differentiation. An additional challenge associated with batch-based culture systems is the lack of ability to longitudinally observe and track individual samples throughout culture. Observation of differentiation and morphogenesis at multiple time points in the same samples is of interest, for example, in order to understand how processes evolve through time and to observe population heterogeneity. Culture platforms which physically isolate cell aggregates provide the ability to longitudinally monitor samples. Examples of such platforms include multiwell plates,22, 23 microwells,24–27 droplets28, and hanging drop29 culture. However, with each of these methods, it is labor- and time-intensive to manipulate aggregates individually, to exchange media, and to apply chemical factors. In particular, studies that require delivery of soluble factors with time scales on the order of seconds to hours are challenging. Additionally, high-resolution imaging, such as confocal microscopy, cannot be performed directly with these culture platforms for live and end-point imaging of sample phenotypes.
In contrast to macroscale culture systems, microfluidic-based platforms offer the ability to address challenges related to culture and handling individual cellular aggregates. Microfluidic systems provide spatiotemporal control over the culture microenvironment due to the nature of low Reynolds number flow and the ability to precisely control media exchange rates.30 Additionally, microscale features can be designed to position and culture cellular aggregates within individual chambers. Finally, microfluidic devices made with traditional soft lithography techniques,31 fabricated from polydimethylsiloxane (PDMS), and bonded to glass coverslips are compatible with high resolution microscopy for live imaging-based assessment of aggregate differentiation.
While previously a few microfluidic approaches for culturing cellular aggregates have been demonstrated, there are technical demands that these systems do not fully address. In one subset of approaches, aggregates are formed on-chip by seeding single cells in wells,32–35 traps,36 hanging drops,37–40, or droplets41, followed by culture of the resulting aggregates. Forming aggregates within devices allows for control of initial size, as with many off-chip methods, and circumvents the challenge of transferring pre-formed aggregates into devices. However, many of the methods that use microwells within microfluidics to form aggregates are not compatible with high-resolution imaging during culture, due to limitations of objective working distances through the device material. In situ imaging is typically desired to assess expression of fluorescent reporters in live samples during culture. Additionally, having the ability to perform off-chip aggregate formation and pre-treatment steps is desired in many protocols, which these existing platforms do not currently possess. Examples of the utilities of this capability include formation of aggregates from multiple cell types, incorporation of microparticles within aggregates, and Matrigel embedding steps. Thus, a platform that is designed for loading of pre-formed aggregates has greater experimental flexibility and can be used more broadly. While previous work has shown the ability to load pre-formed aggregates into devices with wells or traps,42–46 the majority of existing platforms are low-throughput, accommodating 20 samples or less, and are not inherently scalable (e.g. the fluidics required for trapping are not compact or the trapping mechanism is not deterministic). In addition, many existing platforms use trapping mechanisms that physically constrain aggregates, which in addition to limiting space for size increases during culture can also impose exogenous mechanical cues or induce diffusion limitations in traps.
In this work, we have created a culture platform for stem cell aggregates that satisfies the following criteria: can be deterministically loaded with pre-formed aggregates, allows aggregate growth over multiple days of culture without physically constraining aggregates, is compatible with high resolution live and end point imaging, and has a compact design that can be scaled up to increase higher throughput. We show that this microfluidic platform provides the ability to culture and perform multi-functional, on-chip phenotypic analyses of individual stem cell aggregates at high spatial density. This is facilitated by the abilities to deterministically trap individual pre-formed aggregates, easily exchange fluids, and couple devices directly with high-resolution microscopy. This platform also reduces heterogeneity in cultures in terms of aggregate size and morphology. We envision applications of this technology in standardizing the generation of PSC-derived tissue models and in expanding abilities for phenotypic analysis.
Materials and Methods
Device Fabrication
Microfluidic devices were fabricated in polydimethylsiloxane (PDMS) (Dow Corning Sylgard 184, Midland, MI) by soft lithography.31 Briefly, a master mold was fabricated by standard UV photolithography with the negative photoresist SU8-2100 (Microchem, Newton, MA) to create three layers of respective heights 200 μm, 100 μm, and 200 μm. Before PDMS molding, the master was treated with tridecafluoro-1,1,2,2-tetrahydrooctyl-1-trichlorosilane vapor (United Chemical Technologies, Bristol, PA) to allow release of the PDMS. To make devices, PDMS was mixed in a 10:1 ratio of pre-polymer and crosslinker, degassed to remove air bubbles, poured on the master mold, degassed a second time to remove remaining bubbles, and cured for 2 hours at 70 °C. Following curing, devices were cut and inlet and outlet holes for fluidic connections were punched with 18 gauge blunt needles (McMaster-Carr, Elmhurst, IL). Devices were then bonded onto glass coverslips by oxygen plasma bonding.
Mouse embryonic stem cell (mESC) culture
Mouse ESCs (D3 cell line) were maintained in tissue culture-treated polystyrene dishes (Corning Inc., Corning, NY) coated with 0.1% gelatin (Millipore, EmbryoMax). Undifferentiated culture media consisted of Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech, Herndon, VA) supplemented with 15% fetal bovine serum (Hyclone, Logan, UT), 100 U/mL penicillin, 100 μg/mL streptomycin, 0.25 μg/mL amphotericin (Mediatech), 2 mM L-glutamine (Mediatech), 1× MEM non-essential amino acid solution (Mediatech), 0.1 mM 2-betamercaptoethanol (Fisher Scientific, Fairlawn, NJ), and 103 U/mL leukemia inhibitory factor (LIF) (ESGRO, Chemicon, Temecula, CA). Cells were passaged at approximately 70% confluence (typically every 2–3 days) and seeded at a density of 1 million cells for a 100 mm plate. Culture media was completely exchanged every other day.
Aggregate formation
A single cell suspension of undifferentiated mESCs was obtained by dissociating monolayer cultures with 0.05% trypsin-EDTA (Mediatech). Aggregates were formed by centrifugation (200 rcf) of mESCs into 400 μm agarose (OmniPur®, EMD Millipore) microwells placed within a 6-well tissue culture polystyrene plate (Corning, Inc.).16 To yield either 500 cell or 1000 cell aggregates, volumes of 1 mL or 2 mL, respectively, of the single cell suspension (concentration: 3 million cells/mL) were added to each well of the 6-well plate. Cells were incubated in the wells in culture media without LIF for approximately 18–24 hours to allow aggregates to form.
Batch suspension culture
Following formation, aggregates were cultured in 100 mm bacteriological grade polystyrene Petri dishes (BD, Franklin Lakes, NJ) with approximately 2000 aggregates per plate in 10 mL of culture media (without LIF). Plates were maintained on rotary orbital shakers (Benchmark Scientific, Edison, NJ) at a frequency of 45 rpm.17, 47 Media was exchanged every other day, with 90% of the media replenished with fresh media.
Multiwell culture
Following formation, aggregates were cultured in 96-well U-bottom multiwell plates (Corning). Plates were prepared by coating with 2% Pluronic (ThermoFisher) for 1 hour to prevent cell attachment to the surface. To achieve one aggregate per well, aggregates were initially suspended in media without LIF at a concentration of 400 aggregates/mL and diluted twice (at 1:10 and 1:12.5) to yield a final concentration of 5 aggregates/mL before volumes of 200 μL were pipetted into each well. The final volume of each well was 200 μL, and no media exchange was performed for the duration of culture.
Device loading and operation
Prior to each experiment devices, fittings (blunt 16 gauge needles; McMaster-Carr), and tubing (1/32″ ID silicone tubing; Cole Parmer) were sterilized by autoclaving. Devices were connected to a second identical device during operation to increase the overall fluidic resistance and thereby decrease the flow rate during loading. Prior to aggregate loading, devices were first primed with 70% ethanol using a syringe to remove air bubbles. Next, devices were rinsed with sterile phosphate-buffered saline (PBS) and then incubated with a 1 mg/ml bovine serum albumin (BSA; Millipore) solution for 30 minutes to passivate the surfaces.33, 48 A 200 μL pipette tip filled with cell culture medium was placed at the device inlet for aggregate loading and perfusion. Aggregates were collected and resuspended at a concentration of approximately 600 aggregates/mL in a solution of 1.05 g/mL Percoll (Sigma Aldrich) in phosphate-buffered saline (PBS) to prevent rapid settling. Then, aggregates were pipetted into the inlet and loaded into devices via gravity-driven flow. Once aggregates were loaded, device outlets were connected to a syringe pump (PHD 2000; Harvard Apparatus), and the entire setup was placed in a humidified incubator (HERAcell 240i, Thermo Scientific). During culture, devices were continuously perfused at a defined flow rate by withdrawing culture media without LIF from a 200 μL pipette tip reservoir at the inlet. Additionally, devices were kept in a shallow, sterile water bath to limit evaporation. Phase contrast images of loaded devices were acquired using an EVOS microscope (LifeTechnologies).
LIVE/DEAD assay
A LIVE/DEAD assay (Invitrogen) was performed to evaluate cell viability at day 4 of differentiation. For aggregates cultured in devices, all steps were performed in devices. For aggregates cultured in batch suspension culture, all steps were performed in 1.5 mL microcentrifuge tubes. Aggregates were rinsed with PBS and then incubated with 2 μM calcein AM and 4 μM Ethidium-D in PBS with calcium and magnesium for 45 minutes. Samples were imaged on a confocal microscope (Zeiss LSM 700 Confocal Microscope). Device samples were imaged in devices; batch suspension culture samples were transferred to glass-bottom 24-well plates (MatTek Corporation) for imaging. Z stacks were acquired up to a depth of approximately 50 μm with a spacing of 3 μm.
Immunofluorescent staining and imaging
Immunofluorescent staining was performed in devices by perfusing all solutions through the pipette tip inlets by gravity-driven flow. For aggregates cultured in batch suspension, all steps were performed in 1.5 mL microcentrifuge tubes, and samples were continuously rotated on a tube rotator. For multiwell culture, all steps were performed in multiwell plates. First, samples were washed with PBS and fixed in 10% formalin at room temperature for 40 minutes. Following a wash in block buffer (2% BSA, 0.1% Tween 20, in PBS), samples were permeabilized in 1.5% Triton X 100 in PBS for 30 minutes. After washing in serum block buffer (2% donkey serum, 0.1% Tween 20, in PBS) for 30 minutes, samples were incubated overnight at room temperature in the primary antibody solution (1:200 Santa Cruz Biotechnology Oct-3/4 (N-19): sc-8628 in serum block buffer). The next day, samples were washed in serum block buffer for 30 minutes and then incubated in the secondary antibody solution (1:200 AlexaFluor®546 donkey anti-goat) at room temperature for 4 hours. For the final 15 minutes of incubation, nuclear stain Hoechst 3342 was added (final concentration: 10 μg/mL). Samples were washed a final time with serum block buffer for 30 minutes, stored overnight at 4°C, and imaged the next day. Device samples were imaged in devices; batch suspension culture samples were transferred to glass-bottom 24-well plates (MatTek Corporation) for imaging. Samples were imaged on a confocal microscope (Zeiss LSM 700 Confocal Microscope) with a 20× objective. Z stacks were acquired up to a depth of 30 μm with spacings of 3–5 μm. For larger aggregates that did not fit in a single field of view, 2×2 tile scan images with 10% overlap were acquired and stitched together with the microscope software (ZEN, Zeiss).
Statistical analysis
Experiments with replicate data are represented as the mean +/− standard deviation. Sample sizes (n) and number of independent experiments are detailed in the corresponding figure legends. Statistical analyses were performed using GraphPad Prism software. Statistical tests were performed using either a Mann-Whitney U test or non-parametric one-way ANOVA (with Kruskal-Wallis) combined with Dunn’s multiple comparison’s test for comparison of individual samples. P values less than 0.05 were considered significant.
Results and Discussion
Design of the microfluidic device
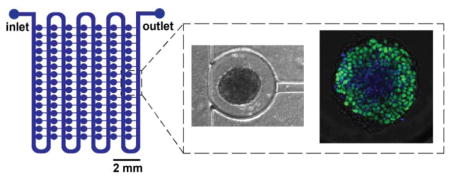
The design of the microfluidic device consists of an array of 105 traps for culture of individual aggregates (Figure 1). Although the master is a multi-layer master, the devices can be fabricated using a standard single-layer PDMS micromolding process. The traps are connected by a main channel for aggregate loading and perfusion of media and reagents. Loading of individual aggregates into traps is accomplished by a previously demonstrated hydrodynamic loading mechanism49–51 that is dependent on the ratio of two flow paths in the device (Fig. 1e). Briefly, the presence of a resistance channel at the back of each trap creates a secondary flow path that directs aggregates into traps. Once an aggregate enters a trap it blocks most of the flow to that trap so that subsequent aggregates preferentially continue to flow down the main channel and enter subsequent traps. Isolating individual aggregates in separate traps has a number of advantages, namely, preventing physical interactions between aggregates and enabling longitudinal tracking of individual aggregates during culture.
Figure 1.

Overview of microfluidic platform. a) Schematics illustrate the design of the device, with inset detailing the main channel, trap, and resistance channel. b,c) Diagrams show key device geometrical features. d) Photograph of a dye-filled device. e) Image shows the hydrodynamic loading mechanism, which is dependent on the balance of flow through the main channel versus flow through the traps and resistance channels.
Designing a device that allows for both initial loading of aggregates and subsequent culture of aggregates with significant size changes presents an engineering challenge. For reproducible loading of single aggregates in individual traps, there are two primary design requirements. First, the dimensions of the resistance channels must be small enough such that aggregates are not able to pass through the resistance channels during loading. To address this, we designed the resistance channel width (60 μm, Fig. 1b) to be less than half of the average diameter (~160 μm) of aggregates used for this study. Second, the relative fluidic resistances of the two flow paths in the device must be properly balanced for efficient loading of traps.49, 50 If flow through the traps and resistance channels is too low (fluidic resistance is too high), then few aggregates will be trapped; however, if the flow is too high then multiple aggregates will accumulate within traps, also resulting in inefficient trapping. These requirements were balanced in the final device dimensions (Fig. 1 b,c). We found that a ratio of trap to main channel resistance of ~20 resulted in good loading. To address the challenge of designing a device that physically accommodates growing aggregates over multiple days of culture, a pinched-off circular trap geometry was used (Fig. 1 b,c). The trap dimensions (500 μm width and height) allow for initial loading of relatively small aggregates (< 190 μm in diameter) without constricting growing aggregates over time (up to a maximum diameter of 500 μm).
Device operation and validation
To demonstrate the capabilities of our platform for aggregate culture, we differentiated aggregates by removing LIF from the culture media. Aggregates were pre-formed in microwells (D0) and then loaded into devices the following day (D1) (Fig. 2a). For comparison, aggregates were also differentiated in batch suspension culture. When devices were loaded, typically ~50% of traps were loaded with single aggregates, with the remaining traps containing no or multiple aggregates. We observed that multiple aggregate loading often occurred when clumps of aggregates entered a trap together, or when the first aggregate to enter a trap did not block the restriction channel before a second aggregate entered. The loading efficiency of this device reflects the tradeoff of scaling the traps to be small to allow effective loading and scaling to be large so that they can allow the aggregate size changes of up to five-fold over the course of culture. This is a unique challenge, in contrast to examples of previous work that trap objects such as cells and embryos, which do not grow bigger during the course of experiment and thus can be loaded using traps that fit tightly around the object. In our case, the criterion of not constraining aggregates within traps means that the deterministic loading mechanism results in an unavoidable imperfect loading efficiency. To compensate for the 50% loading efficiency, one could simply scale up the number of traps to increase the number of successful samples, which is well within experimental control. A scaled-up device would occupy a larger footprint, but there are no inherent design or operational challenges inherent to scaling up the platform other than making larger masters.
Figure 2.

Validation of device culture. a) Experimental design. b) Phase contrast images show representative aggregates at days 1 and 4 of differentiation. c) Representative confocal images of day 4 aggregates treated with a LIVE/DEAD stain (live cells: green, dead cells: red); (i) Maximum projection images; (ii) single channel z planes.
Aggregates remained trapped over multiple days of culture, as seen in the representative phase contrast images (Fig. 2b). Importantly, aggregates maintained a three-dimensional, spherical morphology during device culture, comparable to that of batch suspension culture, as opposed to plating down on the glass surface and forming morphologies resembling monolayer culture. In further support of the notion of little to no cell adhesion to the glass culture surface, we observe that day 4 aggregates can move within the device under flow perturbations. Devices were continuously perfused at a flow rate of 10 μL/hr, which was approximately equivalent to exchanging the volume of the device once per hour. This flow rate was chosen as it was comparable to the media exchange rate of batch suspension culture on the basis of volume of media per aggregate per time. To confirm that this perfusion rate supported cell viability aggregates were treated with a LIVE/DEAD stain. Representative confocal microscopy images (Fig. 2c; Supplemental Fig. 2) demonstrated that there was high viability and minimal cell death in aggregates cultured in devices for 4 days and that viability was comparable to that of batch culture.
Growth and size control
A disadvantage of batch-based aggregate culture methods is that samples can physically interact with one another, which introduces a source of heterogeneity into cultures. We hypothesized that culture of aggregates in individual traps would maintain better uniformity compared to batch suspension culture in aspects such as size and morphology by preventing physical interactions between multiple aggregates. To assess how device culture impacted aggregate morphology and size during differentiation, phase contrast images of aggregates were acquired at day 1 (immediately following loading) and day 4 of differentiation, and the batch and on-chip cultures were compared (Fig. 3a). In batch cultures at day 4, a variety of aggregate sizes and morphologies were observed. The presence of small aggregates in the cultures (comparable to day 1 sizes) was likely the result of single cells shedding off from aggregates and forming new, smaller aggregates (Fig. 3a, indicated by red arrows). Aggregates with budding morphologies, i.e. aggregates with small, spherical protrusions, were also observed in batch cultures (Fig. 3a, indicated by white arrows). In contrast, aggregate sizes were more uniform and the budding morphology was observed less frequently in device cultures.
Figure 3.
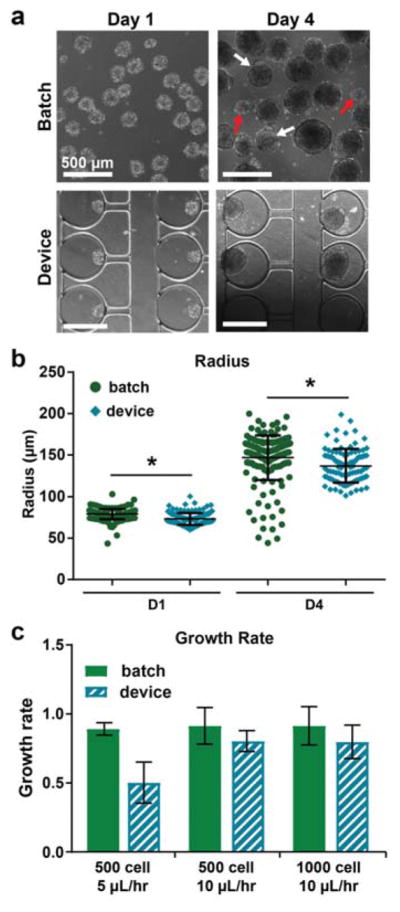
Device culture reduces variability in aggregate size. a) Phase contrast images showing size and morphology at days 1 and 4. Red arrows indicate subpopulations of much smaller aggregates. White arrows indicate aggregates with budding morphologies. b) Aggregate radii were quantified from phase contrast images at day 1 (before loading into devices) and day 4 of differentiation. Two independent samples are shown for each condition, with n ≥ 80 aggregates for batch and n ≥ 40 aggregates for devices. * P < 0.0001. c) Aggregate growth rates for different starting sizes and device perfusion rates. Growth rate was defined as change in size between days 1 and 4, normalized by initial size. n ≥ 2 batch samples or devices, respectively, per condition. All samples: n.s.
Aggregate size was quantified from the phase contrast images. At day 4, the mean size was similar for batch and device cultures, but there was a subpopulation of smaller aggregates present in the batch cultures, consistent with qualitative assessments (Fig. 3b). The size distribution of aggregates was significantly larger for batch culture compared to device culturing, demonstrating the utility of the microfluidic platform for controlling aggregate size. It was also observed that device culture better controlled aggregate size compared to batch for different initial sizes of aggregates (500 cell starting size) (Supplemental Fig. 3).
Previous work has shown that microfluidic perfusion culture can modulate the soluble microenvironment, thereby affecting cell phenotype.11 To characterize basic characteristics of the soluble environment, we calculated the Peclet number and found it to be ~90 (Supplemental Methods), suggesting that the platform operated under a convection-dominated regime. Based on the perfusion rate used, soluble molecules secreted by cells likely had a short enough residence time (~ minutes) to have a minimal effect on aggregates in neighboring traps. Additionally, it is possible that an aggregate’s position within a device (i.e. closer to the inlet versus outlet) could have an impact on its phenotype. From the size data, however, there was no evidence that aggregate phenotypes differed depending on position in the device (Supplemental Fig. 3c–e and 5a–b).
We compared the growth rate of aggregates cultured in devices to those maintained in batch culture. The metric for growth rate was defined as the change in average radii between days 1 and 4 of culture, normalized by the initial radii. We found that there was no significant difference in growth rate for aggregates cultured in devices, compared to batch, for two different starting sizes of aggregates (500 cell and 1000 cell) and at a device perfusion rate of 10 μL/hr (Fig. 3c). From this, we conclude that at these parameters, aggregates receive sufficient nutrients and grow comparably. Interestingly, aggregates cultured at a perfusion rate of 5 μL/hr exhibited a noticeably lower growth rate than batch (although not statistically significant) (Fig. 3c). Importantly, we did not observe significant differences in viability for this condition (Supplemental Fig. 2), suggesting that the reduced growth rate in devices could be due to limited mass transfer of nutrients. For this reason, the following experiments were conducted at 10 μL/hr perfusion.
On-chip immunofluorescent staining and image analysis
To demonstrate the utility of our microfluidic platform for performing various phenotypic assays on-chip in conjunction with multi-day culture, we fixed and immunofluorescently stained aggregates at day 4 of differentiation (Fig. 4). We examined expression of the pluripotency marker Oct4, which is highly expressed in pluripotent stem cells and decreases in expression over the first week of undirected aggregate differentiation.52 Oct4 expression was quantified in day 4 aggregates cultured in devices, batch, or round bottom 96-well multiwell plates53 (Fig. 5). We performed a multiwell culture condition in order to compare on-chip differentiation to a platform where aggregates are also physically isolated. We hypothesized that the differences we saw in size and morphology between aggregates cultured in devices versus batch may manifest in differences in Oct4 expression. However, similar expression levels were observed across the three conditions. Aggregates cultured in devices had slightly higher Oct4 expression levels than both batch and multiwell culture formats (Fig. 5b, Supplemental Fig. 5e). Although aggregate size can play a role in phenotype and rate of differentiation,15, 26, 54, 55 we did not find a correlation between Oct4 expression and aggregate size (Supplemental Fig. 5a–c). This data suggests that microfluidic culture can be leveraged to modulate aggregate differentiation, in this case, by maintaining pluripotency of aggregates at a higher level following LIF removal from the media. Previous work has demonstrated that microfluidic perfusion modulates the residence time of soluble secreted factors in the cellular microenvironment,56 and this may play a role in promoting pluripotency, although the specific mechanism in this case is unknown.
Figure 4.

On-chip immunostaining and quantification of protein expression. (1) Immunostaining is performed on-chip by perfusing all reagents through the device via gravity-driven flow. (2) Confocal microscopy is used to image samples. (3) Image processing is used to quantify single-cell level protein expression within aggregates.
Figure 5.

Device facilitates studying individual sample phenotypes. a) Representative immunostaining images. b) Mean aggregate Oct4 expression for different culture platforms. Two independent experiments are shown for batch and device conditions (n ≥ 25 each) and one experiment is shown for multiwell condition (n = 8). * P < 0.00001 vs. batch and P < 0.0001 vs. multiwell. c) Inter-aggregate heterogeneity in Oct4 expression for device and batch cultures.
Next, we calculated the coefficient of variation of Oct4 expression to examine inter- and intra-aggregate heterogeneity in expression levels, across a given culture platform (Fig. 5c, Supplemental Fig. 5f). There were no significant differences in inter-aggregate heterogeneity (Fig. 5c), suggesting that each of the culture platforms perform equally well in this particular respect. However, there were subtle differences in intra-aggregate heterogeneity, indicating that there was more diversity in the Oct4 expression levels among cells of a single aggregate in device samples. Although these studies focused on a limited number of parameters to assess cell phenotypes, we anticipate that future studies will be valuable to assess how microfluidic culture modulates heterogeneity in terms of phenotypes seen during early differentiation beyond size.
Conclusions
Here we present the development of a microfluidic platform for culture, longitudinal tracking, and imaging of stem cell aggregates. Physical isolation of individual aggregates in individual traps prevents aggregate-aggregate interactions that contribute to heterogeneity in cultures and also enables correlation of live imaging and end point assay phenotypic information. Culture of aggregates in a high-density array format permits culture and analysis of ~50 samples at once, and this platform can be readily scaled up or parallelized to increase throughput. Finally, the design of the device permits high-resolution imaging for assessment of sample phenotypes, including incorporating techniques such as tissue clearing and multiphoton microscopy to image through denser, larger tissue constructs. Due to these combined capabilities, we anticipate that this platform can be applied for the generation of stem cell derived microtissues for drug screening and mechanistic studies of stem cell differentiation and tissue morphogenesis. Future applications are not limited to stem cells, and this platform could readily be applied to studying other cell aggregate systems, such as tumor spheroids. With the combined advantages of standardizing the culture environment and enabling single sample level analysis, we envision this platform as a tool in generating PSC aggregate derived cell and tissue models.
Supplementary Material
Acknowledgments
Funding for this work was provided by the NSF Emergent Behaviors of Integrated Cellular Systems Science and Technology Center (CBET 0939511). HL is partially supported by NIH (NS096581, GM088333, EB021676, EB020424, AG05030, and GM108962). TCM is supported by CIRM (LA1_C14-08015). EJH is supported by a NSF Stem Cell Biomanufacturing IGERT (DGE 0965945) and a NSF Graduate Research Fellowship (DGE-1650044). The authors acknowledge Dr. Melissa Kemp for feedback on the manuscript and Dr. Thomas Levario for helpful discussions. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship Program under Grant No. DGE-1650044. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation.
Notes and references
- 1.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 2.Martin GR. Proceedings of the National Academy of Sciences. 1981;78:7634–7638. doi: 10.1073/pnas.78.12.7634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Evans MJ, Kaufman MH. Nature. 1981;292:154–156. doi: 10.1038/292154a0. [DOI] [PubMed] [Google Scholar]
- 4.Itskovitz-Eldor J, Schuldiner M, Karsenti D, Eden A, Yanuka O, Amit M, Soreq H, Benvenisty N. Molecular medicine (Cambridge, Mass) 2000;6:88–95. [PMC free article] [PubMed] [Google Scholar]
- 5.Dvash T, Mayshar Y, Darr H, McElhaney M, Barker D, Yanuka O, Kotkow KJ, Rubin LL, Benvenisty N, Eiges R. Human Reproduction. 2004;19:2875–2883. doi: 10.1093/humrep/deh529. [DOI] [PubMed] [Google Scholar]
- 6.Sasai Y. Cell Stem Cell. 2013;12:520–530. doi: 10.1016/j.stem.2013.04.009. [DOI] [PubMed] [Google Scholar]
- 7.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eiraku M, Takata N, Ishibashi H, Kawada M, Sakakura E, Okuda S, Sekiguchi K, Adachi T, Sasai Y. Nature. 2011;472:51–56. doi: 10.1038/nature09941. [DOI] [PubMed] [Google Scholar]
- 9.Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, Clevers H. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- 10.Kinney MA, Hookway TA, Wang Y, McDevitt TC. Annals of biomedical engineering. 2014;42:352–367. doi: 10.1007/s10439-013-0953-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Przybyla L, Voldman J. Annu Rev Anal Chem (Palo Alto Calif) 2012;5:293–315. doi: 10.1146/annurev-anchem-062011-143122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zandstra PW, Le HV, Daley GQ, Griffith LG, Lauffenburger DA. Biotechnol Bioeng. 2000;69:607–617. [PubMed] [Google Scholar]
- 13.Yu H, Alexander CM, Beebe DJ. Lab Chip. 2007;7:726–730. doi: 10.1039/b618793e. [DOI] [PubMed] [Google Scholar]
- 14.Blagovic K, Kim LY, Voldman J. PLoS ONE. 2011;6:e22892. doi: 10.1371/journal.pone.0022892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bauwens CL, Peerani R, Niebruegge S, Woodhouse KA, Kumacheva E, Husain M, Zandstra PW. Stem cells. 2008;26:2300–2310. doi: 10.1634/stemcells.2008-0183. [DOI] [PubMed] [Google Scholar]
- 16.Ungrin MD, Joshi C, Nica A, Bauwens C, Zandstra PW. PLoS ONE. 2008;3:e1565. doi: 10.1371/journal.pone.0001565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kinney MA, Saeed R, McDevitt TC. Integr Biol (Camb) 2012;4:641–650. doi: 10.1039/c2ib00165a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doetschman TC, Eistetter H, Katz M, Schmidt W, Kemler R. Journal of embryology and experimental morphology. 1985;87:27–45. [PubMed] [Google Scholar]
- 19.Zandstra PW, Bauwens C, Yin T, Liu Q, Schiller H, Zweigerdt R, Pasumarthi KB, Field LJ. Tissue engineering. 2003;9:767–778. doi: 10.1089/107632703768247449. [DOI] [PubMed] [Google Scholar]
- 20.Cameron CM, Hu WS, Kaufman DS. Biotechnol Bioeng. 2006;94:938–948. doi: 10.1002/bit.20919. [DOI] [PubMed] [Google Scholar]
- 21.Sargent CY, Berguig GY, Kinney MA, Hiatt LA, Carpenedo RL, Berson RE, McDevitt TC. Biotechnol Bioeng. 2010;105:611–626. doi: 10.1002/bit.22578. [DOI] [PubMed] [Google Scholar]
- 22.Ng ES, Davis RP, Azzola L, Stanley EG, Elefanty AG. Blood. 2005;106:1601–1603. doi: 10.1182/blood-2005-03-0987. [DOI] [PubMed] [Google Scholar]
- 23.Koike M, Kurosawa H, Amano Y. Cytotechnology. 2005;47:3–10. doi: 10.1007/s10616-005-3743-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakazawa K, Yoshiura Y, Koga H, Sakai Y. Journal of bioscience and bioengineering. 2013;116:628–633. doi: 10.1016/j.jbiosc.2013.05.005. [DOI] [PubMed] [Google Scholar]
- 25.Moeller HC, Mian MK, Shrivastava S, Chung BG, Khademhosseini A. Biomaterials. 2008;29:752–763. doi: 10.1016/j.biomaterials.2007.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hwang YS, Chung BG, Ortmann D, Hattori N, Moeller HC, Khademhosseini A. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:16978–16983. doi: 10.1073/pnas.0905550106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Azarin SM, Lian X, Larson EA, Popelka HM, de Pablo JJ, Palecek SP. Biomaterials. 2012;33:2041–2049. doi: 10.1016/j.biomaterials.2011.11.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alessandri K, Sarangi BR, Gurchenkov VV, Sinha B, Kießling TR, Fetler L, Rico F, Scheuring S, Lamaze C, Simon A, Geraldo S, Vignjević D, Doméjean H, Rolland L, Funfak A, Bibette J, Bremond N, Nassoy P. Proceedings of the National Academy of Sciences. 2013;110:14843–14848. doi: 10.1073/pnas.1309482110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maltsev VA, Wobus AM, Rohwedel J, Bader M, Hescheler J. Circulation Research. 1994;75:233–244. doi: 10.1161/01.res.75.2.233. [DOI] [PubMed] [Google Scholar]
- 30.El-Ali J, Sorger PK, Jensen KF. Nature. 2006;442:403–411. doi: 10.1038/nature05063. [DOI] [PubMed] [Google Scholar]
- 31.Whitesides GM, Ostuni E, Takayama S, Jiang X, Ingber DE. Annual Review of Biomedical Engineering. 2001;3:335–373. doi: 10.1146/annurev.bioeng.3.1.335. [DOI] [PubMed] [Google Scholar]
- 32.Sakai Y, Hattori K, Yanagawa F, Sugiura S, Kanamori T, Nakazawa K. Biotechnology journal. 2014;9:971–979. doi: 10.1002/biot.201300559. [DOI] [PubMed] [Google Scholar]
- 33.Kim C, Lee KS, Bang JH, Kim YE, Kim MC, Oh KW, Lee SH, Kang JY. Lab Chip. 2011;11:874–882. doi: 10.1039/c0lc00516a. [DOI] [PubMed] [Google Scholar]
- 34.Occhetta P, Centola M, Tonnarelli B, Redaelli A, Martin I, Rasponi M. Sci Rep. 2015;5:10288. doi: 10.1038/srep10288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patra B, Chen YH, Peng CC, Lin SC, Lee CH, Tung YC. Biomicrofluidics. 2013;7:54114. doi: 10.1063/1.4824480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu CY, Tseng SY, Yang SM, Hsu L, Liu CH, Chang HY. Biofabrication. 2014;6:015009. doi: 10.1088/1758-5082/6/1/015009. [DOI] [PubMed] [Google Scholar]
- 37.de Groot TE, Veserat KS, Berthier E, Beebe DJ, Theberge AB. Lab Chip. 2016;16:334–344. doi: 10.1039/c5lc01353d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frey O, Misun PM, Fluri DA, Hengstler JG, Hierlemann A. Nat Commun. 2014;5:4250. doi: 10.1038/ncomms5250. [DOI] [PubMed] [Google Scholar]
- 39.Lee WG, Ortmann D, Hancock MJ, Bae H, Khademhosseini A. Tissue engineering Part C, Methods. 2010;16:249–259. doi: 10.1089/ten.tec.2009.0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rismani Yazdi S, Shadmani A, Burgel SC, Misun PM, Hierlemann A, Frey O. Lab Chip. 2015;15:4138–4147. doi: 10.1039/c5lc01000d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chan HF, Zhang Y, Ho Y-P, Chiu Y-L, Jung Y, Leong KW. 2013;3:3462. doi: 10.1038/srep03462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Astolfi M, Peant B, Lateef MA, Rousset N, Kendall-Dupont J, Carmona E, Monet F, Saad F, Provencher D, Mes-Masson AM, Gervais T. Lab Chip. 2016;16:312–325. doi: 10.1039/c5lc01108f. [DOI] [PubMed] [Google Scholar]
- 43.Bergstrom G, Christoffersson J, Schwanke K, Zweigerdt R, Mandenius CF. Lab Chip. 2015;15:3242–3249. doi: 10.1039/c5lc00449g. [DOI] [PubMed] [Google Scholar]
- 44.Hribar KC, Finlay D, Ma X, Qu X, Ondeck MG, Chung PH, Zanella F, Engler AJ, Sheikh F, Vuori K, Chen SC. Lab Chip. 2015;15:2412–2418. doi: 10.1039/c5lc00159e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khoury M, Bransky A, Korin N, Konak LC, Enikolopov G, Tzchori I, Levenberg S. Biomed Microdevices. 2010;12:1001–1008. doi: 10.1007/s10544-010-9454-x. [DOI] [PubMed] [Google Scholar]
- 46.Ruppen J, Cortes-Dericks L, Marconi E, Karoubi G, Schmid RA, Peng R, Marti TM, Guenat OT. Lab Chip. 2014;14:1198–1205. doi: 10.1039/c3lc51093j. [DOI] [PubMed] [Google Scholar]
- 47.Carpenedo RL, Sargent CY, McDevitt TC. Stem cells. 2007;25:2224–2234. doi: 10.1634/stemcells.2006-0523. [DOI] [PubMed] [Google Scholar]
- 48.Chen YH, Peng CC, Tung YC. Biomicrofluidics. 2015;9:054111. doi: 10.1063/1.4931638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chung K, Kim Y, Kanodia JS, Gong E, Shvartsman SY, Lu H. Nat Methods. 2011;8:171–176. doi: 10.1038/nmeth.1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chung K, Rivet CA, Kemp ML, Lu H. Analytical chemistry. 2011;83:7044–7052. doi: 10.1021/ac2011153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Levario TJ, Zhan M, Lim B, Shvartsman SY, Lu H. Nature protocols. 2013;8:721–736. doi: 10.1038/nprot.2013.034. [DOI] [PubMed] [Google Scholar]
- 52.White DE, Sylvester JB, Levario TJ, Lu H, Streelman JT, McDevitt TC, Kemp ML. Integr Biol (Camb) 2015;7:825–833. doi: 10.1039/c5ib00072f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vrij EJ, Espinoza S, Heilig M, Kolew A, Schneider M, van Blitterswijk CA, Truckenmuller RK, Rivron NC. Lab Chip. 2016;16:734–742. doi: 10.1039/c5lc01499a. [DOI] [PubMed] [Google Scholar]
- 54.Bauwens CL, Song H, Thavandiran N, Ungrin M, Masse S, Nanthakumar K, Seguin C, Zandstra PW. Tissue engineering Part A. 2011;17:1901–1909. doi: 10.1089/ten.TEA.2010.0563. [DOI] [PubMed] [Google Scholar]
- 55.Mohr JC, Zhang J, Azarin SM, Soerens AG, de Pablo JJ, Thomson JA, Lyons GE, Palecek SP, Kamp TJ. Biomaterials. 2010;31:1885–1893. doi: 10.1016/j.biomaterials.2009.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Przybyla LM, Voldman J. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:835–840. doi: 10.1073/pnas.1103100109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.