

Abstract
The pharmacological manipulation of Liver X Receptors (LXRs) has been an attractive therapeutic strategy for atherosclerosis treatment as they control reverse cholesterol transport and inflammatory response. In this study, we present the development and efficacy of nanoparticles (NPs) incorporating the synthetic LXR agonist GW3965 (GW) in targeting atherosclerotic lesions. Collagen IV (Col IV) targeting ligands were employed to functionalize the NPs to improve targeting to the atherosclerotic plaque, and formulation parameters such as the length of the polyethylene glycol (PEG) coating molecules were systematically optimized. In vitro studies indicated that the GW-encapsulated NPs upregulated the LXR target genes and downregulated pro-inflammatory mediator in macrophages. The Col IV-targeted NPs encapsulating GW (Col IV-GW-NPs) successfully reached atherosclerotic lesions when administered for 5 weeks to mice with preexisting lesions, substantially reducing macrophage content (~30%) compared to the PBS group, which was with greater efficacy vs. non-targeting NPs encapsulating GW (GW-NPs) (~18%). In addition, mice administered the Col IV-GW-NPs did not demonstrate increased hepatic lipid biosynthesis or hyperlipidemia during the treatment period, unlike mice injected with the free GW. These findings suggest a new form of LXR-based therapeutics capable of enhanced delivery of the LXR agonist to atherosclerotic lesions without altering hepatic lipid metabolism.
1. Introduction
Atherosclerosis is the most common cause of coronary artery disease, which accounts for the largest number of deaths in the US. This pathology is characterized by deposition of cholesterol and triglyceride (TG)-rich lipoproteins within the arterial wall, leading to an inflammatory response. Consequently, circulating monocytes are recruited into the arterial wall and undergo differentiation into macrophages, which can progressively transform into cholesterol-laden foam cells. The continuous accumulation of foam cells and extracellular materials generates the atherosclerotic plaques that can rupture and cause acute local occlusive thrombosis, and this eventually results in myocardial infarction or stroke [1].
Liver X receptors (LXRα and LXRβ) are a family of nuclear receptors that control the expression of key proteins regulating lipid metabolism by dimerizing with retinoid X receptors (RXRs) [2]. In macrophages, LXR activation increases the expression of the cholesterol transporter ATP binding cassette transporter subfamilies A member 1 (ABCA1) and G member 1 (ABCG1), promoting cholesterol efflux to apolipoprotein A1 (ApoA1) and high-density lipoprotein (HDL), respectively, which transport cholesterol to the liver for further elimination. LXR activation also dampens the gene expression of pro-inflammatory genes by various mechanisms including SUMOylation-mediated transrepression and histone acetylation modification, among others [3]. Hence, synthetic LXR agonists such as GW3965 (referred to as GW) simultaneously promote cholesterol efflux and hamper pro-inflammatory signals in plaque macrophages, offering a seemingly attractive pharmacological alternative to treat atherosclerosis. Unfortunately, their beneficial effects on atherosclerosis are accompanied by an undesirable increase in plasma levels and hepatic content of lipids, particularly TG, which has hindered Food and Drug Administration (FDA) approval of the LXR agonists that have entered clinical trials [4–6]. Therefore, innovative strategies that reduce the adverse side effects of LXR activation while maintaining efficacy, such as tissue-selective LXR-based therapeutics, are needed.
Nanomedicines have shown successful patient outcomes in clinical trials by improving the pharmacokinetic profile of cytotoxic drugs and decreasing their toxicity [7–9]. Nanoparticles (NPs) designed to accumulate in desired tissues, the next generation of NP-based therapies, are also currently in clinical trials [10,11]. While targeted NPs have been thoroughly investigated for cancer treatment, only a few studies of nanotherapeutics actively targeting atherosclerosis have been reported (e.g., [12,13]). Our group previously developed polymeric NPs encapsulating GW to enhance the pro-resolving activity and mitigate the toxicity of GW [14,15]. Those strategies, however, did not include a systematic method for the formulation and optimization of targeted nanoplatforms in the treatment of inflammatory diseases. Herein, we report the development of NPs encapsulating GW designed to have selective advantage in targeting atherosclerotic lesions over the liver, and their therapeutic efficacy in the Ldlr−/− mouse model of atherosclerosis. We chose a collagen IV (Col IV)–targeting heptapeptide ligand identified previously by our group [16], to deliver our NP formulation to atherosclerotic lesions, as the Col IV-targeted NPs previously developed for the delivery of peptide Ac2-26 and IL-10 to plaques exhibited superior targeting abilities to non-targeting NPs via the specific binding with Col IVs overexpressed at sites of atherosclerosis [17–19].
The NPs in the present study comprise three distinct functional components: i) a biodegradable hydrophobic polymeric core (poly(D,L-lactide) (PLA) with terminal ester groups); ii) a lipid layer of methoxy(polyethylene glycol) (mPEG)-functionalized phospholipids and 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC); and, iii) a Col IV targeting peptide conjugated to a 1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE)-PEG with a molecular weight of 2000 kDa (Col IV-DSPE-PEG2000 conjugate) as part of the surface layer. The lipid-polymer hybrid NPs have been reported as promising platforms for small-molecule drug delivery [20], protein delivery [21], and gene delivery [22] owing to the complementary characteristics of polymeric NPs and liposomes that enhance their physical stability and biocompatibility. To obtain ideal targeted NPs for in vivo use, optimization of the weight ratio of polymer to lipids, the molar ratio of the lipids making up the surface layer, and the length of the PEG-coating molecules on the surface of the NPs are all critical. Here, we report the results of the systematic optimization of the formulation parameters above to construct the Col IV-targeting NPs and their GW encapsulation. Notably, we demonstrate that these NPs have anti-atherogenic effects on macrophages in vivo without altering plasma or hepatic lipid homeostasis. This new targeted NP system for the delivery of GW to atherosclerotic plaques suggests a new modality for combating inflammation in advanced atherosclerosis.
2. Results and Discussion
2.1. Formulation parameters of PLA-DSPE-mPEGs-DLPC-NPs
PEGylation has been a key process in reducing the formation of protein corona on the surface of NPs, which can hinder the recognition of their receptor targets in the blood stream. PEG is a promising antifouling polymer for use in engineering NPs. However, it is also important to choose the right density and length of the PEG-coating molecules in designing targeted NPs with optimal binding specificity, as PEG interferes with the binding of the targeting ligand to its target [23]. The biorecognition of targeted liposomes and inorganic NPs was maximized when the length of the PEG-coating molecules was properly shortened relative to the ligand linker [23–26]. On the other hand, the optimal length of targeted lipid-polymer hybrid NPs was not yet been identified. To examine the effects of the length of PEG-coating molecules on lipid-polymer NPs, a series of NPs with various lengths of methoxy-capped PEGylated phospholipids (DSPE-mPEGs) with diverse molecular weights (DSPE-mPEG550, DSPE-mPEG1000, and DSPE-mPEG2000) were prepared using a modified emulsion method (Figure 1A). An organic phase mixture with an 80:20 (v/v) ratio of ethyl acetate and benzyl alcohol was used to solubilize the lipids (DSPE-mPEGs and DLPC) as well as the ester-terminated PLA (PLA-OCH3) prior to rapid mixing with the aqueous phase. After overnight evaporation of the organic solvents, self-assembled NPs consisting of a hydrophobic polymer core (PLA-OCH3) surrounded by a lipid layer of DLPC/DSPE-mPEG550, DLPC/DSPE-mPEG1000 or DLPC/DSPE-mPEG2000 were obtained: NPs(550), NPs(1000), and NPs(2000), respectively. Our studies revealed that all lipids could be simply dissolved in an organic phase in which polymers were solubilized without an additional heating step that was frequently needed to dissolve the lipids in the aqueous phase previously [27,28].
Figure 1.
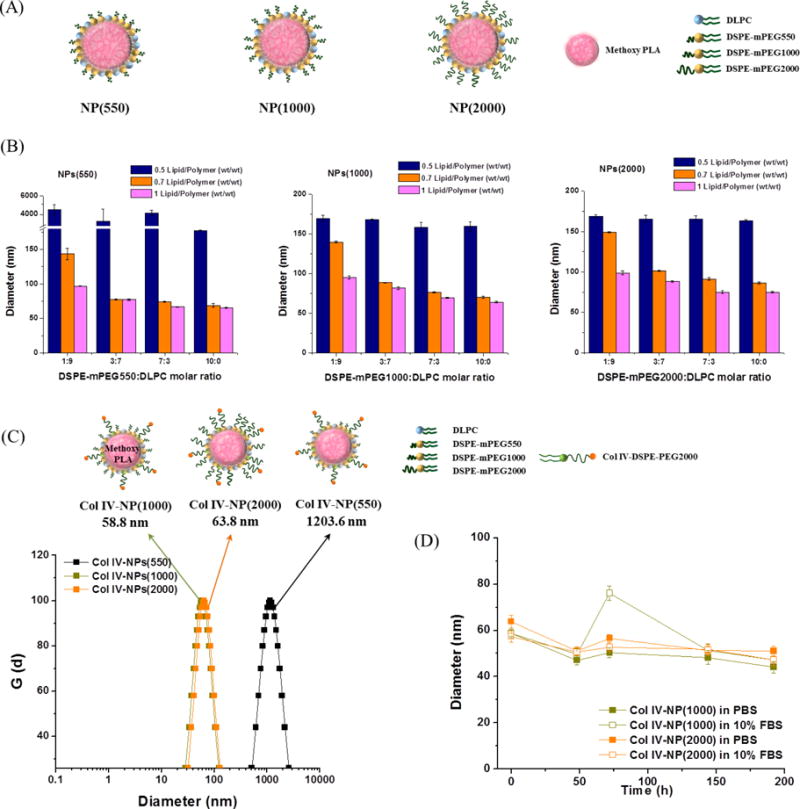
Development of the lipid-polymer hybrid NPs with various lengths of methoxy-capped PEGylated phospholipids (DSPE-mPEGs) with diverse molecular weights (DSPE-mPEG550, DSPE-mPEG1000, and DSPE-mPEG2000), and engineering of the Col IV-targeting NPs. (A) Schematics of the NP(550), NP(1000), and NP(2000) comprising PLA cores surrounded by DLPC/DSPE-mPEG550, DLPC/DSPE-mPEG1000, and DLPC/DSPE-mPEG2000, respectively. (B) NPs(550), NPs(1000), and NPs(2000) with variations in the formulation parameters such as weight ratio of total lipids to PLA (0.5, 0.7, and 1, wt/wt) and molar ratio of DSPE-mPEGs to DLPC (1:9, 3:7, 7:3, and 10:0) were measured by DLS in distilled water. (C) Hydrodynamic size measurement of the Col IV-NPs in PBS using DLS. Col IV-NPs(550), Col IV-NPs(1000), and Col IV-NPs(2000) were synthesized with DSPE-mPEGs, DLPC, and Col IV-DSPE-PEG2000 (67.5:27.5:5. molar ratio) at 70% of the PLA weight. DSPE-mPEG550, DSPE-mPEG1000, and DSPE-mPEG2000 were used for the preparation of Col IV-NPs(550), Col IV-NPs(1000), and Col IV-NPs(2000), respectively. (D) In vitro stabilities of Col IV-NPs in PBS and 10% FBS at 37°C. Data are expressed as mean ± SEM.
To investigate the effects of the molar ratio of lipid-PEG/lipid (DSPE-mPEGs/DLPC)–containing lipid layer and the weight ratio of total lipids/polymer (DSPE-mPEGs + DLPC/PLA-OCH3) on NP size, lipid-PEG/lipid molar ratios of 1:9, 3:7, 7:3, and 10:0 were tested against total lipids/polymer weight ratios of 0.5:1, 0.7:1, and 1:1. In all formulations of NPs(550), NPs(1000), and NPs(2000), increasing the total lipids/polymer weight ratio yielded smaller NPs (Figure 1B). At the total lipids/polymer weight ratio of 0.5:1, an increase in lipid-PEG/lipid molar ratio from 1:9 to 10:0 did not alter NP size, whereas an increase in the lipid-PEG/lipid molar ratio from 1:9 to 3:7 substantially reduced the NP size at total lipids/polymer weight ratios of 0.7:1 and 1:1. The results indicate that NP sizes can be finely tuned given the presence of sufficient amounts of total lipids covering the hydrophobic PLA core. We chose formulations with diameters <100 nm in distilled water as suitable for further modification and characterization. Thus, a total lipids/polymer weight ratio of 0.7:1 and a lipid-PEG/lipid molar ratio of 3:7 were considered minimum requirements for optimal NP formulation with minimal PEG content.
Next, we examined the stability of the NPs over time by monitoring changes in their size. We incubated NPs(550), NPs(1000), and NPs(2000) with a total lipids/polymer weight ratio of 0.7:1 and lipid-PEG/lipid molar ratios of 3:7 or 7:3 in PBS and PBS containing 10% fetal bovine serum (FBS) at 37 °C. As shown in Figure S1, most of the NPs were stable in both solutions for 7 days, maintaining their size between 35 - 45 nm. Particularly, the hydrodynamic sizes of the NPs in PBS and PBS containing 10% FBS were slightly smaller than those measured in distilled water because of the increased ionic strength on the surface of NPs in PBS solution [29]. In contrast, NPs(550) with a lipid-PEG/lipid molar ratio of 3:7 significantly increased in size in PBS containing 10% FBS within 2 days, suggesting that the higher density of PEG-coating molecules is required to achieve stealth function when they are short in length. Hence, a lipid-PEG/lipid molar ratio of 7:3 was selected as the final formulation parameter for further modification of the NPs(550), NPs(1000), and NPs(2000).
2.2. Preparation of Col IV-NPs with various PEG lengths
The length of PEG used for conjugating with Col IV targeting ligand was fixed by selecting a DSPE-PEG with molecular weight of 2000 kDa, since it was the most commonly used in developing targeted lipid-polymer hybrid NPs [30–32]. To engineer Col IV–targeting lipid-polymer NPs (Col IV-NPs) with various lengths of PEG-coating molecules, the Col IV-DSPE-PEG2000 conjugate was first synthesized by conjugating the KLWVLPK peptide to maleimide-functionalized DSPE-PEG2000 (DSPE-PEG-MAL) via the free thiol of the C-terminal GGGC linker using maleimide chemistry, and purified by HPLC. MALDI TOF results indicate that the DSPE-PEG-MAL (average MW: 2941.6 Da) was conjugated to the peptide (MW: 1157.4 Da), yielding a Col IV-DSPE-PEG2000 conjugate (average MW. 4076.9 Da), which was successfully purified (Figure S2). Multiple peaks in the MALDI results are attributable to the polydispersity of PEG. Next, a series of Col IV-NPs, i.e., Col IV-NPs(550), Col IV-NPs(1000) and Col IV-NPs(2000), were prepared by pairing the Col IV-DSPE-PEG2000 with DSPE-mPEG550/DLPC, mPEG1000/DLPC, and mPEG2000/DLPC. As shown in Figure 1C, the hydrodynamic sizes of the Col IV-NPs(1000) and Col IV-NPs(2000) were 58.8 ± 0.6 nm and 63.8 ± 0.7 nm, respectively. Whereas, Col IV-NPs(550) was larger than ~ 1 μm, indicating that at least PEG1000 is needed for stabilizing the Col IV–targeting ligand-modified surface as well as successful coating of the NPs. The hydrodynamic sizes of Col IV-NPs(1000) and Col IV-NPs(2000) were stable throughout the 192-h study in both PBS and PBS containing 10% FBS (Figure 1D).
Binding studies for the Col IV-NPs were then performed via SPR measurement. The NPs(1000) and NPs(2000) were employed as control NPs to demonstrate the targeting abilities of Col IV–targeting peptides decorated on the Col IV-NPs(1000) and Col IV-NPs(2000), respectively. We found that the Col IV-NPs(1000) specifically bound to Col IV proteins even at a low concentration (0.3 mg/mL) and remained on the Col IV surface with no appreciable dissociation owing to the multivalent interactions of the NPs (Figure S3A) [33]. In contrast, no binding was observed in Col IV-NPs(2000), in which the length of the PEG-coating molecules (DSPE-mPEG2000) used to construct the NPs was similar to that of the targeting ligand displaying lipid-PEG (Col IV-DSPE-PEG2000), as well as in non-targeting NPs(1000) and NPs(2000) (Figure S3B–D). These results suggest that the PEG conjugated with the Col IV targeting ligand should be longer than the PEG coated on the surface of lipid-polymer hybrid NPs, similar to the PEGylated liposomal or inorganic targeted NPs [23,26]. Consequently, the Col IV-NPs(1000) surrounded by a lipid layer of DSPE-mPEG1000/DLPC/Col IV-DSPE-PEG2000 were considered optimal and suitable for further use in cell culture and in vivo studies. The optimized NPs(1000) and Col IV-NPs(1000) maintained their size between 41 – 64 nm in serum (100% FBS) for 48 h without generating aggregates (Figure S4).
2.3. Synthesis and characterization of GW-encapsulated Col IV-NPs (Col IV-GW-NPs)
Next, GW was encapsulated into the Col IV-NPs through the modified emulsion method (Figure 2A). A nanoemulsion process using GW in an organic phase mixture and a formulation of 0.7:1 of total lipids/polymer weight ratio, 67.5:27.5:5 of DSPE-mPEG1000/DLPC/Col IV-DSPE-PEG2000 molar ratio, and 0.26 – 0.54 PLA-OCH3 polymer inherent viscosity core yielded Col IV-GW-NPs with a drug loading amount of 10.8 wt. % (encapsulation efficiency/EE: 45%). Likewise, empty NPs (which have no drug) and non-targeting GW-NPs were prepared. The loading amount of GW in the GW-NPs was 12.0 wt. % (EE: 57%). The GW loading amounts (10.8 ~ 12.0 wt. %) in the lipid-polymer hybrid NPs were comparable to or higher than the previously developed GW-encapsulated polymeric NPs (2.5 ~ 10.9 wt. %), whereas their hydrodynamic sizes were smaller than the previous ones (100 ~ 156 nm). The hydrodynamic sizes of the empty NPs, GW-NPs, and Col IV-GW-NPs in PBS were 41.9 ± 0.4 nm, 73.8 ± 1.3 nm, and 83.9 ± 2.6 nm, respectively (Figure 2B). The increase in the hydrodynamic sizes of GW-NPs and Col IV-GW-NPs implies successful drug encapsulation and functionalization with Col IV targeting ligands. To demonstrate their colloidal stability in serum, we dispersed the NPs in serum (100% FBS) and measured them. The hydrodynamic sizes of the empty NPs, GW-NPs, and Col IV-GW-NPs in serum were 41.1 ± 0.5 nm, 80.3 ± 3.5 nm, and 86.7 ± 1.2 nm, respectively (Figure 2B), indicating negligible or little increase in their size due to protein corona formation. These results demonstrate the high colloidal stability of the stealth-like NPs even in the presence of serum proteins [34]. Additionally, we performed nanoparticle tracking analysis (NTA) for the NPs, GW-NPs, and Col IV-GW-NPs, and the hydrodynamic sizes of the groups were 77.0 ± 0.5 nm, 89.7 ± 0.4 nm, and 99.0 ± 1.4 nm, respectively, indicating a size increase trend similar to that observed in dynamic light scattering (DLS) (Figure S5).
Figure 2.
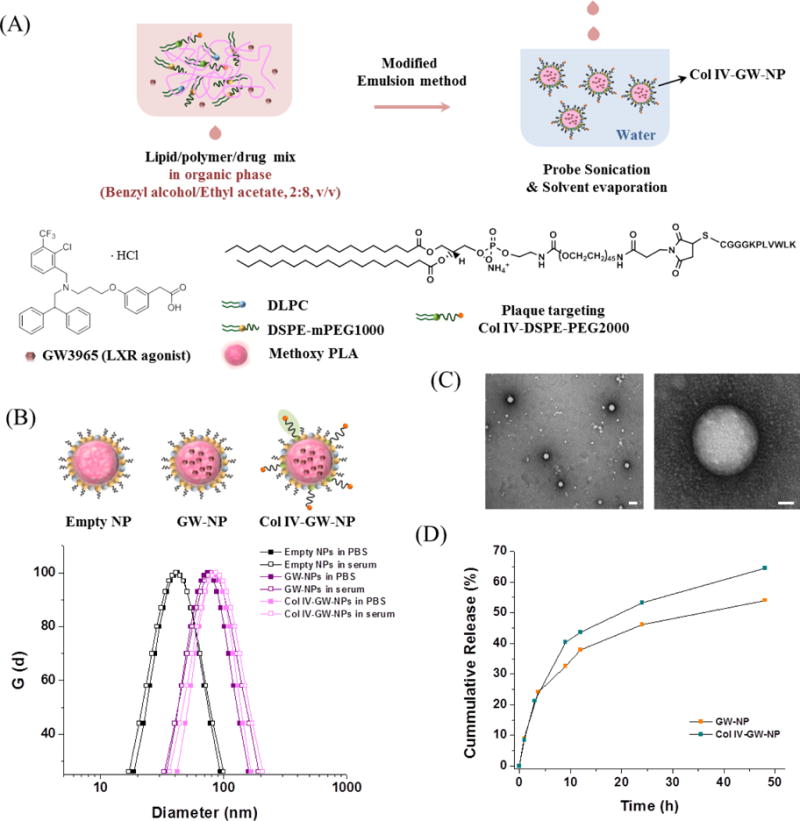
Formulation and characterization of GW-encapsulated NPs. (A) Schematic of Col IV-GW-NP design. The lipids/polymer/drug mixture were dissolved in benzyl alcohol/ethyl acetate co-solvent (2:8, v/v), and the NPs were formed by modified single-emulsion method in water via a single self-assembly process. (B) The mean hydrodynamic diameters of empty NPs, GW-NPs, and Col IV-GW-NPs in PBS and serum (100% FBS), respectively, determined by DLS. (C) Representative TEM images of Col IV-GW-NPs. The scale bars of the left image and the right high-magnification image are 100 nm and 20 nm, respectively. (D) In vitro drug release profiles of GW-NPs and Col IV-GW-NPs incubated at 100 rpm and 37 °C in PBS.
These results show that the modified emulsion method yielded small-sized lipid-polymer hybrid NPs with a higher drug-loading capacity compared to nanoprecipitation or previous emulsion methods using polyvinyl alcohol. The surface charges of the empty NPs, GW-NPs, and Col IV-GW-NPs were −11.4 ± 4.1 mV, −4.4 ± 3.1 mV, and 1.8 ± 0.8 mV, respectively. Since the ends of the Col IV targeting peptide ligands have positively charged lysine groups, the surface charge of the GW-NPs was increased after the modification with Col IV targeting ligands. Transmission electron microscopy (TEM) images revealed that the encapsulation of GW did not cause morphological changes, and the NPs were homogeneously dispersed in aqueous solution with a well-defined spherical shape (Figure S6 and Figure 2C). The size ranges of the empty NPs, GW-NPs, and Col IV-GW-NPs shown in TEM were 35 – 40 nm, 60 – 75 nm, and 65 – 85 nm, respectively, indicating similar trend observed in DLS results. The DLS and TEM results indicated that the drug loading did not generate any aggregated precipitates, as the GW was evenly distributed by encapsulation throughout the hydrophobic core of the NPs.
Release kinetics studies of GW were then performed by incubating NPs in PBS at 37°C and measuring the remaining GW concentrations in the NPs at different time intervals using HPLC. The cumulative release curves of the GW-NPs and Col IV-GW-NPs exhibit an initial fast release of GW (~23 %) for the first ~ 3.5 h, followed by sustained release, indicating half-lives of ~40 h and ~23 h, respectively (Figure 2D). The incorporation of a 5% molar ratio of Col IV-DSPE-PEG2000 onto the lipid layer of the NPs slightly accelerated the rate of drug release. The GW release kinetics was similar to the release graphs of hydrophobic small-molecule drugs from previously developed lipid-polymer hybrid NPs [20], suggesting that shorter PEG-coating molecules (DSPE-mPEG1000) covering the surface of NPs did not significantly alter the drug release profile.
2.4. Col IV-GW-NPs efficiently deliver GW to primary cultured macrophages
Next, we evaluated whether the GW, either encapsulated in the NPs (GW-NPs or Col IV-GW-NPs) or dissolved directly in the medium (free GW), could stimulate the LXRs in primary cultured macrophages and control transcription of several genes involved in the cholesterol efflux pathway, including ABCA1 [35,36]. We first assessed the uptake of the rhodamine-labeled NPs and Col IV-NPs in murine thioglycollate-elicited peritoneal macrophages. Confocal images and quantification results demonstrated comparable uptake of both NPs by macrophages (Figure 3A and 3B). We then evaluated the cellular internalization of the NPs and Col IV-NPs in the presence of different inhibitors such as filipin and chlorpromazine (CPZ), which represent caveolae- and clathrin-mediated endocytic pathways, respectively, or cytochalasin D (CytD) and Latrunculin A (LatA), which represent phagocytosis pathway. We observed ~25 %, ~35–40 %, and ~10–38 % reductions of both NPs uptake upon treatment with CPZ, CytD, and LatA, respectively, suggesting the role of clathrin-mediated endocytosis and phagocytosis in both NPs uptake (Figure 3C). Then, we isolated thioglycollate-elicited macrophages from wild-type mice and exposed them to 1 μM of free GW or equivalent GW dose of GW-NPs and Col IV-GW-NPs, respectively. After the corresponding treatments for 18 h, we isolated total mRNA from the treated macrophages and investigated the expression levels of two classical LXR target genes: ABCA1 and sterol regulatory element-binding transcription factor 1 (SREBP-1c). Notably, cells treated with the GW-NPs and Col IV-GW-NPs revealed markedly elevated mRNA levels of ABCA1 compared to those treated with PBS, which were used as a negative control (Figure 3D). These results were comparable to the findings in cells treated with free GW, demonstrating that GW-NPs and Col IV-GW-NPs efficiently delivered GW to macrophages and activated LXR signaling to a similar extent as did free GW. Following the same trend, the expression of SREBP1c was also increased by all the GW treatments (Figure 3E), indicating that LXR target genes were activated through the treatments with GW-NPs or Col IV-GW-NPs as well as with free GW.
Figure 3.
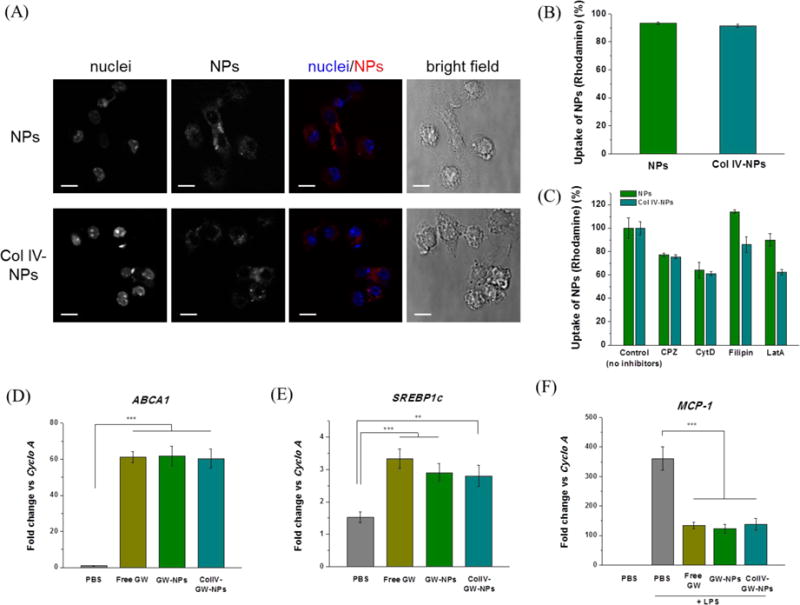
Effects of GW-NPs or Col IV-GW-NPs on LXR target gene expression in in vitro macrophages. To determine the uptake of the NPs, murine thioglycollate-elicited peritoneal macrophages were exposed to rhodamine-labeled NPs or Col IV-NPs for 4 hours. (A) Confocal microscopy shows the internalization of the NPs (red) into macrophages. Nuclei are represented in blue. Individual images are represented in greyscale. Cell shape is shown using the bright field. White bar; 10μm. (B) Quantification of the NPs uptake by macrophages using flow cytometry. (C) NPs uptake in the presence of specific endocytic or phagocytic inhibitors. (D, E) Murine thioglycollate-elicited peritoneal macrophages were exposed to the indicated treatments for 18 h at a concentration equivalent to 1 μM of GW. (F) After this period, cells were exposed to 100 ng/ml of LPS for 6 h. After treatment, RNA was isolated and gene expression analyzed by qRT-PCR. Cyclophilin A (Cyclo A) was used as housekeeping control. Experiments were performed in triplicate. (** p<0.01 and *** p<0.001). Data are expressed as mean ± SEM.
In addition to the beneficial effects of LXR activation on cholesterol efflux, LXR signaling mediates potent anti-inflammatory effects [3]. One of the best-characterized mechanisms is the trans-repression of the Toll-like receptor 4 (TLR4) - lipopolysaccharide (LPS) signaling pathway, which is mediated by SUMOylation of the LXR receptor [4]. Recently, upregulation of ABCA1 also revealed anti-inflammation as a critical mediator [37]. To evaluate whether the GW-NPs or Col IV-GW-NPs could repress inflammatory gene expression in macrophages, we pre-incubated peritoneal macrophages for 18 h with 1 μM of free GW, GW-NPs, or Col IV-GW-NPs, followed by stimulation with 100 ng/ml of LPS for 6 h. After the treatments, we isolated total mRNA from the treated macrophages and examined gene expression of the pro-inflammatory marker, monocyte chemoattractant protein-1 (MCP-1). As shown in Figure 3F, LPS stimulation substantially increased the mRNA levels of MCP-1 in cultured macrophages. In contrast, pre-incubation with free GW, GW-NPs, or Col IV-GW-NPs elicited significantly more trans-repression of MCP-1 expression compared to LPS treatment alone. Consistent with the observation in the LXR target gene expressions, our findings indicate that mRNA levels of MCP-1 in the cells treated with free GW and GW encapsulated in the NPs (GW-NPs or Col IV-GW-NPs) were similar, suggesting that the NPs successfully release the encapsulated drug molecules in cultured macrophages (as also shown in the in vitro GW release results), causing LXR activation of the macrophages and mediating anti-inflammation. Microscopic visualization after incubation with the aforementioned treatments, together with the measurement of lactate dehydrogenase (LDH) levels in the medium, exhibited that any experimental condition had a significant effect on cellular viability (data not shown).
2.5. Col IV-GW-NPs treatment promotes a decrease in CD68+ cells in atherosclerotic lesions
We next assessed the drug-delivery capability of Col IV-GW-NPs to in vivo target atherosclerotic plaques, and compared it to that of GW-NPs. To monitor the GW and NPs in the Ldlr−/− mouse model of atherosclerosis, we labeled GW with BODIPY (GW-BODIPY) (Figure S7) and encapsulated it in the NPs or Col IV-NPs, in which the core of the NPs were labeled by incorporating the fluorescent polymer Cy5.5-conjugated PLA (PLA-Cy5.5) (Figure 4A). The resulting GW-BODIPY encapsulated NPs (GW-BODIPY-NPs) and Col IV-NPs (Col IV-GW-BODIPY-NPs) were injected into Ldlr−/− mice fed a western diet for 14 weeks (8 mg of GW-BODIPY/kg for the NPs). We then sacrificed mice 4 h post-injection and collected the descending aortas, livers, and plasma to quantify the GW-BODIPY and PLA-Cy5.5 in tissues using the IVIS 200 in vivo imaging system.
Figure 4.
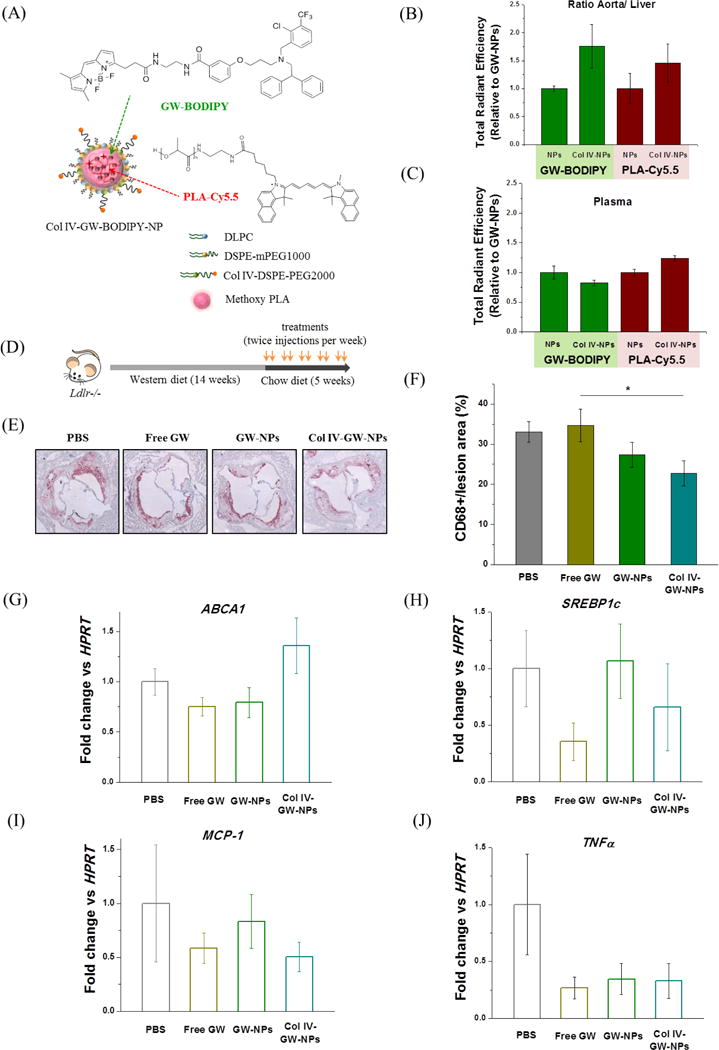
Plaque-targeting abilities and therapeutic efficacies of GW-NPs and Col IV-GW-NPs in the Ldlr−/− mouse model of atherosclerosis. (A) Schematic of the fluorescently labeled Col IV-targeting NP. Fluorescently labeled GW (GW-BODIPY) was encapsulated in the polymeric core of the NP labeled with PLA-Cy5.5 to obtain Col IV-GW-BODIPY-NP. The Ldlr−/− mice (n=2/group) fed a western diet for 14 weeks were injected intravenously with 8 mg of GW/kg body weight in a single dose of fluorescently labeled NPs 4 hours prior to tissue collection. (B) Relative accumulation of GW-BODIPY and PLA-Cy5.5 in the aorta in comparison to the liver. (C) Relative amounts of GW-BODIPY and PLA-Cy5.5 in plasma. (D) Ldlr−/− mice were fed on a western diet for 14 weeks then switched to a chow diet for another 5 weeks. While on the chow diet, the mice were intravenously injected twice a week with 8 mg/kg of body weight (based on the amount of free GW) of free GW, GW-NPs or Col IV-GW-NPs or PBS (control) to study the effect of the different treatments on plaque macrophage accumulation. (E) Representative pictures of aortic roots showing CD68+ cells stained by immunohistochemistry. (F) Quantification of the percentage of CD68+ cells in lesion area. (G–I) CD68+ cells were isolated by laser capture microscopy to analyze changes on gene expression by qRT-PCR. (PBS, n=8; free GW, n=6; GW-NPs, n=6; Col IV-GW-NPs, n=8) (* p<0.05, Col IV-GW-NP group vs free GW group). Data are expressed as mean ± SEM.
Quantitative assessment of the aorta-to-liver fluorescent ratios and plasma fluorescence intensity in the mice revealed that both GW-BODIPY and PLA-Cy5.5 preferentially accumulated in the aortas of mice treated with the Col IV-GW-BODIPY-NPs to a greater extent than mice that received GW-BODIPY-NPs (Figure 4B and Figure S8). However, the amounts of GW-BODIPY and PLA-Cy5.5 in plasma remained relatively unaltered in both groups (Figure 4C). These results indicate that the presence of the Col IV–targeting peptides on the surface of the NPs enhanced the delivery capability of GW to atherosclerotic lesions compared to the non-targeting NPs.
We next tested the relative benefits of Col IV-GW-NPs on atherosclerosis. For this purpose, Ldlr−/− mice fed a western diet for 14 weeks to develop advanced atherosclerosis lesions were switched to a chow diet and injected intravenously with free GW, GW-NPs, or Col IV-GW-NPs (8 mg of GW/kg based on the amount of free GW) twice a week for 5 weeks (Figure 4D). As a control, another group of mice were injected with equal volumes of PBS. After the treatment period, mice were sacrificed and aortic roots were harvested to measure the plaque macrophage (CD68+ cells) content in the atherosclerotic lesion area, which reflects local inflammation known to contribute to atherosclerotic disease progression in both mice and humans [38]. While treatment with free GW and GW-NPs elicited no decrease in CD68+ cells in the aortic lesion, mice treated with Col IV-GW-NPs had significantly reduced macrophage (CD68+ cell) content (~30%) compared to the PBS group, which was with greater efficacy vs. GW-NPs (~18%) (Figure 4E and 4F). These and the fluorescent accumulation (Figure 4B) results argue that the Col IV–targeting peptides employed on the surface of the NPs led to more efficient delivery of GW to atherosclerotic plaques compared to the non-targeting GW-NPs or free GW, subsequently reversing inflammation in the atherosclerotic mice.
To evaluate the phenotypic alterations in the lesional plaque macrophages treated with free GW, GW-NPs, and Col IV-GW-NPs, we performed laser capture microdissection (LCM) on CD68+ areas, isolated total mRNA from the macrophages, and quantified gene expression via RT-PCR. We found an upregulated mRNA level of ABCA1 in the CD68+ macrophages of the mice treated with Col IV-GW-NPs compared to other treatment groups, suggesting that GW released from the NPs mediated increased LXR activation in the lesional plaque macrophages (P=0.064 versus free GW) (Figure 4G). On the other hand, SREBP1c expression was not meaningfully increased in any treatment group, which indicates that SREBP1c processing can be pharmacologically separated from ABCA1 regulation and is independently regulated in plaque macrophages (Figure 4H). Meanwhile, pro-inflammatory target genes such as MCP-1 or TNFα were downregulated in macrophages treated with free GW, GW-NPs, and Col IV-GW-NPs in advanced atherosclerosis compared to PBS-treated macrophages, demonstrating that GW treatments in general reduced inflammation in atherosclerotic lesions (Figure 4I and 4J). Taken together, these results indicate that the Col IV targeting ligands of the Col IV-GW-NPs improved the delivery of GW to plaque macrophages, which in turn resulted in activation of LXR signaling and benefits on the content and inflammatory state of plaque macrophages in the Ldlr−/− mouse model of atherosclerosis.
2.6. Free GW, but no Col IV-GW-NPs, alters hepatic lipid metabolism in mice
A major caveat to the pharmacological use of LXR agonists in the treatment of atherosclerosis is that, despite increasing cholesterol efflux in plaque macrophages and foam cells, these compounds alter hepatic lipid metabolism by promoting liver steatosis and hypertriglyceridemia [39].
To determine whether the NPs mitigate the deleterious effects of GW in mice utilized for therapeutic efficacy studies (Figure 4D), we measured circulating or hepatic TG and cholesterol levels. While circulating TG and cholesterol levels were significantly elevated in mice treated with free GW, mice treated with GW-NPs or Col IV-GW-NPs demonstrated levels comparable to those in the PBS-treated group (Figure 5A and 5B). Similarly, the TG and cholesterol contents in the liver were not altered in mice treated with GW-NPs or Col IV-GW-NPs (Figure 5C and 5D). In contrast, free GW treatments elicited a remarkable increase in TG and cholesterol in the liver. These results suggest that the GW encapsulated in the NPs (GW-NPs and Col IV-GW-NPs) does not alter circulating lipid levels or hepatic lipid metabolism while exerting more control over the drug load and release compared to free GW [40], avoiding the development of hepatic steatosis in mice undergoing LXR agonist treatment.
Figure 5.
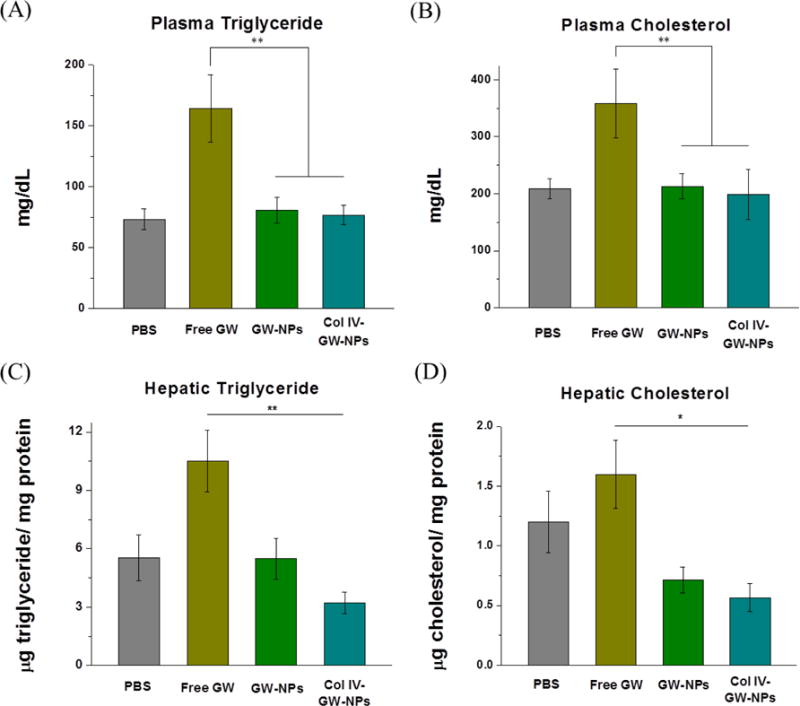
Effects of GW-NPs or Col IV-GW-NPs on circulating and hepatic triglyceride (TG) and cholesterol levels in mice. Ldlr−/− mice were fed a western diet for 14 weeks and then switched to a chow diet for another 5 weeks. While on the chow diet, the mice were intravenously injected twice a week with 8 mg/kg of body weight (based on the amount of free GW compound) of free GW, GW-NPs, or Col IV-GW-NPs or PBS (control) to study the effect of the different treatments on circulating and hepatic lipid metabolism. (A) Plasma TG, (B) and plasma cholesterol levels. (C) Hepatic TG, (D) and hepatic cholesterol levels were calculated from total lipid homogenate. Values refer to the protein content in the homogenate. (PBS, n=8; free GW, n=6; GW-NPs, n=9; Col IV-GW-NPs, n=7) (* p<0.05 and **p<0.01). Data are expressed as mean ± SEM.
We next analyzed changes in LXR-responsive gene expression in liver homogenates to determine the effects of different GW treatments on lipid metabolism. While free GW-treated mice demonstrated significant upregulation of hepatic ABCA1 and ABCG1 compared to mice treated with GW-NPs or Col IV-GW-NPs (Figure 6A and 6B), no changes in hepatic ABCG5 were noted in any of the treatment groups (Figure 6C). We also measured changes in lipogenic transcription factors such as SREBP1c and fatty acid synthase (FASN), whose expression is regulated by both LXRs and SREBP1c [41]. Similarly, mice treated with free GW showed a substantial increase in hepatic SREBP1c and FASN compared to mice treated with GW-NPs or Col IV-GW-NPs (Figure 6D and 6E). Lastly, we measured hepatic mRNA levels of the LXR target cholesterol 7alpha-hydroxylase (CYP7a1), which encodes a key enzyme in the pathway converting cholesterol to bile acid [42]. CYP7a1 levels were higher in mice treated with free GW compared to those treated with Col IV-GW-NPs (Figure 6F).
Figure 6.
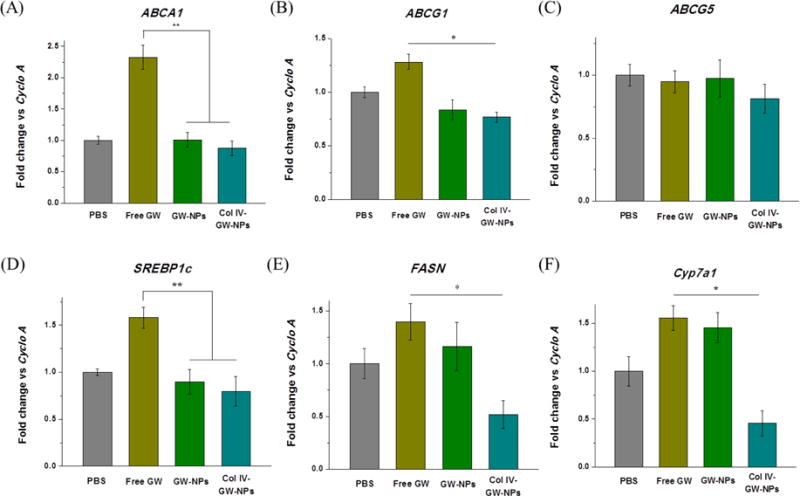
Effects of GW-NPs or Col IV-GW-NPs on hepatic mRNA expression in mice. Ldlr−/− mice (n=6 to 9/group) were fed a western diet for 14 weeks and then switched to a chow diet for another 5 weeks. While on the chow diet, the mice were intravenously injected twice a week with 8 mg/kg of body weight (based on the amount of free GW) of free GW, GW-NPs, or Col IV-GW-NPs or PBS (control) to study the effects of the different treatments on hepatic lipid expression. Total mRNA was isolated and retrotranscribed to determine the expression levels of selected genes. (A) Expression of the ATP-binding cassette A1 (ABCA1), (B) ATP-binding cassette G1 (ABCG1), (C) ATP-binding cassette G5 (ABCG5), (D) sterol regulatory element binding protein (SREBP-1c), (E) fatty acid synthase (FASN), and (F) cytochrome P450 7A1 (Cyp7a1). Cyclo A was used as a housekeeping control. (PBS, n=8; free GW, n=6; GW-NPs, n=9; Col IV-GW-NPs, n=7) (* p<0.05 and ** p<0.01). Data are expressed as mean ± SEM.
Collectively, the studies outlined above indicate that free GW promoted hyperlipidemia and elevated cholesterol and TG accumulation in the liver by altering liver lipid metabolism, which poses a significant obstacle to the development of LXR agonists as human therapeutics. However, NPs had no such negative effect, with targeted Col IV-GW-NPs tending to be even more protective against steatosis. For three of the LXR target genes examined, ABCG5, FASN, and Cyp7a1, the stimulatory effects of the Col IV-GW-NPs tended to be markedly lower than free GW and also lower than GW-NPs, suggesting that the preferential accumulation of GW in atherosclerotic aortic lesions in mice treated with Col IV-GW-NPs remarkably reduced the adverse side effects of LXR activation.
3. Conclusion
The unique roles of LXRs as master regulators of the reverse cholesterol transport pathway in lipid homeostasis and inflammation have attracted much attention in the field of drug discovery for the treatment of atherosclerosis. Unfortunately, no LXR agonists have received FDA approval due to the liver toxicity caused by increased hepatic lipogenesis, steatosis, and hyperlipidemia during the treatment. These adverse effects merit innovative strategies to safely activate the LXR pathway. Fortunately, the targeted delivery of LXR agonists using NPs can facilitate effective LXR-mediated therapy while also protecting off-target organs until delivery at the target site. Here we developed GW-encapsulated targeted NPs consisting of PLA core and a lipid layer of DSPE-mPEG1000/DLPC/Col IV-DSPE-PEG2000 with PEG-coating molecules of optimal length on the surface of the NPs to achieve the best combination with the Col IV-targeting ligand linker (Col IV-DSPE-PEG2000). The Col IV-GW-NPs developed here improved GW anti-atherogenic efficacy without increasing either plasma or liver lipid levels in the Ldlr−/− mouse model of atherosclerosis. Our findings indicate that the formulation strategy for atherosclerotic lesion–targeted NPs encapsulating GW may have potential clinical benefits as a LXR-based treatment for atherosclerosis.
4. Experimental Section
Materials
Poly(D,L-lactide) (PLA) with terminal ester groups (PLA-OCH3, inherent viscosity 0.26–0.54 dL/g in chloroform) was purchased from Durect Lactel® Absorbable Polymers (Pelham, AL, USA). Poly(D,L-lactide) with terminal amine groups (PLA-NH2, Mn 10~15 kDa) was purchased from Polyscitech® (IN, USA). DSPE-mPEGs (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)]) with PEG molecular weight 550 kDa (DSPE-mPEG550), 1000 kDa (DSPE-mPEG1000), and 2000 kDa (DSPE-mPEG2000), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG2000-MAL), 1,2-dilauroyl-sn-glycero-3-phosphocholine (DLPC), were obtained from Avanti Polar Lipids. Cy5.5-NHS monoester was purchased from GE Healthcare Life Sciences. Dimethyl sulfoxide (DMSO), dimethylformamide (DMF), benzyl alcohol, ethyl acetate, acetonitrile (ACN), trifluoroacetic acid (TFA), 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU), N,N-diisopropylethylamine (DIPEA), and GW3965 (GW) were purchased from Sigma–Aldrich. BODIPY® FL EDA, 4,4-Difluoro-5,7-Dimethyl-4-Bora-3a,4a-Diaza-s-Indacene-3-Propionyl ethylenediamine, hydrochloride (BODIPY-NH2) was purchased from ThermoFisher Scientific (Carlsbad, NY). The Collagen type IV (Col IV)-targeting peptide (KLWVLPKGGGC) was purchased from AnyGen Co., Ltd. (South Korea). 1H NMR spectra were recorded on a Bruker AVANCE-400 NMR spectrometer. The nanoparticle (NP) sizes and ζ-potentials were obtained by quasi-electric laser light scattering using a ZetaPALS dynamic light-scattering (DLS) detector (15 mW laser, incident beam ¼ 676 nm; Brookhaven Instruments). The NP diameters measured by DLS were displayed as the effective diameters, which were average of the intensity-, volume-, and number-weighted size distributions, calculated based on the Lognormal distribution. Nanoparticle Tracking Analysis (NTA) for the NPs was performed using Nanosight (Malvern Instruments). Transmission electron microscopy (TEM) was performed on a JEOL 2011 at 200 kV. MALDI TOF Mass Spectrometry was performed on a Bruker Daltons. Biacore3000 instrument (Biacore) was used for surface plasmon resonancy (SPR) measurement.
Preparation and Characterization of Polymer-Lipid Hybrid NPs
PLA-lipid hybrid NPs with different PEG molecular weights were synthesized from PLA-OCH3, DSPE-mPEGs (DSPE-mPEG550, DSPE-mPEG1000, DSPE-mPEG2000), and DLPC using a modified single-emulsion technique. PLA-OCH3, DSPE-mPEGs, and DLPC were dissolved in an organic phase (a mixture of ethyl acetate and benzyl alcohol) and combined with a water phase undergoing high-energy emulsification with a ultrasonic probe sonicator (SONICATOR 4000, MISONIX). NP550, NP1000, and NP2000 were optimized by varying molar ratio of DSPE-mPEGs to DLPC (1:3, 3:7, 7:3, and 10:0) and weight ratio of DSPE-mPEGs/DLPC to PLA (0.5, 0.7, and 1). The PLA/DSPE-mPEGs/DLPC solution with diverse molar ratios of DSPE-mPEG to DLPC and weight ratios of DSPE-mPEGs/DLPC to PLA was added into the water solution drop-wise under gentle stirring followed by probe sonication. Then the NPs were allowed to self-assemble overnight with continuous stirring while the organic phase was allowed to evaporate. The remaining organic solvent and free molecules were removed by washing the NP solution three times using an Amicon Ultra-15 centrifugal filter (EMD Millipore, Germany) with a molecular weight cut-off of 100 kDa and then re-suspended in either water or phosphate saline buffer (PBS) to obtain a final desired concentration. The NPs were used immediately or stored at 4 °C. The stability of the NPs in PBS and in 10% fetal bovine serum (FBS)-containing PBS at 37 °C was tested under shaking (100 rpm) with various incubation times (0 to 7 days) and the changes in the hydrodynamic size of the NPs were measured by DLS. Samples for TEM were stained with 0.75% uranyl formate or 1% uranyl acetate and measured on a coated copper grid.
Synthesis and Purification of Col IV-DSPE-PEG2000 Conjugate
DSPE-PEG2000-MAL (50 mg, 0.017 mmol) dissolved in chloroform was added the Col IV-targeting peptide (KLWVLPKGGGC, 50 mg, 0.043 mmol) that was previously dissolved in dry DMF. The reaction was stirred at room temperature for 24 h, and then the final product of Col IV-DSPE-PEG2000 conjugate was purified by high-performance liquid chromatography (HPLC; Agilent 1100 (Agilent Technologies, CA)) with a C4 column: 2.2 × 25 cm, gradient: 20–20%/5′-100%/50′, A: 0.05% TFA and B: 0.043% TFA, 80% ACN) and characterized by MALDI TOF Mass Spectrometry (average MW found, 4076.9 Da).
Preparation and Characterization of Col IV-NPs
Col IV-NPs were synthesized with a modified single-emulsion method. PLA-OCH3 was first dissolved in a mixture of ethyl acetate and benzyl alcohol (40 mg/mL). DSPE-mPEGs, DLPC, and Col IV-DSPE-PEG2000 (67.5:27.5:5. molar ratio) were dissolved in a mixture of ethyl acetate and benzyl alcohol (40 mg/mL) at 70% of the PLA weight. DSPE-mPEG550, DSPE-mPEG1000, and DSPE-mPEG2000 were used for the preparation of Col IV-NP(550), Col IV-NP(1000), and Col IV-NP(2000), respectively. The PLA/lipid mixture in a co-solvent of ethyl acetate and benzyl alcohol was then added into the aqueous solution (Hyclone, molecular biology–grade water) drop-wise under gentle stirring followed by probe sonication. Then the NPs were purified by washing with Amicon Ultra-15 centrifugal filter (molecular weight cut-off of 100 kDa). Stability of the NPs in PBS and in 10% FBS at 37 °C was tested under shaking (100 rpm) with various incubation times (0 to 7 days), and changes in the hydrodynamic size of the NPs were measured by DLS.
Preparation and Characterization of GW-NPs and Col IV-GW-NPs
PLA-OCH3 was dissolved in a co-solvent of ethyl acetate and benzyl alcohol (40mg/mL). For the preparation of GW-NP and Col IV-GW-NP, DSPE-mPEG1000/DLPC (7:3, molar ratio) and DSPE-mPEG1000/DLPC/Col IV-DSPE-PEG2000 (67.5:27.5:5. molar ratio), respectively, were dissolved in a co-solvent of ethyl acetate and benzyl alcohol (40 mg/mL) at 70% of the PLA weight. GW dissolved in DMSO was added to the PLA/lipid mixture solution at 20% of the total weight. The PLA/lipid/drug mixture in organic phase was then added into the aqueous solution drop-wise under gentle stirring followed by probe sonication. Then the NPs were purified by washing the NP solution 4 times using an Amicon Ultra-15 centrifugal filter (molecular weight cut-off of 100 kDa). Hydrodynamic size and surface charge were analyzed by DLS. Drug-loading amount of the NPs was analyzed using HPLC.
Nanoparticle SPR Measurements
The binding of NPs to Col IV was measured on a Biacore3000 instrument using CM5 sensor chips. The surface carboxyl groups of CM5 chip were activated with 1:1 0.4M 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC)/0.1M NHS. 25 μg/ml solution of human collagen IV in acetate buffer pH 5.0 was manually flowed to make 600 RU, 1600 RU, 3000 RU for several minutes at a rate of 5 μl/min over 2, 3 and 4 channel (1 is reference without Col IV). The remaining NHS-ester groups on the sensor surface were quenched with a 7-min injection of 1.0 M ethanolamine. Binding of NPs was assessed as a function of concentration in PBS-P+ buffer (sample injection: 40 μg/min, 60 μL). Serial dilutions of the NPs were performed to prepare samples with different concentrations (0.3~5mg/mL) before measurement. EDTA regeneration was used to wash bound NPs before starting the next set of experiments.
Drug Release Kinetics Study
NPs were suspended in water and aliquoted into several semipermeable mini-dialysis tubes (Slide-A-Lyzer, MWCO: 10 kDa, ThermoFisher Scientific). The dialysis tubes were placed in 2 L of PBS (pH 7.4) at 37°C. At defined time intervals, NPs in each tube were collected and prepared in ACN:water (1:1, v/v) and 0.01 mM NaOH for HPLC measurement. The remaining drug in NPs at different time points was quantified by HPLC (C18 column: 4.6 × 15 cm, gradient: 20–80%/5′–80%/30′, A: water and B: ACN).
Synthesis of GW-BODIPY
The liver X receptor (LXR) synthetic agonist GW (25 mg, 40 μmol) was dissolved in dry DMF (500 μL); to this was added HATU (30 mg, 80 μmol) in dry DMF (100 μL), and the reaction was stirred for 20 min. Subsequently, BODIPY-NH2 (5 mg, 13.5 μmol) in dry DMF (500 μL) and DIPEA (54 μmol) were added. The reaction was stirred overnight at room temperature. The GW-BODIPY conjugate was purified using HPLC (C18 column, gradient: 0–0%10′–100%/60′, A: 0.05% TFA and B: 0.043% TFA, 80% ACN) and characterized by 1H NMR and MALDI TOF Mass Spectrometry. 1H NMR (400 MHz, CDCl3): δ 8.72, 8.68 (1H, 2 x –NH-C=O), 8.49–7.74 (BODIPY and GW rings), 7,80- 6.36 (BODIPY and GW rings), 5.28 (s, 1H, -C=C-H), 3.4–3.18 (m, 8H, -CH2-), 2.65–2.57 (-CH2-CH2-N- and -N-CH2-CH-), 2.45 (-CH2-CH2-C=O), 2.34 (sharp s, 6H, -CH3 x 2), 1.84 (m, 6H, -O-CH2-CH2-CH2-) ppm. HRMS (MALDI) calculated for C48H48BClF5N5O3 [M]+, 883.35; found, 884.766.
Synthesis of PLA-Cy5.5
PLA-NH2 (6.8 mg, 0.65 μmol) dissolved in dry DMF (10 mg/mL) was added to Cy5.5-NHS mono ester (1 mg, 1.3 μmol) that was dissolved in dry DMF (10 mg/mL). The reaction was stirred overnight at RT and purified by dialysis membrane (MWCO 3.5 kDa).
Cell Culture Experiments
Peritoneal macrophages were obtained from C57BL/6 mice by inducing sterile peritonitis with intraperitoneal injection of 3 mL of 4% Brewers thioglycollate medium 72 h before harvest. Mice were humanely euthanized and macrophages were extracted from the peritoneal cavity by lavage with 10 mL sterile PBS (pH 7.4) three times. The resulting exudate was pelleted, and cells were washed twice with PBS. Cells were maintained in Dulbecco’s Minimal Essential Medium (DMEM) supplemented with 10% FBS (Gemini, CA), penicillin/streptomycin (100 U/mL; 100 μg/mL, Sigma)), and 4 mM L-glutamine (ATCC, Manassas, VA) at 37°C in a humidified 5% CO2 atmosphere. After plating in 12-well plates, cells were incubated for 1 hour to allow macrophages to adhere, washed three times with PBS, and cultured in complete medium. Peritoneal macrophages were used to determine the efficacy of the different NP formulations on gene expression. Cells were exposed for 18 h to GW-NP, Col IV-GW-NP, or free GW dissolved in DMSO at an equivalent concentration of 1 μM of GW. PBS was used as a negative control.
Flow cytometry
Peritoneal macrophages were grown as described above and treated with rhodamine-labeled NPs or Col IV-NPs (20 μg Rhodamine/mL) for 4 hours. Macrophages were identified by staining with BV650 anti-mouse CD11b (Biolegend, San Diego, CA) and PE/Cy7 anti-mouse F4/80 (Biolegend). Rhodamine (Excitation/Emission; 560nm/583nm) intensities within macrophages were quantified using the LSRII analyzer system (BD biosciences, Franklin Lakes, NJ).
Nanoparticle uptake
To determine the internalization pathway of the NPs by macrophages, we incubated peritoneal macrophages with the corresponding inhibitors (filipin – 1 μg/mL; chlorpromazine (CPZ) – 10 μg/mL; cytochalasin D (CytD) – 0.5 μg/mL; Latrunculin A (LatA) – 0.025 μg/mL) for one hour prior adding the rhodamine-labeled NPs for one extra hour. After the incubation, cells were washed twice with fluorescence-activated cell sorting (FACS) buffer (PBS + 1% BSA) and stained with the corresponding antibodies to determine the uptake efficiency.
Confocal microscopy
Peritoneal macrophages were grown on glass coverslips as described above. Cells were treated with rhodamine-labeled NPs or Col IV-NPs (20 μg Rhodamine/mL) for 4 hours. After incubation, cells were fixed with 10% buffered formalin (Sigma) and washed with PBS. DAPI was used to stain nuclei. Confocal images were acquired with a Leica TCS SP5 II confocal microscope using a 405 diode laser (excitation 405 nm) and two HeNe lasers (excitation 543 and 633 nm) with a 63× Apochromat, numerical aperture 1.40- Oil objective.
Animals and Diets
All mice used in this study were on the C57BL/6 background and obtained from Jackson Laboratories (Bar Harbor, Maine). Mice were allowed free access to food and water, and all in vivo studies were performed in accordance with the New York University Institutional Animal Care and Use Committee as well as the National Institutes of Health Animal guidelines. Atherosclerosis studies were performed using low-density lipoprotein receptor knockout mice (Ldlr−/−), which develop complex atherosclerotic lesions in the vasculature when fed a high-cholesterol diet. Mice were weaned 4 weeks after birth onto a western diet (21% fat supplemented with 0.15% cholesterol wt/wt; Dyets, Bethlehem, PA). After 14 weeks on the western diet, mice were switched to a standard chow diet (5% fat and 0.01% of cholesterol wt/wt; LabDiet, St. Louis, MO) and free GW, GW-NPs, and Col IV-GW-NPs were injected intravenously through the retro-orbital sinus with 8 mg/kg of body weight (based on the concentration of GW compound). Treatments were administered twice a week over a span of 5 weeks. The last injection was administered 4 h prior to sacrifice. Mice were euthanized at the completion of treatment with a peritoneal injection of ketamine/xylazine, and blood was collected in EDTA-containing tubes via cardiac puncture for plasma analyses. Mice were then perfused with 10% sucrose in saline solution (0.9% NaCl in water), after which organs were harvested. Aortic roots were embedded in optimal cutting temperature (OCT) compound (Sakura, Torrance, CA), and OCT blocks immediately frozen at −80°C. Portions of liver were snap-frozen in liquid nitrogen and subsequently stored at −80°C.
In Vivo Biodistribution
Ldlr−/− mice were weaned 4 weeks after birth onto a western diet to induce atherosclerotic lesions in the aorta. After 14 weeks, mice were injected with the GW-BODIPY-NPs or Col IV-GW-BODIPY-NPs (8 mg of GW-BODIPY/kg) and sacrificed at 4 h post injection. After perfusion, the whole aorta (from the aortic root to the iliac bifurcation), liver, and plasma were collected and visualized using the IVIS 200 in vivo imaging system (Perkin Elmer, MA). The fluorescence intensities from the NPs (Cy5.5; excitation/emission 675nm/720nm) and the GW (BODIPY; excitation/emission 460nm/520nm) were quantified as total radiant efficiency (TRE), following manufacturer’s instructions. The values are represented as the ratio of the TRE in the aorta normalized by the TRE present in the liver for each fluorophore.
Immunohistochemistry
OCT blocks containing the aortic roots were sectioned at 6 μm thickness and mounted on glass slides. To detect CD68+ cells in the atherosclerotic lesions, sections were fixed in 100% acetone and stained for CD68+ using primary rat anti-mouse CD68 antibody (Bio-Rad, CA), followed by biotinylated anti-rat IgG secondary antibody (Vector Laboratories, CA), and further visualized using a Vectastain ABC kit (Vector Laboratories). Specimens were subsequently counterstained with hematoxylin/eosin (Sigma), dehydrated using xylene (Fisher Scientific, NH) and mounted with coverslips using Permount (Fisher Scientific). Morphometric analyses were performed using ImageProPlus 7 (Micro Optical Solutions LCC, MA) on bright field images of stained sections at 10× magnification. Total lesion area and CD68+ area were used to determine the percentage of CD68+ staining in the atherosclerotic lesions.
Laser Capture Microdissection (LCM)
RNA from CD68+ cells was isolated from atherosclerotic plaques using LCM, as previously described [43]. Briefly, aortic root sections were stained with hematoxylin/eosin in RNAse-free conditions to further isolate the CD68+ cells using consecutive guiding sections previously stained for CD68+ cells as described above. CD68+ cells from the same sample were pooled to isolate and purify the RNA using the PicoPure Kit (ThermoFisher Scientific). The quality and quantity of the RNA samples was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies). RNA was converted to cDNA and amplified using the WT-Ovation Pico RNA Amplification Kit (NuGEN, San Carlos, CA). Real-time PCR analyses were performed using 5 ng of amplified cDNA in the Quantstudio 7 Flex (Applied Biosystems) (see below).
RNA Isolation in Cell Culture and Liver
Total RNA from tissue samples or cells was obtained using TRIzol reagent (Thermo Scientific) following the manufacturer’s instructions. Once isolated and purified, the RNA concentration was determined with NanoDrop (Thermo Scientific) and reverse-transcribed using the Verso cDNA kit (Thermo Scientific, Carlsbad, CA).
Real-Time PCR Analyses
Quantitative real-time PCR was performed with Taqman Gene Expression Master Mix (Applied Biosystems, Foster City, CA), and Taqman primer/probe mixes for ATP binding cassette transporter subfamily A member 1 (ABCA1), ATP binding cassette transporter subfamily G member 1 (ABCG1), sterol-regulatory binding protein 1c (SREBP1c), monocyte chemoattractant protein 1 (MCP-1), ATP binding cassette transporter subfamily G member 5 (ABCG5), fatty acid synthase (FAS), and cytochrome P450 7A1 (CYP7A1) were used. Gene expression was assessed using the ΔΔCt calculation method using hypoxanthine-guanine phosphoribosyltransferase (HPRT) as a housekeeping gene for LCM-isolated CD68+ cells, and cyclophilin A (Cyclo A) was used as housekeeping gene for the rest of the experiments.
Lactate Dehydrogenase Toxicity Assay
The toxicity of the different treatments in cell culture experiments was tested by colorimetric quantification of lactate dehydrogenase (LDH) release into medium following manufacturer’s instructions (Thermo Scientific, Carlsbad, CA)
Cholesterol and Triglyceride Measurements
Liver samples were weighed and homogenized in PBS. A fraction of the total homogenate was incubated with 3 ml of isopropanol (Sigma) overnight at 4°C to extract the total lipid content. The solvent/lipid mixture was centrifuged at 3000×g for 10 minutes, the supernatant was then collected in glass tubes, dried under a stream of nitrogen, and re-suspended in equal volumes of isopropanol. Total triglyceride and cholesterol measurements were performed using standard colorimetric assays (Wako) following manufacturer’s directions. Total protein in the homogenate was measured by a Lowry assay (BioRad) to normalize the triglyceride and cholesterol levels.
Statistical Analysis
Data are expressed as mean ± SEM and analyzed by one-way ANOVA as appropriate for multiple comparisons. A P value less than 0.05 was considered significant.
Supplementary Material
Acknowledgments
This research was supported by the NHLBI Program of Excellence in Nanotechnology (PEN) contract HHSN268201000045C (E.A.F., O.C.F.) from the National Heart, Lung, and Blood Institute, National Institute of Health (NIH). O.C.F. acknowledges NIH support from grants HL127464; the National Research Foundation of Korea K1A1A2048701; and the David Koch-Prostate Cancer Foundation Award in Nanotherapeutics. These studies were also supported by NIH grants HL117226, and HL084312, and Department of Defense grant PR151468 (E.A.F.). J.A. is a recipient of a Scientist Development Grant from the American Heart Association (16SDG27550012). A.M. was supported by an NYU training grant T32HL098129. In compliance with the Brigham and Women’s Hospital and Harvard Medical School institutional guidelines, O.C.F. discloses his financial interest in Selecta Biosciences, Tarveda Therapeutics, and Placon Therapeutics. These companies did not support the aforementioned research and currently have no rights to any technology or intellectual property developed as part of this research.
Footnotes
The rest of the authors declare no conflicts of interest.
Contributor Information
Dr. Mikyung Yu, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA.
Dr. Jaume Amengual, Division of Cardiology, Department of Medicine, Marc and Ruti Bell Program in Vascular Biology, New York University School of Medicine, New York, NY 10016, USA.
Dr. Arjun Menon, Division of Cardiology, Department of Medicine, Marc and Ruti Bell Program in Vascular Biology, New York University School of Medicine, New York, NY 10016, USA
Dr. Nazila Kamaly, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA Technical University of Denmark. Department of Micro and Nanotechnology, DTU Nanotech, 2800 Kgs. Lyngby, Denmark.
Felix Zhou, Division of Cardiology, Department of Medicine, Marc and Ruti Bell Program in Vascular Biology, New York University School of Medicine, New York, NY 10016, USA.
Dr. Xiaoding Xu, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA
Dr. Phei Er Saw, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA
Dr. Seung-Joo Lee, Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, 240 Longwood Ave., Boston, MA 02115, USA
Kevin Si, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA.
Carleena Angelica Ortega, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA.
Dr. Won Il Choi, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA Center for Convergence Bioceramic Materials, Convergence R&D Division, Korea Institute of Ceramic Engineering and Technology, 202, Osongsaengmyeong 1-ro, Osong-eup, Heungdeok-gu, Cheongju, Chungbuk 28160, Republic of Korea.
Dr. In-Hyun Lee, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA
Yazan Bdour, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA.
Dr. Jinjun Shi, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA
Dr. Morteza Mahmoudi, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA
Dr. Sangyong Jon, KAIST Institute for the BioCentury, Department of Biological Sciences, Korea Advanced Institute of Science and Technology (KAIST), 291 Daehak-ro, Daejeon 34141, Republic of Korea
Prof. Edward A. Fisher, Division of Cardiology, Department of Medicine, Marc and Ruti Bell Program in Vascular Biology, New York University School of Medicine, New York, NY 10016, USA
Prof. Omid. C. Farokhzad, Center for Nanomedicine and Department of Anesthesiology, Brigham and Women’s Hospital, Harvard Medical School, Boston, MA 02115, USA King Abdulaziz University, Jeddah 21589, Saudi Arabia.
References
- 1.Libby P, Tabas I, Fredman G, Fisher EA. Circ Res. 2014;114:1867. doi: 10.1161/CIRCRESAHA.114.302699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee SD, Tontonoz P. Atherosclerosis. 2015;242:29. doi: 10.1016/j.atherosclerosis.2015.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tall AR, Yvan-Charvet L. Nat Rev Immunol. 2015;15:104. doi: 10.1038/nri3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grefhorst A, Elzinga BM, Voshol PJ, Plösch T, Kok T, Bloks VW, van der Sluijs FH, Havekes LM, Romijn JA, Verkade HJ, Kuipers F. J Biol Chem. 2002;277:34182. doi: 10.1074/jbc.M204887200. [DOI] [PubMed] [Google Scholar]
- 5.Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, Chen M, Noh G, Goodman J, Hagger GN, Tran J, Tippin TK, Wang X, Lusis AJ, Hsueh WA, Law RE, Collins JL, Willson TM, Tontonoz P. Proc Natl Acad Sci U S A. 2002;99:7604. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. Genes Dev. 2000;14:2831. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orditura M, Quaglia F, Morgillo F, Martinelli E, Lieto E, De Rosa G, Comunale D, Diadema MR, Ciardiello F, Catalano G, De Vita F. Oncol Rep. 2004;12:549. [PubMed] [Google Scholar]
- 8.O’Brien ME, Wigler N, Inbar M, Rosso R, Grischke E, Santoro A, Catane R, Kieback DG, Tomczak P, Ackland SP, Orlandi F, Mellars L, Alland L, Tendler C. Ann Oncol. 2004;15:440. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 9.Lancet JE, Uy GL, Cortes JE, Newell LF, Lin TL, Ritchie EK, Stuart KR, Strickland SA, Hogge D, Solomon SR, Stone RM, Bixby DL, Kolitz JE, Schiller GJ, Wieduwilt MJ, Ryan DH, Hoering A, Chiarella M, Louie AC, Medeiros BC, Lee H. J Clin Oncol. 2016;34(suppl) doi: 10.1200/JCO.2017.77.6112. Abstr. 7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.US National Library of Medicine. ClinicalTrials.gov. :2016. https://clinicaltrials.gov/ct2/show/NCT02213744?term.
- 11.US National Library of Medicine. ClinicalTrials.gov. :2014. https://clinicaltrials.gov/ct2/show/NCT00964080?term.
- 12.Mulder WJ, Jaffer FA, Fayad ZA, Nahrendorf M. Sci Transl Med. 2014;6:239sr1. doi: 10.1126/scitranslmed.3005101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tarin C, Carril M, Martin-Ventura JL, Markuerkiaga I, Padro D, Llamas-Granda P, Moreno JA, García I, Genicio N, Plaza-Garcia S, Blanco-Colio LM, Penades S, Egido J. Sci Rep. 2015;5:17135. doi: 10.1038/srep17135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gadde S, Even-Or O, Kamaly N, Hasija A, Gagnon PG, Adusumilli KH, Erakovic A, Pal AK, Zhang XQ, Kolishetti N, Shi J, Fisher EA, Farokhzad OC. Adv Healthc Mater. 2014;3:1448. doi: 10.1002/adhm.201300688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang XQ, Even-Or O, Xu X, van Rosmalen M, Lim L, Gadde S, Farokhzad OC, Fisher EA. Adv Healthc Mater. 2015;4:228. doi: 10.1002/adhm.201400337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan JM, Zhang L, Tong R, Ghosh D, Gao W, Liao G, Yuet KP, Gray D, Rhee JW, Cheng J, Golomb G, Libby P, Langer R, Farokhzad OC. Proc Natl Acad Sci U S A. 2010;107:2213. doi: 10.1073/pnas.0914585107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kamaly N, Fredman G, Subramanian M, Gadde S, Pesic A, Cheung L, Fayad ZA, Langer R, Tabas I, Farokhzad OC. Proc Natl Acad Sci U S A. 2013;110:6506. doi: 10.1073/pnas.1303377110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fredman G, Kamaly N, Spolitu S, Milton J, Ghorpade D, Chiasson R, Kuriakose G, Perretti M, Farokhzad O, Tabas I. Sci Transl Med. 2015;7:275ra20. doi: 10.1126/scitranslmed.aaa1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kamaly N, Fredman G, Fojas JJ, Subramanian M, Choi WI, Zepeda K, Vilos C, Yu M, Gadde S, Wu J, Milton J, Carvalho Leitao R, Rosa Fernandes L, Hasan M, Gao H, Nguyen V, Harris J, Tabas I, Farokhzad OC. ACS Nano. 2016;10:5280. doi: 10.1021/acsnano.6b01114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan JM, Zhang L, Yuet KP, Liao G, Rhee JW, Langer R, Farokhzad OC. Biomaterials. 2009;30:1627. doi: 10.1016/j.biomaterials.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 21.Wu J, Kamaly N, Shi J, Zhao L, Xiao Z, Hollett G, John R, Ray S, Xu X, Zhang X, Kantoff PW, Farokhzad OC. Angew Chem Int Ed Engl. 2014;53:8975. doi: 10.1002/anie.201404766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu X, Xu Y, Solis LM, Tao W, Wang L, Behrens C, Xu X, Zhao L, Liu D, Wu J, Zhang N, Wistuba II, Farokhzad OC, Zetter BR, Shi J. Proc Natl Acad Sci U S A. 2015;112:7779. doi: 10.1073/pnas.1505629112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai Q, Walkey C, Chan WC. Angew Chem Int Ed Engl. 2014;53:5093. doi: 10.1002/anie.201309464. [DOI] [PubMed] [Google Scholar]
- 24.Stefanick JF, Ashley JD, Kiziltepe T, Bilgicer B. ACS Nano. 2013;7:2935. doi: 10.1021/nn305663e. [DOI] [PubMed] [Google Scholar]
- 25.Stefanick JF, Ashley JD, Bilgicer B. ACS Nano. 2013;7:8115. doi: 10.1021/nn4033954. [DOI] [PubMed] [Google Scholar]
- 26.Saw PE, Park J, Lee E, Ahn S, Lee J, Kim H, Kim J, Choi M, Farokhzad OC, Jon S. Theranostics. 2015;5:746. doi: 10.7150/thno.10732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang F, You M, Chen T, Zhu G, Liang H, Tan W. Chem Commun. 2014;50:3103. doi: 10.1039/c3cc49003c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, Chan JM, Gu FX, Rhee JW, Wang AZ, Radovic-Moreno AF, Alexis F, Langer R, Farokhzad OC. ACS Nano. 2008;2:1696. doi: 10.1021/nn800275r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pfeiffer C, Rehbock C, Hühn D, Carrillo-Carrion C, de Aberasturi DJ, Merk V, Barcikowski S, Parak WJ. J R Soc Interface. 2014;11:20130931. doi: 10.1098/rsif.2013.0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang L, Zhu D, Dong X, Sun H, Song C, Wang C, Kong D. Int J Nanomedicine. 2015;10:2101. doi: 10.2147/IJN.S77667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo Y, Wang L, Lv P, Zhang P. Oncol Lett. 2015;9:1065. doi: 10.3892/ol.2014.2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao J, Xia Y, Chen H, Yu Y, Song J, Li W, Qian W, Wang H, Dai J, Guo Y. Nanomedicine (Lond) 2014;9:279. doi: 10.2217/nnm.13.20. [DOI] [PubMed] [Google Scholar]
- 33.Tassa C, Duffner JL, Lewis TA, Weissleder R, Schreiber SL, Koehler AN, Shaw SY. Bioconjug Chem. 2010;21:14. doi: 10.1021/bc900438a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelsch A, Tomcin S, Rausch K, Barz M, Mailänder V, Schmidt M, Landfester K, Zentel R. Biomacromolecules. 2012;13:4179. doi: 10.1021/bm301453g. [DOI] [PubMed] [Google Scholar]
- 35.Hong C, Tontonoz P. Nat Rev Drug Discov. 2014;13:433. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- 36.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. Genes Dev. 2000;14:2819. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN, Gratton E, Parks J, Tontonoz P. eLife. 2015;4:e08009. doi: 10.7554/eLife.08009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, Pittet MJ. J Clin Invest. 2007;117:195. doi: 10.1172/JCI29950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. Genes Dev. 2000;14:2831. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bombelli FB, Webster CA, Moncrieff M, Sherwood V. Lancet Oncol. 2014;15:e22. doi: 10.1016/S1470-2045(13)70333-4. [DOI] [PubMed] [Google Scholar]
- 41.Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, Collins JL, Osborne TF, Tontonoz P. J Biol Chem. 2002;277:11019. doi: 10.1074/jbc.M111041200. [DOI] [PubMed] [Google Scholar]
- 42.Peet DJ, Turley SD, Ma W, Janowski BA, Lobaccaro JM, Hammer RE, Mangelsdorf DJ. Cell. 1998;93:693. doi: 10.1016/s0092-8674(00)81432-4. [DOI] [PubMed] [Google Scholar]
- 43.Feig JE, Fisher EA. Methods Mol Biol. 2013;1027:123. doi: 10.1007/978-1-60327-369-5_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.