

Abstract
Exosomes, the smallest sized extracellular vesicles (30 ~ 150 nm) packaged with lipids, proteins, functional messenger RNAs and microRNAs, and double-stranded DNA from their cells of origin, have emerged as key players in intercellular communication. Their presence in bodily fluids, where they protect their cargo from degradation, makes them attractive candidates for clinical application as innovative diagnostic and therapeutic tools. But routine isolation and analysis of high purity exosomes in clinical settings is challenging, with conventional methods facing a number of drawbacks including low yield and/or purity, long processing times, high cost, and difficulties in standardization. Here we review a promising solution, microfluidic-based technologies that have incorporated a host of separation and sensing capabilities for exosome isolation, detection, and analysis, with emphasis on point of care and clinical applications. These new capabilities promise to advance fundamental research while paving the way toward routine exosome-based liquid biopsy for personalized medicine.
Graphical Abstract
Introduction
Extracellular vesicles (EVs) have emerged as an area of intense interest owing to the significant role they play in orchestrating intercellular communication and molecular exchange.1–4 EVs are actively secreted by most, if not all, cells into a variety of bodily fluids (e.g., blood, urine, saliva, synovial fluid, amniotic fluid, ascites, milk) where the diverse array of proteins, lipids, and nucleic acids packaged within them act to relay signals between the cell of origin and recipient cells. In addition to protecting their cargo from degradation in cell-to-cell signaling, EVs canserve as shuttles for infectious agents, not only by mediating the spread of pathogens but also by providing a mechanism for them to escape the host immune response.5–9 Consequently, increasing efforts have been directed toward understanding how to unlock the incredible potential of EVs to function as new disease biomarkers, vaccine candidates, and therapeutic agents.5,10–13
As shown in Figure 1, EVs can be broadly classified into three major sub-populations based on their size and biogenesis:2,14 apoptotic bodies (> 1000 nm), microvesicles (100 ~ 1000 nm), and exosomes (30 ~ 150 nm). Apoptotic bodies are secreted by apoptotic cells, whereas production of microvesicles is typically induced by imbalances in the distribution of lipids on the plasma membrane. These imbalances can emerge as a consequence of cell stimulation, endocytosis, apoptosis, or cytosolic Ca2+ increases.2 Exosomes, meanwhile, primarily originate from endosome-multivesicular body (MVB) complexes, which fuse with the plasma membrane to release exosomes into the extracellular environment.14,15
Figure 1.

Exosomes represent a subset of extracellular vesicles with characteristic size in the 30 ~ 150 nm range. Emerging from endosome-multivesicular body (MVB) complexes within the cell, their surface protein markers and nucleic acid cargo play key roles in mediating intercellular communication. These signaling pathways are of intense interest as important new diagnostic and therapeutic targets for personalized medicine.
Numerous studies have explored the immense diagnostic and therapeutic potential of exosomes, the smallest sized and most homogeneous EVs, owing to their valuable cargo of proteins, functional messenger RNAs (mRNAs) and microRNAs (miRNAs), and double-stranded DNA (dsDNA), along with their role in conveying such information between cells.16–23 This potential becomes evident by considering that many conventional methods to elucidate the state of a tissue involve direct analysis of cellular material obtained from biopsied samples. Exosomes, on the other hand, can be collected from bodily fluid samples via a minimally invasive liquid biopsy.24,25 And since the material packaged within exosomes originates directly from the parent cell, analysis of this cargo may enable a snapshot of the host cell state to be obtained in a much simpler manner than conventional physical biopsy, making it feasible for this kind of analysis to be broadly implemented as a routine diagnostic platform. The potential for exosomes to contain individualized information related to disease state, therapeutic response, exposure to environmental cues, and a myriad of other health factors, all contained in a vesicular package that can be collected from bodily fluid samples, has generated considerable excitement as a pathway to enable personalized medicine.26
A key challenge continues to be a lack of efficient and standard techniques for isolation and analysis of clinical grade exosomes,27–29 difficulties that are compounded when dealing with raw biological fluids that inherently contain a high proportion of proteins, other EVs, and cells with similar physical and/or biomolecular characteristics as exosomes. Current isolation methods rely either on size differences between EVs or on targeting specific surface markers.30 Conventional techniques based on these principles include ultracentrifugation, precipitation, filtration, chromatography, and immuoaffinity-based approaches.
Ultracentrifugation
Centrifugation approaches exploit size differences between cells, subpopulations of EVs, and proteins. Differential centrifugation is currently the gold standard for exosome isolation, involving a sequence of centrifugation steps at progressively higher spin speeds.30,31 Large components including cells, apoptotic bodies, and larger vesicles can be separated using standard centrifugation (< 20,000 g), whereas ultracentrifugation (> 100,000 g) must be used to purify exosomes from protein contaminates. The high spin speeds and long operation times required (~5 h) are major drawbacks. Additionally, differential centrifugation often results in lower exosome recovery and contamination with co-precipitated protein aggregates. Thus, alternative protocols, such as sucrose gradient centrifugation, have been developed to improve exosome isolation efficiency.32–35
Precipitation
To eliminate the requirement of ultracentrifugation, several commercial exosome isolation kits have been developed (e.g., Exo-spin™, ExoQuick™ Exosome precipitation, Total Exosome Isolation Reagent from Invitrogen™, PureExo® Exosome Isolation kit, and miRCURY™ Exosome Isolation kit). These commercial products use special reagents (e.g., polymeric additives) to induce precipitation of exosomes, enabling isolation to be performed within ~ 30 min using a standard centrifuge (~10,000 g). Several studies have compared the efficiency of exosome precipitation methods with conventional ultracentrifugation, and found that these commercial kits generally produce higher yields and purity.36–41 However, for therapeutic applications, the need to add precipitation reagents can inhibit recovery of intact exosomes from the polymer matrix. This is a critical drawback because these residual precipitation matrices could influence exosome biological activities and characteristics.42,43
Filtration
Commercial membrane filters (e.g., polyvinylidene difluoride (PVDF) or polycarbonate, pore size50 ~450 nm) can be used to isolate cells and large EVsin biological samples. Filtration methods are often combined with ultracentrifugation, where membranes are used to sieve cells and large EVs, after which separation of exosomes from proteins is achieved via ultracentrifugation.40,44–46 In order to eliminate the need for ultracentrifugation, a few research groups have explored commercial ultrafiltration (e.g., Amicon filter, 100kD MWCO) as a means toseparate exosomes from protein contaminates.38,47–49 Although filtration is generally faster than centrifugation, optimization of the operating procedure is critical to reduce detrimental clogging effects that can lead to lower exosome yields.30,38
Size-exclusion chromatography (SEC)
Chromatographic methods have also been used to separate exosomes from protein aggregates. Generally, centrifugation or filtration is first applied to remove larger cells or EVs, after which commercial size-exclusion columns can be employed to isolate exosomes.38,46,47,50,51 Small analytes, such as proteins, are retained in the stationary phase while larger species (i.e., exosomes) elute more quickly. Exosomes can therefore be isolated by collecting the eluted fraction at a specific time. Although some studies have reported that exosomes isolated using SEC contained fewer impurities, selection of the appropriate matrix is essential to achieve optimal efficiency.50
Immunoaffinity-based approaches
Exosomes may contain different markers specific to the cell of origin that can allow immunoisolation to be performed. A common immunoaffinity-based approach utilizes antibody coated magnetic beads to capture exosomes that contain the specific markers in bodily fluids. This method allows specific subpopulation of exosomes to be isolated, but is generally not suited for isolating exosomes from large quantities of biological samples.30
In practice, conventional exosome isolation protocols often combine more than one of the methods described above. Depending on the source of the biological samples, each protocol needs to be optimized to achieve a high yield of exosomes with minimal impurities. Downstream analysis of exosomes typically involves some combination of size characterization, surface marker and protein analysis, and characterization of nucleic acid content. Transmission electron microscopy (TEM), scanning electron microscopy (SEM), and atomic force microscopy (AFM) have been widely used to directly observe the morphology of individual exosomes, but it is difficult to quantify exosomal size distributions and concentrations using these techniques.52,53 Commercial instruments for nanoparticle analysis, including dynamic light scattering (DLS) and nanoparticle tracking analysis (NTA), have been used to measure size distributions of exosomes.50 Compared to DLS, NTA offers the additional ability to characterize exosomal concentration.54 Surface markers and protein content of exosomes are generally characterized using classic immunoassays, including enzyme-linked immunosorbent assays (ELISA), western blots, total protein analysis (e.g., bicinchoninic acid (BCA), Bradford assays), and flow cytometry.44,50,55–58 Other detection techniques, such as plasmonic and electrochemical sensing platforms, have also been applied in conjunction with immunoassays to achieve high throughput, easy-to-use, and label-free protein analysis.44,59–61 Finally, nucleic acid content can be probed by first applying conventional isolation methods, after which analysis is performed by either polymerase chain reaction (PCR) or sequencing techniques.62–64
These conventional isolation and characterization methods require dedicated laboratory instruments and complex multi-step workflows, making it challenging to implement exosome-based analysis as a routine diagnostic tool in clinical settings. Microfluidics offers incredible potential to overcome these drawbacks, potentially enabling clinical grade exosomes to be rapidly isolated and analyzed for diagnostic and therapeutic applications. In addition to providing a versatile platform for exosome separation and isolation, the lab-on-a-chip format can also be leveraged to integrate multiple processes in a single instrument, simplifying operation and reducing the risk of cross -contamination.
Microfluidic-based Exosome Isolation
As the clinical importance of exosomes as biomarkers for a host of medical conditions becomes increasingly evident, researchers have developed a variety of microfluidic systems for exosome isolation, detection, and analysis from bodily fluids. Previous reviews have traced the development of miniaturized solutions to address various aspects of the exosomal analysis workflow, and we encourage readers to consult these references for a deeper perspective on the rapid evolution of this field.24,65–69 These liquid biopsy platforms employ a range of exosome isolation approaches such as immunoaffinity, membrane-based filtration, trapping on nanowires, acoustic nanofiltration, and deterministic lateral displacement (DLD) sorting (Figure 2). In acoustics and DLD research, microfluidic-based exosome isolation represented a first-of-a-kind application for separation of biocolloids at the nanoscale using these technologies. These breakthroughs have been particularly significant because they opened new frontiers and illustrated how the demands of exosome isolation push the boundaries of separation science and technology.
Figure 2.

Timeline of progress toward exosome isolation in microfluidic platforms.
Table 1 summarizes isolation methods that have been implemented in microfluidic platforms for exosome analysis (including both devices designed exclusively for exosome isolation and integrated analysis systems) along with their respective performance parameters. Before discussing specific approaches in the literature, it is important to consider the nature of the sample being analyzed. Exosomes obtained from cell cultures and purified via conventional methods (e.g., filtration, ultracentrifugation, immunocapture via targeted markers) are typically employed as “clean” standard samples for device development and initial characterization. Once this initial characterization is performed, clinical utility is assessed by challenging systems with bodily fluids that have received little or no pre-processing (serum, plasma, whole blood). Therefore, in order to focus on potential applications in point-of-care (POC) and clinical settings, Tables 1 – 3 and the accompanying discussion only include results obtained from analysis of bodily fluids where possible. Later, in our discussion of exosomal proteins (Table 4) we provide a comprehensive mapping of microfluidic-based analysis performed using both cell culture and bodily fluid derived samples. Finally, to help facilitate comparison among methods, we computed isolation throughput in Table 1 from the ratio of the sample volume (isolation capacity) to the time required for complete exosome isolation. This parameter emerged as a performance indicator, with values spanning several orders of magnitude from 0.0002 to 70 μL/min.
Table 1.
Exosome isolation in microfluidic platforms.
| Exosome isolation approach | Sample (pre-treatment) * | Isolation capacity [μL] | Recovery yield [%] | Isolation throughput [μL/min] ** | Isolated size*** [nm] | Reference |
|---|---|---|---|---|---|---|
| Immunoaffinity (targeted marker) | ||||||
| Functionalized (CD63) channel with herringbone groves | Serum (0.8 μm filter) | 400 | 42–94 | 13.1 | ~20–135 | Chen 2010 [70] |
| Capture on modified (CD41) mica surface | Plasma (5X dilution) | 150 | NA | 1.2 | ~10–110 | Ashcroft 2012 [71] |
| ExoChip: Functionalized (CD63) multi-chamber/channel device | Serum | 400 | NA | 4 | ~30–300 | Kanwar 2014 [72] |
| Reusable nPLEX: Functionalized (CD24, CD63, EpCAM) gold surface with nanohole arrays | Ascites fluid (0.2 μm filter) | 150 | NA | 8.3 | ~20–260 | Im 2014 [44] |
| Capture on immunomagnetic (EpCAM, IGF-1R, CA125, CD9, CD63, CD81) microbeads | Plasma (pre-mixed with capture beads) | 30 | NA | 2 | ~40–150 | He 2014 [57] |
| Capture on functionalized (CD9, HER2) gold electrodes enhanced by nanoshearing | Serum | 500 | NA | 4.2 | ~30–350 | Vaidyanathan 2014 [73] |
| RInSE: Inertial solution exchange for continuous isolation of affinity-capture (EpCAM) microbeads | Blood (RBC lysis, centrifugation, incubation with capture beads and label) | Continuous flow | NA | 70 | ~30–120 | Dudani 2015 [77] |
| iMER: Isolation using immunomagnetic (EGFR) microbeads | Serum (0.8 μm filter) | 100 | >93 | 4 | ~ 100 | Shao 2015 [79] |
| ExoSearch: Continuous capture on immunomagnetic (CD9) microbeads | Plasma | 20 | 72 | 0.8 | ~50–250 | Zhao 2016 [80] |
| Nano-IMEX: Improved capture efficiency on Y-shaped microposts with (CD81) nanostructured coating | Plasma (10X dilution) | 20 | NA | 0.05 | <150 | Zhang 2016 [74] |
| Functionalized (CD9, CD63) gold surface | Serum | 250 | NA | 5 | ~30–300 | Sina 2016 [75] |
| μMED: Negative (CD45, CD61) and positive (CD81) enrichment with microbeads of different size | Mouse serum | ~100 | NA | ~<10/3 | ~117 | Ko 2016 [81] |
| Capture on immunomagnetic (CD63) particles | Plasma (incubation with capture particles) | ~1000 | NA | 2 | <100 | Fang 2017 [78] |
| Membrane-based filtration | ||||||
| Pressure-driven filtration | Mouse whole blood | 3 | >1.5 | 0.075 | ~150 | Davies 2012 [58] |
| Electrophoresis-driven filtration | Mouse whole blood | 240 | 1.5 | 2 | ~150 | Davies 2012 [58] |
| Electrophoretic isolation on nanoporous membrane | Mouse plasma (2X dilution) | 1000 | 65 | 20 | ~10–400 | Cho 2016 [83] |
| Exodisc: Double filtration | Urine | 1000 | >95 | 36 | 20–600 | Woo 2017 [85] |
| Double filtration | Urine (centrifugation, 0.22 μm filter) | 8000 | 74.2 | 33 | 155 | Liang 2017 [84] |
| Nanowire trapping | ||||||
| Vesicle trapping on array of ciliated (nanowires) micropillars | Mixture of BSA, liposomes, beads | 100 | ~10 | 10 | ~83–120 | Wang 2013 [88] |
| Acoustics | ||||||
| Continuous contact-free acoustic nanofilter | Packed RBC units | 10 | >80 | ~0.24 | ~30–200 | Lee 2015 [98] |
| Lateral displacement | ||||||
| Nano-DLD sorting using pillar array | Commercial urine-derived exosomes | 0.72 | NA | 0.0002 | <100 | Wunsch 2016 [104] |
| Viscoelastic flow | ||||||
| Continuous viscoelasticity-based and field-free microfluidic sorting | Fetal bovine serum | 100 | 93.6 | 10/3 | <200 | Liu 2017 [106] |
NA: not available. RBC: red blood cell. BSA: bovine serum albumin.
Sample is of human origin unless otherwise stated.
Estimated throughput accounting for dilutions and time for other isolation-related steps (e.g., incubation, rinsing, etc.) whenever information is available.
Size of isolated particles is typical and it might not correspond precisely to the sample and operating conditions shown.
Table 3.
Integrated microfluidic platforms for exosome analysis.
| On-chip integrated analysis | Sample (pre- treatment) * | Overall throughput [μL/min] ** | Limit of detection | Measurement in isolated exosomes | Targeted disease | Reference |
|---|---|---|---|---|---|---|
| Overall exosome levels | ||||||
Sequential ExoChip stages:
|
Serum | 3.1 | 0.5 pM fluorescence sensitivity | Stained CD63(+)exosomes | Pancreatic cancer | Kanwar 2014 [72] |
Sequential nano-IMEX stages:
|
Plasma (10X dilution) | 0.029 | ~50 exosomes/μL | Overall levels of EpCAM, CD81, CD9 in CD81(+)exosomes | Ovarian cancer | Zhang 2016 [74] |
Sequential Exodisc stages:
|
Urine | 16.7 | NA | Overall levels of CD9 and CD81 | Bladder cancer | Woo 2017 [85] |
Sequential stages:
|
Urine (centrifugation, 0.22 μm filter) | 17.2 | NA | Overall levels of CD63 | Bladder cancer | Liang 2017 [84] |
| Exosome subpopulations | ||||||
Reusable and continuous nPLEX:
|
Ascites fluid (0.2 μm filter) | 8.3 | ~3000 exosomes (670 aM) | EpCAM, CD24 levels relative to CD63(+) exosomes | Ovarian cancer | Im 2014 [44] |
Multiplexed sequential stages:
|
Serum | 2.9 | 2760 exosomes/μL | HER2(+), CD9(+)exosomes | Breast cancer | Vaidyanathan 2014 [73] |
Inline RInSE cytometer stages:
|
Blood (RBC lysis, centrifugation, incubation with capture beads and label) | 70 | NA | CD81 in EpCAM(+)exosomes | Breast cancer | Dudani 2015 [77] |
Continuous ExoSearch stages:
|
Plasma | 0.5 | 750 exosomes/μL | CA-125, EpCAM, CD24 in CD9(+)exosomes | Ovarian cancer | Zhao 2016 [80] |
Sequential stages:
|
Serum | 2.7 | ~2070 exosomes/μL | HER2 in CD9(+)or CD63(+)exosomes | Breast cancer | Sina 2016 [75] |
Sequential μMED stages:
|
Mouse serum | ~1.7 | 10000 exosomes/μL | GluR2 in CD81(+)exosomes | Concussion | Ko 2016 [81] |
Continuous stages:
|
Plasma (incubation with capture particles) | <2 | NA | EpCAM, HER2 in CD63(+) exosomes | Breast cancer | Fang 2017 [78] |
| Intravesicular proteins | ||||||
Sequential stages:
|
Plasma (pre-mixed with capture beads) | 0.35 | 0.281 pg/mL (IGF-1R), 0.383 pg/mL (p-IGF-1R) | IGF-1R, p-IGF-1R levels in EpCAM(+) exosomes | NSCLC | He 2014 [57] |
| Exosomal mRNAs | ||||||
Sequential iMER stages:
|
Serum (0.8 μm filter) | 0.83 | NA | mRNA levels of EPHA2, EGFR, PDPN, MGTM, APNG in EGFR(+)and EGFRvIII(+)exosomes | GBM | Shao 2015 [79] |
NA: not available. NSCLC: non-small-cell lung cancer. GBM: Glioblastoma multiforme. RBC: red blood cell.
Sample is of human origin unless otherwise stated.
Estimated throughput accounting for dilutions and time for processing steps (e.g., isolation, detection, incubation, rinsing, etc.) whenever information is available.
Table 4.
Exosomal proteins used for on-chip exosome capture (C), on-chip detection (D), or both (&) in microfluidic platforms.

|
Exosomal protein was used as common
 , disease-associated
, disease-associated
 , or non-disease
, or non-disease
 marker.
marker.
Expression of exosomal marker was higher relative to control and signature for disease
 , higher relative to control
, higher relative to control
 , or comparable to control
, or comparable to control
 .
.
NSCLC: non-small-cell lung cancer.
Intravesicular protein.
Immunoaffinity capture
Specific capture of exosomes by antibodies immobilized on solid surfaces represents the most commonly used isolation approach, although individual microfluidic platforms exhibit unique characteristics and their performance varies over a wide range (Figure 2 and Table 1). The targeted exosomal marker, that is, the exosome’s extravesicular protein targeted by capture antibodies, depends on the particular application and whether a specific subpopulation of exosomes is being isolated (the topic of exosomal markers will be addressed later). Here we identify two main types of platforms based on the surface functionalized for exosome capture: (1) devices with modified inner surface(s) and (2) devices that employ capture beads.
Microfluidic devices with inner capture surface(s) are specifically designed to enhance the interaction between targeted exosomes and the functionalized surface(s). Instances of these platforms include the pioneering work of Chen et al. who used herringbone groves to increase the capture efficiency in a straight flow surface-modified channel while achieving relatively high throughput and good recovery yield.70 Ashcroft et al. implemented a detachable microfluidic circuit on top of a modified mica surface to increase the concentration of captured microparticles later examined by AFM.71 For the ExoChip platform, Kanwar et al. designed a device with multiple circular capture chambers interconnected by narrow-channels that increased exosomes’ retention time, and arranged multiple parallel channels in a geometry compatible with a standard plate reader for exosome quantification.72 Recent efforts to achieve improved sensitivity in microfluidic systems implemented nanoshearing effects73 and nanostructured coatings74 to enhance the immunocapture efficiency of targeted exosomes while suppressing non-specific capture of non-targeted species. For instance, Zhang et al. incorporated a nanostructured graphene oxide/polydopamine (GO/PDA) interface on the inner surfaces of the nano-IMEX chip, which also featured an array of Y-shaped microposts providing a larger capture surface and improved mixing (Figure 3A).74 Lastly, surface plasmon resonance (SPR)-based sensing platforms rely on functionalization of gold surfaces for exosome capture, and therefore also fall into this category.44,75
Figure 3.

Examples of microfluidic approaches for exosome isolation. (A) Enhanced immunoaffinity capture of exosomes using nanostructured coatings on the nano-IMEX chip. Adapted from Ref. [74] with permission from the Royal Society of Chemistry. (B) Filtration-based capture of extracellular vesicles on nanoporous membrane assisted with electrophoretic migration. Reprinted from Ref. [83], Copyright 2016, with permission from Elsevier. (C) Trapping of exosome-like lipid vesicles on nanowire-on-micropillar arrays. Reproduced from Ref. [88] with permission from the Royal Society of Chemistry. (D) Acoustic nanofilter for exosome isolation. Adapted with permission from Ref.[98]. Copyright 2015 American Chemical Society. (E) Nano-DLD for exosome sorting using nanopillar array. Adapted by permission from Macmillan Publishers Ltd: Nature Nanotechnology Ref. [104], copyright 2016.
When using functionalized capture beads, microfluidic devices can perform a variety of tasks leveraging sample-bead interactions and subsequent separation of the beads. In the simplest case, the sample is mixed and incubated with capture beads off-chip, so that only downstream bead separation, washing, and analysis steps take place on-chip.57,76–78 For instance, Dudani et al. developed a microfluidic platform based on rapid inertial solution exchange (RInSE) that facilitated continuous-flow, high throughput, and 100% transfer efficiency of exosome capture beads from biofluids into a wash buffer.77 In the iMER fluidic chip, Shao et al. performed on-chip incubation of the sample with immunomagnetic microbeads that captured a specific subpopulation of exosomes with high yield, facilitating subsequent bead separation using a magnet.79 Recent platforms have also implemented on-chip mixing, incubation, and bead trapping, thus minimizing sample pre-treatment.80,81 Ko et al. developed a smartphone-enabled microfluidic system, μMED, that mixed mouse plasma with negative and positive enrichment microbeads of different sizes.81 After on-chip incubation of the mixture, fluid flow was established in the device and two consecutive porous membranes facilitated filtration and trapping of the smaller positive capture beads for subsequent on-chip analysis. In the ExoSearch chip, Zhao et al. implemented passive continuous-flow mixing of serum with immunomagnetic beads in a serpentine channel, achieving good recovery yield of exosomes and enabling bead retention by a magnet in a downstream detection chamber.80
These advances illustrate how antibody-based capture of exosomes enables the development of microfluidic platforms with great potential for integrated analysis in POC and clinical settings. We will provide a more in-depth discussion of integrated platforms later in this review. It is also worth noting that high specificity and affinity for exosome capture is not exclusive of antibodies, with aptamers offering an interesting alternative that, in addition to affinity capture, can produce a signal upon exosome binding thus simplifying their detection.82 However, a major limitation of essentially all previously discussed platforms is the sample pre-treatment (e.g., centrifugation during serum or plasma preparation) required for whole blood analysis. Some of the additional isolation approaches described next offer possible solutions to this particular issue.
Membrane-based filtration
Exosome isolation directly from complex biofluids (e.g., whole blood) is highly desirable to eliminate any sample pre-treatment, especially for microfluidic-enabled POC applications. Davies et al. demonstrated two membrane-based filtration approaches, pressure-and electrophoresis-driven, capable of separating EVs directly from mouse whole blood.58 In both cases, the microfluidic device used an in situ prepared and tunable nanoporous membrane that allowed small EVs to pass through while removing cells and other debris. Although pressure-driven filtration achieved a higher yield, device clogging occurred after extracting 4 μL of filtrate. The electrophoresis mode eliminated this problem, offering significantly higher throughput (Table 1) and higher purity by removing some soluble proteins. Cho et al. also used electrophoretic migration of species, but this time a dialysis membrane (30 nm pore size) captured EVs from diluted mouse plasma while allowing proteins to pass through (Figure 3B).83 This system achieved high throughput, relatively high yield, and removed ~84% of protein impurities. In an expected advancement, two integrated filtration -based platforms have been recently developed for analysis of EVs and, interestingly, they both feature a double filtration approach.84,85 Liang et al. employed a 200-nm pore size membrane to retain larger EVs and impurities from urine samples, while isolating and enriching small EVs by means of a second membrane with a pore size of 30 nm that allows proteins to pass through.84 This platform achieved high throughput and good recovery of 30 – 200 nm EVs relative to ultracentrifugation. The “lab-on-a-disc” Exodisc system developed by Woo et al. was also based on a double filtration architecture, although the membranes used (20 and 600 nm pore sizes) imply exosome co-isolation with larger EVs that may decrease exosome purity.85 Nonetheless, the Exodisc achieved high throughput, high recovery yield of EVs, removed >95% of protein contaminants, and isolated EVs whose mRNA concentrations were >100-fold higher relative to EVs isolated by ultracentrifugation. Finally, the development of integrated platforms employing immuno-filtration approaches for specific exosome capture is anticipated.86
Exosome trapping on nanowires
Microfluidic immobilization and isolation of exosomes without using antibodies is an additional approach initially explored using a PEG-lipid-modified surface.87 Later, Wang et al. developed a microfluidic platform for multi-scale filtration using a ciliated nanowire-on-micropillar structure capable of trapping exosome-like lipid vesicles (Figure 3C).88 After creating an array of micropillars using conventional microfabrication techniques, porous silicon nanowires were etched onto the micropillars’ sidewalls. Then, the nanowire forest selectively trapped liposomes while filtering proteins and larger 500 nm beads. The captured liposomes were subsequently recovered via dissolution of the nanowires in PBS for 24 h. Along these lines, a 3D nanowire network structure originally designed for DNA separation89 was described to isolate exosomes with higher efficiency than ultracentrifugation.90 Similarly, trapping of EVs using PDMS-anchored ZnO nanowires on a microfluidic device was also reported.91 Therefore, physically trapping exosomes using nanowires seems to be a promising approach, especially because of its relatively high throughput and potentially high capture efficiency. Since exosomes are likely captured along with other small EVs, such platforms might require additional immunoaffinity labeling for the specific detection of exosomes,92 but they nevertheless appear well suited for analysis of exosomal content via lysis and integration with downstream analysis platforms.
Acoustic isolation
An acoustic standing field established in a fluid medium produces differential acoustic radiation forces acting on particles immersed in the fluid depending on particle properties such as size, density, and compressibility.93 This principle has been used to produce acoustic trapping of microparticles (100 – 1000 nm EVs) on seed microbeads against the flow direction inside a capillary, although no size discrimination in the exosome size range was reported.94,95 Meanwhile, particle manipulation in microfluidic devices using surface acoustic waves (SAW) offers many advantages including biocompatibility, contact-free manipulation, fast fluidic actuation, and simple fabrication and integration with microfluidic components.96,97 In a recent demonstration, Lee et al. performed continuous and versatile sorting of microvesicles (< 1 μm), isolating vesicles smaller than 200 nm.98 To achieve this, interdigitated transducer (IDT) electrodes produced a symmetric standing SAW perpendicular to the flow direction, moving larger particles toward side outlets while facilitating the collection of small particles at the center outlet (Figure 3D). The technology offered high separation yields and in-situ electronic control of the particle size cutoff, facilitating fractionation of different types of microvesicles. Improved throughput and integration with immunoaffinity approaches for isolation of specific exosome subpopulations are anticipated, along with implementations with different SAW-fluid interaction geometries for exosome isolation.99
Nano-DLD sorting
DLD is a continuous-flow particle sorting technique implemented in microfluidic devices embedding a gradient of pillar arrays whose geometry determines a critical cutoff diameter DC. Particles with a diameter larger than DC will be displaced laterally following a bumping mode throughout the array, while smaller particles travel following the streamlines of the fluid and, on average, do not displace laterally, thus enabling particle sorting.100,101 Though DLD separation of cancer-cell-derived microvesicles from EVs in microfluidic devices has been reported,102 sorting of nanoparticles has remained a challenge as diffusional transport becomes an important factor. Recently, by considering the modulation of nanoparticle-pillar electrostatic interactions by the ionic strength of the buffer solution, separation of 51 nm and 190 nm particles with a gap of 2 μm between pillars was achieved, although no demonstration of biocolloid sorting at the nanoscale was made.103 Wunsch et al. reported the first nano-DLD platform for separation of colloids and exosomes as small as 20 nm, made possible by fabrication of pillar arrays with gap sizes from 25 to 235 nm (Figure 3D).104 The exosome fractionation histogram in (Figure 3E) was obtained with a nanopillar gap of 235 nm during a 60-h run, after surface-modification of the device to avoid clogging. This demonstration opens up the possibility for analysis of EVs of selected size ranges with high resolution, facilitating study of exosomes relative to other small EVs of similar size, although higher throughput and evaluation of the robustness of the platform with various bodily fluids are necessary.
Viscoelastic flow sorting
Another label-free passive microfluidic approach recently implemented for sorting of EVs is viscoelastic microfluidics, where particle separation is determined by elastic lift forces acting on particles of different sizes in a viscoelastic medium.105,106 The microfluidic chip, implemented by Liu et al., uses a diluted poly-(oxyethylene) (PEO) solution as a viscoelastic sheath fluid to align the EV sample along the sidewalls of the channel.106 Then, the optimized channel geometry, PEO concentration, and flow condition fine tune elastic lift forces in the system to continuously migrate large EVs toward the channel centerline where they are collected from an outlet in the middle, while exosomes (<200 nm) can be isolated from two-sided outlets. This device achieves >90% purity and >80% recovery of exosomes from untreated fetal bovine serum (FBS) and EVs isolated from cell culture media. This demonstration of viscoelastic microfluidics offers great potential for exosome isolation in POC and clinical settings because of its simplicity and throughput (200 μL/h), although further validation with clinical samples will be necessary.
Microfluidic-based Exosome Detection and Analysis
The aforementioned isolation technologies can be employed in a stand-alone manner, functioning as preparative front ends that interface with conventional downstream detection and analysis. But as shown in Figure 4, microfluidic platforms also make it possible to enhance detection and analysis of exosomes by virtue of offering an attractive combination of high throughput and sensitivity with low reagent consumption and the potential for portability. Immunocapture-based approaches have been successfully implemented in a microfluidic format in combination with a variety of detection methods, with fluorescence detection being the most widely used(Figure 5 and Table 2). Finally, we note that there is variability among the literature in terms of how sensitivity and detection limits are quantified. Therefore, we have expressed sensitivity on an exosomes/μL basis where possible in Table 2 to facilitate comparison among methods.
Figure 4.

Timeline of progress toward exosome analysis in microfluidic platforms.
Figure 5.

Examples of microfluidic approaches for exosome detection. (A) RInSE system leverages inertial focusing to perform continuous bead-based isolation and fluorescence analysis via flow cytometry. Reprinted from Ref. [77], with the permission of AIP Publishing. (B) Enhanced immunocapture via electrohydrodynamic nanoshearing using on-chip electrodes enables colorimetric detection of exosomes. Adapted with permission from Ref. [73]. Copyright 2014 American Chemical Society. (C) μNMR-based detection using on-chip microcoil arrays is achieved by immunoafinity labeling using magnetic nanoparticles. Adapted by permission from Macmillan Publishers Ltd: Nature Medicine Ref. [108], copyright 2012. (D) An aptamer-based electrochemical approach enables label-free detection. Reprinted from Ref. [82], Copyright 2016, with permission from Elsevier.
Table 2.
Exosome detection and analysis in microfluidic platforms.
| Exosome detection approach | Sample (pre-treatment) * | Sensitivity/detection limit | EV size** [nm] | Reference |
|---|---|---|---|---|
| Fluorescence | ||||
| ExoChip: Fluorescent staining, detection via conventional plate reader | Serum | 0.5 pM fluorescence sensitivity | ~30–300 | Kanwar 2014 [72] |
| ELISA-based (IGF-1R, p-IGF-1R) chemifluorescence imaging | Plasma (pre-mixed with capture beads) | 0.281 pg/mL (IGF-1R), 0.383 pg/mL (p-IGF-1R) | ~40–150 | He 2014 [57] |
| RInSE: Inertial solution exchange for continuous isolation of affinity-capture (EpCAM) microbeads | Blood (RBC lysis, centrifugation, incubation with capture beads and label) | NA | ~30–120 | Dudani 2015 [77] |
| ExoSearch: Fluorescently labeled antibodies (CA-125, EpCAM, CD24), multiplex fluorescence imaging | Plasma | 750 exosomes/μL | ~50–250 | Zhao 2016 [80] |
| Nano-IMEX: Enhanced capture and detection via nanostructured surface coating, ELISA-based (CD9, CD81, EpCAM) fluorescence imaging | Plasma (10X dilution) | ~50 exosomes/μL | <150 | Zhang 2016 [74] |
| μMed: ELISA-based (GluR2) optofluidic detection via smartphone | Mouse serum | 10000 exosomes/μL | ~117 | Ko 2016 [81] |
| Nano flow cytometer, imaging of single fluorescently labeled vesicles passing through nanochannels | Cell culture supernatant (centrifugation, 0.2 μm filter, ultracentrifugation, fluorescence labeling) | Single particle sensitivity, 170 fM LOD for vesicle concentration | ~<300 | Friedrich 2017 [107] |
| Fluorescently labeled antibodies (EpCAM, HER2), fluorescence imaging_ | Plasma (incubation with capture particles) | NA | <100 | Fang 2017 [78] |
| Colorimetric | ||||
| Nanoshearing-enhanced capture and detection, ELISA-based (CD9, HER2) colorimetric sensing | Serum | 2760 exosomes/μL | ~30–350 | Vaidyanathan 2014 [73] |
| Exodisc: ELISA-based (CD9, CD81) colorimetric detection | Urine | NA | 20–600 | Woo 2017 [85] |
| ELISA-based (CD63) colorimetric detection aided with smartphone imaging | Urine (centrifugation, 0.22 μm filter) | NA | 155 | Liang 2017 [84] |
| Other immunoaffinity-based | ||||
| μNMR: Labeling with target-specific (CD63, EGFR, EGFRvIII, IDH1 R132H, PDPN) magnetic nanoparticles, on-chip NMR detection | Plasma (0.2 μm filter, differential centrifugation) | 10000 microvesicles | 50–150 | Shao 2012 [108] |
| nPLEX: Functionalized gold surface with nanohole arrays (EpCAM, CD24, CD63), portable SPR detection | Ascites fluid (0.2 μm filter) | ~3000 exosomes (670 aM) | ~20–260 | Im 2014 [44] |
| Microcapillary electrophoresis of immunolabeled EVs (CD63, CD44), laser dark-field microimaging | Mouse plasma | NA | ~50–450 | Akagi 2015 [109] |
| SPR detection of clinically relevant exososmes (HER2) | Serum | ~2070 exosomes/μL | ~30–300 | Sina 2016 [75] |
| Other | ||||
| Continuous electrical detection during transit through a nanoconstriction | Mouse plasma (2.5X dilution, centrifugation, pre-mixed with calibration particles) | Single particle sensitivity | ~50–100 | Fraikin 2011 [111] |
| Electrical detection of RNA by binding to complementary miR-550 probes on an ion exchange nanomembrane surface | Cell media | 2 pM of miRNA | ~20–50 | Taller 2015 [112] |
| iMER: Detection of mRNAs (EPHA2, EGFR, PDPN, MGTM, APNG) via on-chip RT-qPCR | Serum (0.8 μm filter) | NA | ~ 100 | Shao 2015 [79] |
| Aptamer probes immobilized on gold electrodes (CD63), electrochemical detection | Cell culture supernatant (0.22 μm filter, centrifugation, exosome isolation kit) | 1000 particles/μL | ~100 | Zhou 2016 [82] |
NA: not available. RBC: red blood cell. BSA: bovine serum albumin.
Sample is of human origin unless otherwise stated.
Size of isolated particles is typical and it might not correspond precisely to the sample and operating conditions shown.
Fluorescence detection
The ExoChip developed by Kanwar et al. incorporated a PDMS-based microchannel array functionalized with exosome capture antibodies.72 Immobilized exosomes were then fluorescently stained, and the chip was imaged using a plate reader for detection and quantification with 0.5 pM sensitivity. Bead-based immunoassays have also been adapted to the microfluidic format, as shown by He et al. who achieved integrated immunoisolation and enrichment of exosomes from plasma using magnetic beads.57,76 Chemi-fluorescence detection was then performed via ELISA analysis of target proteins with sub pg/mL sensitivity. The bead-based approach also yielded narrower exosomal size distributions as compared with conventional ultracentrifugation methods, suggesting higher specificity. The optofluidic μMED platform developed by Ko et al. was applied for detection of enzyme amplified biomarkers from brain-derived exosomes.81 Target exosomes were captured on immobilized beads, after which a horseradish peroxidase modified antibody based immunoassay was performed to enable fluorescence detection using a smartphone-based mobile fluorimeter.
A novel approach for high throughput continuous flow analysis in a bead-based immunocapture format was the RInSE platform demonstrated by Dudani et al. (Figure 5A).77 Functionalized beads were used for isolation of exosomes, after which they were injected into a microchannel designed to induce inertial focusing and buffer exchange. This approach enabled the beads to be precisely positioned for continuous fluorescence analysis via flow cytometry. Friedrich et al. also employed a cytometry-based approach consisting of an array of parallel nanochannels. Quantification was performed by tracking the transit of individual fluorescently labeled EVs through the nanochannel array, yielding a 170 fM detection limit.107 Zhang et al. described a nano-IMEX platform capable of performing enhanced microfluidic ELISA-based exosome detection by employing a GO/PDA nanostructured coating.74 This coating increases the surface area for immobilization of capture antibodies, enabling improved detection and profiling of colon cancer exosomes from a cell culture medium with a detection limit of ~50 exosomes/μL. Zhao et al. demonstrated the ExoSearch approach involving continuous-flow immunocapture using magnetic beads for multiplex detection.80 The collected species were stained by injection of fluorescently labeled antibodies to enable detection via multi-color fluorescence imaging, with a detection limit of 750 exosomes/μL. Fang et al. also employed immunocapture via magnetic beads, after which antibody-based fluorescence labeling was performed on chip to enable microscope-based imaging and quantification.78
Colorimetric detection
Progress has also been made toward development of approaches that can enable colorimetric detection of exosomes by direct visualization, potentially simplifying the instrument design to a format more amenable for POC applications. Vaidyanathan et al. demonstrated a novel approach to augment immunocapture via a nanoshearing methodology whereby an alternating current electrohydrodynamic flow generated in the vicinity of on-chip functionalized electrodes enhanced both sensitivity and specificity (Figure 5B).73 Detection and quantification were performed both visually and via absorbance measurements from colorimetric solution, with a 3-fold enhancement in detection sensitivity reported in comparison to other hydrodynamic flow based assays. Liang et al. also employed a smartphone camera, but to perform colorimetric ELISA detection of exosomes captured against a filtration membrane.84 This system was applied to identify elevated concentrations of urinary EVs in bladder cancer patients as compared with healthy controls. Finally, Woo et al. reported ELISA-based detection of exosomes associated with bladder cancer in their Exodisc platform.85 Following isolation using a two-stage filtration arrangement, on-disc ELISA was performed, after which the sample was transferred to a detection chamber where the OD at 450 nm was measured. The “lab-on-a-disc” format provided an efficient platform for automation of sequential capture, labeling, and detection operations.
Other immunoaffinity-based sensing approaches
Detection methods that do not rely on fluorescence or colorimetric sensing include a microfluidic system incorporating miniaturized nuclear magnetic resonance (μNMR)-based detection, explored by Shao et al. (Figure 5C).108 Magnetic nanoparticle immunolabeling was achieved by targeting proteins on the microvesicles, rendering them superparamagnetic. Embedded on-chip microcoil arrays enabled subsequent NMR detection. Im et al. demonstrated SPR detection of ovarian cancer exosomes using the nPLEX microfluidic chip incorporating an array of plasmonic nanoholes.44 The nanoholes were arranged to generate an electromagnetic field distribution near the sensing surface tailored to the size range of exosomes of interest. Exosomes were immobilized in the vicinity of the nanoholes via immunocapture on the functionalized surface, and detected by transmission mode SPR using a portable imaging system that achieved 670 aM sensitivity. Another adaptation of SPR detection was employed by Sina et al. as part of a two-step isolation and detection process.75 Custom-made SPR chips were employed for analysis of exosomes from breast cancer patient samples, where characteristics of the resulting spectral shifts enabled identification and quantification of exosome populations from serum samples. Electrokinetic methods have also been employed for analysis of exosomal materials, as demonstrated by Akagi et al. who applied a microscale capillary electrophoresis chip and laser dark-field microimaging system to detect shifts in the zeta potential distribution resulting from binding with antibody markers. These shifts were then correlated with over-expression of glycoprotein biomarkers for breast cancer analysis.109,110
Other detection methods
Other methods have also been explored that make it possible to detect, quantify, and perform analysis of exosomes with minimal labeling and washing steps. Fraikin et al. demonstrated a microfluidic platform for high throughput detection and sizing of nanoparticle species in multicomponent mixtures.111 Analysis was performed via electrical detection of species transiting through a nanoconstriction, where coupled fluidic and electric circuits were arrayed in a manner that enabled the measured voltage change during transit to be correlated to the species size with single particle sensitivity. Although particle and bacteriophage species were the primary focus of this study, the authors found that the system was able to reveal the presence of plasma-borne nanoparticles, suggesting potential for applications involving exosome analysis. The iMER microchip platform for analysis of exosomal mRNA was demonstrated by Shao et al. 79 This platform achieved enrichment of exosomes via antibody functionalized bead capture, lysis, and multiplexed detection and quantification of the target mRNA by real-time quantitative PCR (RT-qPCR). Another mRNA-based approach reported by Taller et al. involved application of a surface acoustic wave method for lysis of exosomes followed by detection with a nanomembrane sensor using two separate chips.112,113 Lysis of raw cell media was performed, after which target mRNA was detected via measurement of a voltage shift proportional to RNA binding to complementary probes on the functionalized membrane surface. Aptamer-based electrochemical detection was explored by Zhou et al. using a microchannel network containing gold electrodes with immobilized aptamers specific to exosome transmembrane protein CD63 (Figure 5D).82 Upon binding of exosomes to complementary aptamers, probing strands pre-labeled with redox moieties were released, generating a decrease in the corresponding electrochemical signal.
Integrated Microfluidic Platforms for Exosome Analysis
Microfluidic systems capable of performing integrated on-chip exosome isolation and analysis represent a significant step toward realizing the vision of exosome-based diagnostics in POC and clinical settings. Over the past few years, researchers have developed integrated microfluidic platforms for analysis of overall exosome levels, detection of disease-specific subpopulations of exosomes, and quantification of intravesicular proteins and mRNAs (Figure 6). These integrated platforms offer several advantages compared to conventional time-consuming protocols, especially when applied toward scenarios involving high throughput processing of small sample volumes and low exosome concentrations. In addition, increased diagnostic accuracy can be achieved by implementing on-chip multiplexed assays.
Figure 6.

Timeline of progress toward development of integrated microfluidic platforms for exosome analysis.
Integrated microfluidic platforms summarized in Table 3 are classified according to their intended ultimate exosome analysis. To maintain a focus on POC and clinical applications, we attain note that relevant operating conditions employed in the analysis of bodily fluids (serum, plasma, whole blood) are presented. Notice, however, that most of these platforms can also process cell culture derived exosomes (Table 4), albeit at the expense of additional sample pre -treatment. Operation of integrated platforms in both continuous and discontinuous stages has enabled great sensitivity to be achieved, with sample-to-answer throughputs ranging from 0.029 to 70 μL/min (a parametric basis we have selected to help enable comparison among different platforms). Most of these systems employed immunoaffinity isolation of exosomes, detected and quantified exosomal biomarkers including proteins and mRNAs, and primarily targeted cancer diagnostics. Therefore, these platforms are extremely valuable for fundamental studies, while simultaneously offering enormous potential to enable exosome-based liquid biopsy for early disease diagnosis, prognosis, and therapy monitoring.
Integrated analysis of overall exosome levels
Common exosomal surface markers (i.e., extravesicular proteins expressed in exosomes regardless of their origin, such as CD63, CD9, and CD81) facilitate specific isolation, detection, and quantification of overall exosome populations in biofluids. Exosome quantification can potentially serve as a biomarker for disease diagnosis,114 as for example where counts of CD9(+) and CD81(+) exosomes from human plasma were found to be elevated in both non-small-cell lung cancer (NSCLC) and ovarian cancer patients relative to healthy donors.57 Kanwar et al. reported the first integrated microfluidic platform, the ExoChip, developed for specific quantification of exosomes from serum samples.72 This system employed CD63 for immunoisolation of exosomes on the device’s multiple circular chambers, followed by staining of their membranes with a fluorescent dye (DiO), and plate reader quantification. The ExoChip device measured a statistically significant 2.34-fold increase in exosome levels in pancreatic cancer patients relative to healthy individuals. Zhang et al. used the ultrasensitive nano-IMEX microfluidic chip (Figure 2A), to probe plasma samples from ovarian cancer patients and healthy individuals.74 Instead of quantifying total exosomes or performing molecular profiling, the platform measured combined expression levels of a specific disease marker, EpCAM, and common exosomal markers, CD9 and CD81, in CD81(+) exosomes using a fluorescence immunoassay. The nano-IMEX platform successfully discriminated patients from controls and showed a ~10-fold decrease in the overall levels of the three markers following treatment.
More recently, two high throughput integrated platforms employed a double filtration approach for isolation of EVs from urine of bladder cancer patients and healthy controls, while using colorimetric ELISA for quantification of pan exosomal markers.84,85 Liang et al. isolated and enriched small EVs (30 – 200 nm) followed by quantification of overall CD63 levels, measuring significantly higher concentrations of urine-derived EVs in cancer patients relative to controls while achieving great diagnostic accuracy (area under curve, AUC = 0.96).84 The easy read out of the colorimetric assay aided with smartphone imaging makes this a promising approach for POC settings. Meanwhile, the Exodisc platform developed by Woo et al. isolated and enriched small and medium sized EVs (20 – 600 nm), followed by quantification of CD9 and CD81 expression levels that were found to be higher in patients relative to controls (Figure 7A).85 The tabletop-sized Exodisc used an innovating and fully automated “lab-on-a-disc” approach potentially suitable for clinical settings.
Figure 7.

Examples of integrated microfluidic platforms for exosome analysis. (A) Exodisc platform for overall EV quantification. Adapted with permission from Ref. [85]. Copyright 2017 American Chemical Society. (B) Smartphone-enabled μMED platform for POC analysis of disease-specific exosomes. Adapted from Ref. [81] with permission from Nature Publishing Group. (C) Integrated platform for intravesicular protein analysis. Reproduced from Ref. [57] with permission from the Royal Society of Chemistry. (D) iMER chip for integrated and multiplexed quantification of exosomal mRNA levels. Reproduced from Ref. [79] with permission from Nature Publishing Group.
Some additional integrated platforms can also quantify overall exosome levels from clinical samples;44,73,78 however, they go one-step further to quantify disease-specific exosomes, and are described next along with other integrated systems for detection of specific exosome subpopulations.
Integrated detection of specific exosome subpopulations
Most integrated microfluidic systems for exosome analysis seek to identify disease-associated extravesicular markers that provide high diagnostic accuracy.115 This became evident from an early attempt at an integrated system for exosome analysis, the μNMR platform developed by Shao et al. 108 Despite requiring off-chip exosome isolation and lysis, this platform integrated on-chip operations such as immunomagnetic labeling of exosomes, microfiltration, and NMR sensing for analysis of clinical samples from glioblastoma multiforme (GBM) patients and healthy volunteers. Thus, μNMR enabled identification of four signature biomarkers for GBM detection (EGFR, EGFRvIII, PDPN, and IDH1 R132H) in patient samples (Table 4), offering >90% combined accuracy (AUC = 0.95). In fully integrated systems, identification of disease-specific markers is accomplished either by targeting exosomal surface markers of interest during the immunoaffinity isolation stage, or by isolating bulk exosomes first using pan exosomal markers or filtration approaches, and later adding specific detection antibodies. These integrated platforms typically achieve high sensitivity and throughput, and cover a wide range of operation and detection schemes (e.g., continuous/discontinuous flow, SPR/fluorescence/colorimetric sensing, and multiplexed/singleplexed analysis). More importantly, they provide clear examples that demonstrate the advantages of multiplexing and show the utility of such platforms for POC applications.
The SPR-based nPLEX, developed by Im et al., constituted the first microfluidic platform for exosome analysis with high-level integration and multiplexing capabilities.44 Continuous and simultaneous immunoaffinity isolation and label-free detection of specific exosomes was performed by monitoring binding events on gold nanohole arrays in real-time using transmission SPR. To achieve multiplexed analysis, the chip employed a configuration with parallel fluidic channels functionalized to permit capture and detection of up to 12 different subpopulations of exosomes. In addition, elution of exosomes attached on the sensor surface facilitated their recovery for downstream analysis and repeated use of the device. The clinical potential of nPLEX was demonstrated by analyzing ascites fluids from ovarian cancer and non-cancer patients and quantifying EpCAM and CD24 protein levels, measured relative to CD63(+) exosome counts. Results from this multiplexed analysis showed a 97% diagnostic accuracy for combined EpCAM and CD24 levels (AUC = 0.995). More recently, Sina et al. used a custom-built SPR-based platform for analysis of exosome subpopulations, although isolation and detection of relevant exosomes occurred sequentially and without multiplexing.75 Exosomes were first isolated on a gold surface using ubiquitous markers (CD9 or CD63), followed by label-free immunoaffinity detection of cancer-specific exosomes expressing HER2. Both processes produced characteristic spectral shifts that could be monitored in real-time. Analysis of serum from HER2(+) breast cancer patients revealed that 14 – 35% of the isolated bulk exosome population expressed HER2.
In addition to the SPR-based platforms just described, continuous-flow operation was also implemented using fluorescence for specific detection of exosome subpopulations.77,80 Dudani et al. used RInSE to isolate capture microbeads from pre-processed blood, with off-chip incubation steps lasting up to 4.5 h (Table 3).77 Then, inertial focusing was employed to align the beads during continuous flow, with captured EpCAM(+) exosomes labeled with phycoerythrin-CD81 conjugate, permitting inline fluorescence detection using a custom-built flow cytometer. Although it achieved very high throughput, this platform could further benefit from on-chip integration between upstream and downstream systems to minimize sample pre-treatment and perform exosome quantification, respectively. In the ExoSearch chip, Zhao et al. implemented a continuous-flow integrated platform for multiplexed analysis of plasma from ovarian cancer patients and healthy individuals.80 After continuously isolating CD9(+) exosomes on immunomagnetic microbeads, a switchable magnet retained the beads as an aggregate inside a probing microchamber while a washing buffer was applied. Then, a mixture of fluorescently labeled antibodies targeting exosomal tumor markers CA-125, EpCAM, and CD24 was introduced at a slow flow rate, followed by additional washing, multiplexed imaging, and release of the retained beads. By combining these three tumor specific markers, the multiplexed platform achieved excellent diagnostic accuracy (AUC = 1.0). Furthermore, the ExoSearch chip could process up to 10 mL of sample in a single continuous run. Fang et al. recently developed an integrated system, appearing to operate in continuous-flow and similar to the ExoSearch chip, for immunomagnetic capture of CD63(+) exosomes and detection of EpCAM and HER2 exosomal tumor markers in plasma samples.78 This platform revealed that EpCAM levels were significantly higher in exosomes from breast cancer patients relative to healthy controls, while exosomal expression of HER2, in a different set of patients, was in good agreement with HER2 expression in tissues from the primary tumor.
Another important aspect of microfluidic platforms is their potential to function in POC settings. Characteristics of such systems include portability, low-cost, fast and simple operation, and easy read-out, many of which can be readily found in integrated platforms for exosome analysis.44,73,79,81,84 Among those designed for identification of specific exosome subpopulations, Vaidyanathan et al. developed a multiplexed microfluidic device based on nanoshearing effects induced by alternating current electrohydrodynamics.73 This approach enhanced specificity when capturing CD9(+) or disease-specific HER2(+) exosomes, after which an on-chip colorimetric immunoassay facilitated qualitative naked eye detection and/or quantitative measurement of targeted exosomes via a spectrophotometer. The analysis of serum from HER2(+) and HER2(−) breast cancer patients illustrated the selective and specific capture of HER2(+) exosomes.
Other integrated platforms such as n-PLEX and μMED have incorporated custom miniaturized optics to enable the portability necessary for POC analysis of patient samples.44,81 The smartphone-enabled μMED by Ko et al. represents a well-suited technology for use in POC settings (Figure 7B).81 A compact and low-cost 3D printed smartphone attachment housed a disposable chip for exosome analysis and embedded optical components that used the phone’s bright LED and camera for fluorescence detection. Inside the disposable chip, CD81(+) exosomes isolated on positive enrichment microbeads were probed in a fluorescence immunoassay targeting brain-derived GluR2(+) exosomes, with data analysis performed on the smartphone via a dedicated app. In addition, μMED could operate via capillary action or vacuum pack to establish flow inside the chip without external active flow control. This integrated platform demonstrated rapid analysis of a specific brain-derived exosome subpopulation from the serum of mice with mild traumatic brain injury.
Integrated analysis of intravesicular proteins
In addition to identifying specific subpopulations of exosomes from clinical samples, further integrated capabilities have been demonstrated including exosome lysis and additional stages for capture and analysis of the released exosomal contents. He et al. reported an integrated microfluidic platform for high-sensitivity detection of relevant intravesicular protein markers from a specific subpopulation of exosomes isolated from clinical samples (Figure 7C).57,76 First, immunomagnetic microbeads with captured EpCAM(+) exosomes were retained in a chamber by a magnet with > 99.9% efficiency, washed with PBS buffer, and incubated with a lysis buffer to release exosomal proteins. This lysate was then flushed into a serpentine microchannel for mixing with immunomagnetic beads injected from two side channels, providing enhanced mixing and sufficient incubation residence time for specific capture of intravesicular proteins. A magnet once more retained the beads in a second chamber where the final chemiluminescence-based immunoassay took place, achieving detection of total IGF-1R and intravesicular p-IGF-1R. The results showed overexpression of IGF-1R in EpCAM(+) exosomes of NSCLC patients relative to controls, while no correlation between p-IGF-1R and disease state or IGF-1R levels was found. Given the significant interest in these proteins for cancer diagnosis and treatment,116–118 the pioneering exosome-based analysis performed could be important for establishing IGF-1R and p-IGF-1R as exosomal biomarkers. Although μNMR and nPLEX also performed on-chip detection of intravesicular proteins, such as HSP70, HSP90, and IDH1 R132H, some samples were from cell cultures and off-chip exosome isolation and/or lysis was required.44,108
Integrated analysis of exosomal mRNAs
The discovery that exosomes contain functional mRNAs and microRNAs,17 as well as both single and double-stranded DNA,19,119 adds to the significant value of exosomes for disease diagnosis and therapy monitoring. Although microfluidic-based approaches have facilitated exosome isolation for subsequent off-chip RNA analysis,44,58,70,72,77,83 on-chip analysis of exosomal mRNAs and microRNAs has also been recently implemented.79,112 The integrated and multiplexed iMER platform developed by Shao et al. performed analysis of mRNA levels in tumor exosomes isolated from serum of GBM patients and healthy individuals (Figure 7D).79 Following magnetic separation of capture microbeads with EGFR(+) and EGFRvIII(+) exosomes, a lysis buffer released exosomal contents that were injected through a filter of densely packed glass microbeads achieving a high extraction yield of RNA. After sequential washing steps, water eluted the RNA on the glass beads and transported it into a chamber for reverse transcription. Then, transcribed DNA was dispensed into four different chambers for multiplexed analysis of targeted mRNAs by RT-qPCR. For iMER operation, the chip used a custom-made PCR set-up with a thermo-cycler and a portable fluorescence detector, while torque-actuated valves controlled fluidic flow. The iMER chip revealed that combined exosomal mRNA levels of EPHA2, EGFR, and PDPN have a diagnostic accuracy of 90% for GBM (AUC = 0.945). Furthermore, serial measurements of mRNA levels of MGMT and APNG, two important enzymes involved in repairing DNA damaged by the drug temozolomide (TMZ), demonstrated the feasibility of drug resistance monitoring during treatment using iMER’s integrated exosome analysis. Therefore, additional microfluidic-based approaches targeting exosomal nucleic acids are anticipated, although not limited to cancer but also including conditions such as cardiovascular disease.120
Exosomal Proteins in Microfluidic-based Exosome Analysis
Exosomes represent a particular type of small (< ~ 150 nm in diameter) EV of endosomal origin. As such, their endosome-associated proteins are typically employed for exosome characterization.2,14,15 When using microfluidic platforms for exosome analysis, a wide range of exosomal proteins have enabled capture and/or detection of small vesicles from various biofluids with cancer as the main targeted disease. Table 4 presents a comprehensive summary of exosomal proteins employed in microfluidic systems classified according to their intended use as follows: common exosomal markers (e.g., CD9, CD63, and CD81) used to identify overall exosome populations, disease-associated exosomal markers(e.g., CD24, EpCAM, and HER2) employed to recognize specific subpopulations of exosomes from diseased cells, and non-disease exosomal markers (e.g., CD41 and CD45) intended to identify exosomes from healthy cells. But, a major challenge when using protein markers for exosome analysis is to properly discriminate exosomes from other types of EVs because of overlap in protein composition. It has recently been pointed out that many of the proteins often used as exosomal markers (e.g., flotillin-1, HSP70, MHCII, and CD63) are also present in multiple types of EVs.121 The lack of purity when isolating and analyzing the exosomal population (or subpopulations) hinders the ability to obtain clinically relevant insights.
Conventional techniques for exosome isolation and analysis have made initial progress toward addressing the need for universal exosome-specific protein markers capable of distinguishing them from other EVs.121,122 In what appears to be a promising approach, Kowal et al. proposed an isolation protocol for categorization of EVs, along with markers that could be used to discriminate the suggested isolation-dependent categories. It was proposed that endosome-derived exosomes, which are enriched in all three CD9, CD63, and CD81 tetraspanins, can be distinguished from many other different types of EVs by using syntenin-1 and TSG101 as specific exosomal markers.121 However, it is still too early to tell whether the proposed approach and results obtained are sufficiently robust and reproducible to be clinically meaningful. Fortunately, there is awareness about the need for standardization of EV research for improved evaluation and replication of experiments when employing conventional methods. The International Society for Extracellular Vesicles (ISEV), founded in 2012, provides minimum experimental requirements for definition of EVs and their functions.123 This has facilitated the creation of databases such as ExoCarta, intended as a resource for exosomal cargo,124 and the recently introduced crowdsourcing EV-TRACK platform.125 Efforts like these are essential to bring consensus about experimental methods for categorization of EVs and the potential identification of universal exosome-specific protein markers.
Conclusions and Outlook
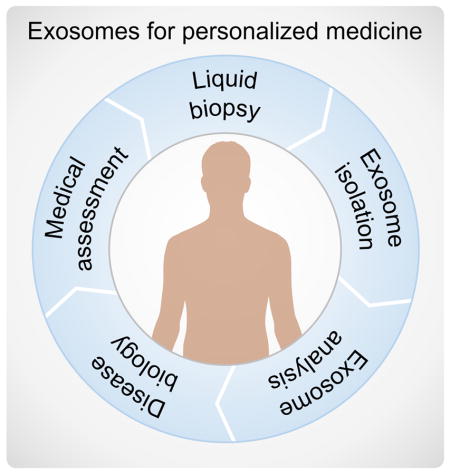
Analysis of exosomes and other extracellular materials is a cornerstone of emerging liquid biopsy techniques, whose importance in diagnostics and personalized medicine is only beginning to be understood (Figure 8). But development of technologies that can make routine analysis of liquid biopsy samples feasible is also critically important, and presents a tremendous opportunity for microfluidic solutions. It is notable that most of the advancements highlighted here have occurred within the past 5 years, indicating that this field is still in its infancy. This body of literature lays a foundation for future progress by introducing new methods for exosome capture, detection, and analysis. These advancements also suggest directions for future work needed to realize the vision of exosome-based POC applications.
Figure 8.

Microfluidic technologies promise to enable liquid biopsy and exosome analysis to become a routine tool for POC diagnostics and personalized medicine.
Antibody-free processing
Many of the methods that have been developed for exosomal capture and analysis rely on antibodies specific to a target of interest. In terms of immunocapture, antibodies specific to proteins on the exosome’s lipid membrane are leveraged for collection and enrichment on solid surfaces, whereas antibody labeling provides a basis for many detection and analysis methods. Despite the success of these antibody-based approaches, especially to isolate and identify disease-specific exosomes, there is interest in antibody-free methods that can potentially streamline workflows by reducing or eliminating incubation times and washing steps required in both immunocapture and immunolabeling, which could degrade and contaminate the sample. The ability to use antibody-free techniques also ensures that the biological function of exosomes is not impacted by the presence of probe species during downstream analysis. Detection methods that do not rely on fluorescence imaging are also of interest because they can help to reduce the instrument footprint for portable applications.
Integration with downstream biological analysis
Many microfluidic platforms have focused on exosomal analysis via detection and quantification. But additional research is needed to fully understand the biological significance of exosomes in mediating intercellular signaling and therapeutic response. Studies aimed at addressing these fundamental questions critically depend on the ability to harvest intact exosomes for subsequent biological and biophysical characterization. Microfluidic integration provides an ideal platform to streamline this workflow by minimizing handling steps and enabling highly multiplexed analysis. Development of microfluidic platforms that inherently consume small amounts of sample and reagents, require little to no operator-dependent processing, and deliver rapid sample-to-answer times promises to enable robust, reproducible, and isolation-independent categorization of EVs while identifying unique exosomal proteins.
Standardization
It is currently challenging to extract common operational parameters and performance metrics (e.g., throughput, limit of detection) associated with systems developed by different research groups, highlighting the increasing need for standardization. Databases such as ExoCarta (http://www.exocarta.org), EV-TRACK (http://evtrack.org), EVpedia (http://evpedia.info), and Vesiclepedia (http://www.microvesicles.org) that follow ISEV guidelines are currently available for conventional exosome analysis, but comparable resources aimed at promoting standardized reporting of microfluidic-based exosomal sample preparation, collection, and analysis methods are lacking. This information is critically important, as differences in sample collection and handling can impact the results of exosome-based analysis.27,126–128 Independent databases focused on microfluidic platforms are critically needed to enable comparison of results obtained by different investigators, making it possible to achieve consensus in the scientific community about how to unlock the full potential of exosome analysis in fundamental research and clinical applications.
Commercialization opportunities
Although it is typically claimed that microfluidic technologies represent a “cost-effective” alternative relative to conventional benchtop-scale instruments for exosome isolation and analysis, this largely remains a subjective classification because it is challenging to obtain consistent cost information across multiple methodologies. Despite this and thanks to recent progress and growing interest in the field, microfluidic platforms appear well positioned to enable routine exosome-based analysis of liquid biopsy samples at the POC and in clinical settings, which eventually will lead to commercialization opportunities. However, anumber of challenges must be overcome to position such microfluidic systems in the clinical market. First, most of the platforms described have only been employed in research settings, and the performance and capabilities enabled by those systems already subjected to clinical testing still require validation in larger cohorts of patients. Second, without proper standardization it is not possible to guarantee robustness and reproducibility of the results obtained using microfluidic technologies and unlock the real potential of exosome-based analysis as previously discussed. Finally, additional hurdles come in the form of health care regulations and approvals. Thus, microfluidic technologies for exosome analysis are more likely to be commercialized in the academic research market first, with early examples seen in the adoption of commercially available microfluidic-based instruments for exosome analysis.129–133
Acknowledgments
This work was supported in part by the US National Institutes of Health (NIH) under grant 1R03AI113585, and the Cancer Prevention and Research Institute of Texas (CPRIT) under grant RP150421.
References
- 1.El Andaloussi S, et al. Nat Rev Drug Discov. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 2.Raposo G, Stoorvogel W. J Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hosseini-Beheshti E, et al. Mol Cell Proteomics. 2012;11:863–885. doi: 10.1074/mcp.M111.014845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katsuda T, Kosaka N, Ochiya T. Proteomics. 2014;14:412–425. doi: 10.1002/pmic.201300389. [DOI] [PubMed] [Google Scholar]
- 5.Schorey JS, Bhatnagar S. Traffic. 2008;9:871–881. doi: 10.1111/j.1600-0854.2008.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bhatnagar S, Schorey JS. J Biol Chem. 2007;282:25779–25789. doi: 10.1074/jbc.M702277200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bhatnagar S, et al. Blood. 2007;110:3234–3244. doi: 10.1182/blood-2007-03-079152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pelchen-Matthews A, Raposo G, Marsh M. Trends Microbiol. 2004;12:310–316. doi: 10.1016/j.tim.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Robbins PD, Morelli AE. Nat Rev Immunol. 2014;14:195–208. doi: 10.1038/nri3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nawaz M, et al. Nat Rev Urol. 2014;11:688–701. doi: 10.1038/nrurol.2014.301. [DOI] [PubMed] [Google Scholar]
- 11.Lawson C, et al. J Endocrinol. 2016;228:R57–R71. doi: 10.1530/JOE-15-0201. [DOI] [PubMed] [Google Scholar]
- 12.Zhang HG, Grizzle WE. Clin Cancer Res. 2011;17:959–964. doi: 10.1158/1078-0432.CCR-10-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Théry C, Zitvogel L, Amigorena S. Nat Rev Immunol. 2002;2:569–579. doi: 10.1038/nri855. [DOI] [PubMed] [Google Scholar]
- 14.Colombo M, Raposo G, Théry C. Annu Rev Cell Dev Biol. 2014;30:255–289. doi: 10.1146/annurev-cellbio-101512-122326. [DOI] [PubMed] [Google Scholar]
- 15.Kowal J, Tkach M, Théry C. Curr Opin Cell Biol. 2014;29:116–125. doi: 10.1016/j.ceb.2014.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Taylor DD, Gercel-Taylor C. Gynecol Oncol. 2008;110:13–21. doi: 10.1016/j.ygyno.2008.04.033. [DOI] [PubMed] [Google Scholar]
- 17.Valadi H, et al. Nat Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 18.Properzi F, Logozzi M, Fais S. Biomarkers Med. 2013;7:769–778. doi: 10.2217/bmm.13.63. [DOI] [PubMed] [Google Scholar]
- 19.Thakur BK, et al. Cell Res. 2014;24:766–769. doi: 10.1038/cr.2014.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thind A, Wilson C. J Extracell Vesicles. 2016;5:31292. doi: 10.3402/jev.v5.31292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnsen KB, et al. Biochim Biophys Acta. 2014;1846:75–87. doi: 10.1016/j.bbcan.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 22.Vlassov AV, et al. Biochim Biophys Acta. 2012;1820:940–948. doi: 10.1016/j.bbagen.2012.03.017. [DOI] [PubMed] [Google Scholar]
- 23.Alexander M, et al. Nat Commun. 2015;6:7321. doi: 10.1038/ncomms8321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He M, Zeng Y. J Lab Autom. 2016;21:599–608. doi: 10.1177/2211068216651035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yoshioka Y, et al. Nat Commun. 2014;5:3591. doi: 10.1038/ncomms4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma A, Khatun Z, Shiras A. Nanomedicine. 2016;11:421–437. doi: 10.2217/nnm.15.210. [DOI] [PubMed] [Google Scholar]
- 27.Van Deun J, et al. J Extracell Vesicles. 2014;3:24858. doi: 10.3402/jev.v3.24858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cvjetkovic A, Lötvall J, Lässer C. J Extracell Vesicles. 2014;3:23111. doi: 10.3402/jev.v3.23111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abramowicz A, Widlak P, Pietrowska M. Mol ByoSyst. 2016;12:1407–1419. doi: 10.1039/c6mb00082g. [DOI] [PubMed] [Google Scholar]
- 30.Momen-Heravi F, et al. Biol Chem. 2013;394:1253–1262. doi: 10.1515/hsz-2013-0141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Théry C, et al. Current Protocols in Cell Biology. ch. 3. John Wiley & Sons, Inc; Hoboken, NJ: 2006. pp. 03.22.01–03.22.29. [Google Scholar]
- 32.Bard MP, et al. Am J Respir Cell Mol Biol. 2004;31:114–121. doi: 10.1165/rcmb.2003-0238OC. [DOI] [PubMed] [Google Scholar]
- 33.Andre F, et al. Lancet. 2002;360:295–305. doi: 10.1016/S0140-6736(02)09552-1. [DOI] [PubMed] [Google Scholar]
- 34.Keller S, et al. J Transl Med. 2011;9:86. doi: 10.1186/1479-5876-9-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tauro BJ, et al. Methods. 2012;56:293–304. doi: 10.1016/j.ymeth.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 36.Lane RE, et al. Sci Rep. 2015;5:7639. doi: 10.1038/srep07639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor DD, Zacharias W, Gercel-Taylor C. Serum/Plasma Proteomics: Methods and Protocols. ch. 15. Springer; New York, NY: 2011. pp. 235–246. [Google Scholar]
- 38.Lobb RJ, et al. J Extracell Vesicles. 2015;4:27031. doi: 10.3402/jev.v4.27031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Helwa I, et al. PLoS ONE. 2017;12:e0170628. doi: 10.1371/journal.pone.0170628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alvarez ML, et al. Kidney Int. 2012;82:1024–1032. doi: 10.1038/ki.2012.256. [DOI] [PubMed] [Google Scholar]
- 41.Sáenz-Cuesta M, et al. Front Immunol. 2015;6:00050. [Google Scholar]
- 42.Paolini L, et al. Sci Rep. 2016;6:23550. doi: 10.1038/srep23550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gámez-Valero A, et al. Sci Rep. 2016;6:33641. doi: 10.1038/srep33641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Im H, et al. Nat Biotechnol. 2014;32:490–495. doi: 10.1038/nbt.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanaka Y, et al. Cancer. 2013;119:1159–1167. doi: 10.1002/cncr.27895. [DOI] [PubMed] [Google Scholar]
- 46.Muller L, et al. J Immunol Methods. 2014;411:55–65. doi: 10.1016/j.jim.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nordin JZ, et al. Nanomedicine: NBM. 2015;11:879–883. doi: 10.1016/j.nano.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 48.Gercel-Taylor C, et al. Anal Biochem. 2012;428:44–53. doi: 10.1016/j.ab.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 49.Arslan F, et al. Stem Cell Res. 2013;10:301–312. doi: 10.1016/j.scr.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 50.Baranyai T, et al. PLoS ONE. 2015;10:e0145686. doi: 10.1371/journal.pone.0145686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Böing AN, et al. J Extracell Vesicles. 2014;3:23430. doi: 10.3402/jev.v3.23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu Y, Deng W, Klinke DJ., II Analyst. 2015;140:6631–6642. doi: 10.1039/c5an00688k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma S, et al. ACS Nano. 2010;4:1921–1926. doi: 10.1021/nn901824n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van der Pol E, et al. J Thromb Haemost. 2013;11:36–45. doi: 10.1111/jth.12254. [DOI] [PubMed] [Google Scholar]
- 55.Pospichalova V, et al. J Extracell Vesicles. 2015;4:25530. doi: 10.3402/jev.v4.25530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orozco AF, Lewis DE. Cytometry A. 2010;77:502–514. doi: 10.1002/cyto.a.20886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He M, et al. Lab Chip. 2014;14:3773–3780. doi: 10.1039/c4lc00662c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Davies RT, et al. Lab Chip. 2012;12:5202–5210. doi: 10.1039/c2lc41006k. [DOI] [PubMed] [Google Scholar]
- 59.Jeong S, et al. ACS Nano. 2016;10:1802–1809. doi: 10.1021/acsnano.5b07584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liang K, et al. Nat Biomed Eng. 2017;1:0021. doi: 10.1038/s41551-016-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yadav S, et al. ChemElectroChem. 2017;4:967–971. [Google Scholar]
- 62.Eldh M, et al. Mol Immunol. 2012;50:278–286. doi: 10.1016/j.molimm.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 63.Li M, et al. Phil Trans R Soc B. 2014;369:20130502. [Google Scholar]
- 64.Zarovni N, et al. Methods. 2015;87:46–58. doi: 10.1016/j.ymeth.2015.05.028. [DOI] [PubMed] [Google Scholar]
- 65.Gholizadeh S, et al. Biosens Bioelectron. 2017;91:588–605. doi: 10.1016/j.bios.2016.12.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ko J, Carpenter E, Issadore D. Analyst. 2016;141:450–460. doi: 10.1039/c5an01610j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liga A, et al. Lab Chip. 2015;15:2388–2394. doi: 10.1039/c5lc00240k. [DOI] [PubMed] [Google Scholar]
- 68.Pariset E, Agache V, Millet A. Adv Biosys. 2017;1:1700040. doi: 10.1002/adbi.201700040. [DOI] [PubMed] [Google Scholar]
- 69.Yang F, et al. Biotechnol J. 2017;12:1600699. [Google Scholar]
- 70.Chen C, et al. Lab Chip. 2010;10:505–511. doi: 10.1039/b916199f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ashcroft BA, et al. Biomed Microdevices. 2012;14:641–649. doi: 10.1007/s10544-012-9642-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kanwar SS, et al. Lab Chip. 2014;14:1891–1900. doi: 10.1039/c4lc00136b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vaidyanathan R, et al. Anal Chem. 2014;86:11125–11132. doi: 10.1021/ac502082b. [DOI] [PubMed] [Google Scholar]
- 74.Zhang P, He M, Zeng Y. Lab Chip. 2016;16:3033–3042. doi: 10.1039/c6lc00279j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sina AAI, et al. Sci Rep. 2016;6:30460. doi: 10.1038/srep30460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.He M, Godwin A, Zeng Y. In: Microfluidic Methods for Molecular Biology. 1. Lu C, Verbridge SS, editors. ch. 6. Springer International Publishing; Cham, Switzerland: 2016. pp. 113–139. [Google Scholar]
- 77.Dudani JS, et al. Biomicrofluidics. 2015;9:014112. doi: 10.1063/1.4907807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fang S, et al. PLoS ONE. 2017;12:e0175050. doi: 10.1371/journal.pone.0175050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shao H, et al. Nat Commun. 2015;6:6999. doi: 10.1038/ncomms7999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhao Z, et al. Lab Chip. 2016;16:489–496. doi: 10.1039/c5lc01117e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ko J, et al. Sci Rep. 2016;6:31215. doi: 10.1038/srep31215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhou Q, et al. Methods. 2016;97:88–93. doi: 10.1016/j.ymeth.2015.10.012. [DOI] [PubMed] [Google Scholar]
- 83.Cho S, et al. Sens Actuator B-Chem. 2016;233:289–297. [Google Scholar]
- 84.Liang L-G, et al. Sci Rep. 2017;7:46224. doi: 10.1038/srep46224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Woo H-K, et al. ACS Nano. 2017;11:1360–1370. doi: 10.1021/acsnano.6b06131. [DOI] [PubMed] [Google Scholar]
- 86.Kang YT, et al. Proceedings of the 20th International Conference on Miniaturized Systems for Chemistry and Life Sciences; Dublin, Ireland. 2016. [Google Scholar]
- 87.Akagi T, et al. Proceedings of the 16th International Conference on Miniaturized Systems for Chemistry and Life Sciences; Okinawa, Japan. 2012. [Google Scholar]
- 88.Wang Z, et al. Lab Chip. 2013;13:2879–2882. doi: 10.1039/c3lc41343h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rahong S, et al. Sci Rep. 2014;4:5252. doi: 10.1038/srep05252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yasui T, et al. Proceedings of the 18th International Conference on Miniaturized Systems for Chemistry and Life Sciences; San Antonio, Texas, USA. 2014. [Google Scholar]
- 91.Yasui T, et al. Proceedings of the 20th International Conference on Miniaturized Systems for Chemistry and Life Sciences; Dublin, Ireland. 2016. [Google Scholar]
- 92.Konakade Y, et al. Proceedings of the 18th International Conference on Miniaturized Systems for Chemistry and Life Sciences; San Antonio, Texas. 2014. [Google Scholar]
- 93.Bruus H. Lab Chip. 2012;12:1014–1021. doi: 10.1039/c2lc21068a. [DOI] [PubMed] [Google Scholar]
- 94.Evander M, et al. Lab Chip. 2015;15:2588–2596. doi: 10.1039/c5lc00290g. [DOI] [PubMed] [Google Scholar]
- 95.Rezeli M, et al. Anal Chem. 2016;88:8577–8586. doi: 10.1021/acs.analchem.6b01694. [DOI] [PubMed] [Google Scholar]
- 96.Ding X, et al. Lab Chip. 2013;13:3626–3649. doi: 10.1039/c3lc50361e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Destgeer G, Sung HJ. Lab Chip. 2015;15:2722–2738. doi: 10.1039/c5lc00265f. [DOI] [PubMed] [Google Scholar]
- 98.Lee K, et al. ACS Nano. 2015;9:2321–2327. doi: 10.1021/nn506538f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sehgal P, Hartman J, Kirby BJ. Proceedings of the 20th International Conference on Miniaturized Systems for Chemistry and Life Sciences; Dublin, Ireland. 2016. [Google Scholar]
- 100.Huang LR, et al. Science. 2004;304:987–990. doi: 10.1126/science.1094567. [DOI] [PubMed] [Google Scholar]
- 101.McGrath J, Jimenez M, Bridle H. Lab Chip. 2014;14:4139–4158. doi: 10.1039/c4lc00939h. [DOI] [PubMed] [Google Scholar]
- 102.Santana SM, et al. Biomed Microdevices. 2014;16:869–877. doi: 10.1007/s10544-014-9891-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zeming KK, et al. Lab Chip. 2016;16:75–85. doi: 10.1039/c5lc01051a. [DOI] [PubMed] [Google Scholar]
- 104.Wunsch BH, et al. Nat Nanotech. 2016;11:936–940. doi: 10.1038/nnano.2016.134. [DOI] [PubMed] [Google Scholar]
- 105.Leshansky AM, et al. Phys Rev Lett. 2007;98:234501. doi: 10.1103/PhysRevLett.98.234501. [DOI] [PubMed] [Google Scholar]
- 106.Liu C, et al. ACS Nano. 2017;11:6968–6976. doi: 10.1021/acsnano.7b02277. [DOI] [PubMed] [Google Scholar]
- 107.Friedrich R, et al. Lab Chip. 2017;17:830–841. doi: 10.1039/c6lc01302c. [DOI] [PubMed] [Google Scholar]
- 108.Shao H, et al. Nat Med. 2012;18:1835–1840. doi: 10.1038/nm.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Akagi T, et al. PLoS ONE. 2015;10:e0123603. doi: 10.1371/journal.pone.0123603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Akagi T, Hanamura N, Ichiki T. J Photopolym Sci Technol. 2015;28:727–730. [Google Scholar]
- 111.Fraikin J-L, et al. Nat Nanotech. 2011;6:308–313. doi: 10.1038/nnano.2011.24. [DOI] [PubMed] [Google Scholar]
- 112.Taller D, et al. Lab Chip. 2015;15:1656–1666. doi: 10.1039/c5lc00036j. [DOI] [PubMed] [Google Scholar]
- 113.Richards KE, Go DB, Hill R. In: MicroRNA Detection and Target Identification: Methods and Protocols. Dalmay T, editor. ch. 5. Springer; New York, NY: 2017. pp. 59–70. [Google Scholar]
- 114.Cappello F, et al. Eur J Pharm Sci. 2017;96:93–98. doi: 10.1016/j.ejps.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 115.Melo SA, et al. Nature. 2015;523:177–182. doi: 10.1038/nature14581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Law JH, et al. Cancer Res. 2008;68:10238–10246. doi: 10.1158/0008-5472.CAN-08-2755. [DOI] [PubMed] [Google Scholar]
- 117.Pollak M. Nat Rev Cancer. 2012;12:159–169. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- 118.Farabaugh SM, Boone DN, Lee AV. Front Endocrinol. 2015;6:00059. doi: 10.3389/fendo.2015.00059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Balaj L, et al. Nat Commun. 2011;2:180. doi: 10.1038/ncomms1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cheng H-L, et al. Proceedings of the 20th International Conference on Miniaturized Systems for Chemistry and Life Sciences; Dublin, Ireland. 2016. [Google Scholar]
- 121.Kowal J, et al. Proc Natl Acad Sci USA. 2016;113:E968–E977. doi: 10.1073/pnas.1521230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Xu R, et al. Methods. 2015;87:11–25. doi: 10.1016/j.ymeth.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 123.Lötvall J, et al. J Extracell Vesicles. 2014;3:26913. doi: 10.3402/jev.v3.26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Keerthikumar S, et al. J Mol Biol. 2016;428:688–692. doi: 10.1016/j.jmb.2015.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Van Deun J, et al. Nat Methods. 2017;14:228–232. doi: 10.1038/nmeth.4185. [DOI] [PubMed] [Google Scholar]
- 126.Yuana Y, et al. J Extracell Vesicles. 2015;4:29260. doi: 10.3402/jev.v4.29260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lacroix R, et al. J Thromb Haemost. 2013;11:1190–1193. [Google Scholar]
- 128.Witwer KW, et al. J Extracell Vesicles. 2013;2:20360. doi: 10.3402/jev.v2i0.20360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Di Noto G, et al. Biosens Bioelectron. 2016;77:518–524. doi: 10.1016/j.bios.2015.09.061. [DOI] [PubMed] [Google Scholar]
- 130.Grasso L, et al. Anal Bioanal Chem. 2015;407:5425–5432. doi: 10.1007/s00216-015-8711-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rupert DLM, et al. Anal Chem. 2014;86:5929–5936. doi: 10.1021/ac500931f. [DOI] [PubMed] [Google Scholar]
- 132.Zhu L, et al. Anal Chem. 2014;86:8857–8864. doi: 10.1021/ac5023056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sitar S, et al. Anal Chem. 2015;87:9225–9233. doi: 10.1021/acs.analchem.5b01636. [DOI] [PubMed] [Google Scholar]