

Abstract
Background
Vilazodone is a novel antidepressant agent that combines selective serotonin (5-HT) reuptake inhibitor (SSRI) activity and 5-HT(1A) receptor partial agonist activity.
Objective
A pilot study was conducted to compare vilazodone (novel compound) and paroxetine (gold standard) on antidepressant effects, tolerability, and inflammation and immune modulation.
Methods
A 12-week, double-blind, randomized clinical trial was conducted with 56 nondemented older adults diagnosed with major depressive disorder (MDD). Between-group differences in mood, tolerability, and safety, as well as genomic markers of inflammation and immune modulation, were examined.
Results
Both treatment groups demonstrated similar improvement in depressed mood. Leukocyte gene expression profiles demonstrated reduction of specific proinflammatory gene transcripts and bioinformatic indications of reduced nuclear factor kappa B (NF-κB), activator protein (AP)-1, and cAMP response element binding (CREB) activity in the vilazodone group compared to the paroxetine group. Transcript origin analyses implicated monocytes and dendritic cells as the primary cellular origins of transcript reductions in the vilazodone-treated group.
Conclusions
Vilazodone's antidepressant effects may be associated with reduction of proinflammatory gene expression and immune modulation. Further research is required.
Keywords: geriatric depression, gene, antidepressant, immune, inflammation
Introduction
Late-life depression (LLD) is a significant health issue and has a rising prevalence given population aging [1]. In addition, existing antidepressant therapies have modest efficacy. A recent meta-analysis of trials found a response rate of 48 % and a remission rate of 33.7 % [2]. Fortunately, there are novel compounds available to investigate.
Vilazodone is a novel antidepressant agent that combines selective serotonin (5-HT) reuptake inhibitor (SSRI) activity and 5-HT(1A) receptor partial agonist activity [3]. It is indicated for the treatment of major depressive disorder (MDD) in adults in the United States of America (USA) [4]. It is administered orally, once daily, with food. At the recommended dosage of 40 mg/day, vilazodone was effective in the short-term treatment of MDD in adults. This was evidenced by significant improvements on multiple measures of depression (i. e., Montgomery-Åsberg Depression Rating Scale [MADRS] and Hamilton Depression Rating Scale [HDRS-17]), in 2 pivotal, 8-week, randomized, double-blind, placebo-controlled, phase III studies [3, 5]. Significant differences between vilazodone and placebo on the MADRS and HDRS-17 were seen after 1 week of treatment (first efficacy time-point) in 1 of the studies [3]. Long-term treatment with vilazodone 40 mg/day was associated with an improvement from baseline in depressive symptoms in a 52-week, noncomparative, phase III study [6]. Vilazodone was generally well tolerated in the short- and long-term treatment of MDD, with diarrhea and nausea being the most frequently occurring treatment-emergent adverse events [6]. Vilazodone had a minimal impact on sexual functioning in the 3 phase III studies [6]. There has not been a trial of vilazodone in older adults.
Paroxetine is an established SSRI antidepressant with documented efficacy and safety in older adults. It can be considered as an established active comparator for a new drug in the geriatric population [7]. Paroxetine is a potent and selective inhibitor of the neuronal reuptake of 5-HT, thereby facilitating serotoninergic transmission; this action appears to account for the antidepressant activity observed with this drug [8]. Results of short-term clinical trials have shown paroxetine to be significantly superior to placebo and comparable to amitriptyline, clomipramine, imipramine, dothiepin, and mianserin in relieving symptoms associated with MDD [8].
In the field of psychiatric immunology, much of the focus on the role of the immune system in depression pathophysiology and biomarker development has been placed on the innate immune response and inflammation [9]. These facets of the immune system are predominantly explored in antidepressant immunopharmacology [10]. The most recent evidence from reviews and meta-analyses suggests that antidepressants may exert effects on the immune system. A meta-analysis from 2011 of 22 studies [10] explored the effect of antidepressants on serum proinflammatory cytokines, tumor necrosis factor alpha (TNF-α), interleukin (IL)-1β, and IL-6 in 603 depressed subjects. A stratified subgroup analysis in this meta-analysis by class of antidepressants indicated that SSRI may reduce levels of IL-6 and TNF-α, whereas other types of antidepressants—while efficacious in antidepressant effects—did not appear to reduce cytokine levels or showed trend evidence in a few studies only. The involvement of immune factors in the pathophysiology of depression is now considered to be far greater than that of only the innate immune system, inflammation, and glia [11]. A complex interaction is suspected to occur in the central nervous system (CNS) between parts of the innate and adaptive immune system [12]. Systemic immune cells, namely macrophages and T-cells, have been found to have variously pro- and anti-neuroplastic, and pro- and anti-inflammatory effects on the brain [11, 13]. A recent systematic review [14] comprehensively reviewed the effects of antidepressant classes on both the innate and adaptive immune system. This review found that effects of antidepressant classes on adaptive immune factors are complex and poorly understood, with few studies conducted. Methodological heterogeneity was found to be high among these studies (e. g., length of study, cohort characteristics, dosage used, and immune marker analysis), and there were no studies exploring the immune effects of vilazodone. This review recommended larger comparative studies, particularly in clinical populations.
The current pilot study was conducted to determine whether there were any differences in mood and tolerability between vilazodone and paroxetine in older depressed adults. We anticipate that the vilazodone will be superior to paroxetine in improving levels of depressive symptoms given broader CNS mechanisms. Secondly, we examined the comparative pharmacogenomics effects of the compounds on inflammation and immune modulation in circulating leukocytes. We hypothesize that vilazodone will have greater anti-inflammatory effects given broader CNS mechanisms.
Methods
Participants
The UCLA Institutional Review Board approved all study procedures (registered ClinicalTrials.gov trial NCT01608295). All participants were outpatients and were recruited via advertisements from UCLA Neuropsychiatric Hospital (NPH) Geriatric Assessment Program, the NPH Geriatric Day Treatment Program, and affiliated clinics at the affiliated clinics at the VA and Olive View Medical Center. Patients were assessed via the UCLA Later Life Mood, Stress and Wellness Program.
208 individuals were assessed for eligibility. Of these, 71 were screened and 56 older adults were randomized to receive vilazodone (n = 26) and paroxetine (n = 30). Twenty of the vilazodone group and 25 of the paroxetine group completed the study (5 vilazodone and 5 paroxetine dropped out). One of the participants in the vilazodone group was lost to follow-up. See ► Fig. 1 for the CONSORT-format flow diagram of participants through the study.
Fig. 1.
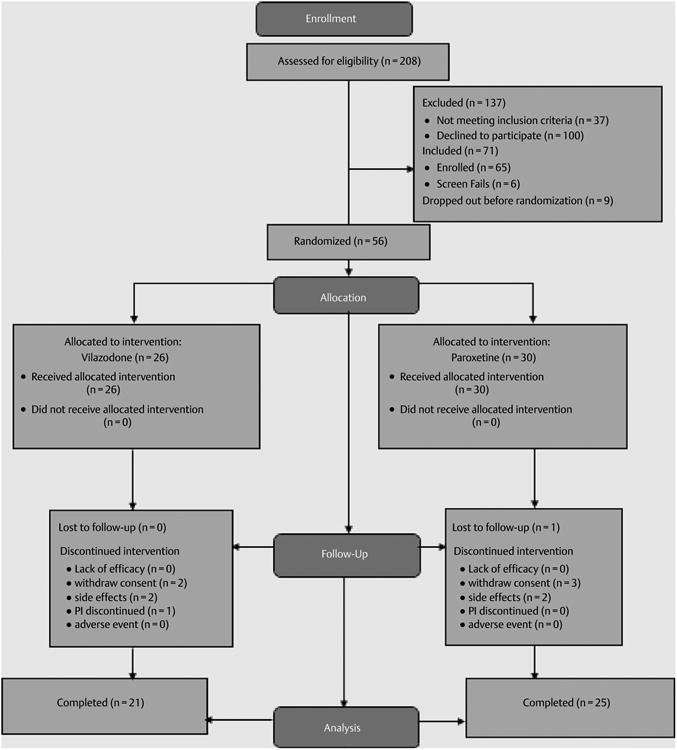
CONSORT flow diagram.
Inclusion and exclusion criteria
Structured Clinical Interview DSM-IV (SCID) was used by HL (primary investigator) and master-level research associates, formerly trained, to make a diagnosis of major depression and/or rule out other diagnosis (e. g., psychosis and dementia) according to the DSM-IV criteria at screening.
Inclusion criteria were the following: (1) the presence of an MDD diagnosed according to the DSM-5 criteria; (2) a 24-item HDRS score of 17 or higher at baseline [15]; and (3) a Mini-Mental State Exam (MMSE) score > 24 [16]. Exclusion criteria included any current and/or lifetime history of other psychiatric disorders (except unipolar depression with or without comorbid generalized anxiety disorder) or recent and/or current unstable medical or neurological disorders; any disabilities preventing participation in the study; diagnosis of mild cognitive impairment/dementia; and those with known allergic reactions to paroxetine or vilazodone.
Procedures
Eligible recruits provided written informed consent prior to enrolling in the study. Using a computer-generated randomization table, a treatment-blinded statistician randomized participants to either vilazodone or paroxetine. Follow-up took place weekly for the first 4 weeks of treatment and then every 2 weeks for the remainder of the study. Each follow-up assessment included measures of efficacy, safety, and compliance.
Outcome measures
The primary outcome measure was HDRS scores. The study also measured symptoms of mood using the Geriatric Depression Scale (GDS) [17], clinical improvement using the Clinical Global Impression – Severity and Improvement Scale (CGI) [18], medical and vascular risk factors using the Cerebrovascular Risk Factor Prediction Chart (CVRF) [19], and the Cumulative Illness Rating Scale for Geriatrics (CIRS-G) [20]. A blinded, master-level rater was used for outcome measure assessments; they were trained by HL.
Intervention procedures
Subjects were randomized to vilazodone or paroxetine. The allowed range of the drugs according to tolerability and efficacy assessments was between 10–40 mg/day for vilazodone and 10–30 mg/day for paroxetine. The dose finding procedures were performed in the first 3 weeks and after this remained stable until the end of the acute trial. Doses of 10–30 mg of paroxetine are standard in geriatric depression studies and in clinical experience. We assessed the optimal dose of vilazodone based on tolerability and safety in this population. Participants were randomized to receive 10–20–40 mg of vilazodone blindly with increment dose titration 10 mg per day for the week 1, 20 mg per day for the week 2, and 40 mg per day for the weeks 3–12. The comparison group received similar blinded titration with paroxetine 10 mg per day (week 1), 20 mg per day (week 2); and 30 mg per day (weeks 3–12). Given of the blinding involved, the dose titration decision was based on balancing tolerability and efficacy of the drugs. The dose ranges for paroxetine and vilazodone were within clinically effective range reported in the literature [21, 22].
Randomization
All eligible subjects were randomized to vilazodone or paroxetine group using a computer-generated random assignment scheme, which assigned subjects in a 1:1 ratio to each group. Randomization was done prior to the subjects being assigned to the groups.
Safety and adherence assessments
Physical examination and vital signs were assessed at baseline; in addition a 12-lead electrocardiography was collected if any cardiac complaints were present. Pulse rate, systolic blood pressure, and body weight were obtained at each visit. Laboratory tests (as listed below) were performed at baseline. The UKU Side Effect Rating Scale [23], a comprehensive rating scale for monitoring adverse events of psychotropic drugs used in clinical trials, was completed at all visits except screening. Treatment compliance was assessed by employing indirect measures of adherence including questioning of the patients, counting returned pills, and measuring drug levels at weeks 3, 8, and 16.
Gene expression profiling and analysis
Genome-wide transcriptional profiles were collected from peripheral blood leukocytes sampled at baseline and week 12 from 56 older adults with major depression who were randomly selected for testing and randomized to either vilazodone or paroxetine. Peripheral blood mononuclear cells were isolated by Ficoll density gradient centrifugation of antecubital venipuncture samples drawn between 10:00 and 11:00 a.m. at baseline and 16 weeks later following the completion of intervention procedures. RNA samples were extracted using the Qiagen RNeasy system, assessed for suitable integrity using an Agilent 2200 TapeStation, and quantitated by Ribogreen assay. Labeled cRNA was synthesized from 100 ng of total RNA using the Ambion Totalprep 96 kit. Amplified and labeled samples were hybridized to Illumina HT-12 v4 expression chips in the UCLA Neuroscience Genomics Core Laboratory according to the standard Illumina protocol. Post-hybridization washing and staining was done using a SciGene Little Dipper robotic processing platform, and arrays were scanned using an Illumina iScan confocal scanner.
Statistical analysis
Data was analyzed using the SAS (v9.2) statistical package. Patients in the 2 treatment groups were compared (using t-tests for continuous variables and chi-square tests for categorical variables) on all demographic and clinical measures at baseline to assess the success of the randomization procedures. Outcome measures (HDRS, GDS, CGI, CIRS-G, and CVRF scores) were analyzed using a mixed-effects general linear model, with group, time, and the group × time interaction as predictors. Post-hoc analyses determined the significance of between and within group differences. The significance threshold for the outcome measure was set at 0.05 (2-tailed). Effect sizes for changes in the outcome measures (Cohen's d) were also estimated for each group.
Gene expression data were quantile-normalized and log2-transformed for analysis by standard linear statistical models estimating the magnitude of differential change over time (i. e., group differences in average values of the change score: follow-up – baseline). “Gene set” bioinformatics analyses were then performed to assess immune system activation using 2 a-priori- defined sets: (1) 19 canonical proinflammatory gene transcripts (e. g., IL-1B, IL-6, IL-8, TNF), and (2) 31 canonical innate antiviral transcripts (e. g., OAS1-3, MX1-2, IFI/IFIT genes, etc.) [24]. Subsequent bioinformatics analyses identified all genes showing 1.2-fold or greater difference across groups in average change over time and used the Telis system [25] to scan the promoters of those genes for differential prevalence of transcription factor-binding motifs indicative of inflammation (e. g., activator protein [AP]-1 and nuclear factor kappa B [NF-κB]), innate antiviral signaling involving type I interferons (e. g., transcription factors such as IRF family factors), and neural/endocrine signaling pathways that might potentially mediate effects of the nervous system on immune cell inflammatory signaling (e. g., the cAMP response element binding [CREB] transcription factor family, which mediates sympathetic nervous system signaling through β-adrenergic receptors, and the glucocorticoid receptor involved in hypothalamopituitary adrenal axis signal transduction). Transcript origin analysis was also applied to determine whether the up- or down-regulated genes might share any specific immune cell subpopulation as their predominant cellular source within the circulating leukocyte pool [26]. Additional linear model analyses tested whether changes in depressive symptoms were associated with changes in expression of the a priori proinflammatory and antiviral gene sets. In all bioinformatics analyses, p-values were derived from standard errors estimated by 200 cycles of bootstrap resampling of residual vectors from linear model analyses (which accounts for potential correlation among residuals across genes) [27].
Results
► Table 1 presents the baseline demographic and clinical characteristics of the vilazodone and paroxetine groups. As noted, there were no statistically significant differences for measures between the groups at baseline. ► Table 2 presents the outcomes measures at both baseline and follow-up. Of note, the antidepressant effects of vilazodone and paroxetine were similar and did not reach statistical significance (for ESs for HDRS scores for vilazodone (baseline (mean (SD)) 17.2 (3.7) and follow-up 7.6 (4.8); ES = 2.25), and paroxetine (baseline (mean (SD)) 16.6 (4.1) and follow-up 7.8 (5.9); ES = 1.31). The time × group interaction was not statistically significant (F(8,43) = 1.3; p = 0.3). The GDS score also showed similar improvements for vilazodone group (baseline (mean (SD)) 17.0 (5.7) and follow-up 10.6 (7.3); ES = 1.18) and the paroxetine group (baseline (mean (SD)) 16.1 (6.8) and follow-up 11.1 (7.1); ES = 0.62). The time × group interaction was not statistically significant (F(8,43) = 1.1; p = 0.4). The ESs were similar with all other measures. There were no statistically significant differences in side effects between the groups.
Table 1.
Baseline characteristics by treatment group.
| Vilazodone group (n = 26) | Paroxetine group (n = 30) | Analysis | |
|---|---|---|---|
| Variables | Mean (SD) | Mean (SD) | t-test |
| Age (years) | 71.5 (7.2) | 71.5 (7.7) | T(54) = 0.0, p = 1.0 |
| Education | 16.3 (3.0) | 16.2 (2.8) | T(53) = 0.1, p = 0.9 |
| MMSE | 28.9 (1.2) | 28.2 (1.4) | T(53) = 1.8, p = 0.07 |
| BMI | 27.0 (5.0) | 25.0 (4.2) | T(50) = 1.6, p = 0.1 |
| Age of Onset | 45.5 (22.4) | 42.3 (25.9) | T(54) = 0.5, p = 0.6 |
| HDRS | 17.2 (3.7) | 16.6 (4.1) | T(54) = 0.5, p = 0.6 |
| N (%) | N (%) | chi-square (p) | |
| Sex | |||
| Male | 13 (50) | 13 (43) | χ (1) = 0.3, p = 0.3 |
| Female | 13 (50) | 17 (57) | |
BMI: body mass index; MMSE: Mini-Mental State Examination; HDRS: Hamilton Depression Rating Scale; SD: standard deviation
Table 2.
Outcome measures at baseline and follow-up.
| Vilazodone group (n = 21) | Paroxetine group (n = 25) | Analysis | |||||
|---|---|---|---|---|---|---|---|
| Clinical measures * | Baseline | Follow-up | Es | Baseline | Follow-up | Es | Time * group interaction |
| HDRS | 17.2 (3.7) | 7.6 (4.8) | 2.25 | 16.6 (4.1) | 7.8 (5.9) | 1.31 | F(8,43) = 1.3, p = 0.3 |
| GDS | 17.0 (5.7) | 10.6 (7.3) | 1.18 | 16.1 (6.8) | 11.1 (7.1) | 0.62 | F(8,43) = 1.1, p = 0.4 |
| CGI severity | 3.3 (0.8) | 2.5 (1.2) | 0.78 | 3.4 (0.6) | 2.7 (1.1) | 0.79 | F(8,43) = 1.1, p = 0.4 |
| CIRS total | 5.1 (3.0) | 5.0 (3.5) | 0.03 | 4.1 (3.2) | 3.9 (3.5) | 0.06 | F(1,43) = 1.0, p = 0.3 |
| CVRF | 12.3 (5.9) | 11.2 (4.4) | 0.21 | 11.3 (5.3) | 10.3 (4.6) | 0.20 | F(1,43) = 0.0, p = 0.9 |
Mean (SD) are presented for HDRS: Hamilton Depression Rating Scale; GDS: Geriatric Depression Scale; CGI: Clinical Global Improvement; CIRS: Cumulative Illness Rating Scale; and CVRF: Cardiovascular Risk Factor. ES: effect size
In analyses of vilazodone and paroxetine effects on leukocyte gene expression, initial analyses focused on a-priori-specified sets of indicator genes for general proinflammatory signaling and activation of the type I interferon system; results showed no notable effects on the interferon system (difference: mean = − 0.015 ± SE 0.046 log2 RNA expression units, p = 0.7467), but a markedly greater decrease over time in expression of proinflammatory indicator genes for vilazodone-treated patients compared to paroxetine-treated patients (difference: mean = − 0.314 ± SE 0.133 log2 RNA expression units, p = 0.0294). These findings using a-priori- defined gene sets were highly consistent with characteristics of the genes showing the most marked relative reduction in the vilazodone-treated group; these included many cardinal proinflammatory genes including those encoding IL-8, IL-1β, TNF, PTGS2 (COX2), FOS/JUN (AP-1), and EGR1, as well as immunologic activation indicators CD83, CD69, and HLA-DR (► Table 3 contains gene-specific results for all transcripts showing ≥ 1.2-fold differential change across groups). There was no apparent biological theme in the smaller list of genes that were relatively increased the vilazodone-treated group.
Table 3.
Genes showing > 1.2-fold differential change over time in vilazodone vs. paroxetine.
| Change in RNA expression (T2-T1 log2 RNA) | |||||
|---|---|---|---|---|---|
| Paroxetine | Vilazodone | Fold- difference | |||
| Up-regulated > 1.2-fold | |||||
| LCN2 | − 0.158 | 0.406 | 1.48 | ||
| CAMP | 0.004 | 0.536 | 1.45 | ||
| LOC653600 | 0.046 | 0.535 | 1.40 | ||
| PGLYRP1 | − 0.045 | 0.372 | 1.33 | ||
| CEACAM8 | − 0.099 | 0.300 | 1.32 | ||
| LTF | − 0.004 | 0.371 | 1.30 | ||
| DEFA4 | 0.055 | 0.408 | 1.28 | ||
| SH3BGRL2 | − 0.160 | 0.193 | 1.28 | ||
| MGC13057 | − 0.128 | 0.216 | 1.27 | ||
| GP9 | − 0.084 | 0.247 | 1.26 | ||
| RNASE3 | 0.003 | 0.333 | 1.26 | ||
| TMEM158 | − 0.080 | 0.249 | 1.26 | ||
| CD24 | − 0.082 | 0.243 | 1.25 | ||
| LOC645128 | − 0.061 | 0.260 | 1.25 | ||
| CLEC1B | − 0.155 | 0.166 | 1.25 | ||
| CEACAM6 | − 0.047 | 0.273 | 1.25 | ||
| PF4V1 | − 0.018 | 0.303 | 1.25 | ||
| HSPA1B | − 0.036 | 0.277 | 1.24 | ||
| MPL | − 0.060 | 0.249 | 1.24 | ||
| SPARC | − 0.125 | 0.183 | 1.24 | ||
| TREML1 | 0.009 | 0.308 | 1.23 | ||
| SDPR | − 0.192 | 0.103 | 1.23 | ||
| OLFM4 | − 0.064 | 0.226 | 1.22 | ||
| ALOX12 | − 0.117 | 0.172 | 1.22 | ||
| TSC22D1 | − 0.060 | 0.227 | 1.22 | ||
| NGFRAP1 | − 0.027 | 0.258 | 1.22 | ||
| ITGA2B | − 0.125 | 0.157 | 1.22 | ||
| GPR18 | − 0.059 | 0.221 | 1.21 | ||
| ID3 | − 0.063 | 0.217 | 1.21 | ||
| NRGN | − 0.152 | 0.128 | 1.21 | ||
| MMD | − 0.108 | 0.166 | 1.21 | ||
| TUBB1 | − 0.208 | 0.064 | 1.21 | ||
| CA2 | − 0.158 | 0.111 | 1.21 | ||
| HIST1H2BJ | 0.080 | 0.347 | 1.20 | ||
| ACRBP | − 0.056 | 0.210 | 1.20 | ||
| Down-regulated > 1.2-fold | |||||
| IL8 | 0.533 | − 0.584 | 0.46 | ||
| EGR1 | 0.681 | − 0.380 | 0.48 | ||
| CCL3 | 0.500 | − 0.393 | 0.54 | ||
| FOSB | 0.472 | − 0.383 | 0.55 | ||
| IL1B | 0.613 | − 0.236 | 0.56 | ||
| CCL3L1 | 0.472 | − 0.326 | 0.57 | ||
| EGR2 | 0.407 | − 0.355 | 0.59 | ||
| CCL3L3 | 0.254 | − 0.456 | 0.61 | ||
| JUN | 0.369 | − 0.340 | 0.61 | ||
| OSM | 0.381 | − 0.295 | 0.63 | ||
| TNF | 0.408 | − 0.266 | 0.63 | ||
| PTGS2 | 0.302 | − 0.333 | 0.64 | ||
| RGS1 | 0.345 | − 0.251 | 0.66 | ||
| JUNB | 0.277 | − 0.279 | 0.68 | ||
| DUSP2 | 0.319 | − 0.200 | 0.70 | ||
| CXCL2 | 0.281 | − 0.223 | 0.71 | ||
| HBEGF | 0.346 | − 0.150 | 0.71 | ||
| LOC728835 | 0.322 | − 0.157 | 0.72 | ||
| CD83 | 0.283 | − 0.196 | 0.72 | ||
| LOC338758 | 0.208 | − 0.270 | 0.72 | ||
| IER2 | 0.313 | − 0.164 | 0.72 | ||
| CCL4L1 | 0.259 | − 0.210 | 0.72 | ||
| IER3 | 0.268 | − 0.195 | 0.73 | ||
| TNFAIP3 | 0.217 | − 0.244 | 0.73 | ||
| CCL20 | 0.369 | − 0.078 | 0.73 | ||
| LOC728830 | 0.355 | − 0.091 | 0.73 | ||
| NFKBIZ | 0.161 | − 0.278 | 0.74 | ||
| NFKBIA | 0.195 | − 0.227 | 0.75 | ||
| HES4 | 0.130 | − 0.277 | 0.75 | ||
| PPP1R15A | 0.316 | − 0.085 | 0.76 | ||
| G0S2 | 0.293 | − 0.103 | 0.76 | ||
| DDIT4 | 0.186 | − 0.184 | 0.77 | ||
| AXUD1 | 0.189 | − 0.181 | 0.77 | ||
| GADD45B | 0.187 | − 0.178 | 0.78 | ||
| CDKN1C | 0.141 | − 0.218 | 0.78 | ||
| CD69 | 0.111 | − 0.248 | 0.78 | ||
| TRIB1 | 0.182 | − 0.167 | 0.78 | ||
| BTG2 | 0.295 | − 0.048 | 0.79 | ||
| DUSP1 | 0.162 | − 0.174 | 0.79 | ||
| ADM | 0.225 | − 0.109 | 0.79 | ||
| HLA-DRB5 | 0.312 | − 0.010 | 0.80 | ||
| HLA-DRB1 | 0.220 | − 0.089 | 0.81 | ||
| ZFP36 | 0.218 | − 0.078 | 0.81 | ||
| FOS | 0.170 | − 0.121 | 0.82 | ||
| C5AR1 | 0.242 | − 0.049 | 0.82 | ||
| NFIL3 | 0.131 | − 0.156 | 0.82 | ||
| RPPH1 | 0.219 | − 0.067 | 0.82 | ||
| LOC643930 | 0.197 | − 0.081 | 0.82 | ||
| SNORA70 | 0.089 | − 0.185 | 0.83 | ||
| CCL4L2 | 0.279 | 0.006 | 0.83 | ||
| PNRC1 | 0.179 | − 0.094 | 0.83 | ||
| DEFA1 | 0.432 | 0.163 | 0.83 | ||
| PID1 | 0.212 | − 0.057 | 0.83 | ||
| NR4A2 | 0.140 | − 0.126 | 0.83 | ||
Results of subsequent promoter-based bioinformatic analyses indicated reduced activity of NF-κB, AP-1, and CREB transcription factors in the vilazodone-treated group relative to the paroxetine-treated group (all p < 0.05). There were no indications of differential glucocorticoid receptor activity (p > 0.48). See ► Fig. 2 for further details.
Fig. 2.

Changes in gene activity in vilazodone group relative to paroxetine group. AP-1: activator protein-1; NF-kB: nuclear factor kappa B; GR: glucocorticoid receptor; CREB: cAMP response element binding protein
Transcript origin analyses implicated monocytes and dendritic cells as the primary cellular origins of genes relatively down-regulated in the vilazodone-treated group (both p < 0.01; see ► Fig. 3). There was also a trend toward contribution from B lymphocytes, but that indication would not reach statistical significance after correction for multiple testing across the 6 cell types analyzed (nominal p = 0.016).
Fig. 3.

Transcript origin analysis of down-regulated genes in vilazodone group. DC: dendritic cell; NK: natural killer cell; B: B cell
Generally similar results emerged for all of these analyses when controlling for patient level characteristics (age, sex, white vs. non-white race, body mass index, alcohol consumption) or when controlling for RNA transcripts that serve as markers of major leukocyte subsets (i. e., CD3, CD19, CD14, etc.).
Follow-up analyses tested whether the magnitude of change in HDRS scores was correlated with the magnitude of change in expression of the a-priori-specified proinflammatory gene composite. Results showed a significant positive association (regression coefficient: + 0.141 ± 0.064 change in log2 RNA expression per SD of HDRS score change, p = 0.0424). No such association emerged for the type I interferon-related gene composite (regression coefficient: + 0.006 ± 0.022 change in log2 RNA expression per SD of HDRS score change, p = 0.8051).
Discussion
This pilot study is the first to explore the clinical effects of vilazodone in geriatric depression, the first to explore the immune effects of vilazodone, and the first to conduct a head-to-head comparison of 2 antidepressants on immune gene expression. The results of this clinical trial suggest vilazodone has similar effects on depressive symptoms in LLD, statistically similar to paroxetine. These antidepressant effects for the vilazodone group may be contributed to by down-regulation of proinflammatory genes (including reduced activity of NF-κB, AP-1, and CREB) and may arise predominately from monocytes and dendritic cells.
The anti-inflammatory effects of vilazodone are not entirely surprising given a systematic review of this area shows antidepressants do appear, in general, reduce proinflammatory cytokine levels, particularly TNF-α, IL-1β, and IL-6 [14]. However, the enhanced anti-depressant effects of vilazodone over paroxetine have not previously been explored or demonstrated and hence are of interest. We suggest this is given vilazodone combines the SSRI activity with partial agonism of 5-HT(1A) receptor, vs. paroxetine which has only SSRI activity. There is only 1 other study that explores the head-to-head anti-inflammatory effects of antidepressants. In adult depressed populations, pretreatment levels of CRP informed later efficacy of escitalopram and nortriptyline—high CRP levels at baseline were associated with greater antidepressant effect of nortriptyline than escitalopram over time [28]. This potentially suggests nortriptyline is more anti-inflammatory than escitalopram. However, preclinical studies may inform this area. For example, Tynan et al. [29] compared the anti-inflammatory effects of SSRIs and serotonin-norepinephrine reuptake inhibitors (SNRIs) on lipopolysaccharide (LPS)-stimulated microglia, in vivo. This study found SSRIs potently inhibited microglial TNF-α and nitric oxide (NO) production much greater than SNRIs. They found cAMP signaling was involved in regulating these anti-inflammatory responses. Interestingly, the anti-inflammatory effects of these drugs differed depending on their concentrations. It would be instructive to compare the anti-inflammatory effects of vilazodone and paroxetine in such preclinical models. It should be mentioned, however, that IL-6 levels were not substantially different between the 2 treatment groups (i. e., < 1.2-fold differential reduction). A larger cohort, however, may have yielded a larger difference.
The anti-inflammatory effects of vilazodone appear to originate from monocytes and dendritic cells. In a chronic stress situation, an innate immune response takes place, mainly consisting of resident microglia and peripherally derived monocytes, macrophages [30], and, possibly, dendritic cells [31]. Therefore, it is not surprising that these data suggest that monocytes and dendritic cells are potential players in depression and hence antidepressant treatment response. There is some evidence that a skew in this balance towards the Th1 phenotype, the associated “proinflammatory” cytokines and interface with M1-type classically activated macrophages detrimental and leads to clinical depression [32, 33]. Hence, anti-inflammatory effects of antidepressants via monocytes is 1 plausible mechanistic pathway by which antidepressants take their antidepressant effects. Evidence from the abovementioned study by Tynan et al. [29] demonstrates that in LPS-stimulated microglial/monocyte study environments, SSRIs potentially inhibit microglial/monocyte TNF-α and NO production, and cAMP signaling may be involved in regulating these anti-inflammatory responses. As for dendritic cells, the role of these cells in the pathophysiology of depression and pharmacology are poorly understood. Dendritic cells are the most potent professional antigen-presenting cells that play a central role in the initiation and modulation of innate and adaptive immune response [34], but their role in the depression context is still largely unexplored. Dendritic cells may play an important role in brain diseases as they can reach CNS from periphery in both human diseases [35] and animal models [36]. We are not aware of any studies exploring the effects of antidepressants on dendritic cells.
A key limitation of this study is the small sample size, which precludes our ability to draw clinical conclusions on this trial regarding the efficacy of vilazodone in a geriatric population. A larger replication should be performed, comparing vilazodone to placebo or “gold standard” treatment, such as paroxetine. It is important to explore the effects of vilazodone in an LLD population given the differing pharmacokinetic and pharmacodynamic profile of medicaments as compared to adult populations. There is some evidence to suggest that adult and LLD often differ in terms of presentation, neuropsychological features, neurobiological factors, and treatment response [37–41]; however, this remains to be accepted by expert peer-group consensus or meta-analysis.
Conclusion
The vilazodone group had similar improvement in depressive symptoms, relative to the paroxetine group, and showed notable reductions in leukocyte proinflammatory gene expression. These results may be consistent with the hypothesis that inflammatory signaling may contribute to the pathophysiology of depression. This pilot trial should inform future larger trials of geriatric depression, and further research should explore the individual biomarkers.
Acknowledgments
SC would like to acknowledge support National Institutes of Health Grant P30 AG017265. HL was supported by a Forest Research Institute grant VII-IT-02 and MH086481, MH077650, and MH097892. We would like to thank the Cousins Center for Psychoneuroimmunology at UCLA for assisting with sample collection. HE, PS, JK, NSC, HY, SC, and HL conceived and/or designed the work that led to the submission, acquired data, and/or played an important role in interpreting the results. HE, MF, HL, SC, and PS drafted or revised the manuscript. All authors approved the final version.
Footnotes
Trial Registration: NCT01608295. Vilazodone for treatment of geriatric depression.
Conflict of Interest: The authors declare no conflict of interest.
References
- 1.Ferrari AJ, Charlson FJ, Norman RE, et al. Burden of depressive disorders by country, sex, age, and year: Findings from the global burden of disease study 2010. PLoS Med. 2013;10:e1001547. doi: 10.1371/journal.pmed.1001547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kok RM, Nolen WA, Heeren TJ. Efficacy of treatment in older depressed patients: A systematic review and meta-analysis of double-blind randomized controlled trials with antidepressants. J Affect Disord. 2012;141:103–115. doi: 10.1016/j.jad.2012.02.036. [DOI] [PubMed] [Google Scholar]
- 3.Rickels K, Athanasiou M, Robinson DS, et al. Evidence for efficacy and tolerability of vilazodone in the treatment of major depressive disorder: A randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70:326–333. doi: 10.4088/jcp.08m04637. [DOI] [PubMed] [Google Scholar]
- 4.FDA. VIlazodone hydrochloride approval for major depressive disorder. FDA; 2011. [Google Scholar]
- 5.Khan A, Cutler AJ, Kajdasz DK, et al. A randomized, double-blind, placebo-controlled, 8-week study of vilazodone, a serotonergic agent for the treatment of major depressive disorder. J Clin Psychiatry. 2011;72:441–447. doi: 10.4088/JCP.10m06596. [DOI] [PubMed] [Google Scholar]
- 6.Robinson DS, Kajdasz DK, Gallipoli S, et al. A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31:643–646. doi: 10.1097/JCP.0b013e31822c6741. [DOI] [PubMed] [Google Scholar]
- 7.Lundmark J, Scheel Thomsen I, Fjord-Larsen T, et al. Paroxetine: pharmacokinetic and antidepressant effect in the elderly. Acta Psychiatr Scand Suppl. 1989;350:76–80. doi: 10.1111/j.1600-0447.1989.tb07177.x. [DOI] [PubMed] [Google Scholar]
- 8.Dechant KL, Clissold SP. Paroxetine. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in depressive illness. Drugs. 1991;41:225–253. doi: 10.2165/00003495-199141020-00007. [DOI] [PubMed] [Google Scholar]
- 9.Muller N, Hofschuster E, Ackenheil M, et al. Investigations of the cellular immunity during depression and the free interval: Evidence for an immune activation in affective psychosis. Prog Neuropsychopharmacol Biol Psychiatry. 1993;17:713–730. doi: 10.1016/0278-5846(93)90055-w. [DOI] [PubMed] [Google Scholar]
- 10.Hannestad J, Dellagioia N, Bloch M. The effect of antidepressant medication treatment on serum levels of inflammatory cytokines: A meta-analysis. Neuropsychopharmacology. 2011;36:2452–2459. doi: 10.1038/npp.2011.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eyre HA, Stuart MJ, Baune BT. A phase-specific neuroimmune model of clinical depression. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:265–274. doi: 10.1016/j.pnpbp.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 12.Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122:1164–1171. doi: 10.1172/JCI58644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eyre H, Baune BT. Neuroplastic changes in depression: A role for the immune system. Psychoneuroendocrinology. 2012;37:1397–1416. doi: 10.1016/j.psyneuen.2012.03.019. [DOI] [PubMed] [Google Scholar]
- 14.Eyre HA, Lavretsky H, Kartika J, et al. Modulatory effects of antidepressant classes on the innate and adaptive immune system in depression. Pharmacopsychiatry. 2016;49:85–96. doi: 10.1055/s-0042-103159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 17.Yesavage JA. Depression in the elderly. How to recognize masked symptoms and choose appropriate therapy. Postgrad Med. 1992;91:255–258. 261. doi: 10.1080/00325481.1992.11701181. [DOI] [PubMed] [Google Scholar]
- 18.Guy W. Clinical Global Impressions (CGI) In: Services UDoHaH, editor. ECDEU Assessment Manual for Psychopharmacology. Rockville: Alcohol drug abuse and mental health administration, NIMPH Psychopharmacology Research Branch; 1976. pp. 218–222. [Google Scholar]
- 19.American Heart Association. Stroke risk factor prediction chart. Dallas: American Heart Association; 1990. [Google Scholar]
- 20.American Heart Association. Stroke risk factor prediction chart. Dallas, TX: 1990. DOI: ■■■. [Google Scholar]
- 21.Schone W, Ludwig M. A double-blind study of paroxetine compared with fluoxetine in geriatric patients with major depression. J Clin Psychopharmacol. 1993;13:34S–39S. doi: 10.1097/00004714-199312002-00006. [DOI] [PubMed] [Google Scholar]
- 22.Schatzberg AF, Kremer C, Rodrigues HE, et al. Double-blind, randomized comparison of mirtazapine and paroxetine in elderly depressed patients. Am J Geriatr Psychiatry. 2002;10:541–550. [PubMed] [Google Scholar]
- 23.Lingjaerde O, Ahlfors UG, Bech P, et al. The UKU side effect rating scale A new comprehensive rating scale for psychotropic drugs and a cross-sectional study of side effects in neuroleptic-treated patients. Acta Psychiatr Scand Suppl. 1987;334:1–100. doi: 10.1111/j.1600-0447.1987.tb10566.x. [DOI] [PubMed] [Google Scholar]
- 24.Fredrickson BL, Grewen KM, Coffey KA, et al. A functional genomic perspective on human well-being. Proc Natl Acad Sci USA. 2013;110:13684–13689. doi: 10.1073/pnas.1305419110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cole SW, Yan W, Galic Z, et al. Expression-based monitoring of transcription factor activity: The TELiS database. Bioinformatics. 2005;21:803–810. doi: 10.1093/bioinformatics/bti038. [DOI] [PubMed] [Google Scholar]
- 26.Cole SW, Hawkley LC, Arevalo JM, et al. Transcript origin analysis identifies antigen-presenting cells as primary targets of socially regulated gene expression in leukocytes. Proc Natl Acad Sci USA. 2011;108:3080–3085. doi: 10.1073/pnas.1014218108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Efron B, Tibshirani RJAn. Introduction to the Bootstrap. New York: Chapman Hall; 1993. [Google Scholar]
- 28.Uher R, Tansey KE, Dew T, et al. An inflammatory biomarker as a differential predictor of outcome of depression treatment with escitalopram and nortriptyline. Am J Psychiatry. 2014;171:1278–1286. doi: 10.1176/appi.ajp.2014.14010094. [DOI] [PubMed] [Google Scholar]
- 29.Tynan RJ, Weidenhofer J, Hinwood M, et al. A comparative examination of the anti-inflammatory effects of SSRI and SNRI antidepressants on LPS stimulated microglia. Brain Behav Immun. 2012;26:469–479. doi: 10.1016/j.bbi.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 30.Prinz M, Priller J. Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci. 2014;15:300–312. doi: 10.1038/nrn3722. [DOI] [PubMed] [Google Scholar]
- 31.Butovsky O, Kunis G, Koronyo-Hamaoui M, et al. Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer's disease model. Eur J Neurosci. 2007;26:413–416. doi: 10.1111/j.1460-9568.2007.05652.x. [DOI] [PubMed] [Google Scholar]
- 32.Rook GA, Lowry CA. The hygiene hypothesis and psychiatric disorders. Trends Immunol. 2008;29:150–158. doi: 10.1016/j.it.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 33.Leonard B, Maes M. Mechanistic explanations how cell-mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci Biobehav Rev. 2012;36:764–785. doi: 10.1016/j.neubiorev.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 34.Banchereau J, Briere F, Caux C, et al. Immunobiology of dendritic cells. Annu Rev Immunol. 2000;18:767–811. doi: 10.1146/annurev.immunol.18.1.767. [DOI] [PubMed] [Google Scholar]
- 35.Pashenkov M, Teleshova N, Link H. Inflammation in the central nervous system: the role for dendritic cells. Brain Pathol. 2003;13:23–33. doi: 10.1111/j.1750-3639.2003.tb00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D'Agostino PM, Gottfried-Blackmore A, Anandasabapathy N, et al. Brain dendritic cells: Biology and pathology. Acta Neuropathol. 2012;124:599–614. doi: 10.1007/s00401-012-1018-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alexopoulos GS. Depression in the elderly. Lancet. 2005;365:1961–1970. doi: 10.1016/S0140-6736(05)66665-2. [DOI] [PubMed] [Google Scholar]
- 38.Disabato BM, Morris C, Hranilovich J, et al. Comparison of brain structural variables, neuropsychological factors, and treatment outcome in early-onset versus late-onset late-life depression. Am J Geriatr Psychiatry. 2014;22:1039–1046. doi: 10.1016/j.jagp.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sachs-Ericsson N, Corsentino E, Moxley J, et al. A longitudinal study of differences in late- and early-onset geriatric depression: Depressive symptoms and psychosocial, cognitive, and neurological functioning. Aging Ment Health. 2013;17:1–11. doi: 10.1080/13607863.2012.717253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Janssen J, Hulshoff Pol HE, de Leeuw FE, et al. Hippocampal volume and subcortical white matter lesions in late life depression: Comparison of early and late onset depression. J Neurol Neurosurg Psychiatry. 2007;78:638–640. doi: 10.1136/jnnp.2006.098087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rapp MA, Dahlman K, Sano M, et al. Neuropsychological differences between late-onset and recurrent geriatric major depression. Am J Psychiatry. 2005;162:691–698. doi: 10.1176/appi.ajp.162.4.691. [DOI] [PubMed] [Google Scholar]