

I. Immune dysregulation in autism spectrum disorder
Autism spectrum disorder (ASD) constitutes a group of neurodevelopmental disorders with overlapping but variable behavioral and cognitive symptoms, severity, and comorbid conditions. The core diagnostic features of ASD are impaired social interaction, communication challenges, and expression of repetitive or perseverative behaviors (Kanner, 1943; Moldin et al., 2006). Although ASD has a strong genetic basis, its etiology is complex, with several genetic factors likely to be involved as well as interactions with environmental influences (Betancur, 2011; DiCicco-Bloom et al., 2006; Goines and Ashwood, 2013; Risch et al., 1999). ASD therefore represents a collection of conditions with heterogeneous causes, but they may converge on common molecular pathways to bring about the features that characterize this disorder. Immune dysregulation has gained significant attention as a pathway for the pathogenesis of ASD (Depino, 2013; Korvatska et al., 2002).
Immune system abnormalities have been observed widely in the brain and periphery of ASD individuals. Studies have found chronic neuroinflammation in ASD, indicated by increased activation of microglia and astrocytes and production of cytokines and chemokines in the brain (Li et al., 2009; Morgan et al., 2010; Vargas et al., 2005). Transcriptome analyses of ASD brains also have indicated an upregulation of immune response genes (Garbett et al., 2008; Voineagu et al., 2011; Ziats and Rennert, 2011). Similarly, in the periphery, studies have found elevated expression of pro-inflammatory cytokines and other immune factors in the blood and gastrointestinal tract of ASD individuals (Ashwood et al., 2004; Ashwood et al., 2011; Enstrom et al., 2010). Additionally, ASD has been associated with inflammatory disorders and autoimmunity in not only the affected individuals but also their mothers (Atladottir et al., 2009; Buie et al., 2010; Jonakait, 2007). Moreover, prenatal exposure to maternal immune activation (MIA) has been implicated as an environmental risk factor for ASD.
Several epidemiological studies have found that maternal infection during pregnancy correlated with an increased frequency of ASD in the children (Atladottir et al., 2010; Brown et al., 2014; Chess, 1977; Wilkerson et al., 2002). The infection activates a maternal immune reaction in which the molecular signaling cascades are thought to adversely affect neurodevelopment and subsequent behavior manifested as ASD in the offspring. Supporting this idea, animal models have shown that MIA results in offspring with behavioral, neurological, and immunological abnormalities like those observed in ASD (Abrahams and Geschwind, 2010; Depino, 2013). For example, offspring of pregnant mice that had been exposed to viral infection or injected with synthetic double-stranded RNA poly(I:C), mimicking viral infection, exhibited ASD-like behaviors. Notably, they demonstrate abnormal ultrasonic vocalization mirroring communication challenges in ASD, reduced social approach mirroring impaired social interaction in ASD, and increased marble burying mirroring repetitive behaviors in ASD (Choi et al., 2016; Malkova et al., 2012; Schwartzer et al., 2013; Shi et al., 2003; Smith et al., 2007). The offspring have been reported also to exhibit ASD-like alterations in cortical and cerebellar growth and function (Choi et al., 2016; De Miranda et al., 2010; Meyer et al., 2008; Shi et al., 2009) as well as in immune responses (Arrode-Bruses and Bruses, 2012; Garay et al., 2013; Hsiao et al., 2012; Meyer et al., 2006; Meyer et al., 2008). How can MIA exposure lead to ASD in susceptible individuals?
II. Induction of inflammation
A. Modeling MIA in mice
To model MIA as a risk factor for ASD, inflammation has been induced in pregnant mice using several approaches, including direct infection or exposure to lipopolysaccharide (LPS) to mimic bacterial infection or poly(I:C) to mimic viral infection (Choi et al., 2016; Oskvig et al., 2012; Shi et al., 2003; Shi et al., 2009; Smith et al., 2007). The resulting phenotypes in the offspring depend on the trigger and the degree and temporal window that prenatal inflammation is induced. One frequently used protocol is a high single-dose intraperitoneal injection of poly(I:C) in pregnant mice on embryonic day (E)12.5 (Choi et al., 2016; Schwartzer et al., 2013; Shi et al., 2009; Smith et al., 2007). This gestational stage resembles the late first trimester in humans (Clancy et al., 2007), during which viral infections have been correlated with increased incidence of ASD in the offspring (Atladottir et al., 2010). Poly(I:C) evokes a pro-inflammatory antiviral response in the mother that is similar to the immune response occurring after activation of Toll-like receptor 3 (TLR3) by viral infection (Alexopoulou et al., 2001; Datta et al., 2003; Traynor et al., 2006). Mimicking MIA during viral infection in this way results in offspring that demonstrate many ASD-like phenotypes, as described above.
B. IL-6 in inflammation and ASD
The pro-inflammatory cytokine interleukin-6 (IL-6) has been identified as a causal factor in ASD-like phenotypes associated with MIA (Samuelsson et al., 2006; Smith et al., 2007). Increased expression of IL-6 is often noted in ASD (Ashwood et al., 2011; Li et al., 2009; Vargas et al., 2005), and animal models suggest that this increase is pathogenic in MIA-associated ASD. In the poly(I:C) induction model of MIA, ASD-like phenotypes in the offspring can be prevented if pregnant mice are deficient in IL-6, implicating the necessity of IL-6 signaling (Smith et al., 2007). Importantly, injecting pregnant mice with IL-6 alone was sufficient to recapitulate many of the ASD-related behavioral and immunological features in offspring exposed to MIA (Garbett et al., 2012; Smith et al., 2007). Therefore, MIA induces maternal IL-6, which can cross the placenta (Dahlgren et al., 2006; Hsiao and Patterson, 2011) and has been found upregulated in the fetal brain (Meyer et al., 2006; Wu et al., 2015). Besides its function in inflammation, IL-6 has a known role in normal neurodevelopment, including regulation of neural cell proliferation, differentiation, migration, axonal pathfinding, and synaptic connections (Deverman and Patterson, 2009; Islam et al., 2009). The presence of higher IL-6 levels due to MIA can therefore directly alter fetal brain development that results in ASD. However, IL-6 is a pleiotropic signal capable of triggering the expression of other cytokines and immune regulatory genes that may additionally impact the developing brain or are responsible for precipitating ASD. Investigation into the molecular pathways through which IL-6 may act to promote ASD in the offspring following MIA has revealed an important contribution from interleukin-17A (IL-17A) signaling (see Figure 1).
Figure 1.
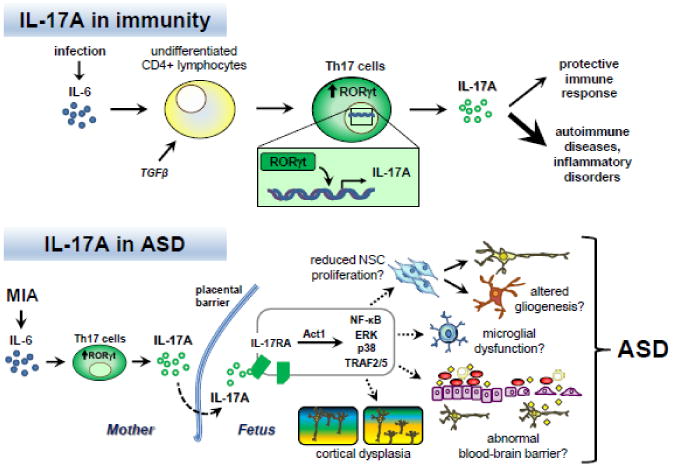
IL-17A and immunity: Infection causes increased expression and secretion of the proinflammatory cytokine interleukin-6 (IL-6). In the presence of the cytokine transforming growth factor β (TGFβ), IL-6 can stimulate the differentiation of naïve CD4+ lymphocytes into T helper 17 (Th17) cells. A key downstream effector of IL-6 involved in Th17 differentiation is the transcription factor retinoic acid receptor-related orphan receptor γt (RORγt). RORγt promotes transcription of the pro-inflammatory cytokine IL-17A. IL-17A signaling plays a protective role in adaptive immunity. Dysregulation of Th17 cells and IL-17A production are associated with autoimmune diseases and inflammatory disorders.
IL-17A and ASD: In the case of maternal immune activation (MIA), IL-6 is induced and stimulates the differentiation of Th17 cells in the mother, leading to increased IL-17A secretion. IL-17A in turn crosses the placental barrier or is induced in the fetus, where it can act on cells expressing the receptor IL-17RA in the developing nervous system. Stimulation of IL-17RA can activate several intracellular signaling pathways mediated by Act1, such as NF-κB, ERK, p38 MAPK, and TRAF2/5. Consequently, various developmental processes can be altered, including neural stem cell (NSC) proliferation, gliogenesis, microglial function, blood-brain barrier activity, and neuronal connectivity. In these ways, inappropriate Th17 cell and IL-17A activity may adversely impact prenatal development, resulting in cortical dysplasia and the manifestation of behaviors associated with ASD in the offspring (Choi et al. 2016).
In the peripheral immune system, major downstream effectors of IL-6 are the transcription factors signal transducer and activator of transcription 3 (STAT3) and retinoic acid receptor-related orphan nuclear receptor γt (RORγt). RORγt is expressed exclusively in lymphoid cells that secrete the pro-inflammatory cytokine IL-17A (Ivanov et al., 2006; Zhou et al., 2007). Most prominent are the Th17 cells, a subset of CD4+ T helper (Th) lymphocytes that produce large quantities of IL-17A to mediate inflammatory processes (Ivanov et al., 2006). IL-6 drives RORγt activity in naïve CD4+ T cells, which is required for their differentiation into the Th17 subset (Zhou et al., 2007; Zhou et al., 2008). Following infections or stimulation by poly(I:C), IL-6 is released early in the acute phase of the immune response to downregulate immunosuppressive regulatory T (Treg) cells and promote Th17 cells (Bettelli et al., 2006; Jonakait, 2007). In the presence of the immunoregulatory cytokine transforming growth factor β (TGFβ), naïve CD4+ T cells co-express RORγt and the transcription factor forkhead box P3 (FOXP3), which inhibits RORγt activity (Zhou et al., 2008). FOXP3 is required for generating Treg cells, but IL-6 suppresses FOXP3 expression and therefore limits Treg differentiation while relieving inhibition of RORγt (Bettelli et al., 2006; Zhou et al., 2008). Additionally, IL-6 activates STAT3 in naïve CD4+ T cells to induce further expression of RORγt, which in turn cooperates with STAT3 to induce IL-17A expression (Chen et al., 2009; Manel et al., 2008; Voo et al., 2009). RORγt and IL-17A are gene expression signatures of the Th17 lineage. Thus, the combination of IL-6 with TGFβ directs T cell development into the Th17 subset. To maintain and amplify the pool of Th17 cells, IL-6 also induces STAT3- and RORγt-dependent expression of IL-1 receptors by these cells, making them responsive to IL-1 (Chung et al., 2009; Ikeda et al., 2014). IL-1 reinforces RORγt expression and downregulates FOXP3 expression, thereby enhancing Th17 differentiation (Chung et al., 2009; Ikeda et al., 2014). STAT3 also induces Th17 production of IL-21, which has an autocrine positive feedback effect on STAT3 activity (Korn et al., 2007; Wei et al., 2007; Zhou et al., 2007). Concurrently, RORγt induces Th17 expression of IL-23 receptors for stabilizing the commitment of these cells to the Th17 lineage (Stritesky et al., 2008). IL-23, enhanced by IL-1, then stimulates the terminal differentiation of Th17 cells through maintenance of lineage signature gene expression and activation of effector genes such as IL-22 in mature Th17 cells (Chung et al., 2009; Stritesky et al., 2008).
C. Th17 cells
Th17 cells have pro-inflammatory actions that are normally beneficial to host defense (Cai et al., 2016; Chung et al., 2003; Conti et al., 2009; Huang et al., 2004; Huppler et al., 2014; Korn et al., 2009; Ye et al., 2001). Through the secretion of effector cytokines, Th17 cells potently induce tissue inflammation as a protective response against infection. IL-17A, also known as IL-17, is the signature cytokine produced by Th17 cells. In turn, IL-17A promotes the production of cytokines such as granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF), IL-6, IL-1β, and tumor necrosis factor α (TNFα), and chemokines such as CCL20 and CXCL1 (or GROα) from different cell types to recruit neutrophils and mediate inflammation (Fossiez et al., 1996; Huang et al., 2007; Jones and Chan, 2002; Kawaguchi et al., 2001; Laan et al., 1999; Laan et al., 2001; Moseley et al., 2003). In addition to IL-17A, Th17 cells produce IL-17F, another member of the IL-17 cytokine family with the highest homology to IL-17A (Hymowitz et al., 2001; Liang et al., 2007). IL-17A and IL-17F can be secreted as homodimers and heterodimers which have overlapping but distinct proinflammatory functions and differing potency, with more strength and effects attributed to IL-17A (Ishigame et al., 2009; Liang et al., 2007; Wright et al., 2007; Yang et al., 2008). As described earlier, other Th17 effectors are IL-21, which has a self-amplifying effect, and IL-22, which has paracrine effects such as remodeling of endothelial and epithelial barriers, involved in inflammation (Aggarwal et al., 2001; Caruso et al., 2007; Dumoutier et al., 2000; Monteleone et al., 2006). Interestingly, Th17 cells can themselves secrete IL-6 (Langrish et al., 2005), which can result in a positive feedback loop, as well as GM-CSF (Codarri et al., 2011) and TNFα (Langrish et al., 2005), which can synergize with IL-17A to enhance the inflammatory response. Paradoxically, Th17 cells have been reported also to co-express interferon γ (IFNγ), a proinflammatory cytokine that has classically been the signature effector of Th1 lymphocytes and inhibits Th17 differentiation (Acosta-Rodriguez et al., 2007; Chen et al., 2007; Wilson et al., 2007). These Th17 cells with simultaneous expression of IL-17A and IFNγ have been associated with greater resistance to Treg regulation and greater pathogenic potential (Annunziato et al., 2007).
Inappropriate Th17 activity has been linked to the pathogenesis of several autoimmune and inflammatory disorders, including rheumatoid arthritis, asthma, and multiple sclerosis (MS) (Korn et al., 2009). Uncontrolled Th17 activation can lead to excessive IL-17A levels and a hyperinflammatory state in the periphery. In the brain, IL-17A has been shown to disrupt the blood-brain barrier, facilitating infiltration of peripheral factors and even Th17 cells that injure the central nervous system in MS (Kebir et al., 2007). IL-17A is produced by other immune cells as well, such as neutrophils in the periphery (Ferretti et al., 2003) and microglia in the brain (Kawanokuchi et al., 2008). Because these cells can also be stimulated by IL-17A, a propagative cycle could result from an interaction with Th17-released IL-17A. Under normal brain conditions, IL-17A can induce neuroinflammation as a protective response by activating glia to produce inflammatory cytokines and chemokines (Hu et al., 2014; Kawanokuchi et al., 2008). However, at high levels, IL-17A has been associated with brain pathology not only in MS and experimental autoimmune encephalomyelitis (EAE), a mouse model for MS, but also in epilepsy and ischemia (Das Sarma et al., 2009; He et al., 2013; Kawanokuchi et al., 2008; Kebir et al., 2007; Komiyama et al., 2006; Wang et al., 2009). Dysregulation of Th17 cells and subsequent IL-17A signaling has also been implicated in ASD pathogenesis (Figure 1).
III. The role of IL-17A signaling in ASD
A. Th17 cells and IL-17A in ASD
Accumulating evidence support a role for Th17 cells and their effector cytokine IL-17A in ASD. The gene for IL-17A was identified in a genome-wide analysis to have enriched copy number variants associated with ASD (van der Zwaag et al., 2009). In subsets of ASD individuals, IL-17A has been found at elevated levels in the blood and correlated with the severity of behavioral symptoms (Akintunde et al., 2015; Al-Ayadhi and Mostafa, 2012; Suzuki et al., 2011). In the MIA mouse model of ASD, poly(I:C) treatment has been found to increase IL-17A levels in maternal blood and the postnatal brain (Choi et al., 2016; Garay et al., 2013). Poly(I:C) treatment also increases mRNA levels of IL-17A in the placenta (Choi et al., 2016). Correspondingly, lymphocytes isolated from the placenta and offspring of poly(I:C)-treated mothers differentiate significantly more into Th17 cells or secrete more IL-17A upon stimulation (Choi et al., 2016; Hsiao et al., 2012; Mandal et al., 2010; Mandal et al., 2011). To determine whether this concordant increase in IL-17A expression may be symptomatic or pathogenic in ASD, a recent study inhibited IL-17A signaling using antibody blockade of the cytokine in poly(I:C)-treated pregnant mice and found that ASD-like phenotypes in the offspring were prevented (Choi et al., 2016). Specifically, mouse behaviors modeling the core diagnostic features of ASD and cortical malformation mirroring abnormal cortical growth in ASD individuals (Casanova et al., 2013; Stoner et al., 2014) were normalized in the MIA-exposed offspring. The source of aberrant IL-17A levels was ascribed to maternal Th17 cells because genetic removal of the transcription factor RORγt selectively from CD4+ T cells in pregnant mice, thereby suppressing Th17 differentiation, also was able to prevent these ASD-like phenotypes in MIA-exposed offspring (Choi et al., 2016). Thus, IL-17A dysregulation may play a causal role in the development of ASD (Figure 1).
B. IL-17A pathway in MIA-associated ASD
IL-17A sources during pregnancy
Immune challenges in the mother are relayed to the fetus via the placenta. The finding that mRNA levels of IL-17A are elevated in the placenta in response to poly(I:C)-evoked MIA suggests that immune cells are activated within the placenta to produce IL-17A. This is supported by the other finding that lymphocytes in poly(I:C)-treated pregnant mice secrete more IL-17A but was a response specific to cells isolated from the placenta (Choi et al., 2016). During normal pregnancy, the placenta promotes maternal immune tolerance to prevent rejection of the fetus while still retaining the capacity to detect and defend against infection. Treg cells in the placenta play an important role maintaining the immunosuppressive uterine environment to permit fetal development (Zenclussen et al., 2006). However, under normal conditions, Th17 cells are also present in the decidua, the maternally derived component of the placenta (Nakashima et al., 2010; Wu et al., 2014), which indicates a delicate coordination of regulatory and pro-inflammatory T cell activity in the placenta. MIA shifts this balance by altering the cytokine milieu, enhancing IL-6 levels in particular, to favor activation of maternal Th17 cells. Indeed, poly(I:C)-evoked MIA increases Th17 cells in the placenta but not Treg cells (Choi et al., 2016). As a result, Th17 cells in the mother may compromise placental function, with detrimental effects on the developing fetus.
Maternal Th17 cells release effectors like IL-17A which may impact fetal development indirectly by regulating placental function and production of intermediary soluble factors that enter fetal circulation or directly by transport into the fetus. Maternal Th17 cells have been reported also to cross to the fetus themselves (Wienecke et al., 2012). In the case of MIA during pregnancy, IL-17A may be acting directly on the fetal brain at E14.5 to alter neurodevelopment resulting in ASD-like phenotypes. Direct injection of IL-17A cytokine into the fetal brain at this stage in the absence of MIA was shown to phenocopy poly(I:C)-induced MIA effects of ASD-like behaviors and cortical disorganization in the offspring (Choi et al., 2016). IL-17A mRNA was not detected in E14.5 fetal brain, indicating that IL-17A cytokine is not produced locally at this stage (Choi et al., 2016). Maternal sources likely provide the input IL-17A signal to the fetal brain during MIA, but it remains to be demonstrated directly that IL-17A or even Th17 cells from the mother traverse the placenta under these conditions.
Alternatively, maternal IL-17A induces a trans-placental signaling cascade that triggers IL-17A production in the fetus peripherally, which then acts on the developing brain. At this time in the periphery of the fetus, progenitor blood cells are proliferating and maturing in the liver, which is the main site of hematopoiesis or the formation of blood cells such as lymphoid cells during fetal development (Crawford et al., 2010). T cell progenitors are the primary lymphoid cells in the mouse fetal liver around E12.5 but begins decreasing by E13.5 as their main site of hematopoiesis shifts to the thymus (Crawford et al., 2010). Thus, the fetus may be capable of producing Th17 cells and providing a source of IL-17A in the periphery this way. On the other hand, it remains possible that IL-17A is locally produced by resident cells in the fetal brain but at low levels which require more sensitive methods of detection. In contrast with the cytokine, mRNA of the IL-17A receptor IL-17RA is detectable in E14.5 fetal brain and, moreover, is upregulated by poly(I:C)-evoked MIA (Choi et al., 2016). In this way, low levels of IL-17A may be able to exert detrimental effects on neurodevelopment. It is important to note that directly injecting IL-17A into the fetal brain at E14.5 resulted in a thinning of the cortical plate not observed following MIA induced at E12.5 (Choi et al., 2016), which underscores distinctions in the timing and severity of immune challenge, stage of the inflammatory response, and contributions of effectors besides IL-17A to MIA. However, the current data taken together suggest that MIA leading to ASD critically involves induction of maternal IL-17A signaling that is relayed to the fetus via the placenta somehow and disrupts normal brain development.
IL-6 and links to IL-17A signaling
These findings extend earlier studies that support a pathogenic role for IL-6 in MIA-associated ASD by delineating a mechanistic pathway through activation of maternal Th17 cells and subsequent IL-17A signaling. Consistent with IL-17A acting downstream of IL-6, induction of IL-6 is early and acute following poly(I:C) treatment of pregnant mice while IL-17A has a slower onset (Arrode-Bruses and Bruses, 2012; Choi et al., 2016). In particular, strong upregulation of IL-17A cytokine levels in maternal blood and mRNA levels in the placenta was observed 48 hours post-poly(I:C) treatment, on E14.5, whereas IL-6 activity had returned to basal levels at this time point (Choi et al., 2016). Supporting this causal sequence of cytokine activity, IL-17A blocking antibody in pregnant mice prevents ASD-like phenotypes in the offspring induced by maternal IL-6 injection (Choi et al., 2016), which was shown previously to be sufficient to reproduce poly(I:C)-induced MIA effects (Smith et al., 2007). Conversely, pregnant mice lacking IL-6 cannot up-regulate IL-17A (Choi et al., 2016) and have been shown to bear phenotypically normal offspring following poly(I:C)-evoked MIA (Smith et al., 2007), but direct IL-17A injection into the fetal brain causes ASD-like behavior to reappear in these offspring (Choi et al., 2016). Moreover, direct injection of IL-6 cytokine into E14.5 fetal brain in the absence of MIA failed to cause ASD-like phenotypes in the offspring (Choi et al., 2016). Combined, these results indicate that IL-17A but not IL-6 acts directly on the fetal brain, at least during the E14.5 stage, to disrupt neurodevelopment and precipitate ASD-like phenotypes in the offspring following MIA. However, because IL-6 protein and mRNA levels in the placenta and the fetal brain are known to increase rapidly after MIA induction (Hsiao and Patterson, 2011; Meyer et al., 2006; Wu et al., 2015), these findings suggest critical windows for cytokine action or cytokine-specific functions in different phases of neurodevelopment.
IV. IL-17A targets in the brain
A. IL-17A receptors
The IL-17 receptor (IL-17R) family consists of five related subunits (A-E) (Gaffen, 2009). IL-17A cytokine targets the receptor IL-17RA to trigger downstream signaling. Binding of IL-17A induces IL-17RA to recruit the IL-17RC subunit to form the complete receptor complex (Ely et al., 2009; Toy et al., 2006; Yao et al., 1995). Assembly of the subunits is required for IL-17A-dependent functions (Gu et al., 2013; Toy et al., 2006; Yao et al., 1995). In the periphery, IL-17RA is expressed in several different cell types, with the greatest expression in the spleen, kidney and hematopoietic or immune cells (Gu et al., 2013; Yao et al., 1995). In the brain, several cell types have been reported to express IL-17RA but at low or undetectable levels basally (Das Sarma et al., 2009; He et al., 2013; Wang et al., 2009). Expression is strongly increased during inflammation associated with brain injury, disease and dysfunction. Resident cells with enhanced mRNA and protein levels of IL-17RA have been observed in ischemia and the oxygen-glucose deprivation model of ischemia (Wang et al., 2009), in MS and its EAE model (Das Sarma et al., 2009; Kebir et al., 2007), and in focal cortical dysplasia (He et al., 2013). Increased IL-17RA expression in the inflamed brain suggests a heightened responsiveness to IL-17A signaling.
Similarly, poly(I:C)-evoked MIA leads to an increase in mRNA levels of IL-17RA in the fetal brain at E14.5 (Choi et al., 2016). Whether this upregulation is mediated by a positive feedback loop through IL-17A binding in the fetal brain or by another effector mechanism is not known but is prevented by maternal treatment with IL-17A blocking antibody (Choi et al., 2016), which supports the necessity of IL-17A signaling in MIA effects. Consistent with these results, ASD-like phenotypes were prevented in MIA-exposed offspring if IL-17RA expression was reduced in the mother through genetic deletion of the receptor, thereby blocking IL-17A signal transduction (Choi et al., 2016). Interestingly, pregnant mice with reduced IL-17RA expression did not exhibit the increase of blood IL-17A levels following poly(I:C) treatment (Choi et al., 2016), which provides further support not only for a critical role of maternal IL-17A signaling but also for a positive feedback mechanism regulating expression of IL-17A and its receptor in MIA. On the other hand, this finding obscures IL-17A acting on receptors in the mother or the fetus to precipitate MIA effects in the offspring. Because injecting IL-17A directly into the fetal brain was able to reproduce MIA effects, this identifies a contribution from fetal IL-17RA signaling (Choi et al., 2016). Moreover, direct injection of IL-17A into the brain of fetuses lacking IL-17RA was unable to cause ASD-like phenotypes in the offspring (Choi et al., 2016). Therefore, MIA induction of IL-17A signals through the receptor IL-17RA to mediate downstream consequences on the developing fetus (see Figure 1).
B. IL-17RA expression during cortical development
Within the human brain, weak expression of IL-17RA has been reported in neurons, glia, and endothelial cells from control cortical tissue (He et al., 2013; Kebir et al., 2007). From the mouse brain, mRNA and protein expression of IL-17RA has been observed under basal conditions in microglia and astrocytes (Das Sarma et al., 2009; Kawanokuchi et al., 2008), oligodendrocytes (Paintlia et al., 2011), cerebellar granule cells (Hu et al., 2014) but not cortical and hippocampal neurons (Kawanokuchi et al., 2008; Wang et al., 2009), and neural stem cells (Li et al., 2013). Expression then increases dramatically under states of inflammation, including MIA (Choi et al., 2016). In some instances, infiltrating immune cells from the bloodstream partially account for the upregulation of IL-17RA signal in the brain (Das Sarma et al., 2009). In MIA however, the increased mRNA levels of IL-17RA within the fetal brain, specifically the cortex, have not yet been localized to a particular cell type, but there are several possibilities.
At the time of poly(I:C)-evoked MIA on E12.5, cortical neurogenesis has already begun in the mouse fetal brain. Around E10, the neuroepithelial cells lining the cerebral ventricles are generating a few early neurons as well as transforming into radial glia, which function as the primary progenitors or neural stem cells (NSCs) that give rise to glutamatergic neurons and later to astrocytes and oligodendrocytes within the cortex (Noctor et al., 2002; Qian et al., 2000). Radial glia also function as a scaffold for newborn neurons in the ventricular (VZ) and subventricular zones (SVZ) to migrate out toward the pial surface, forming cortical layers II-VI in an inside-out manner whereby each wave of newborn neurons will migrate past existing layers to form more superficial layers (Nadarajah et al., 2003). In parallel to this process, γ-aminobutyric acid (GABA)ergic interneurons are born outside of the cortex from ganglionic eminences in the fetal brain and migrate tangentially into the cortex to be incorporated there (Marin and Rubenstein, 2001). An early stream of interneurons that migrates into the cortex at the pial surface around E11.5 eventually become layer I Cajal-Retzius neurons (Marin and Rubenstein, 2001). By E12.5, cortical neurogenesis and neuronal migration to form the cortical layers are underway and lasts until E17-18, when synaptogenesis begins (Greig et al., 2013; Li et al., 2010; Semple et al., 2013). At E16, neurogenesis begins switching to gliogenesis, in which radial glia generate astrocytes and then oligodendrocytes instead of neurons (Qian et al., 2000; Semple et al., 2013; Stevens, 2008). Therefore, during the period of prenatal exposure to MIA initiated at E12.5 and continued through E14.5 by IL-17A signaling, astrocytes and oligodendrocytes are not yet present whereas NSCs and neurons are proliferating, differentiating, and migrating in the fetal brain.
Microglia also are present in the brain during this developmental period. Microglia are brain-resident macrophages known to have diverse functions, including active surveillance of their microenvironment, secretion of immune and trophic factors for homeostasis as well as repair, clearing dead or dying cells and debris, and shaping synaptic connections (Ferrer et al., 1990; Hanisch, 2002; Luo and Chen, 2012; Paolicelli et al., 2011; Sierra et al., 2010). In the embryonic brain, microglia have been implicated in regulating neurodevelopment. Microglia are derived from yolk sac progenitors of hematopoietic origin that colonize the embryonic mouse brain beginning around E10 (Ginhoux et al., 2010), which coincides with the emergence of radial glia. This timing puts microglia in a position to influence the other resident brain cells that have yet to develop. Indeed, embryonic microglia have been demonstrated to regulate NSC numbers by supporting not only proliferation (Antony et al., 2011) but also normal programmed cell death during cortical development (Cunningham et al., 2013). Additionally, they have been demonstrated to regulate NSC differentiation into astrocytes (Antony et al., 2011; Nakanishi et al., 2007), outgrowth of dopaminergic axons (Squarzoni et al., 2014), laminar positioning of cortical interneurons (Squarzoni et al., 2014), and angiogenesis (Fantin et al., 2010) in the embryonic brain. Microglia persist within the brain throughout life, developing as a unique population that self-renews and is distinct from peripheral macrophages (Bruttger et al., 2015). During embryonic development, microglia proliferate and migrate throughout the brain and become more ramified as they mature (Squarzoni et al., 2014; Swinnen et al., 2013). In the cortex, they accumulate at the pial surface, distribute sparsely near the ventricular walls, and are largely absent from the cortical plate that eventually becomes the cortical layers, prior to E16.5 in mice (Squarzoni et al., 2014; Swinnen et al., 2013).
Also during this developmental period, vasculature is present in the brain. Vascularization of the mouse cortex starts around E11 when endothelial cells invade the neural tissue to form blood vessels (Daneman et al., 2010). As these vessels grow, the blood-brain barrier (BBB) starts forming to create a physical barrier between peripheral blood circulation and the central nervous system so that the brain maintains and develops in a precise internal environment (Bauer et al., 1993; Ek et al., 2012). BBB properties include intercellular tight junctions, low rates of transcytosis, and expression of transporters in endothelial cells that selectively control exchange across the vascular wall (Ek et al., 2012). Tight junctions between endothelial cells can be observed shortly after they grow into the embryonic brain and already restrict the passage of several small molecules (Bauer et al., 1993; Daneman et al., 2010; Ek et al., 2012). In the adult brain, astrocytic end feet are an important component of the BBB, encircling the endothelial cells that form the blood vessels, but astrocytes are not yet generated when angiogenesis initially occurs in the embryonic brain (Daneman et al., 2010). In contrast, pericytes, another important BBB component that surrounds the endothelial cells, are present and have been found to regulate vascular permeability at this stage (Daneman et al., 2010). One way is by downregulating endothelial cell expression of proteins that promote BBB permeability and immune cell infiltration (Daneman et al., 2010). The BBB becomes fully functional where molecules do not leak across the vasculature by E15.5 in the mouse cortex (Ben-Zvi et al., 2014).
Therefore, during the period from E12.5 when MIA is evoked by poly(I:C) to E14.5 when IL-17A is significantly elevated following MIA, neurons, NSCs, microglia, and endothelial cells are present in the fetal brain. All these cell types have been reported to express IL-17RA, although it remains to be demonstrated in the fetal brain. Then any or all of these cells may upregulate IL-17RA expression in response to MIA, contributing to the MIA-induced increase of IL-17RA mRNA levels in the fetal brain (Choi et al., 2016). Furthermore, MIA induction of IL-17A signaling could potentially act on any or all of these cell types to disrupt cortical development, leading to cortical dysplasia and behavioral abnormalities associated with ASD in the offspring (Figure 1).
V. Potential mechanisms of IL-17RA activation on cortical development
A. Cell type-specific effects
How might MIA induction of excessive IL-17A signaling in the fetal brain lead to ASD-like cortical malformation and behaviors in the offspring? One study reported that IL-17RA expression in cultured embryonic neurons increases following ischemic stress after initially being undetectable at basal states, and stimulation with IL-17A exacerbates neuronal death (Wang et al., 2009). In contrast with neurons, IL-17A stimulation of cultured NSCs from embryonic mice does not increase cell death but does reduce NSC numbers along with astrocyte and oligodendrocyte numbers (Li et al., 2013). IL-17A acts instead to inhibit NSC proliferation and their subsequent differentiation into later stages of neural cells (Li et al., 2013). MIA-induced IL-17A signaling may have similar effects on neurons and NSCs in the fetal brain. Indeed, poly(I:C) has been shown to reduce embryonic NSC division and neuronal numbers in the superficial layers of the cortex (De Miranda et al., 2010). In this way, if MIA induces IL-17RA activation in cortical neurons and NSCs of the developing brain, network organization and function may be affected, resulting in the cortical dysplasia and behavioral abnormalities associated with ASD.
If microglia express IL-17RA at embryonic stages, then MIA-induced IL-17A signaling may also activate microglia in the fetus and alter their normal functions, which could contribute to the defects observed in the offspring. Besides carrying out phagocytosis, activated microglia can release a variety of soluble factors including cytokines like IL-6 and TNFα that affect neurodevelopment depending on the stimulus and context (Cacci et al., 2008; Cunningham et al., 2013). For example, addition of microglia-conditioned media to cultured neurons was found to increase neuronal proliferation (Morgan et al., 2004). Similarly, embryonic NSCs cultured from microglia-deficient mice showed impaired proliferation and astrogliogenesis (Antony et al., 2011). Conversely, LPS-activated microglia were found to secrete pro-inflammatory factors that induced NSC apoptosis, reduced neuronal differentiation and enhanced glial differentiation in culture (Cacci et al., 2008). In vivo, microglia activated under conditions of LPS-evoked MIA were found to decrease the NSC pool and thickness of the VZ and SVZ in the embryonic cortex (Cunningham et al., 2013) and induce premature entry of interneurons into the cortical plate, resulting in deficient distribution around layer V (Squarzoni et al., 2014). Embryonic microglia also have been shown to regulate vascularization in the developing brain through close contact with endothelial cells and secretion of soluble factors that stimulate angiogenesis (Fantin et al., 2010). Thus, activated microglia in the embryonic brain may promote excessive growth of some cell types, which may come at the expense of other types, disturb normal cell migration, and alter brain connectivity and vasculature.
One study reported that poly(I:C)-evoked MIA does not affect the density and activation state of microglia during embryonic development, suggesting that embryonic microglia might not be involved in the poly(I:C)-induced MIA effects on the fetal brain (Smolders et al., 2015). However, this result may reflect induction-dependent differences in inflammation, as poly(I:C) was administered intravenously at E11.5 or E15.5 (Smolders et al., 2015). Because the nature, intensity, and timing of MIA affects the resulting phenotypes observed in the offspring, it is possible that embryonic microglia are activated under other conditions of poly(I:C)-evoked MIA. Microglia also may be found to participate in poly(I:C)-evoked MIA effects on the fetal brain using other measures of microglial activity, such as the production of specific factors. Microglial cultures stimulated with IL-17A have been shown to increase production of cytokines such as IL-6, chemokines such as CXCL2, and neurotrophic factors such as brain-derived neurotrophic factor (Das Sarma et al., 2009; Kawanokuchi et al., 2008). MIA-induced IL-17A may similarly stimulate microglia in the fetal brain, which in turn affect neurodevelopmental processes that lead to the ASD-like phenotypes in offspring. Interestingly, microglia not only are a target of IL-17A, but also have been shown to produce IL-17A (Kawanokuchi et al., 2008), which can result in a positive feedback loop. At E14.5 however, IL-17A mRNA was not detected in the embryonic brain, which suggests that resident microglia are not producing IL-17A at this stage (Choi et al., 2016). It remains to be determined if and how IL-17A stimulation of microglia directly through IL-17RA activation or indirectly alters their functions during fetal brain development.
Activation of IL-17RA on endothelial cells in the fetal brain may also play a role in mediating ASD-like phenotypes associated with MIA. The BBB physically prevents the translocation of lymphocytes, including Th17 cells, into the brain where they can influence inflammation and exert harmful effects. For example, increased neuronal death was observed when neurons were co-cultured with Th17 cells, which secreted the cytolytic molecules perforin and granzyme B (Kebir et al., 2007). From E12.5 to E14.5, the BBB in the mouse cortex is not yet fully sealed but is capable of barrier functions that exclude many peripheral molecules and cells from the developing brain. However, IL-17A stimulation of endothelial IL-17RA have been shown to promote BBB permeability by causing the disruption of tight junctions and release of signals that recruit macrophages to the area (Huppert et al., 2010; Kebir et al., 2007). If endothelial cells in the fetal brain respond similarly to IL-17A signaling induced by poly(I:C)-evoked MIA, then this enables the infiltration of peripheral factors that can not only disturb neurodevelopment but also injure the brain, thereby precipitating defects in the offspring. Additionally, activated microglia can increase BBB permeability (Prat et al., 2001) and may thus intensify these effects in the fetal brain. Future studies investigating IL-17RA expression in the fetal brain can help identify the cell type-specific targets and effects of IL-17A following MIA.
B. IL-17RA signal transduction pathways
Act1 functions downstream of the IL-17A receptor
A critical effector of intracellular signaling downstream from IL-17RA is Act1 (also known as CIKS) (Leonardi et al., 2000; Li et al., 2000). In fact, loss of Act1 leads to many similar phenotypes as loss of IL-17RA (Boisson et al., 2013; Corneth et al., 2014). For example, Act1-deficient mice display attenuated EAE (Kang et al., 2010) as mice lacking IL-17RA do (Gonzalez-Garcia et al., 2009). Act1 mediates IL-17RA signal transduction through several identified protein domains, including a helix-loop-helix (HLH) domain, a pair of TNF receptor associated-factor (TRAF) domains, a U-Box E3 ligase domain, and finally a SEF/IL-17R (SEFIR) domain (Conti et al., 2009; Swaidani et al., 2009). Once IL-17A binds and activates IL-17RA to form the complete receptor complex with IL-17RC, Act1 is recruited via the SEFIR and U-box domains (Chang et al., 2006; Liu et al., 2009). Following Act1 recruitment to the IL-17A receptor, a number of signaling cascades have been shown to be activated. In the peripheral immune system, these cascades include the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway and mitogen-activated protein kinase (MAPK) pathways, which consist of the extracellular signal-regulated kinase (ERK), p38, and JUN N-terminal kinase (JNK) pathways (Arthur and Ley, 2013; Huang et al., 2009a). Act1 is expressed in cells throughout the nervous system (Kang et al., 2010; Kang et al., 2013) and based on its role mediating IL-17RA signal transduction in peripheral systems, is likely to function in this capacity in the brain as well. Indeed, IL-17A induction of glial cell death in EAE was found to function through Act1 (Kang et al., 2013).
NF-κB: a major effector IL-17RA/Act1 signaling
One of the primary signaling cascades activated via Act1 is the NF-κB pathway (Li et al., 2000). NF-κB is a transcription factor that is centrally involved in the production of chemokine, cytokine, and immune effectors for inflammation (Barnes, 1997; Ghosh and Dass, 2016). NF-κB normally resides in the cytoplasm unable to enter the nucleus because it is bound to an inhibitory regulatory protein called IkBα (Jacobs and Harrison, 1998). Act1 recruits Traf6 via its TRAF interaction domains and subsequently ubiquitinates the protein via its U-box E3 ligase domain. Ubiquitinated Traf6 activates TGFβ-activated kinase 1 (TAK1), which then phosphorylates and activates IkB kinase (IKK). In turn, IKK phosphorylates IκBα, disengaging it from NF-kB in the cytoplasm, which allows NF-κB to translocate into the nucleus to regulate the transcription of numerous gene products directly involved in immune function (Ghosh and Dass, 2016; Gilmore, 2006). For example, NF-κB can induce expression of the chemokine CCL20, which attracts Th17 cells (Fujiie et al., 2001). While its immunological roles are well-described, NF-κB is also known to function in the brain. NF-κB is widely expressed in the nervous system (Bhakar et al., 2002; O'Neill and Kaltschmidt, 1997; Yalcin et al., 2003) and is activated by neurotrophic factors (Carter et al., 1996; Hamanoue et al., 1999) and neurotransmitters (Guerrini et al., 1995; Kaltschmidt et al., 1995). NF-κB also has been shown to regulate different aspects of synaptic function, memory, and cognition in many species (Albensi and Mattson, 2000; Engelmann and Haenold, 2016; Freudenthal et al., 1998; Heckscher et al., 2007; Kaltschmidt et al., 2006; Levenson et al., 2004; Meffert et al., 2003; Yeh et al., 2002). Therefore, IL-17A may act through NF-κB to regulate brain function.
In MIA-associated ASD, aberrant IL-17A signaling might then influence the expression of cortical and behavioral abnormalities in the offspring through NF-κB dysregulation in the fetal brain. In agreement with this idea, NF-κB has been shown to play a role in neurodevelopment. In cultured sensory neurons from the embryonic and postnatal mouse, NF-κB signaling was required for normal size and complexity of neuritic arbors (Gutierrez et al., 2005). Similarly, in organotypic brain slices obtained from neonatal mice, neurons also required NF-κB function for normal dendritic arbors. Blocking NF-κB activity was found to cause significant reductions in branch point numbers and overall dendritic length of pyramidal neurons in the slices (Gutierrez et al., 2005). Some studies, however, have reported finding little or no nascent NF-κB activity in neurons (Jarosinski et al., 2001; Listwak et al., 2013). Other studies provide evidence for NF-κB activity in glial cell populations rather than neurons (Mao et al., 2009; Moerman et al., 1999) or from activation of transcriptional enhancers associated with NF-κB rather than NF-κB itself (Mao et al., 2007). Finally, reports of neuronal NF-κB studies being confounded by poor reagent specificity also cast some doubt on the role of NF-κB in the nervous system (Herkenham et al., 2011). Therefore, while Act1-mediated NF-κB signaling is a promising pathway through which IL-17A may influence brain development and function, more work is needed to firmly establish a link between IL-17A and NF-κB in the nervous system.
IL-17RA and ERK signaling
Another Act1-dependent intracellular signal cascade activated by IL-17A is the ERK pathway. Interestingly, Act1 activation of the ERK pathway is mediated by its U-box E3 ligase activity (Liu et al., 2009), which indicates that the ubiquitination crucial for NF-κB signaling may carry over to multiple pathways downstream of IL-17RA. IL-17A promotes ERK activation in a variety of epithelial cells, chondrocytes, fibroblasts, and stem cells (Andoh et al., 2002; Huang et al., 2009b; Jovanovic et al., 1998; Laan et al., 2001; Lee et al., 2008; Sebkova et al., 2004; Song et al., 2015). IL-17A also has been found to promote ERK activation in cells of the nervous system, such as neurons cultured from dorsal root ganglia (Richter et al., 2012; Segond von Banchet et al., 2013), oligodendrocyte precursors (Rodgers et al., 2015), and astrocytes (Zhang et al., 2016). ERK signaling features prominently in neuronal growth (Gomez and Cohen, 1991; Samuels et al., 2008; Wu and Bradshaw, 1996), differentiation (Rebay and Rubin, 1995; Samuels et al., 2008), process outgrowth (Perron and Bixby, 1999; Wu and Bradshaw, 1996; Yamazaki et al., 2001), neuronal and axonal migration (Hirai et al., 2002), synaptic plasticity (English and Sweatt, 1997; Impey et al., 1998), and cognition (Cesarini et al., 2009; Samuels et al., 2008; Schafe et al., 2000; Walz et al., 2000). Thus, MIA induction of excessive IL-17A activity in the fetal brain may precipitate ASD through the dysregulation of this critical signaling pathway. Indeed, ERK-related mutations and genetic variation have been found in ASD patient groups (Wen et al., 2016). Eccentric ERK activation has also been observed in peripheral lymphocytes from ASD patients (Erickson et al., 2017) and in IPSC-derived neurons from patients with 22q11.2 deletions, an intellectual disability disorder with features related to ASD (Zhao et al., 2015). In animal models, dysregulation of ERK signaling has been shown to promote the expression of ASD-like phenotypes (Faridar et al., 2014; Yufune et al., 2015). Given the enormous role for ERK-related signaling in brain development and post-developmental function, IL-17A dysregulation of this pathway may be a key mechanism linking inflammation to ASD.
Other downstream pathways of IL-17RA
IL-17RA/Act1 signaling can activate an additional MAPK pathway through p38. In the study where IL-17A stimulation of embryonic NSCs was found to reduce their proliferation and differentiation, these effects were in part rescued by the p38 MAPK inhibitor SB203580 (Li et al., 2013). This suggests that p38 functions downstream of IL-17RA in NSCs and may also mediate MIA-induced effects of excessive IL-17A signaling in these cells. Finally, Act1 also plays a role in mRNA stabilization. Upon binding IL-17A, IL-17RA can form a multi-protein complex with Act1, TRAF2, TRAF5, and arginine/serine-rich splicing factor (ASF) (Bulek et al., 2011; Sun et al., 2011). This complex can stabilize CXCL1 or CXCL5 mRNAs by preventing ASF cleavage of the mRNA and enhancing expression of the encoded protein (Herjan et al., 2013). Act1 may perform a similar function in neurons and other brain-resident cells to stabilize mRNAs with related structure to CXCL1/5. Related to its role in mRNA stabilization, a new role for Act1 in the nucleus also has been reported (Velichko et al., 2016). Thus, Act1 activity may function to modulate translation of specific subsets of mRNA critical to neural processes underlying ASD.
VI. Future directions
What are the relevant cellular targets for IL-17A signaling in ASD?
IL-17A is expressed by a variety of tissues in the body and can function in a broad spectrum of contexts, ranging from the familiar immunological effects to also having pronounced effects on cortical development. How IL-17A exerts these effects is complex, which depends on cell type expression of IL-17RA, downstream effectors, and parallel signaling pathways that may act in concert with IL-17A (e.g. TNFα). An important question moving forward in the context of MIA is the identification of the specific cell types that are affected by IL-17A signaling to promote ASD-related deficits. Does aberrant IL-17A signaling directly promote neuronal alterations that lead to ASD or are the effects secondary, resulting from some other physiological change such as blood-brain barrier activity or activation of microglia? It will also be important to determine how MIA-induced IL-17A signaling changes over the course of development. This review limited the discussion of potential IL-17A targets in the developing cortex relevant during the window from E12.5 to E14.5. This corresponds to the time when MIA was evoked by poly(I:C) or direct injection of IL-17A into the fetal brain could reproduce MIA-like effects in the offspring. It is not known how long IL-17A upregulation continues after E14.5 but it is likely to persist after this time based on what is seen with other pro-inflammatory cytokines such as TNFα and IL-1β (Choi et al., 2016). Continued IL-17A activity during cortical development may have effects that additionally contribute to ASD in the offspring. In the case of astrocytes and oligodendrocytes, which do not develop until after E14.5, IL-17A can already disrupt these cell types by adversely affecting their precursors. However, IL-17A is also known to affect astrocytes and oligodendrocytes directly. In the postnatal brain, astrocytes are a major source of soluble factors, including cytokines and chemokines during inflammation (Dong and Benveniste, 2001). IL-17A stimulation of astrocytes can enhance IL-6 signaling (Ma et al., 2010), release of nitric oxide, which increases glutamate release and may therefore promote excitotoxicity (Trajkovic et al., 2001), and infiltration of peripheral immune cells (Rodgers et al., 2010). With oligodendrocytes, IL-17A stimulation can exacerbate apoptosis (Paintlia et al., 2011). Therefore, prolonged IL-17A in the fetal brain could have other detrimental effects on astrocytes and oligodendrocytes besides their initial development. Future studies can address these questions.
Do known IL-17RA signaling pathways also function in MIA-associated ASD?
Intracellularly, IL-17RA activation can promote signaling through the NF-kB and MAPK pathways. Both can exert powerful effects on the developing nervous system. It is entirely possible that aberrant IL-17A signaling in the brain act through these pathways to promote the development of ASD. There is evidence for the involvement of both pathways in ASD pathophysiology. NF-κB signaling has been implicated by in silico studies identifying highly expressed ASD candidate genes (Ziats and Rennert, 2011). A study examining the blood of autistic patients revealed significantly elevated NF-KB activity (Naik et al., 2011). In the valproic acid model of ASD, excess NF-KB activity may be involved in reducing NSC loss at a developmentally critical time, thus contributing to autistic-related behavioral phenotypes (Go et al., 2011). Direct genetic evidence linking ASD and NF-KB homeostatic function has also been recently identified (Manzini et al., 2014). For MAPK, elevated activity has been observed in the brain and lymphocytes from the BTBR mouse model of autism-related behaviors (Faridar et al., 2014). Interestingly, blockade of MAPK with the MEK inhibitor SL237 causes ASD-like phenotypes in mouse models (Yufune et al., 2015). Future studies will be important to determine the molecular signaling pathways downstream of IL-17RA activation in MIA-associated ASD.
What role might postnatal IL-17A signaling play in ASD?
Cells in the brain develop primarily after birth, encompassing a wide range of activities that include gliogenesis, microglial proliferation and maturation, synaptogenesis, and activity-dependent circuit formation. Therefore, critically important is the question of what contribution, if any, postnatal IL-17A signaling has in the manifestation of ASD. Elevated IL-17A levels have been found in a subset of ASD patients (Akintunde et al., 2015; Al-Ayadhi and Mostafa, 2012; Suzuki et al., 2011), suggesting that IL-17A regulation may be perturbed and influence neural processes after prenatal development. Similarly, MIA-exposed offspring in animal models have been reported to show increased IL-17A levels postnatally (Garay et al., 2013). Together with other chronic symptoms of inflammation observed in the brain and periphery of ASD patients and animal models, these data indicate persistent immune dysregulation in ASD (Akintunde et al., 2015; Ashwood et al., 2004; Ashwood et al., 2011; Vargas et al., 2005). This feature may represent a self-propagating inflammatory cycle initiated from developmental stages or a long-term maladaptation to early environmental insults. Prenatal immune challenge may set up an environment in which the fetus develops abnormally, affecting subsequent function of both the postnatal immune and nervous systems. Additionally, both immune and neural cells may be primed by this prenatal event, resulting in exaggerated neuroimmune responses to homeostatic perturbations at postnatal stages. Given the importance of early life experience in shaping the developing brain, increased IL-17A activity due to maternal inflammation may represent a critical trigger for life-long maladaptation of neuroimmune function that in some patients manifests as ASD. Moving forward it will be important to determine how aberrant postnatal IL-17A signaling may further contribute to ASD phenotypes.
In conclusion, ASD is a highly complex neurodevelopmental disorder with equally complex etiology. It is clear that immune dysfunction can play a significant role in the etiology of this disorder. While much remains to be discovered about how the immune system contributes to ASD, a growing number of human and animal model studies implicate a specific role for IL-17A signaling in the manifestation of at least some ASD cases. To better understand IL-17A mechanisms in ASD, we believe that key areas of future investigation will be identifying (1) the brain-specific cellular targets and downstream effectors of IL-17A and (2) the pre- and postnatal contributions of aberrant IL-17A signaling in ASD-related phenotypes. Improved understanding of IL-17A cellular targets, signaling pathways, and developmental influences may allow for targeted therapies and more effective interventions that can counter the effects of immune dysregulation in ASD.
Highlights.
Prenatal exposure to maternal immune activation (MIA) has been linked to ASD risk.
Offspring in MIA mouse models exhibit ASD-like behaviors and cortical dysplasia.
The pro-inflammatory cytokine IL-17A may play a causal role in MIA-associated ASD.
IL-17A injection into the fetal brain can phenocopy MIA-induced effects.
Potential mechanisms of IL-17A signaling effects on neurodevelopment are proposed.
Acknowledgments
We would like to thank SG Hoeffer, J Levenga, J Huh, and DR Littman, for useful discussions and comments. We would also like to thank our generous funding support: NIH NS086933-01 (CAH), The Linda Crnic Institute (CAH), SFARI grant 27444 (DRL, CAH, JH), Alzheimer's Association grant MNIRGDP-12-258900 (CAH), and NIH T32 MH016880 (HW).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrahams BS, Geschwind DH. Connecting genes to brain in the autism spectrum disorders. Arch Neurol. 2010;67:395–399. doi: 10.1001/archneurol.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol. 2007;8:942–949. doi: 10.1038/ni1496. [DOI] [PubMed] [Google Scholar]
- Aggarwal S, Xie MH, Maruoka M, Foster J, Gurney AL. Acinar cells of the pancreas are a target of interleukin-22. J Interferon Cytokine Res. 2001;21:1047–1053. doi: 10.1089/107999001317205178. [DOI] [PubMed] [Google Scholar]
- Akintunde ME, Rose M, Krakowiak P, Heuer L, Ashwood P, Hansen R, Hertz-Picciotto I, Van de Water J. Increased production of IL-17 in children with autism spectrum disorders and co-morbid asthma. J Neuroimmunol. 2015;286:33–41. doi: 10.1016/j.jneuroim.2015.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Ayadhi LY, Mostafa GA. Elevated serum levels of interleukin-17A in children with autism. Journal of neuroinflammation. 2012;9:158. doi: 10.1186/1742-2094-9-158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albensi BC, Mattson MP. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse. 2000;35:151–159. doi: 10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature. 2001;413:732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- Andoh A, Shimada M, Bamba S, Okuno T, Araki Y, Fujiyama Y, Bamba T. Extracellular signal-regulated kinases 1 and 2 participate in interleukin-17 plus tumor necrosis factor-alpha-induced stabilization of interleukin-6 mRNA in human pancreatic myofibroblasts. Biochim Biophys Acta. 2002;1591:69–74. doi: 10.1016/s0167-4889(02)00250-1. [DOI] [PubMed] [Google Scholar]
- Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, Romagnani S. Phenotypic and functional features of human Th17 cells. J Exp Med. 2007;204:1849–1861. doi: 10.1084/jem.20070663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony JM, Paquin A, Nutt SL, Kaplan DR, Miller FD. Endogenous microglia regulate development of embryonic cortical precursor cells. J Neurosci Res. 2011;89:286–298. doi: 10.1002/jnr.22533. [DOI] [PubMed] [Google Scholar]
- Arrode-Bruses G, Bruses JL. Maternal immune activation by poly(I:C) induces expression of cytokines IL-1beta and IL-13, chemokine MCP-1 and colony stimulating factor VEGF in fetal mouse brain. Journal of neuroinflammation. 2012;9:83. doi: 10.1186/1742-2094-9-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13:679–692. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- Ashwood P, Anthony A, Torrente F, Wakefield AJ. Spontaneous mucosal lymphocyte cytokine profiles in children with autism and gastrointestinal symptoms: mucosal immune activation and reduced counter regulatory interleukin-10. J Clin Immunol. 2004;24:664–673. doi: 10.1007/s10875-004-6241-6. [DOI] [PubMed] [Google Scholar]
- Ashwood P, Krakowiak P, Hertz-Picciotto I, Hansen R, Pessah I, Van de Water J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav Immun. 2011;25:40–45. doi: 10.1016/j.bbi.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atladottir HO, Pedersen MG, Thorsen P, Mortensen PB, Deleuran B, Eaton WW, Parner ET. Association of family history of autoimmune diseases and autism spectrum disorders. Pediatrics. 2009;124:687–694. doi: 10.1542/peds.2008-2445. [DOI] [PubMed] [Google Scholar]
- Atladottir HO, Thorsen P, Ostergaard L, Schendel DE, Lemcke S, Abdallah M, Parner ET. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. Journal of autism and developmental disorders. 2010;40:1423–1430. doi: 10.1007/s10803-010-1006-y. [DOI] [PubMed] [Google Scholar]
- Barnes PJ. Nuclear factor-kappa B. Int J Biochem Cell Biol. 1997;29:867–870. doi: 10.1016/s1357-2725(96)00159-8. [DOI] [PubMed] [Google Scholar]
- Bauer HC, Bauer H, Lametschwandtner A, Amberger A, Ruiz P, Steiner M. Neovascularization and the appearance of morphological characteristics of the blood-brain barrier in the embryonic mouse central nervous system. Brain Res Dev Brain Res. 1993;75:269–278. doi: 10.1016/0165-3806(93)90031-5. [DOI] [PubMed] [Google Scholar]
- Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, Gu C. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature. 2014;509:507–511. doi: 10.1038/nature13324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Bhakar AL, Tannis LL, Zeindler C, Russo MP, Jobin C, Park DS, MacPherson S, Barker PA. Constitutive nuclear factor-kappa B activity is required for central neuron survival. J Neurosci. 2002;22:8466–8475. doi: 10.1523/JNEUROSCI.22-19-08466.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, Belkadi A, Picard C, Abel L, Fieschi C, Puel A, Li X, Casanova JL. An ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. 2013;39:676–686. doi: 10.1016/j.immuni.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AS, Sourander A, Hinkka-Yli-Salomaki S, McKeague IW, Sundvall J, Surcel HM. Elevated maternal C-reactive protein and autism in a national birth cohort. Mol Psychiatry. 2014;19:259–264. doi: 10.1038/mp.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruttger J, Karram K, Wortge S, Regen T, Marini F, Hoppmann N, Klein M, Blank T, Yona S, Wolf Y, Mack M, Pinteaux E, Muller W, Zipp F, Binder H, Bopp T, Prinz M, Jung S, Waisman A. Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity. 2015;43:92–106. doi: 10.1016/j.immuni.2015.06.012. [DOI] [PubMed] [Google Scholar]
- Buie T, Campbell DB, Fuchs GJ, 3rd, Furuta GT, Levy J, Vandewater J, Whitaker AH, Atkins D, Bauman ML, Beaudet AL, Carr EG, Gershon MD, Hyman SL, Jirapinyo P, Jyonouchi H, Kooros K, Kushak R, Levitt P, Levy SE, Lewis JD, Murray KF, Natowicz MR, Sabra A, Wershil BK, Weston SC, Zeltzer L, Winter H. Evaluation, diagnosis, and treatment of gastrointestinal disorders in individuals with ASDs: a consensus report. Pediatrics. 2010;125(Suppl 1):S1–18. doi: 10.1542/peds.2009-1878C. [DOI] [PubMed] [Google Scholar]
- Bulek K, Liu C, Swaidani S, Wang L, Page RC, Gulen MF, Herjan T, Abbadi A, Qian W, Sun D, Lauer M, Hascall V, Misra S, Chance MR, Aronica M, Hamilton T, Li X. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol. 2011;12:844–852. doi: 10.1038/ni.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacci E, Ajmone-Cat MA, Anelli T, Biagioni S, Minghetti L. In vitro neuronal and glial differentiation from embryonic or adult neural precursor cells are differently affected by chronic or acute activation of microglia. Glia. 2008;56:412–425. doi: 10.1002/glia.20616. [DOI] [PubMed] [Google Scholar]
- Cai CW, Blase JR, Zhang X, Eickhoff CS, Hoft DF. Th17 Cells Are More Protective Than Th1 Cells Against the Intracellular Parasite Trypanosoma cruzi. PLoS Pathog. 2016;12:e1005902. doi: 10.1371/journal.ppat.1005902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BD, Kaltschmidt C, Kaltschmidt B, Offenhauser N, Bohm-Matthaei R, Baeuerle PA, Barde YA. Selective activation of NF-kappa B by nerve growth factor through the neurotrophin receptor p75. Science. 1996;272:542–545. doi: 10.1126/science.272.5261.542. [DOI] [PubMed] [Google Scholar]
- Caruso R, Fina D, Peluso I, Fantini MC, Tosti C, Del Vecchio Blanco G, Paoluzi OA, Caprioli F, Andrei F, Stolfi C, Romano M, Ricci V, MacDonald TT, Pallone F, Monteleone G. IL-21 is highly produced in Helicobacter pylori-infected gastric mucosa and promotes gelatinases synthesis. J Immunol. 2007;178:5957–5965. doi: 10.4049/jimmunol.178.9.5957. [DOI] [PubMed] [Google Scholar]
- Casanova MF, El-Baz AS, Kamat SS, Dombroski BA, Khalifa F, Elnakib A, Soliman A, Allison-McNutt A, Switala AE. Focal cortical dysplasias in autism spectrum disorders. Acta Neuropathol Commun. 2013;1:67. doi: 10.1186/2051-5960-1-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesarini L, Alfieri P, Pantaleoni F, Vasta I, Cerutti M, Petrangeli V, Mariotti P, Leoni C, Ricci D, Vicari S, Selicorni A, Tartaglia M, Mercuri E, Zampino G. Cognitive profile of disorders associated with dysregulation of the RAS/MAPK signaling cascade. Am J Med Genet A. 2009;149A:140–146. doi: 10.1002/ajmg.a.32488. [DOI] [PubMed] [Google Scholar]
- Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- Chen C, Liu X, Wan B, Zhang JZ. Regulatory properties of copolymer I in Th17 differentiation by altering STAT3 phosphorylation. J Immunol. 2009;183:246–253. doi: 10.4049/jimmunol.0900193. [DOI] [PubMed] [Google Scholar]
- Chen Z, Tato CM, Muul L, Laurence A, O'Shea JJ. Distinct regulation of interleukin-17 in human T helper lymphocytes. Arthritis Rheum. 2007;56:2936–2946. doi: 10.1002/art.22866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chess S. Follow-up report on autism in congenital rubella. J Autism Child Schizophr. 1977;7:69–81. doi: 10.1007/BF01531116. [DOI] [PubMed] [Google Scholar]
- Choi GB, Yim YS, Wong H, Kim S, Kim H, Kim SV, Hoeffer CA, Littman DR, Huh JR. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science. 2016;351:933–939. doi: 10.1126/science.aad0314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung DR, Kasper DL, Panzo RJ, Chitnis T, Grusby MJ, Sayegh MH, Tzianabos AO. CD4+ T cells mediate abscess formation in intra-abdominal sepsis by an IL-17-dependent mechanism. J Immunol. 2003;170:1958–1963. doi: 10.4049/jimmunol.170.4.1958. [DOI] [PubMed] [Google Scholar]
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clancy B, Finlay BL, Darlington RB, Anand KJ. Extrapolating brain development from experimental species to humans. Neurotoxicology. 2007;28:931–937. doi: 10.1016/j.neuro.2007.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, Suter T, Becher B. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560–567. doi: 10.1038/ni.2027. [DOI] [PubMed] [Google Scholar]
- Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, Ho AW, Hai JH, Yu JJ, Jung JW, Filler SG, Masso-Welch P, Edgerton M, Gaffen SL. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–311. doi: 10.1084/jem.20081463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corneth OB, Mus AM, Asmawidjaja PS, Klein Wolterink RG, van Nimwegen M, Brem MD, Hofman Y, Hendriks RW, Lubberts E. Absence of interleukin-17 receptor a signaling prevents autoimmune inflammation of the joint and leads to a Th2-like phenotype in collagen-induced arthritis. Arthritis Rheumatol. 2014;66:340–349. doi: 10.1002/art.38229. [DOI] [PubMed] [Google Scholar]
- Crawford LW, Foley JF, Elmore SA. Histology atlas of the developing mouse hepatobiliary system with emphasis on embryonic days 9.5-18.5. Toxicol Pathol. 2010;38:872–906. doi: 10.1177/0192623310374329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CL, Martinez-Cerdeno V, Noctor SC. Microglia regulate the number of neural precursor cells in the developing cerebral cortex. J Neurosci. 2013;33:4216–4233. doi: 10.1523/JNEUROSCI.3441-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlgren J, Samuelsson AM, Jansson T, Holmang A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr Res. 2006;60:147–151. doi: 10.1203/01.pdr.0000230026.74139.18. [DOI] [PubMed] [Google Scholar]
- Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature. 2010;468:562–566. doi: 10.1038/nature09513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Sarma J, Ciric B, Marek R, Sadhukhan S, Caruso ML, Shafagh J, Fitzgerald DC, Shindler KS, Rostami A. Functional interleukin-17 receptor A is expressed in central nervous system glia and upregulated in experimental autoimmune encephalomyelitis. Journal of neuroinflammation. 2009;6:14. doi: 10.1186/1742-2094-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SK, Redecke V, Prilliman KR, Takabayashi K, Corr M, Tallant T, DiDonato J, Dziarski R, Akira S, Schoenberger SP, Raz E. A subset of Toll-like receptor ligands induces cross-presentation by bone marrow-derived dendritic cells. J Immunol. 2003;170:4102–4110. doi: 10.4049/jimmunol.170.8.4102. [DOI] [PubMed] [Google Scholar]
- De Miranda J, Yaddanapudi K, Hornig M, Prilliman G, Serge R, Lipkin WI. Induction of Toll-Like Receptor 3-Mediated Immunity during Gestation Inhibits Cortical Neurogenesis and Causes Behavioral Disturbances. MBio. 2010 doi: 10.1128/mBio.00176-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depino AM. Peripheral and central inflammation in autism spectrum disorders. Mol Cell Neurosci. 2013;53:69–76. doi: 10.1016/j.mcn.2012.10.003. [DOI] [PubMed] [Google Scholar]
- Deverman BE, Patterson PH. Cytokines and CNS development. Neuron. 2009;64:61–78. doi: 10.1016/j.neuron.2009.09.002. [DOI] [PubMed] [Google Scholar]
- DiCicco-Bloom E, Lord C, Zwaigenbaum L, Courchesne E, Dager SR, Schmitz C, Schultz RT, Crawley J, Young LJ. The developmental neurobiology of autism spectrum disorder. J Neurosci. 2006;26:6897–6906. doi: 10.1523/JNEUROSCI.1712-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Dumoutier L, Van Roost E, Colau D, Renauld JC. Human interleukin-10-related T cell-derived inducible factor: molecular cloning and functional characterization as an hepatocyte-stimulating factor. Proc Natl Acad Sci U S A. 2000;97:10144–10149. doi: 10.1073/pnas.170291697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ek CJ, Dziegielewska KM, Habgood MD, Saunders NR. Barriers in the developing brain and Neurotoxicology. Neurotoxicology. 2012;33:586–604. doi: 10.1016/j.neuro.2011.12.009. [DOI] [PubMed] [Google Scholar]
- Ely LK, Fischer S, Garcia KC. Structural basis of receptor sharing by interleukin 17 cytokines. Nat Immunol. 2009;10:1245–1251. doi: 10.1038/ni.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelmann C, Haenold R. Transcriptional Control of Synaptic Plasticity by Transcription Factor NF-kappaB. Neural Plast. 2016;2016:7027949. doi: 10.1155/2016/7027949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English JD, Sweatt JD. A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J Biol Chem. 1997;272:19103–19106. doi: 10.1074/jbc.272.31.19103. [DOI] [PubMed] [Google Scholar]
- Enstrom AM, Onore CE, Van de Water JA, Ashwood P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain Behav Immun. 2010;24:64–71. doi: 10.1016/j.bbi.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson CA, Ray B, Wink LK, Bayon BL, Pedapati EV, Shaffer R, Schaefer TL, Lahiri DK. Initial analysis of peripheral lymphocytic extracellular signal related kinase activation in autism. J Psychiatr Res. 2017;84:153–160. doi: 10.1016/j.jpsychires.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S, Peri F, Wilson SW, Ruhrberg C. Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction. Blood. 2010;116:829–840. doi: 10.1182/blood-2009-12-257832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faridar A, Jones-Davis D, Rider E, Li J, Gobius I, Morcom L, Richards LJ, Sen S, Sherr EH. Mapk/Erk activation in an animal model of social deficits shows a possible link to autism. Mol Autism. 2014;5:57. doi: 10.1186/2040-2392-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Bernet E, Soriano E, del Rio T, Fonseca M. Naturally occurring cell death in the cerebral cortex of the rat and removal of dead cells by transitory phagocytes. Neuroscience. 1990;39:451–458. doi: 10.1016/0306-4522(90)90281-8. [DOI] [PubMed] [Google Scholar]
- Ferretti S, Bonneau O, Dubois GR, Jones CE, Trifilieff A. IL-17, produced by lymphocytes and neutrophils, is necessary for lipopolysaccharide-induced airway neutrophilia: IL-15 as a possible trigger. J Immunol. 2003;170:2106–2112. doi: 10.4049/jimmunol.170.4.2106. [DOI] [PubMed] [Google Scholar]
- Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, Blanchard D, Gaillard C, Das Mahapatra B, Rouvier E, Golstein P, Banchereau J, Lebecque S. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med. 1996;183:2593–2603. doi: 10.1084/jem.183.6.2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freudenthal R, Locatelli F, Hermitte G, Maldonado H, Lafourcade C, Delorenzi A, Romano A. Kappa-B like DNA-binding activity is enhanced after spaced training that induces long-term memory in the crab Chasmagnathus. Neurosci Lett. 1998;242:143–146. doi: 10.1016/s0304-3940(98)00059-7. [DOI] [PubMed] [Google Scholar]
- Fujiie S, Hieshima K, Izawa D, Nakayama T, Fujisawa R, Ohyanagi H, Yoshie O. Proinflammatory cytokines induce liver and activation-regulated chemokine/macrophage inflammatory protein-3alpha/CCL20 in mucosal epithelial cells through NF-kappaB [correction of NK-kappaB] Int Immunol. 2001;13:1255–1263. doi: 10.1093/intimm/13.10.1255. [DOI] [PubMed] [Google Scholar]
- Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garay PA, Hsiao EY, Patterson PH, McAllister AK. Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun. 2013;31:54–68. doi: 10.1016/j.bbi.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, Mirnics K, Persico AM. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis. 2008;30:303–311. doi: 10.1016/j.nbd.2008.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbett KA, Hsiao EY, Kalman S, Patterson PH, Mirnics K. Effects of maternal immune activation on gene expression patterns in the fetal brain. Transl Psychiatry. 2012;2:e98. doi: 10.1038/tp.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Dass JF. Study of pathway cross-talk interactions with NF-kappaB leading to its activation via ubiquitination or phosphorylation: A brief review. Gene. 2016;584:97–109. doi: 10.1016/j.gene.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Gilmore TD. Introduction to NF-kappaB: players, pathways, perspectives. Oncogene. 2006;25:6680–6684. doi: 10.1038/sj.onc.1209954. [DOI] [PubMed] [Google Scholar]
- Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330:841–845. doi: 10.1126/science.1194637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go HS, Seo JE, Kim KC, Han SM, Kim P, Kang YS, Han SH, Shin CY, Ko KH. Valproic acid inhibits neural progenitor cell death by activation of NF-kappaB signaling pathway and up-regulation of Bcl-XL. J Biomed Sci. 2011;18:48. doi: 10.1186/1423-0127-18-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goines PE, Ashwood P. Cytokine dysregulation in autism spectrum disorders (ASD): possible role of the environment. Neurotoxicol Teratol. 2013;36:67–81. doi: 10.1016/j.ntt.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez N, Cohen P. Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature. 1991;353:170–173. doi: 10.1038/353170a0. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Garcia I, Zhao Y, Ju S, Gu Q, Liu L, Kolls JK, Lu B. IL-17 signaling-independent central nervous system autoimmunity is negatively regulated by TGF-beta. J Immunol. 2009;182:2665–2671. doi: 10.4049/jimmunol.0802221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig LC, Woodworth MB, Galazo MJ, Padmanabhan H, Macklis JD. Molecular logic of neocortical projection neuron specification, development and diversity. Nature reviews. 2013;14:755–769. doi: 10.1038/nrn3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine. 2013;64:477–485. doi: 10.1016/j.cyto.2013.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrini L, Blasi F, Denis-Donini S. Synaptic activation of NF-kappa B by glutamate in cerebellar granule neurons in vitro. Proc Natl Acad Sci U S A. 1995;92:9077–9081. doi: 10.1073/pnas.92.20.9077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez H, Hale VA, Dolcet X, Davies A. NF-kappaB signalling regulates the growth of neural processes in the developing PNS and CNS. Development. 2005;132:1713–1726. doi: 10.1242/dev.01702. [DOI] [PubMed] [Google Scholar]
- Hamanoue M, Middleton G, Wyatt S, Jaffray E, Hay RT, Davies AM. p75-mediated NF-kappaB activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol Cell Neurosci. 1999;14:28–40. doi: 10.1006/mcne.1999.0770. [DOI] [PubMed] [Google Scholar]
- Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–155. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- He JJ, Li S, Shu HF, Yu SX, Liu SY, Yin Q, Yang H. The interleukin 17 system in cortical lesions in focal cortical dysplasias. J Neuropathol Exp Neurol. 2013;72:152–163. doi: 10.1097/NEN.0b013e318281262e. [DOI] [PubMed] [Google Scholar]
- Heckscher ES, Fetter RD, Marek KW, Albin SD, Davis GW. NF-kappaB, IkappaB, and IRAK control glutamate receptor density at the Drosophila NMJ. Neuron. 2007;55:859–873. doi: 10.1016/j.neuron.2007.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herjan T, Yao P, Qian W, Li X, Liu C, Bulek K, Sun D, Yang WP, Zhu J, He A, Carman JA, Erzurum SC, Lipshitz HD, Fox PL, Hamilton TA, Li X. HuR is required for IL-17-induced Act1-mediated CXCL1 and CXCL5 mRNA stabilization. J Immunol. 2013;191:640–649. doi: 10.4049/jimmunol.1203315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Rathore P, Brown P, Listwak SJ. Cautionary notes on the use of NF-kappaB p65 and p50 antibodies for CNS studies. Journal of neuroinflammation. 2011;8:141. doi: 10.1186/1742-2094-8-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai S, Kawaguchi A, Hirasawa R, Baba M, Ohnishi T, Ohno S. MAPK-upstream protein kinase (MUK) regulates the radial migration of immature neurons in telencephalon of mouse embryo. Development. 2002;129:4483–4495. doi: 10.1242/dev.129.19.4483. [DOI] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Chow J, Mazmanian SK, Patterson PH. Modeling an autism risk factor in mice leads to permanent immune dysregulation. Proc Natl Acad Sci U S A. 2012;109:12776–12781. doi: 10.1073/pnas.1202556109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, Patterson PH. Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun. 2011;25:604–615. doi: 10.1016/j.bbi.2010.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu MH, Zheng QF, Jia XZ, Li Y, Dong YC, Wang CY, Lin QY, Zhang FY, Zhao RB, Xu HW, Zhou JH, Yuan HP, Zhang WH, Ren H. Neuroprotection effect of interleukin (IL)-17 secreted by reactive astrocytes is emerged from a high-level IL-17-containing environment during acute neuroinflammation. Clin Exp Immunol. 2014;175:268–284. doi: 10.1111/cei.12219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Kao CY, Wachi S, Thai P, Ryu J, Wu R. Requirement for both JAK-mediated PI3K signaling and ACT1/TRAF6/TAK1-dependent NF-kappaB activation by IL-17A in enhancing cytokine expression in human airway epithelial cells. J Immunol. 2007;179:6504–6513. doi: 10.4049/jimmunol.179.10.6504. [DOI] [PubMed] [Google Scholar]
- Huang G, Shi LZ, Chi H. Regulation of JNK and p38 MAPK in the immune system: signal integration, propagation and termination. Cytokine. 2009a;48:161–169. doi: 10.1016/j.cyto.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Kim HJ, Chang EJ, Lee ZH, Hwang SJ, Kim HM, Lee Y, Kim HH. IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: implications for bone remodeling. Cell Death Differ. 2009b;16:1332–1343. doi: 10.1038/cdd.2009.74. [DOI] [PubMed] [Google Scholar]
- Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis. 2004;190:624–631. doi: 10.1086/422329. [DOI] [PubMed] [Google Scholar]
- Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, Bechmann I, Becher B, Luhmann HJ, Waisman A, Kuhlmann CR. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010;24:1023–1034. doi: 10.1096/fj.09-141978. [DOI] [PubMed] [Google Scholar]
- Huppler AR, Conti HR, Hernandez-Santos N, Darville T, Biswas PS, Gaffen SL. Role of neutrophils in IL-17-dependent immunity to mucosal candidiasis. J Immunol. 2014;192:1745–1752. doi: 10.4049/jimmunol.1302265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hymowitz SG, Filvaroff EH, Yin JP, Lee J, Cai L, Risser P, Maruoka M, Mao W, Foster J, Kelley RF, Pan G, Gurney AL, de Vos AM, Starovasnik MA. IL-17s adopt a cystine knot fold: structure and activity of a novel cytokine, IL-17F, and implications for receptor binding. EMBO J. 2001;20:5332–5341. doi: 10.1093/emboj/20.19.5332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda S, Saijo S, Murayama MA, Shimizu K, Akitsu A, Iwakura Y. Excess IL-1 signaling enhances the development of Th17 cells by downregulating TGF-beta-induced Foxp3 expression. J Immunol. 2014;192:1449–1458. doi: 10.4049/jimmunol.1300387. [DOI] [PubMed] [Google Scholar]
- Impey S, Smith DM, Obrietan K, Donahue R, Wade C, Storm DR. Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci. 1998;1:595–601. doi: 10.1038/2830. [DOI] [PubMed] [Google Scholar]
- Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y. Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:108–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- Islam O, Gong X, Rose-John S, Heese K. Interleukin-6 and neural stem cells: more than gliogenesis. Mol Biol Cell. 2009;20:188–199. doi: 10.1091/mbc.E08-05-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Jacobs MD, Harrison SC. Structure of an IkappaBalpha/NF-kappaB complex. Cell. 1998;95:749–758. doi: 10.1016/s0092-8674(00)81698-0. [DOI] [PubMed] [Google Scholar]
- Jarosinski KW, Whitney LW, Massa PT. Specific deficiency in nuclear factor-kappaB activation in neurons of the central nervous system. Lab Invest. 2001;81:1275–1288. doi: 10.1038/labinvest.3780341. [DOI] [PubMed] [Google Scholar]
- Jonakait GM. The effects of maternal inflammation on neuronal development: possible mechanisms. Int J Dev Neurosci. 2007;25:415–425. doi: 10.1016/j.ijdevneu.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Jones CE, Chan K. Interleukin-17 stimulates the expression of interleukin-8, growth-related oncogene-alpha, and granulocyte-colony-stimulating factor by human airway epithelial cells. Am J Respir Cell Mol Biol. 2002;26:748–753. doi: 10.1165/ajrcmb.26.6.4757. [DOI] [PubMed] [Google Scholar]
- Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, Mineau F, Pelletier JP. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. 1998;160:3513–3521. [PubMed] [Google Scholar]
- Kaltschmidt B, Ndiaye D, Korte M, Pothion S, Arbibe L, Prullage M, Pfeiffer J, Lindecke A, Staiger V, Israel A, Kaltschmidt C, Memet S. NF-kappaB regulates spatial memory formation and synaptic plasticity through protein kinase A/CREB signaling. Mol Cell Biol. 2006;26:2936–2946. doi: 10.1128/MCB.26.8.2936-2946.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt C, Kaltschmidt B, Baeuerle PA. Stimulation of ionotropic glutamate receptors activates transcription factor NF-kappa B in primary neurons. Proc Natl Acad Sci U S A. 1995;92:9618–9622. doi: 10.1073/pnas.92.21.9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Z, Altuntas CZ, Gulen MF, Liu C, Giltiay N, Qin H, Liu L, Qian W, Ransohoff RM, Bergmann C, Stohlman S, Tuohy VK, Li X. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–425. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang Z, Wang C, Zepp J, Wu L, Sun K, Zhao J, Chandrasekharan U, DiCorleto PE, Trapp BD, Ransohoff RM, Li X. Act1 mediates IL-17-induced EAE pathogenesis selectively in NG2+ glial cells. Nat Neurosci. 2013;16:1401–1408. [Google Scholar]
- Kanner L. Autistic disturbances of affective contact. Nervous Child. 1943;2:217–250. [PubMed] [Google Scholar]
- Kawaguchi M, Kokubu F, Kuga H, Matsukura S, Hoshino H, Ieki K, Imai T, Adachi M, Huang SK. Modulation of bronchial epithelial cells by IL-17. J Allergy Clin Immunol. 2001;108:804–809. doi: 10.1067/mai.2001.119027. [DOI] [PubMed] [Google Scholar]
- Kawanokuchi J, Shimizu K, Nitta A, Yamada K, Mizuno T, Takeuchi H, Suzumura A. Production and functions of IL-17 in microglia. J Neuroimmunol. 2008;194:54–61. doi: 10.1016/j.jneuroim.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A. Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med. 2007;13:1173–1175. doi: 10.1038/nm1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y. IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol. 2006;177:566–573. doi: 10.4049/jimmunol.177.1.566. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- Korvatska E, Van de Water J, Anders TF, Gershwin ME. Genetic and immunologic considerations in autism. Neurobiol Dis. 2002;9:107–125. doi: 10.1006/nbdi.2002.0479. [DOI] [PubMed] [Google Scholar]
- Laan M, Cui ZH, Hoshino H, Lotvall J, Sjostrand M, Gruenert DC, Skoogh BE, Linden A. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162:2347–2352. [PubMed] [Google Scholar]
- Laan M, Lotvall J, Chung KF, Linden A. IL-17-induced cytokine release in human bronchial epithelial cells in vitro: role of mitogen-activated protein (MAP) kinases. Br J Pharmacol. 2001;133:200–206. doi: 10.1038/sj.bjp.0704063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Wang P, Kattah MG, Youssef S, Steinman L, DeFea K, Straus DS. Differential regulation of chemokines by IL-17 in colonic epithelial cells. J Immunol. 2008;181:6536–6545. doi: 10.4049/jimmunol.181.9.6536. [DOI] [PubMed] [Google Scholar]
- Leonardi A, Chariot A, Claudio E, Cunningham K, Siebenlist U. CIKS, a connection to Ikappa B kinase and stress-activated protein kinase. Proc Natl Acad Sci U S A. 2000;97:10494–10499. doi: 10.1073/pnas.190245697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenson JM, Choi S, Lee SY, Cao YA, Ahn HJ, Worley KC, Pizzi M, Liou HC, Sweatt JD. A bioinformatics analysis of memory consolidation reveals involvement of the transcription factor c-rel. J Neurosci. 2004;24:3933–3943. doi: 10.1523/JNEUROSCI.5646-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Cui Z, Niu Y, Liu B, Fan W, Yu D, Deng J. Synaptogenesis in the developing mouse visual cortex. Brain Res Bull. 2010;81:107–113. doi: 10.1016/j.brainresbull.2009.08.028. [DOI] [PubMed] [Google Scholar]
- Li X, Chauhan A, Sheikh AM, Patil S, Chauhan V, Li XM, Ji L, Brown T, Malik M. Elevated immune response in the brain of autistic patients. J Neuroimmunol. 2009;207:111–116. doi: 10.1016/j.jneuroim.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Commane M, Nie H, Hua X, Chatterjee-Kishore M, Wald D, Haag M, Stark GR. Act1, an NF-kappa B-activating protein. Proc Natl Acad Sci U S A. 2000;97:10489–10493. doi: 10.1073/pnas.160265197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Li K, Zhu L, Kan Q, Yan Y, Kumar P, Xu H, Rostami A, Zhang GX. Inhibitory effect of IL-17 on neural stem cell proliferation and neural cell differentiation. BMC Immunol. 2013;14:20. doi: 10.1186/1471-2172-14-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang SC, Long AJ, Bennett F, Whitters MJ, Karim R, Collins M, Goldman SJ, Dunussi-Joannopoulos K, Williams CM, Wright JF, Fouser LA. An IL-17F/A heterodimer protein is produced by mouse Th17 cells and induces airway neutrophil recruitment. J Immunol. 2007;179:7791–7799. doi: 10.4049/jimmunol.179.11.7791. [DOI] [PubMed] [Google Scholar]
- Listwak SJ, Rathore P, Herkenham M. Minimal NF-kappaB activity in neurons. Neuroscience. 2013;250:282–299. doi: 10.1016/j.neuroscience.2013.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Qian W, Qian Y, Giltiay V, Lu Y, Swaidani S, Misra S, Deng L, Chen ZJ, Li XV. Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci Signal. 2009;2:ra63. doi: 10.1126/scisignal.2000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XG, Chen SD. The changing phenotype of microglia from homeostasis to disease. Transl Neurodegener. 2012;1:9. doi: 10.1186/2047-9158-1-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Reynolds SL, Baker BJ, Li X, Benveniste EN, Qin H. IL-17 enhancement of the IL-6 signaling cascade in astrocytes. J Immunol. 2010;184:4898–4906. doi: 10.4049/jimmunol.1000142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkova NV, Yu CZ, Hsiao EY, Moore MJ, Patterson PH. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav Immun. 2012;26:607–616. doi: 10.1016/j.bbi.2012.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal M, Marzouk AC, Donnelly R, Ponzio NM. Preferential development of Th17 cells in offspring of immunostimulated pregnant mice. Journal of reproductive immunology. 2010:97–100. doi: 10.1016/j.jri.2010.06.156. [DOI] [PubMed] [Google Scholar]
- Mandal M, Marzouk AC, Donnelly R, Ponzio NM. Maternal immune stimulation during pregnancy affects adaptive immunity in offspring to promote development of TH17 cells. Brain Behav Immun. 2011;25:863–871. doi: 10.1016/j.bbi.2010.09.011. [DOI] [PubMed] [Google Scholar]
- Manel N, Unutmaz D, Littman DR. The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol. 2008;9:641–649. doi: 10.1038/ni.1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzini MC, Xiong L, Shaheen R, Tambunan DE, Di Costanzo S, Mitisalis V, Tischfield DJ, Cinquino A, Ghaziuddin M, Christian M, Jiang Q, Laurent S, Nanjiani ZA, Rasheed S, Hill RS, Lizarraga SB, Gleason D, Sabbagh D, Salih MA, Alkuraya FS, Walsh CA. CC2D1A regulates human intellectual and social function as well as NF-kappaB signaling homeostasis. Cell Rep. 2014;8:647–655. doi: 10.1016/j.celrep.2014.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Yang SH, Simpkins JW, Barger SW. Glutamate receptor activation evokes calpain-mediated degradation of Sp3 and Sp4, the prominent Sp-family transcription factors in neurons. J Neurochem. 2007;100:1300–1314. doi: 10.1111/j.1471-4159.2006.04297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao XR, Moerman-Herzog AM, Chen Y, Barger SW. Unique aspects of transcriptional regulation in neurons--nuances in NFkappaB and Sp1-related factors. Journal of neuroinflammation. 2009;6:16. doi: 10.1186/1742-2094-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nature reviews. 2001;2:780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D. NF-kappa B functions in synaptic signaling and behavior. Nat Neurosci. 2003;6:1072–1078. doi: 10.1038/nn1110. [DOI] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Engler A, Urwyler A, Schedlowski M, Knuesel I, Yee BK, Feldon J. The time of prenatal immune challenge determines the specificity of inflammation-mediated brain and behavioral pathology. J Neurosci. 2006;26:4752–4762. doi: 10.1523/JNEUROSCI.0099-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008;22:469–486. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- Moerman AM, Mao X, Lucas MM, Barger SW. Characterization of a neuronal kappaB-binding factor distinct from NF-kappaB. Brain Res Mol Brain Res. 1999;67:303–315. doi: 10.1016/s0169-328x(99)00091-1. [DOI] [PubMed] [Google Scholar]
- Moldin SO, Rubenstein JL, Hyman SE. Can autism speak to neuroscience? J Neurosci. 2006;26:6893–6896. doi: 10.1523/JNEUROSCI.1944-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteleone G, Caruso R, Fina D, Peluso I, Gioia V, Stolfi C, Fantini MC, Caprioli F, Tersigni R, Alessandroni L, MacDonald TT, Pallone F. Control of matrix metalloproteinase production in human intestinal fibroblasts by interleukin 21. Gut. 2006;55:1774–1780. doi: 10.1136/gut.2006.093187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan JT, Chana G, Pardo CA, Achim C, Semendeferi K, Buckwalter J, Courchesne E, Everall IP. Microglial activation and increased microglial density observed in the dorsolateral prefrontal cortex in autism. Biol Psychiatry. 2010;68:368–376. doi: 10.1016/j.biopsych.2010.05.024. [DOI] [PubMed] [Google Scholar]
- Morgan SC, Taylor DL, Pocock JM. Microglia release activators of neuronal proliferation mediated by activation of mitogen-activated protein kinase, phosphatidylinositol-3-kinase/Akt and delta-Notch signalling cascades. J Neurochem. 2004;90:89–101. doi: 10.1111/j.1471-4159.2004.02461.x. [DOI] [PubMed] [Google Scholar]
- Moseley TA, Haudenschild DR, Rose L, Reddi AH. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003;14:155–174. doi: 10.1016/s1359-6101(03)00002-9. [DOI] [PubMed] [Google Scholar]
- Nadarajah B, Alifragis P, Wong RO, Parnavelas JG. Neuronal migration in the developing cerebral cortex: observations based on real-time imaging. Cereb Cortex. 2003;13:607–611. doi: 10.1093/cercor/13.6.607. [DOI] [PubMed] [Google Scholar]
- Naik US, Gangadharan C, Abbagani K, Nagalla B, Dasari N, Manna SK. A study of nuclear transcription factor-kappa B in childhood autism. PloS one. 2011;6:e19488. doi: 10.1371/journal.pone.0019488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi M, Niidome T, Matsuda S, Akaike A, Kihara T, Sugimoto H. Microglia-derived interleukin-6 and leukaemia inhibitory factor promote astrocytic differentiation of neural stem/progenitor cells. Eur J Neurosci. 2007;25:649–658. doi: 10.1111/j.1460-9568.2007.05309.x. [DOI] [PubMed] [Google Scholar]
- Nakashima A, Ito M, Yoneda S, Shiozaki A, Hidaka T, Saito S. Circulating and decidual Th17 cell levels in healthy pregnancy. Am J Reprod Immunol. 2010:104–109. doi: 10.1111/j.1600-0897.2009.00771.x. [DOI] [PubMed] [Google Scholar]
- Noctor SC, Flint AC, Weissman TA, Wong WS, Clinton BK, Kriegstein AR. Dividing precursor cells of the embryonic cortical ventricular zone have morphological and molecular characteristics of radial glia. J Neurosci. 2002;22:3161–3173. doi: 10.1523/JNEUROSCI.22-08-03161.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA, Kaltschmidt C. NF-kappa B: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–258. doi: 10.1016/s0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- Oskvig DB, Elkahloun AG, Johnson KR, Phillips TM, Herkenham M. Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain Behav Immun. 2012;26:623–634. doi: 10.1016/j.bbi.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paintlia MK, Paintlia AS, Singh AK, Singh I. Synergistic activity of interleukin-17 and tumor necrosis factor-alpha enhances oxidative stress-mediated oligodendrocyte apoptosis. J Neurochem. 2011;116:508–521. doi: 10.1111/j.1471-4159.2010.07136.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, Gross CT. Synaptic pruning by microglia is necessary for normal brain development. Science. 2011;333:1456–1458. doi: 10.1126/science.1202529. [DOI] [PubMed] [Google Scholar]
- Perron JC, Bixby JL. Distinct neurite outgrowth signaling pathways converge on ERK activation. Mol Cell Neurosci. 1999;13:362–378. doi: 10.1006/mcne.1999.0753. [DOI] [PubMed] [Google Scholar]
- Prat A, Biernacki K, Wosik K, Antel JP. Glial cell influence on the human blood-brain barrier. Glia. 2001;36:145–155. doi: 10.1002/glia.1104. [DOI] [PubMed] [Google Scholar]
- Qian X, Shen Q, Goderie SK, He W, Capela A, Davis AA, Temple S. Timing of CNS cell generation: a programmed sequence of neuron and glial cell production from isolated murine cortical stem cells. Neuron. 2000;28:69–80. doi: 10.1016/s0896-6273(00)00086-6. [DOI] [PubMed] [Google Scholar]
- Rebay I, Rubin GM. Yan functions as a general inhibitor of differentiation and is negatively regulated by activation of the Ras1/MAPK pathway. Cell. 1995;81:857–866. doi: 10.1016/0092-8674(95)90006-3. [DOI] [PubMed] [Google Scholar]
- Richter F, Natura G, Ebbinghaus M, von Banchet GS, Hensellek S, Konig C, Brauer R, Schaible HG. Interleukin-17 sensitizes joint nociceptors to mechanical stimuli and contributes to arthritic pain through neuronal interleukin-17 receptors in rodents. Arthritis Rheum. 2012;64:4125–4134. doi: 10.1002/art.37695. [DOI] [PubMed] [Google Scholar]
- Risch N, Spiker D, Lotspeich L, Nouri N, Hinds D, Hallmayer J, Kalaydjieva L, McCague P, Dimiceli S, Pitts T, Nguyen L, Yang J, Harper C, Thorpe D, Vermeer S, Young H, Hebert J, Lin A, Ferguson J, Chiotti C, Wiese-Slater S, Rogers T, Salmon B, Nicholas P, Petersen PB, Pingree C, McMahon W, Wong DL, Cavalli-Sforza LL, Kraemer HC, Myers RM. A genomic screen of autism: evidence for a multilocus etiology. Am J Hum Genet. 1999;65:493–507. doi: 10.1086/302497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JM, Robinson AP, Rosler ES, Lariosa-Willingham K, Persons RE, Dugas JC, Miller SD. IL-17A activates ERK1/2 and enhances differentiation of oligodendrocyte progenitor cells. Glia. 2015;63:768–779. doi: 10.1002/glia.22783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JM, Zhou L, Miller SD. Act1, scene brain: astrocytes play a lead role. Immunity. 2010;32:302–304. doi: 10.1016/j.immuni.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008;28:6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson AM, Jennische E, Hansson HA, Holmang A. Prenatal exposure to interleukin-6 results in inflammatory neurodegeneration in hippocampus with NMDA/GABA(A) dysregulation and impaired spatial learning. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1345–1356. doi: 10.1152/ajpregu.00268.2005. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of pavlovian fear conditioning. J Neurosci. 2000;20:8177–8187. doi: 10.1523/JNEUROSCI.20-21-08177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartzer JJ, Careaga M, Onore CE, Rushakoff JA, Berman RF, Ashwood P. Maternal immune activation and strain specific interactions in the development of autism-like behaviors in mice. Transl Psychiatry. 2013;3:e240. doi: 10.1038/tp.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebkova L, Pellicano A, Monteleone G, Grazioli B, Guarnieri G, Imeneo M, Pallone F, Luzza F. Extracellular signal-regulated protein kinase mediates interleukin 17 (IL-17)-induced IL-8 secretion in Helicobacter pylori-infected human gastric epithelial cells. Infect Immun. 2004;72:5019–5026. doi: 10.1128/IAI.72.9.5019-5026.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segond von Banchet G, Boettger MK, Konig C, Iwakura Y, Brauer R, Schaible HG. Neuronal IL-17 receptor upregulates TRPV4 but not TRPV1 receptors in DRG neurons and mediates mechanical but not thermal hyperalgesia. Mol Cell Neurosci. 2013;52:152–160. doi: 10.1016/j.mcn.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. 2013:106–107. 1–16. doi: 10.1016/j.pneurobio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Fatemi S, Sidwell R, Patterson P. Maternal Influenza Infection Causes Marked Behavioral and Pharmacological Changes in the Offspring. Journal of Neuroscience. 2003:297. doi: 10.1523/JNEUROSCI.23-01-00297.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Smith SE, Malkova N, Tse D, Su Y, Patterson PH. Activation of the maternal immune system alters cerebellar development in the offspring. Brain Behav Immun. 2009;23:116–123. doi: 10.1016/j.bbi.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell. 2010;7:483–495. doi: 10.1016/j.stem.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smolders S, Smolders SM, Swinnen N, Gartner A, Rigo JM, Legendre P, Brone B. Maternal immune activation evoked by polyinosinic:polycytidylic acid does not evoke microglial cell activation in the embryo. Front Cell Neurosci. 2015;9:301. doi: 10.3389/fncel.2015.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Dai D, He X, Zhu S, Yao Y, Gao H, Wang J, Qu F, Qiu J, Wang H, Li X, Shen N, Qian Y. Growth Factor FGF2 Cooperates with Interleukin-17 to Repair Intestinal Epithelial Damage. Immunity. 2015;43:488–501. doi: 10.1016/j.immuni.2015.06.024. [DOI] [PubMed] [Google Scholar]
- Squarzoni P, Oller G, Hoeffel G, Pont-Lezica L, Rostaing P, Low D, Bessis A, Ginhoux F, Garel S. Microglia modulate wiring of the embryonic forebrain. Cell Rep. 2014;8:1271–1279. doi: 10.1016/j.celrep.2014.07.042. [DOI] [PubMed] [Google Scholar]
- Stevens B. Neuron-astrocyte signaling in the development and plasticity of neural circuits. Neurosignals. 2008;16:278–288. doi: 10.1159/000123038. [DOI] [PubMed] [Google Scholar]
- Stoner R, Chow ML, Boyle MP, Sunkin SM, Mouton PR, Roy S, Wynshaw-Boris A, Colamarino SA, Lein ES, Courchesne E. Patches of disorganization in the neocortex of children with autism. N Engl J Med. 2014;370:1209–1219. doi: 10.1056/NEJMoa1307491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stritesky GL, Yeh N, Kaplan MH. IL-23 promotes maintenance but not commitment to the Th17 lineage. J Immunol. 2008;181:5948–5955. doi: 10.4049/jimmunol.181.9.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF) Nat Immunol. 2011;12:853–860. doi: 10.1038/ni.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Matsuzaki H, Iwata K, Kameno Y, Shimmura C, Kawai S, Yoshihara Y, Wakuda T, Takebayashi K, Takagai S, Matsumoto K, Tsuchiya KJ, Iwata Y, Nakamura K, Tsujii M, Sugiyama T, Mori N. Plasma cytokine profiles in subjects with high-functioning autism spectrum disorders. PloS one. 2011;6:e20470. doi: 10.1371/journal.pone.0020470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swaidani S, Bulek K, Kang Z, Liu C, Lu Y, Yin W, Aronica M, Li X. The critical role of epithelial-derived Act1 in IL-17- and IL-25-mediated pulmonary inflammation. J Immunol. 2009;182:1631–1640. doi: 10.4049/jimmunol.182.3.1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen N, Smolders S, Avila A, Notelaers K, Paesen R, Ameloot M, Brone B, Legendre P, Rigo JM. Complex invasion pattern of the cerebral cortex bymicroglial cells during development of the mouse embryo. Glia. 2013;61:150–163. doi: 10.1002/glia.22421. [DOI] [PubMed] [Google Scholar]
- Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, Tocker J, Peschon J. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- Trajkovic V, Stosic-Grujicic S, Samardzic T, Markovic M, Miljkovic D, Ramic Z, Mostarica Stojkovic M. Interleukin-17 stimulates inducible nitric oxide synthase activation in rodent astrocytes. J Neuroimmunol. 2001;119:183–191. doi: 10.1016/s0165-5728(01)00391-5. [DOI] [PubMed] [Google Scholar]
- Traynor TR, Majde JA, Bohnet SG, Krueger JM. Sleep and body temperature responses in an acute viral infection model are altered in interferon type I receptor-deficient mice. Brain Behav Immun. 2006;20:290–299. doi: 10.1016/j.bbi.2005.08.008. [DOI] [PubMed] [Google Scholar]
- van der Zwaag B, Franke L, Poot M, Hochstenbach R, Spierenburg HA, Vorstman JA, van Daalen E, de Jonge MV, Verbeek NE, Brilstra EH, van 't Slot R, Ophoff RA, van Es MA, Blauw HM, Veldink JH, Buizer-Voskamp JE, Beemer FA, van den Berg LH, Wijmenga C, van Amstel HK, van Engeland H, Burbach JP, Staal WG. Gene-network analysis identifies susceptibility genes related to glycobiology in autism. PloS one. 2009;4:e5324. doi: 10.1371/journal.pone.0005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas DL, Nascimbene C, Krishnan C, Zimmerman AW, Pardo CA. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann Neurol. 2005;57:67–81. doi: 10.1002/ana.20315. [DOI] [PubMed] [Google Scholar]
- Velichko S, Zhou X, Zhu L, Anderson JD, Wu R, Chen Y. A Novel Nuclear Function for the Interleukin-17 Signaling Adaptor Protein Act1. PloS one. 2016;11:e0163323. doi: 10.1371/journal.pone.0163323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380–384. doi: 10.1038/nature10110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voo KS, Wang YH, Santori FR, Boggiano C, Wang YH, Arima K, Bover L, Hanabuchi S, Khalili J, Marinova E, Zheng B, Littman DR, Liu YJ. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proc Natl Acad Sci U S A. 2009;106:4793–4798. doi: 10.1073/pnas.0900408106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walz R, Lenz G, Roesler R, Vianna MM, Martins V, Brentani R, Rodnight R, Izquierdo I. Time-dependent enhancement of inhibitory avoidance retention and MAPK activation by post-training infusion of nerve growth factor into CA1 region of hippocampus of adult rats. Eur J Neurosci. 2000;12:2185–2189. doi: 10.1046/j.1460-9568.2000.00123.x. [DOI] [PubMed] [Google Scholar]
- Wang DD, Zhao YF, Wang GY, Sun B, Kong QF, Zhao K, Zhang Y, Wang JH, Liu YM, Mu LL, Wang DS, Li HL. IL-17 potentiates neuronal injury induced by oxygen-glucose deprivation and affects neuronal IL-17 receptor expression. J Neuroimmunol. 2009;212:17–25. doi: 10.1016/j.jneuroim.2009.04.007. [DOI] [PubMed] [Google Scholar]
- Wei L, Laurence A, Elias KM, O'Shea JJ. IL-21 is produced by Th17 cells and drives IL-17 production in a STAT3-dependent manner. J Biol Chem. 2007;282:34605–34610. doi: 10.1074/jbc.M705100200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Alshikho MJ, Herbert MR. Pathway Network Analyses for Autism Reveal Multisystem Involvement, Major Overlaps with Other Diseases and Convergence upon MAPK and Calcium Signaling. PloS one. 2016;11:e0153329. doi: 10.1371/journal.pone.0153329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienecke J, Hebel K, Hegel KJ, Pierau M, Brune T, Reinhold D, Pethe A, Brunner-Weinzierl MC. Pro-inflammatory effector Th cells transmigrate through anti-inflammatory environments into the murine fetus. Placenta. 2012;33:39–46. doi: 10.1016/j.placenta.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Wilkerson DS, Volpe AG, Dean RS, Titus JB. Perinatal complications as predictors of infantile autism. Int J Neurosci. 2002;112:1085–1098. doi: 10.1080/00207450290026076. [DOI] [PubMed] [Google Scholar]
- Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- Wright JF, Guo Y, Quazi A, Luxenberg DP, Bennett F, Ross JF, Qiu Y, Whitters MJ, Tomkinson KN, Dunussi-Joannopoulos K, Carreno BM, Collins M, Wolfman NM. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J Biol Chem. 2007;282:13447–13455. doi: 10.1074/jbc.M700499200. [DOI] [PubMed] [Google Scholar]
- Wu HX, Jin LP, Xu B, Liang SS, Li DJ. Decidual stromal cells recruit Th17 cells into decidua to promote proliferation and invasion of human trophoblast cells by secreting IL-17. Cell Mol Immunol. 2014;11:253–262. doi: 10.1038/cmi.2013.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu WL, Adams CE, Stevens KE, Chow KH, Freedman R, Patterson PH. The interaction between maternal immune activation and alpha 7 nicotinic acetylcholine receptor in regulating behaviors in the offspring. Brain Behav Immun. 2015;46:192–202. doi: 10.1016/j.bbi.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YY, Bradshaw RA. Induction of neurite outgrowth by interleukin-6 is accompanied by activation of Stat3 signaling pathway in a variant PC12 cell (E2) line. J Biol Chem. 1996;271:13023–13032. doi: 10.1074/jbc.271.22.13023. [DOI] [PubMed] [Google Scholar]
- Yalcin A, Koulich E, Mohamed S, Liu L, D'Mello SR. Apoptosis in cerebellar granule neurons is associated with reduced interaction between CREB-binding protein and NF-kappaB. J Neurochem. 2003;84:397–408. doi: 10.1046/j.1471-4159.2003.01540.x. [DOI] [PubMed] [Google Scholar]
- Yamazaki M, Chiba K, Mohri T, Hatanaka H. Activation of the mitogen-activated protein kinase cascade through nitric oxide synthesis as a mechanism of neuritogenic effect of genipin in PC12h cells. J Neurochem. 2001;79:45–54. doi: 10.1046/j.1471-4159.2001.00533.x. [DOI] [PubMed] [Google Scholar]
- Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, Cohen JI, Spriggs MK. Herpesvirus Saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity. 1995;3:811–821. doi: 10.1016/1074-7613(95)90070-5. [DOI] [PubMed] [Google Scholar]
- Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, Shellito JE, Bagby GJ, Nelson S, Charrier K, Peschon JJ, Kolls JK. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519–527. doi: 10.1084/jem.194.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh SH, Lin CH, Lee CF, Gean PW. A requirement of nuclear factor-kappaB activation in fear-potentiated startle. J Biol Chem. 2002;277:46720–46729. doi: 10.1074/jbc.M206258200. [DOI] [PubMed] [Google Scholar]
- Yufune S, Satoh Y, Takamatsu I, Ohta H, Kobayashi Y, Takaenoki Y, Pages G, Pouyssegur J, Endo S, Kazama T. Transient Blockade of ERK Phosphorylation in the Critical Period Causes Autistic Phenotypes as an Adult in Mice. Sci Rep. 2015;5:10252. doi: 10.1038/srep10252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenclussen AC, Gerlof K, Zenclussen ML, Ritschel S, Zambon Bertoja A, Fest S, Hontsu S, Ueha S, Matsushima K, Leber J, Volk HD. Regulatory T cells induce a privileged tolerant microenvironment at the fetal-maternal interface. Eur J Immunol. 2006;36:82–94. doi: 10.1002/eji.200535428. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Huang R, Zhang Y, Yi H, Bai Y, Chao J, Yao H. IL-17 induces MIP-1alpha expression in primary mouse astrocytes via TRPC channel. Inflammopharmacology. 2016;24:33–42. doi: 10.1007/s10787-015-0256-x. [DOI] [PubMed] [Google Scholar]
- Zhao D, Lin M, Chen J, Pedrosa E, Hrabovsky A, Fourcade HM, Zheng D, Lachman HM. MicroRNA Profiling of Neurons Generated Using Induced Pluripotent Stem Cells Derived from Patients with Schizophrenia and Schizoaffective Disorder, and 22q11.2 Del. PloS one. 2015;10:e0132387. doi: 10.1371/journal.pone.0132387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol. 2007;8:967–974. doi: 10.1038/ni1488. [DOI] [PubMed] [Google Scholar]
- Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR. TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature. 2008;453:236–240. doi: 10.1038/nature06878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziats MN, Rennert OM. Expression profiling of autism candidate genes during human brain development implicates central immune signaling pathways. PloS one. 2011;6:e24691. doi: 10.1371/journal.pone.0024691. [DOI] [PMC free article] [PubMed] [Google Scholar]