

Abstract
In 2013 we unveiled the cryo-electron microscopy (CryoEM) method of MicroED, or three-dimensional (3D) electron diffraction of microscopic crystals. Here tiny 3D crystals of biological material are used in an electron microscope for diffraction data collection under cryogenic conditions. The data is indexed, integrated, merged and scaled using standard X-ray crystallography software to determine structures at atomic resolution. In this review we provide an overview of the MicroED method and compare it with other CryoEM methods.
Introduction
Whether being used to obtain images of individual biomolecules or using crystalline samples to collect high-resolution data through diffraction methods, cryo-electron microscopy, or CryoEM, is revolutionizing structural biology. CryoEM can be separated into four techniques for molecular structure determination (Table 1): cryo-tomography, single particle reconstruction, 2D electron crystallography, and Micro-electron diffraction, or MicroED. Cryo-tomography is generally used to study intact biomolecular complexes within the cellular environment. This provides mechanistic insights that are difficult to obtain by other methods. However, the structural data produced by cryo-tomography are relatively low resolution (1–5 nm resolution for thin samples). Single particle reconstructions are capable of producing biomolecular structures at near atomic resolution without the need of crystals as long as the samples are large (>200 kDa) homogenous and stable [1]. Both the 2D electron crystallography and MicroED techniques use crystalline material; however due to the significant differences in samples (2D crystalline arrays vs. 3D crystals), differences in data collection (still diffraction vs. continuous rotation), and data processing (MRC suite of software vs. standard X-ray processing software), they should be considered distinct branches of CryoEM. In this review, we will focus on these two crystal-based CryoEM techniques, and describe recent progress in MicroED.
Table 1.
Techniques within molecular cryo-electron microscopy
| Cryo-tomography | Single particle reconstructions | Electron crystallography | MicroED | |
|---|---|---|---|---|
| Samples typically used | Whole cells or organelles | Purified biomolecules | 2D crystals | 3D crystals |
| Strengths | Directly observe molecular interactions | Material required is significantly less than in crystallography | In the case of membrane proteins, the interactions with lipid bilayer can be seen | Crystals used are several orders of magnitude smaller than those used in standard X-ray crystallography |
| Biomolecules are in the native cellular environment | Multiple conformations can be solved with a single sample | Protein dynamics probed | Data can be processed in standard X-ray crystallography programs | |
| No crystals necessary | No crystals necessary | Highest resolution cryo-EM technique | ||
| Shortcomings | Relatively low resolution | Requires large proteins or protein complexes (size limitation) | Requires 2D crystals | Requires 3D crystals |
| Modest resolution | Incomplete data in the direction parallel to the electron beam |
Electron diffraction in materials and in biology
Electron diffraction is a powerful tool for studying the atomic structures of materials and biological samples. Many methods have been developed over the years by a number of groups focusing on inorganic materials or small organic compounds that are relatively dose insensitive [2–6]. For biological samples that are dose sensitive and should be studied in cryogenic conditions with low dose methods, electron diffraction has traditionally been restricted to the use of two-dimensional (2D) crystals, which consist of a 1–3 layers of biomolecules ordered in a 2D crystalline lattice. These 2D crystals are capable of yielding atomic resolution structures of membrane proteins in the surrounding lipid environment [7,8]; however most structures solved by 2D crystallography are at modest resolutions (4–10 Å). It was not until 2013 that electron diffraction was used for the first time for determining the structure of a protein from 3D crystals by a newly established method called MicroED [9••]. This method has since been used to solve a number of macromolecular structures at cryogenic temperatures and with extremely low electron dose (Table 2).
Table 2.
Structures determined by MicroED
| Sample | Year | Data collection approach | Resolution | PDB | EMDB | SBGRID |
|---|---|---|---|---|---|---|
| Lysozyme | 2013 | Still diffraction | 2.9 Å | 3J4G [9••] | 2945 | |
| 2014 | Continuous rotation | 2.5 Å | 3J6K [42••] | 6342 | 185 | |
| 2016 | Continuous rotation | 1.5 Å | 5K7O | 8217 | ||
| Catalase | 2014 | Continuous rotation | 3.2 Å | 3J7B [45] | 6314 | 186 |
| 2015 | Continuous rotation | 3.2 Å | 3J7U [53] | |||
| Ca2+-ATPase | 2015 | Continuous rotation | 3.4 Å | 3J7T [53] | ||
| α-Synuclein NACore | 2015 | Continuous rotation | 1.4 Å | 4RIL [46••] | 3028 | 193 |
| α-Synuclein preNAC | 2015 | Continuous rotation | 1.4 Å | 4ZNN [46••] | 3001 | |
| Proteinase K | 2016 | Continuous rotation | 1.75 Å | 5I9S [54] | 8077 | 262 |
| 2016 | Continuous rotation | 1.3 Å | 5K7S | 8221 | ||
| Prion Zn-NNQQNY | 2016 | Continuous rotation | 1.0 Å | 5K2E [55] | 8196 | |
| Prion Cd-NNQQNY | 2016 | Continuous rotation | 1.0 Å | 5K2F [55] | 8197 | |
| Prion GNNQQNY1 | 2016 | Continuous rotation | 1.1 Å | 5K2G [55] | 8198 | |
| Prion GNNQQNY2 | 2016 | Continuous rotation | 1.05 Å | 5K2H [55] | 8199 | |
| Tau peptide | 2016 | Continuous rotation | 1.1 Å | 5K7N | 8216 | |
| Xylanase | 2016 | Continuous rotation | 1.9 Å | 5K7P | 8218 | |
| Thaumatin | 2016 | Continuous rotation | 2.11 Å | 5K7Q | 8219 | |
| Trypsin | 2016 | Continuous rotation | 1.5 Å | 5K7R | 8220 | |
| Thermolysin | 2016 | Continuous rotation | 1.6 Å | 5K7T | 8222 |
Although many aspects of electron diffraction are analogous to X-ray crystallography, there are some important distinctions that should be kept in mind when comparing the two techniques. First, electrons interact much stronger with matter than X-rays and deposit less energy onto the sample [10]. This means that electron diffraction can extract meaningful high-resolution data from crystals that are orders of magnitude smaller in volume than what is needed from a conventional X-ray crystallography experiment. Second, the wavelength of electrons produced in an electron microscope is significantly smaller than the wavelengths used for X-ray crystallography. For example, the wavelength of the electrons from a microscope operated at 200 kV (λ = 0.0251 Å) is over 60 times smaller than the X-rays from the commonly used Copper Kα source (λ = 1.54 Å). This leads to an Ewald sphere that is much larger with electron diffraction, and the Ewald sphere is essentially flat at the resolutions seen in macromolecular crystallography. Finally, because the scattered electrons can be focused by the microscope, images of the crystals can also be collected and accurate phase information can be recovered.
Biomolecular electron crystallography requires special set-up of the microscope because of the sensitivity of biological samples when compared with hard materials. Hydration is critical, and in order for the hydrated sample to withstand the high-vacuum within the TEM and still produce high-resolution data, samples are vitrified and loaded into a cryo-transmission electron microscope (cryo-TEM) [11]. Also, when compared to samples used in materials science, biological specimens are extremely radiation sensitive [12]. Therefore, very low doses are used when collecting MicroED data from biological material. We use approximately 0.01 e−/Å2/s for biological samples, orders of magnitude smaller than what is used with more dose resistant samples that are typically exposed to approximately 1000 e−/Å2/s.
Electron crystallography of 2D protein arrays
The best-known examples of 2D electron crystallography involve work with membrane proteins that are crystallized within a lipid bilayer and produce planar 2D-crystals of protein and lipid. The pioneering work of Henderson and Unwin in 1975 produced 7 Å projection maps from naturally occurring 2D crystals of bacteriorhodopsin [13], which revealed how the α-helices are organized within a transmembrane protein for the first time. Later work with bacteriorhodopsin 2D crystals yielded a high-resolution data where a complete atomic model of the protein could be built and refined [14]. Following the work on bacteriorhodopsin, several high-resolution structures of membrane proteins have been solved, including, but not limited to, the plant light-harvesting complex II [15] and several aquaporins [7,8,16,17]. The structure of taxol stabilized αβ tubulin was also solved by this method although the protein is not membrane embedded [18]. In addition to producing high-resolution structures, 2D electron crystallography also provides quality structural information from relatively low-resolution structures, due to the high quality phase information obtained from imaging the crystals [19•,20–23].
2D crystals are typically grown from solutions when detergent solubilized lipids are mixed together with the detergent solubilized membrane protein of interest. Subsequent removal of the detergent by slow dialysis leads to protein reconstitution and crystallization within the newly forming lipid membranes [24]. Crystals are applied to a continuous-carbon coated EM grid, excess liquid is removed by blotting, and the preparation is then frozen in liquid ethane or nitrogen. After being loaded into the cryoEM, either images or diffraction data can be collected. To obtain a 3D dataset, diffraction patterns or images of the crystals must be collected at various tilt angles. Each crystal is imaged or diffracted at a known tilt angle. Radiation damage prohibits the collection of a full tilt series from a single 2D crystal. Therefore, full data sets are constructed by merging data from many crystals covering the tilt range. Because the 2D crystals all lie on the grid in a preferred orientation and the samples cannot be tilted beyond a certain angle, data collected from 2D crystals suffer from a systematic incompleteness often referred to as a ‘missing cone’ [25]. This leads to anisotropic data, with higher resolution data being obtained parallel to the plane of the membrane.
Collecting images of 2D crystals provides both amplitudes and phases, and are sufficient to determine a 3D density map of the protein. However, while the phases are extremely accurate, the amplitudes are affected by the contrast transfer function and are not as high-quality. Additionally, obtaining high resolution images can be difficult especially at high-tilt angles. Imaging difficulties arise in large part due to mechanical and thermal induced drifting of the sample and charging effects that reduce the obtainable resolution [26]. The collection of electron diffraction data is much less sensitive to these same sample drift problems, and can produce high-resolution and high-quality amplitude information. However, with diffraction all phase information is lost. To obtain phases, the high-resolution diffraction data can be combined with the high-quality phase information obtained from imaging. When the diffraction resolution is significantly higher than that of the images, as is often the case, fragment-based phase extension methods can be used [27]. Detailed procedures for performing 2D crystallographic experiments can be found in the many reviews written on the topic [24,28–30].
Electron crystallography of 2D arrays has been used effectively to study protein dynamics. This has been accomplished for a number of membrane proteins including: the light activated bacteriorhodopsin [31], the acetylcholine receptor embedded in tubular arrays [32], and more recently the voltage gated sodium channel [19•].
Electron crystallography goes 3D with MicroED
Although the initial work with 2D crystals was being performed in the 1970s, thin 3D microcrystals (a few hundred nanometers thick) were also being studied by EM, with the best example being bovine liver catalase [33–38]. In work by Dorset and Parsons, it was shown that hydrated catalase microcrystals could provide diffraction data beyond 3 Å and that the diffraction was not affected by multiple scattering [35]. Not long after this work, Unwin and Henderson determined a 9 Å projection structure of catalase from 3D catalase crystals preserved with glucose [33]. More recently 3D crystals of lysozyme were imaged and the mosaic building blocks identified [39•]. Although these were important steps forward for electron microscopy, a full 3D structure was never determined because the existing methodology, specifically how collect and process 3D diffraction data, was not adequate for 3D crystals. Therefore, it was important that a new method for data collection had to be developed.
In 2013, a new technique called Microcrystal Electron Diffraction [9••,40], or MicroED, was developed based on the idea that if multiple diffraction patters were taken from a single crystal, a large enough region of reciprocal space could be sampled, allowing proper indexing and processing of the diffraction data set. The first iteration of MicroED used a series of still diffraction patterns from single crystals (approximately 2 million unit cells in size), with the crystal rotated in discreet angles between exposures. Each diffraction pattern was collected at liquid nitrogen temperature with an ultra-low dose rate of ~0.01 e−/Å2/s. This extremely low dose rate still produced quality diffraction patterns to high resolution (up to 1.7 Å with lysozyme microcrystals initially) and allowed each crystal to be exposed up to 90 times [9••]. High-tilt cryoholders were used which allowed the samples to be tilted up to ±70° (140° total possible rotation range), and the crystals were tilted by 1° between exposures. This led to data sets which produced up to a 90° wedge of data from a single crystal. Because such a large amount of reciprocal space was sampled, the crystal orientation could be determined accurately and the diffraction patterns properly indexed. Each crystal data set was indexed and integrated, followed by merging of all data into one final data set. The data from 3 crystals were integrated and merged using software developed specifically for still diffraction MicroED data [41]. This initial proof of concept work yielded a refined lysozyme structure to 2.9 Å (Figure 1a) with very good refinement statistics, and represented the first time electron diffraction had been used successfully to determine the structure of a protein from 3D microcrystals in cryogenic conditions.
Figure 1.
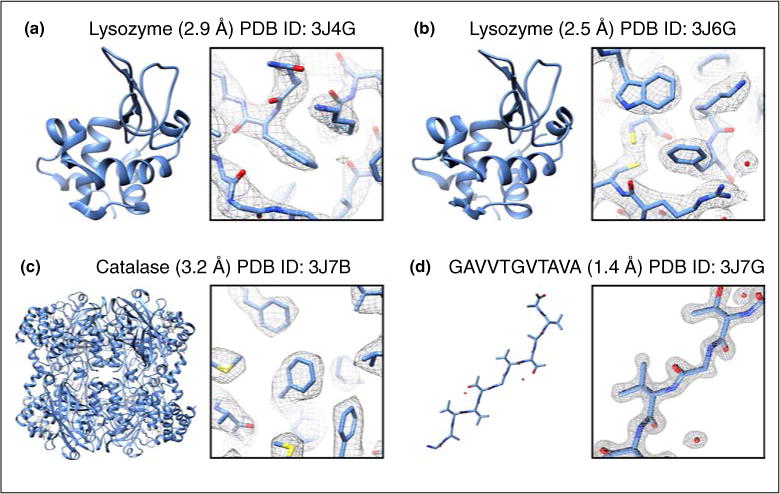
Example structures determined from MicroED. Structures are shown along with their PDB ID, resolution, full model (left) and representative region of the model and density map (right).
Following this first proof of principle work, an improved method of MicroED data collection was developed [42••]. This new method, called ‘continuous-rotation’ MicroED data collection, uses the high frame rates achievable by a CMOS detector or a direct electron detector to collect diffraction data as the crystal is continuously rotated in the electron beam by the sample stage of the microscope. The methodology is similar to other techniques used for non-biological dose insensitive samples studied at ambient temperatures [2,4–6]. Continuous rotation MicroED led to several improvements in data quality and allowed for data processing using standard X-ray crystallographic software [43•]. The first benefit of continuous rotation data collection comes from a reduction in dynamic scattering, a process where the electron beam diffracts multiple times within the sample leading to a redistribution of reflection intensities. As was seen previously with precession electron diffraction, rotating the sample by continuous rotation (or the beam in the case of precession) is able to reduce dynamic scattering and intensity redistribution, which ultimately leads to more accurate intensity measurements [44]. The second advantage comes from the improved sampling of reciprocal space that occurs as the sample is rotated and data is collected. This allows the collection of full reflection intensities as opposed to the partial sampling obtained when the crystal remains still during data collection of individual frames. The third benefit of continuous rotation is that the movement of the crystal in the electron beam is similar to the crystal movements in X-ray crystallography. Because the experimental setups are so similar, MicroED data collected by continuous rotation can be processed by programs developed for X-ray diffraction data. Because of its many advantages over the collection of diffraction stills, ‘continuous-rotation’ data collection has become the standard method of MicroED data collection, and its first use resulted in a 2.5 Å structure of lysozyme using a single nanocrystal (Figure 1b). From this point forward, any mention of MicroED data collection in this review refers to continuous rotation data collection.
Following these studies the MicroED method was tested on a new sample, and for historical reasons mentioned above, we chose to study bovine liver catalase microcrystals [45]. Catalase (P212121; a = 67.8 Å, b =172.1 Å, c = 182.1 Å; α = β = γ = 90°) presented a few unique challenges for MicroED, which included a larger unit cell and lower symmetry compared to lysozyme (P43212; a = b = 76.0 Å, c =37.2 Å; α = β = γ =90°), and crystals which have a preferred orientation on the grid resulting in a missing cone of data in the direction of the c* crystallographic axis. Data from a single nanocrystal (approximately 15 × 7 × 0.2 μm) was collected while the stage rotated from approximately 53° to 0°. Subsequent data processing yielded a final scaled and merged diffraction data set from this single crystal that was 79.4% complete to 3.2 Å. With this amount of data, an acceptable molecular replacement solution could be found and the 3.2 Å resolution structure (Figure 1c) was refined with acceptable statistics (Rwork/Rfree = 26.2%/30.8%). Additional experiments with a molecular replacement search model that did not contain the bound NADP co-factor showed that the missing NADP could be found in the difference density maps. Furthermore, what was more exciting was that the subtle NADPH induced conformational change of phenylalanine-197 could be seen using the difference density maps, indicating the quality of MicroED data is in fact very high.
The first novel structures solved by MicroED were published in late 2015 [46••]. These structures were of peptide fragments that form the toxic core of α-synculein, the protein responsible for Parkinson’s disease and lead to very important insights into how the protein forms toxic aggregates. The two α-synculein peptides formed very small and thin needle like nanocrystals (approximately 1–2 μm long, 20–50 nm wide and thick; Figure 2) and had resisted all other methods of structure determination, including those that utilize X-ray Free Electron Lasers (XFELs). The structures were solved by MicroED at 1.4 Å resolution and at the time represented the highest resolution structures of a biological molecules determined by any cryo-electron microscopy technique published to date (Figure 1d). More recent MicroED studies allowed direct phasing methods to be used for structure determination of 4 different samples at 1 Å resolution [55].
Figure 2.
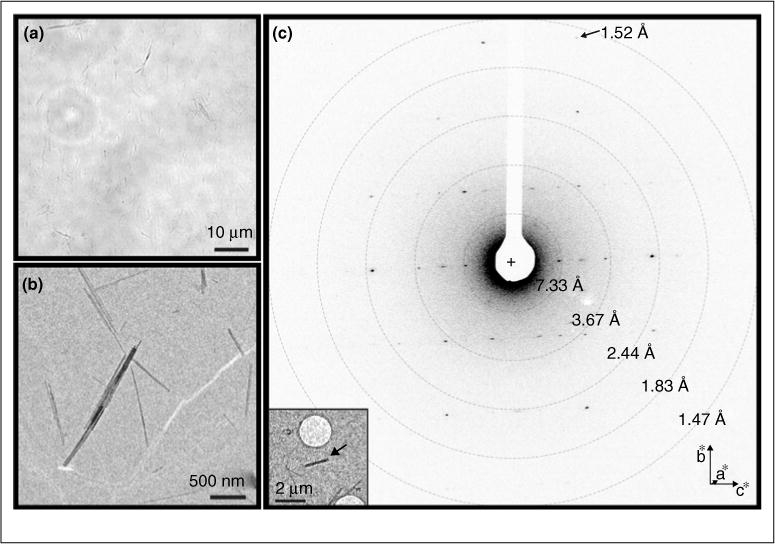
Crystals from the toxic core of α-synuclein. The crystals used to determine the α-synuclein structure were much too small to be seen by light microscopy (a). However, when visualized within the TEM many extremely small microcrystals could be seen (b), which diffracted to approximately 1.4 Å (c).
Source: Adapted from Ref. [46••].
MicroED experimental setup and data collection
Samples prepared for MicroED are typically grown in standard hanging or sitting drop crystallization experiments as for X-ray crystallography. Microcrystals are transferred to a glow discharged grid by pipetting the drop directly onto the carbon surface. The excess sample is blotted and vitrified using automated vitrification devices in a procedure similar to what is used for single-particle or electron tomography cryo-EM work. Alternatively, the grids can be blotted by hand and frozen manually as for 2D crystals. Vitrified samples can be stored under liquid nitrogen until loading into a standard cryo-EM. Once a suitable crystal is found, the crystal is exposed with the beam in focus and diffraction data is continuously collected as the stage is rotated in the beam. See Figure 3 for a flow chart of the data collection process. Detailed protocols for sample preparation data collection have been recently published [47•].
Figure 3.
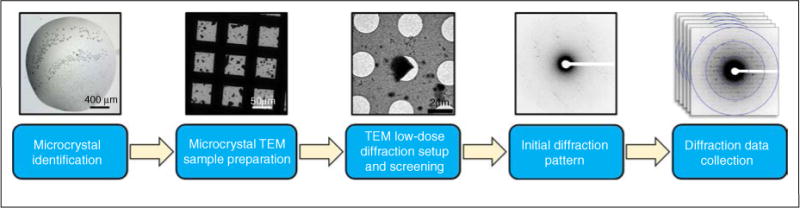
Flow diagram for MicroED data collection protocol.
Source: Adapted from Ref. [47•].
MicroED data processing
Following data collection, the diffraction data set is indexed, integrated, merged and scaled using standard macromolecular crystallography programs originally developed for X-ray crystallography. Initially, data were processed in MOSFLM [48,49]; however, data has since also been processed in HKL2000 [50], DIALS [51], and XDS [52]. In depth descriptions and protocols on MicroED data processing have been previously published [43•], and we encourage readers to refer to this publication for detailed descriptions of the data processing procedures.
MicroED future directions
MicroED is still a relatively new method in CryoEM and there are many exciting areas of future research and improvements. One of the most interesting areas of MicroED research is the development of phasing methods. As was done with first structures solved by X-ray crystallography, the phase could be solved by the use of heavy atoms for isomorphous replacement could potentially be used to obtain phase information. A more unique interesting approach to obtain phases would be to incorporate imaging into MicroED. Images of 3D microcrystals have previously been collected and processed [39•], however integration into diffraction data has not been accomplished. The development of more accurate electron scattering factors promises to greatly improve refinement. There are also many other improvements to sample preparation, data collection, and data processing that will be developed when more samples are investigated with MicroED. It is important to note that many labs have been producing microcrystals for serial femtosecond crystallography (SFX), and many of these can be directly used for MicroED. Currently, the main advantages of MicroED over SFX are the availability of cryo-TEM instrumentation for MicroED relative to the extremely limited access to XFELS for SFX, and the smaller amount of sample required for MicroED (1–10 crystals for MicroED vs. thousands to millions for SFX). As MicroED is further improved and refined, the technique promises to become a widespread tool for the determination of difficult biomolecular samples that form microcrystals. Together with other techniques, we expect that MicroED will play a major role in macromolecular structure determination in the coming decades.
Acknowledgments
The Gonen laboratory is supported by the Howard Hughes Medical Institute.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Bartesaghi A, Merk A, Banerjee S, Matthies D, Wu X, Milne JL, Subramaniam S. 2.2 A resolution cryo-EM structure of beta-galactosidase in complex with a cell-permeant inhibitor. Science. 2015;348:1147–1151. doi: 10.1126/science.aab1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang YB, Su J, Furukawa H, Yun YF, Gandara F, Duong A, Zou XD, Yaghi OM. Single-crystal structure of a covalent organic framework. J Am Chem Soc. 2013;135:16336–16339. doi: 10.1021/ja409033p. [DOI] [PubMed] [Google Scholar]
- 3.Wan W, Sun JL, Su J, Hovmoller S, Zou XD. Three-dimensional rotation electron diffraction: software RED for automated data collection and data processing. J Appl Crystallogr. 2013;46:1863–1873. doi: 10.1107/S0021889813027714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mugnaioli E, Gorelik T, Kolb U. “Ab initio” structure solution from electron diffraction data obtained by a combination of automated diffraction tomography and precession technique. Ultramicroscopy. 2009;109:758–765. doi: 10.1016/j.ultramic.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Jiang JX, Jorda JL, Yu JH, Baumes LA, Mugnaioli E, Diaz-Cabanas MJ, Kolb U, Corma A. Synthesis and structure determination of the hierarchical meso-microporous zeolite ITQ-43. Science. 2011;333:1131–1134. doi: 10.1126/science.1208652. [DOI] [PubMed] [Google Scholar]
- 6.Gemmi M, La Placa MGI, Galanis AS, Rauch EF, Nicolopoulos S. Fast electron diffraction tomography. J Appl Crystallogr. 2015;48:718–727. [Google Scholar]
- 7.Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–638. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gonen T, Sliz P, Kistler J, Cheng Y, Walz T. Aquaporin-0 membrane junctions reveal the structure of a closed water pore. Nature. 2004;429:193–197. doi: 10.1038/nature02503. [DOI] [PubMed] [Google Scholar]
- 9••.Shi D, Nannenga BL, Iadanza MG, Gonen T. Three-dimensional electron crystallography of protein microcrystals. Elife. 2013;2:e01345. doi: 10.7554/eLife.01345. This was the first study to use MicroED and represented the first time 3D microcrystals were used for structure determination in cryoTEM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henderson R. The potential and limitations of neutrons, electrons and X-rays for atomic resolution microscopy of unstained biological molecules. Q Rev Biophys. 1995;28:171–193. doi: 10.1017/s003358350000305x. [DOI] [PubMed] [Google Scholar]
- 11.Dubochet J, Adrian M, Chang JJ, Homo JC, Lepault J, McDowall AW, Schultz P. Cryo-electron microscopy of vitrified specimens. Q Rev Biophys. 1988;21:129–228. doi: 10.1017/s0033583500004297. [DOI] [PubMed] [Google Scholar]
- 12.Glaeser RM, Taylor KA. Radiation damage relative to transmission electron microscopy of biological specimens at low temperature: a review. J Microsc. 1978;112:127–138. doi: 10.1111/j.1365-2818.1978.tb01160.x. [DOI] [PubMed] [Google Scholar]
- 13.Henderson R, Unwin PN. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 14.Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 15.Kuhlbrandt W, Wang DN, Fujiyoshi Y. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 16.Murata K, Mitsuoka K, Hirai T, Walz T, Agre P, Heymann JB, Engel A, Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- 17.Hiroaki Y, Tani K, Kamegawa A, Gyobu N, Nishikawa K, Suzuki H, Walz T, Sasaki S, Mitsuoka K, Kimura K, et al. Implications of the aquaporin-4 structure on array formation and cell adhesion. J Mol Biol. 2006;355:628–639. doi: 10.1016/j.jmb.2005.10.081. [DOI] [PubMed] [Google Scholar]
- 18.Nogales E, Wolf SG, Downing KH. Structure of the alpha beta tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 19•.Kowal J, Chami M, Baumgartner P, Arheit M, Chiu PL, Rangl M, Scheuring S, Schroder GF, Nimigean CM, Stahlberg H. Ligand-induced structural changes in the cyclic nucleotide-modulated potassium channel MloK1. Nat Commun. 2014;5:3106. doi: 10.1038/ncomms4106. Electron crystallography was used to investigate ligand bound and unbound structures of the MloK1 ion channel within a lipid bilayer. These new structural insights led to new hypotheses on how ligands are able modulate MloK1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tsai CJ, Tani K, Irie K, Hiroaki Y, Shimomura T, McMillan DG, Cook GM, Schertler GF, Fujiyoshi Y, Li XD. Two alternative conformations of a voltage-gated sodium channel. J Mol Biol. 2013;425:4074–4088. doi: 10.1016/j.jmb.2013.06.036. [DOI] [PubMed] [Google Scholar]
- 21.Oshima A, Tani K, Hiroaki Y, Fujiyoshi Y, Sosinsky GE. Three-dimensional structure of a human connexin26 gap junction channel reveals a plug in the vestibule. Proc Natl Acad Sci U S A. 2007;104:10034–10039. doi: 10.1073/pnas.0703704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breyton C, Haase W, Rapoport TA, Kuhlbrandt W, Collinson I. Three-dimensional structure of the bacterial protein-translocation complex SecYEG. Nature. 2002;418:662–665. doi: 10.1038/nature00827. [DOI] [PubMed] [Google Scholar]
- 23.Abe K, Tani K, Fujiyoshi Y. Conformational rearrangement of gastric H(+), K(+)-ATPase induced by an acid suppressant. Nat Commun. 2011;2:155. doi: 10.1038/ncomms1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nannenga BL, Iadanza MG, Vollmar BS, Gonen T. Overview of electron crystallography of membrane proteins: crystallization and screening strategies using negative stain electron microscopy. Curr Protoc Protein Sci. 2013;15 doi: 10.1002/0471140864.ps1715s72. Chapter 17:Unit17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glaeser RM, Tong L, Kim SH. Three-dimensional reconstructions from incomplete data: interpretability of density maps at “atomic” resolution. Ultramicroscopy. 1989;27:307–318. doi: 10.1016/0304-3991(89)90021-1. [DOI] [PubMed] [Google Scholar]
- 26.Fujiyoshi Y. The structural study of membrane proteins by electron crystallography. Adv Biophys. 1998;35:25–80. doi: 10.1016/s0065-227x(98)90004-1. [DOI] [PubMed] [Google Scholar]
- 27.Wisedchaisri G, Gonen T. Fragment-based phase extension for three-dimensional structure determination of membrane proteins by electron crystallography. Structure. 2011;19:976–987. doi: 10.1016/j.str.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gonen T. The collection of high-resolution electron diffraction data. Methods Mol Biol. 2013;955:153–169. doi: 10.1007/978-1-62703-176-9_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abeyrathne PD, Chami M, Pantelic RS, Goldie KN, Stahlberg H. Preparation of 2D crystals of membrane proteins for high-resolution electron crystallography data collection. Methods Enzymol. 2010;481:25–43. doi: 10.1016/S0076-6879(10)81001-8. [DOI] [PubMed] [Google Scholar]
- 30.Chou HT, Evans JE, Stahlberg H. Electron crystallography of membrane proteins. Methods Mol Biol. 2007;369:331–343. doi: 10.1007/978-1-59745-294-6_16. [DOI] [PubMed] [Google Scholar]
- 31.Subramaniam S, Henderson R. Crystallographic analysis of protein conformational changes in the bacteriorhodopsin photocycle. Biochim Biophys Acta. 2000;1460:157–165. doi: 10.1016/s0005-2728(00)00136-5. [DOI] [PubMed] [Google Scholar]
- 32.Unwin N. Structure and action of the nicotinic acetylcholine receptor explored by electron microscopy. FEBS Lett. 2003;555:91–95. doi: 10.1016/s0014-5793(03)01084-6. [DOI] [PubMed] [Google Scholar]
- 33.Unwin PN, Henderson R. Molecular structure determination by electron microscopy of unstained crystalline specimens. J Mol Biol. 1975;94:425–440. doi: 10.1016/0022-2836(75)90212-0. [DOI] [PubMed] [Google Scholar]
- 34.Unwin PNT. Beef liver catalase structure — interpretation of electron micrographs. J Mol Biol. 1975;98:235–242. doi: 10.1016/s0022-2836(75)80111-2. [DOI] [PubMed] [Google Scholar]
- 35.Dorset DL, Parsons DF. Electron-diffraction from single, fully-hydrated, ox liver catalase microcrystals. Acta Crystallogr Sect A. 1975;A31:210–215. [Google Scholar]
- 36.Longley W. The crystal structure of bovine liver catalase: a combined study by X-ray diffraction and electron microscopy. J Mol Biol. 1967;30:323–327. doi: 10.1016/s0022-2836(67)80042-1. [DOI] [PubMed] [Google Scholar]
- 37.Matricar Vr, Moretz RC, Parsons DF. Electron-diffraction of wet proteins — catalase. Science. 1972;177:268–&. doi: 10.1126/science.177.4045.268. [DOI] [PubMed] [Google Scholar]
- 38.Dorset DL, Parsons DF. Thickness measurements of wet protein crystals in electron-microscope. J Appl Crystallogr. 1975;8:12–14. [Google Scholar]
- 39•.Nederlof I, Li YW, van Heel M, Abrahams JP. Imaging protein three-dimensional nanocrystals with cryo-EM. Acta Crystallogr D Biol Crystallogr. 2013;69:852–859. doi: 10.1107/S0907444913002734. In this work, images of 3D lysozyme microcrystals were collected, and high-resolution projection maps of the crystals were calculated. The future prospects of directly solving the protein structure through imaging are discussed. [DOI] [PubMed] [Google Scholar]
- 40.Nannenga BL, Gonen T. Protein structure determination by MicroED. Curr Opin Struct Biol. 2014;27:24–31. doi: 10.1016/j.sbi.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Iadanza MG, Gonen T. A suite of software for processing MicroED data of extremely small protein crystals. J Appl Crystallogr. 2014;47:1140–1145. doi: 10.1107/S1600576714008073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42••.Nannenga BL, Shi D, Leslie AG, Gonen T. High-resolution structure determination by continuous-rotation data collection in MicroED. Nat Methods. 2014;11:927–930. doi: 10.1038/nmeth.3043. This study introduced the continuous rotation method for MicroED data collection. This new procedure for data collection greatly improved the quality of the data as well as the subsequent data processing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43•.Hattne J, Reyes FE, Nannenga BL, Shi D, de la Cruz MJ, Leslie AG, Gonen T. MicroED data collection and processing. Acta Crystallogr A Found Adv. 2015;71:353–360. doi: 10.1107/S2053273315010669. This work describes in detail how MicroED data is processed and is an excellent starting point for understanding the data processing procedures. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Midgley PA, Eggeman AS. Precession electron diffraction — a topical review. IUCr J. 2015;2:126–136. doi: 10.1107/S2052252514022283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nannenga BL, Shi D, Hattne J, Reyes FE, Gonen T. Structure of catalase determined by MicroED. Elife. 2014;3:e03600. doi: 10.7554/eLife.03600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46••.Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, Sangwan S, Guenther EL, Johnson LM, Zhang M, et al. Structure of the toxic core of alpha-synuclein from invisible crystals. Nature. 2015 doi: 10.1038/nature15368. http://dx.doi.org/10.1038/nature15368). MicroED was used to determine the structure of the toxic core of α-synuclein from peptide microcrystals that had resisted structure determination from all other methods. The final 1.4 Å structures represent the highest resolution by cryoEM and the first novel structures determined with MicroED. [DOI] [PMC free article] [PubMed]
- 47•.Shi D, Nannenga BL, de la Cruz J, Liu J, Sawtelle S, Calero G, Reyes FE, Hattne J, Gonen T. The collection of MicroED data for macromolecular crystallography. Nat Protocols. 2016;11:895–904. doi: 10.1038/nprot.2016.046. http://dx.doi.org/10.1038/nprot.2016.046. This paper presents the detailed, step by step methods for MicroED sample preparation and data collection. Those interested in using MicroED are greatly encouraged to follow these detailed procedures. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Battye TG, Kontogiannis L, Johnson O, Powell HR, Leslie AG. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr. 2011;67:271–281. doi: 10.1107/S0907444910048675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leslie AGW, Powell HR. Processing diffraction data with MOSFLM. In: Read RJ, Sussman JL, editors. Evolving Methods for Macromolecular Crystallography. Vol. 245. pp. 41–51. [Google Scholar]
- 50.Otwinowski Z, Minor W. [20] Processing of X-ray diffraction data collected in oscillation mode. Macromol Crystallogr Part A. 1997:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 51.Waterman DG, Winter G, Parkhurst JM, Fuentes-Montero L, Hattne J, Brewster AS, Sauter NK, Evans G. The DIALS framework for integration software. CCP4 Newsl. 2013:16–19. [Google Scholar]
- 52.Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yonekura K, Kato K, Ogasawara M, Tomita M, Toyoshima C. Electron crystallography of ultrathin 3D protein crystals: atomic model with charges. Proc Natl Acad Sci USA. 2015;112:3368–3373. doi: 10.1073/pnas.1500724112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hattne J, Shi D, de la Cruz MJ, Reyes FE, Gonen T. Modeling truncated pixel values of faint reflections in MicroED images. J Appl Crystallogr. 2016:49. doi: 10.1107/S1600576716007196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sawaya MR, Rodriguez JA, Cascio D, Collazo M, Shi D, Reyes FE, Hattne J, Gonen T, Eisenberg DS. Proc Natl Acad Sci USA. PNAS; 2016. Ab initio structure determination from prion nanocrystals at atomic resolution by MicroED. in press. published ahead of print September 19, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]