

Abstract
In this review we discuss the current advances relating to structure determination from protein microcrystals with special emphasis on the newly developed method called MicroED. This method uses a transmission electron cryo-microscope to collect electron diffraction data from extremely small 3-dimensional (3D) crystals. MicroED has been used to solve the 3D structure of the model protein lysozyme to 2.9 Å resolution. As the method further matures, MicroED promises to offer a unique and widely applicable approach to protein crystallography using nanocrystals.
Introduction
The 3-dimensional (3D) structures of proteins and other biomolecules are an important source of insight into the function and mechanism of biological processes. Through the ability to visualize the structures of proteins and other biomolecules, structural biology has greatly enhanced our understanding of the biochemical processes of life. Of the techniques used to gather structural information, X-ray crystallography is by far the most successful and highly utilized technique, a fact highlighted by the more than 80 000 structures solved by this technique (www.pdb.org [1]). Despite the great advances in X-ray crystallography, the requirement for large well-ordered crystals remains a formidable barrier. For difficult targets such as membrane proteins and protein complexes, the optimization of initial small crystals found during screening can take significant time and resources and may never yield the large well-ordered crystals needed for traditional X-ray crystallography [2].
While continued development of microfocus beamlines have allowed researchers to obtain data from smaller and smaller crystals [3–7], the inherent problem of radiation damage caused by X-rays is one that is not easily overcome. In this review we discuss new methods for protein crystallography, with special focus on the recently developed MicroED method for diffraction of microcrystals and nanocrystals using a transmission electron microscope (TEM). MicroED can bypass the hurdles of crystal size and radiation damage allowing for the use of extremely small crystals for protein structure determination at atomic resolution [8••].
Bypassing the microcrystal radiation damage barrier
Even when diffraction experiments are conducted at cryogenic temperatures, the radiation damage experienced by a protein crystal during data collection will ultimately lead to a reduction in data quality as the experiment progresses [9–12]. Because the crystal must be sufficiently large to withstand the negative effects of radiation damage, there is a lower limit in size of the smallest crystals that will provide useful data by traditional X-ray crystallography. Therefore, there is a great need to find methods that could potentially overcome this obstacle and facilitate data collection from smaller and potentially more locally well-ordered crystals [13].
In an attempt to alleviate this problem, researchers have turned to femtosecond X-ray crystallography [14••,15•], which can use small microcrystals. The basis of this technique is ‘diffract-before-destruction’ where the diffracting X-ray pulse is so intense and quick that the data are obtained on a time scale faster than that of the radiation damage [16,17]. Since its inception the technique has continually been developed and optimized. Recently the method has been improved to the level that the integrated data was of high enough quality to determine phases experimentally [18]. While this technique shows great promise and has been used to determine the structures of several proteins (e.g. Photosystem I [14••], Lysozyme [15•,18], and Cathepsin B [19]), it requires a large number of crystals and access to X-ray lasers, which are relatively new and not widely available.
Another approach for reducing the effects of radiation damage while obtaining diffraction data is to use electrons instead of X-rays. The wavelength of electrons used for diffraction is approximately 50× shorter than the wavelengths used for X-ray diffraction. While this has effects on the Ewald sphere and what the resulting diffraction patterns look like (Figure 1), the principles are essentially the same between electron diffraction and X-ray diffraction. A key difference between the two diffraction techniques is that electrons deposit 2–3 orders of magnitude less energy into a crystal per useful scattering event [20•]. Additionally, electrons interact with matter much more strongly compared to X-rays; therefore, the requirement of large crystal size is reduced when using electrons as opposed to X-rays. For biological samples, electrons were first used to collect diffraction data from 2-dimensional (2D) crystals of bacteriorhodopsin to a resolution of 7 Å using a TEM [21,22]. This pioneering work by Henderson and Unwin launched the field of 2D electron crystallography which has since been used to solve the structure of many membrane proteins in their lipid environments [23], with the highest resolution structure resolved to 1.9 Å [24].
Figure 1.
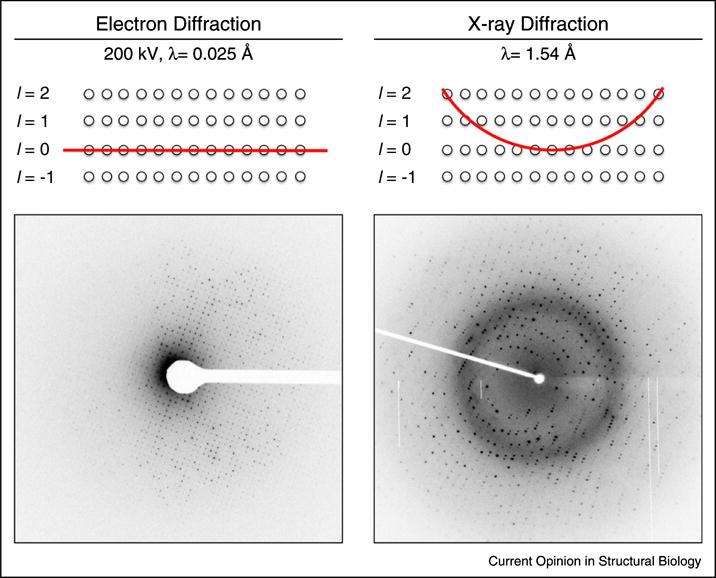
Comparison of diffraction data obtained from lysozyme crystals by electron diffraction and X-ray diffraction. Because the wavelength of the diffracting electrons is so short, the resulting Ewald sphere (left, red line) is essentially a plane when compared to the Ewald sphere for X-ray diffraction (right, red line). Diffraction only occurs when the Ewald sphere contacts a reflection in reciprocal space (top panels, white circles represent reflections in reciprocal space). Therefore, because the Ewald sphere is so flat, the patterns produced from electron diffraction (bottom left) appear as planar 2-dimensional slices through the 3-dimensional volume of reflections, whereas the patterns from X-ray diffraction (bottom right) appear as circular 2-dimensional projections of the sphere on the detector.
Electron diffraction of 3D protein crystals in the TEM has been attempted over the years but none yielded a refined structure [25–28]. One of the major hurdles was the beam damage associated with data collection, which typically allowed only one diffraction pattern to be collected per crystal [28]. By only having one pattern per crystal it is difficult to properly index the reflections and determine the crystallographic orientation especially as the data is affected by the shape function.
MicroED is a new method that was used successfully to determine protein structure by electron diffraction from microcrystals [8••]. In MicroED an extremely low electron dose is used to collect multiple electron diffraction patterns from each crystal at varying angles (Figures 2A and 3). Using this approach, the first complete high-resolution structure of a protein was reported from 3D microcrystals in a TEM. The remaining sections of this review will be devoted to MicroED and discussions on the future directions of this technique.
Figure 2.
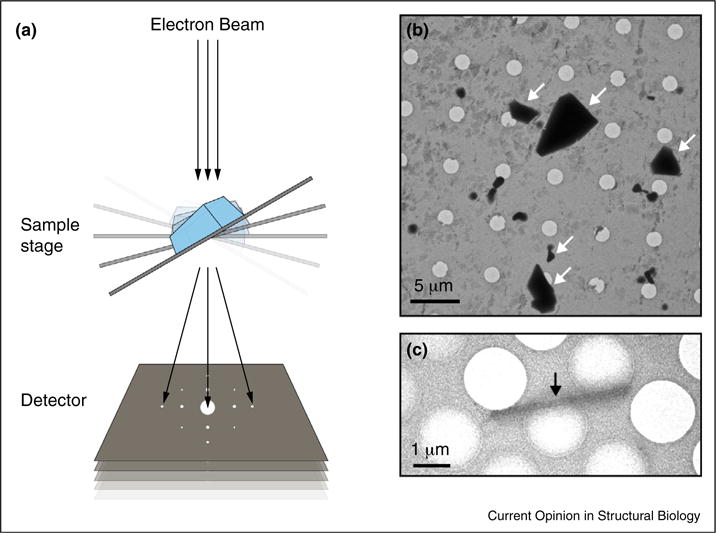
MicroED data collection and microcrystal visualization. (A) Schematic of MicroED data collection by tilting the sample stage between subsequent exposures. Each exposure is of relatively low dose (~0.01 e−/Å2/s) which allows the collection of multiple diffraction patterns from a single crystal that are combined into a single data set. Each data set consists of up to 90 still frames taken at 0.1°–1° intervals. (B,C) Visualizing microcrystals (arrows) in the TEM prior to data collection. The dimensions of microcrystals suitable for MicroED range from approximately 1–10 μm in length and width and 0.1– 1μm in thickness. The crystals of a membrane transporter (B) visualized by negative stain EM, whereas the crystals of a novel designed protein (C) are seen with cryo-EM in over-focused diffraction mode.
Figure 3.
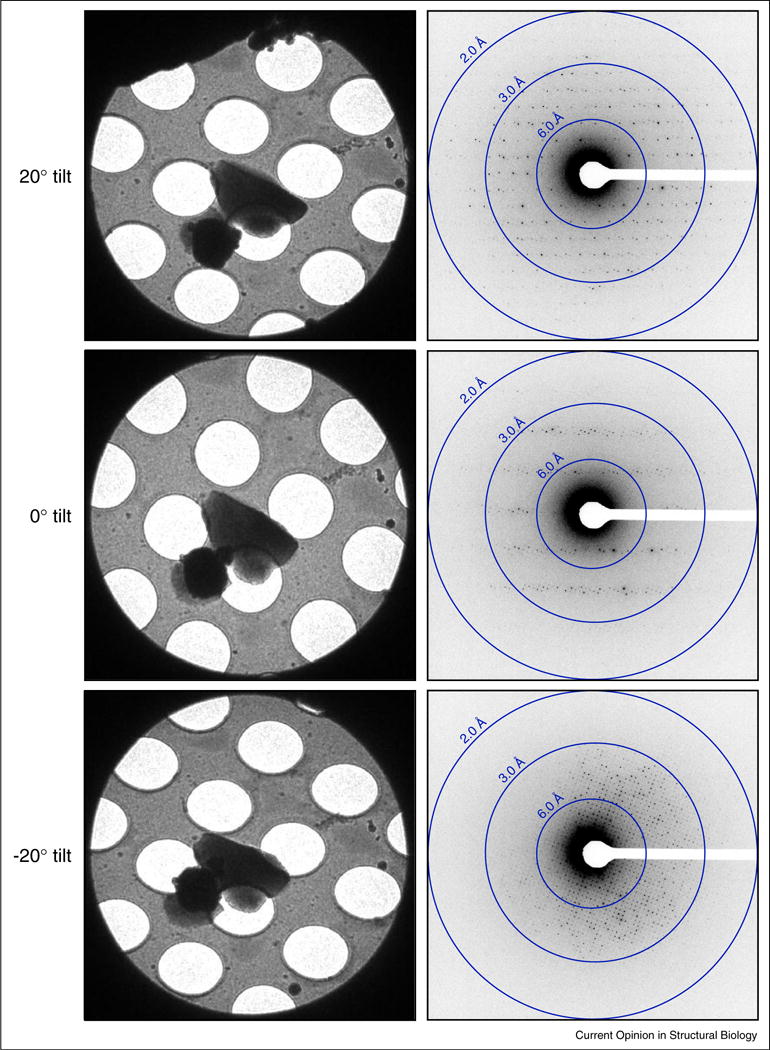
Examples of tilted diffraction data of Lysozyme collected by MicroED. Representative crystal images (left) and diffraction patterns (right) taken at tilts of 20°, 0°, and −20°. All images and diffraction patterns are from the same crystal. The MicroED diffraction data shows distinct reflections with a maximum resolution of 1.7 Å.
MicroED sample preparation and data collection
As with X-ray crystallography, conditions must be optimized in MicroED for sample preparation, cryo protection, and data collection strategy.
In order to prepare samples for MicroED, microcrystals are taken directly from the drops where they were formed and deposited on a carbon coated electron microscopy grid. The sample must have the excess liquid removed by blotting with filter paper, as the electron beam is unable to penetrate thick samples. Therefore, some optimization of blotting is necessary to strike the right balance between leaving enough solution so that the crystals are well-hydrated and preserved, but not so much that the sample is too thick for electron diffraction. Conditions for sample preparation can be screened by negative stain EM [29], however this only provides a rough starting point as conditions can deviate significantly when using stains and will need to be further optimized for cryoEM.
Once the sample is loaded into the microscope, the grid can be screened for microcrystals (Figure 2B and C). When a microcrystal is located, a test diffraction pattern is first collected to determine the quality of the crystal. Factors that are considered when assessing diffraction quality include, but are not limited to, the intensity and sharpness of the recorded reflections and maximum resolution obtained [30••]. Examples of high quality diffraction patterns can be seen in Figure 3. Once a promising micro-crystal of good quality is found, a diffraction data set is collected by exposing the crystal with extremely low dose (~0.01 e−/Å2/s) followed by tilting the crystal using the microscope stage and repeating this cycle of exposure and tilting (Figure 2A) until the crystal succumbs to the effects of radiation damage. The crystal is tilted by a constant value (typically 0.1°–1.0°) between exposures, which creates a data set of still diffraction patterns taken at defined intervals of rotation. Because it is clear how each still pattern relates to all other diffraction patterns within the data set, the orientation of the crystal can be determined and the data set correctly indexed and integrated [31••].
The stage of the electron microscope limits the attainable tilt angle to ±70°. This means that a single crystal can yield a 140° wedge of data. Depending on crystal symmetry, the degree of tilting performed, and the crystal’s orientation in the microscope, there may be missing regions of data, which will need to be filled in by more data sets originating from additional crystals (Figure 4). Each crystal would have to be oriented differently on the grid to allow ample sampling in reciprocal space. Following data integration it is possible to identify the orientation of the crystal and find out how much of the reciprocal space of the crystal was covered during data collection. By using this knowledge, data sets that complement each other can be combined, effectively filling in the data by removing missing wedges and improving completeness. Once a complete data set is obtained, the structure can be phased and refined using standard programs for X-ray crystallography such as PHENIX [32], CNS [33] and the CCP4 program suite [34]. As a proof of principle, this entire process was performed with lysozyme microcrystals giving rise to a refined structure of lysozyme at 2.9 Å resolution (Figure 4) and an Rwork/Rfree of 25.5/27.8 [8••]. Diffraction was recorded to 1.7 Å, however the data was truncated to 2.9 Å to ease processing with in house developed programs. In the future we expect that the resolution will be extended by integrating MicroED data with standard crystallography software and optimization of data collection strategies.
Figure 4.
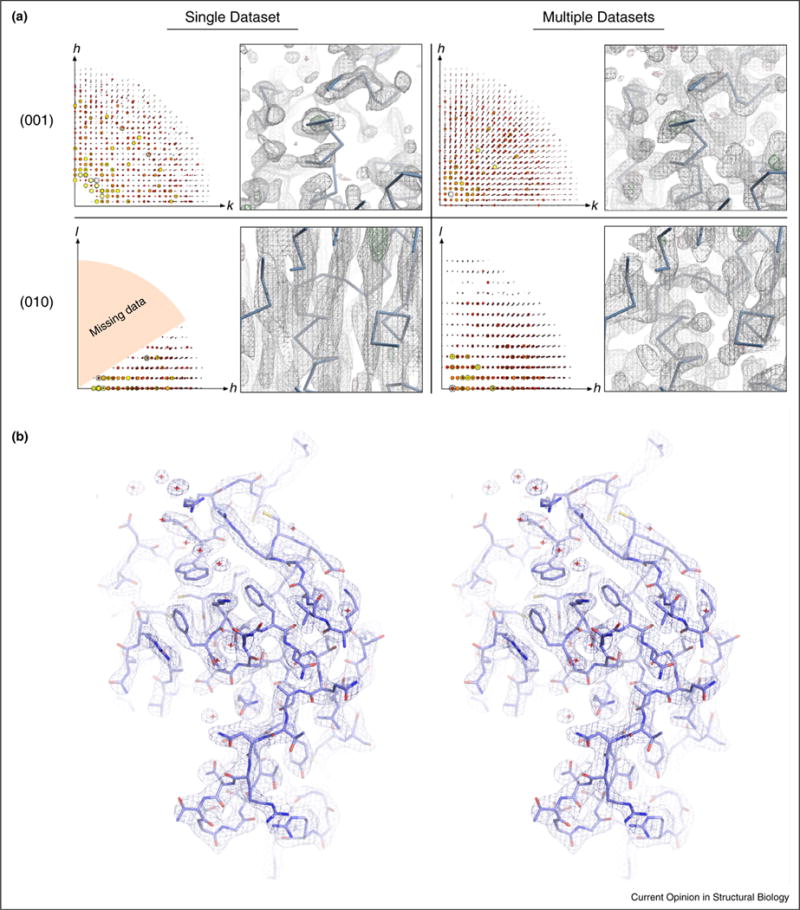
Combining data sets and solving the final structure. (A) The consequences of data completeness (left side of panels) on the resulting density maps (right side of panels). When a single data set from lysozyme having a large missing wedge of data is used (shown as orange in the (0 1 0) single data set panel), the data quality in the direction containing complete data (along (0 0 1)) shows reasonable density, whereas the density in the direction of the missing data (along (0 1 0)) is extremely poor. By combining additional data sets that include the missing reflections from the original single data set, the quality of the density can be recovered and a complete 3D map obtained. The protein backbone of lysozyme (Cα trace) is shown in blue in the density maps. (B) Stereo view of the final 2.9 Å structure of lysozyme solved by MicroED. The 2Fobs−Fcalc map (contoured at 1.5σ) of the representative region shown displays well-defined density around the side chains and nearby waters.
Crystal size in MicroED
As was mentioned above, electrons interact with matter relatively strongly and this places limitations on the upper size limit of 3D crystals that can be used by MicroED. The effects of diffuse scattering and dynamic scattering within the crystal both increase as the crystal thickness increases, and while these are generally assumed to be negligible in X-ray crystallography, their effects cannot be completely ignored in electron diffraction. Diffuse scattering, which is caused by partial disorder within the crystal as well as inelastic scattering, leads to increased background noise and errors in the measurement of reflection intensities [35]. Dynamic scattering occurs when an electron that has been scattered elastically undergoes a second elastic scattering event as it exits the sample. Because both dynamic scattering events follow Bragg’s law, the effect is that a portion of the electrons destined to be counted as intensity for a specific reflection are redistributed to other reflections in the diffraction data set leading to inaccuracies in the resulting integrated data [36,37].
We found that Lysozyme crystals approximately 500 nm or thinner were not completely overcome by the effects described above and could provide usable data, whereas thicker crystals were essentially unusable. It is likely that different crystals will have different thickness limits as the density and the packing within the crystals will affect the mean free path of the electrons through the sample. Therefore, the usable range of crystal size will need to be determined for each microcrystal sample experimentally.
Data processing
In the current implementation of MicroED, the diffraction patterns are collected as still exposures rather than the oscillation method commonly used in X-ray crystallography. Because of this, the recorded reflections within a data set are typically partial reflections. To work with a data set containing only partial intensities, in-house programs and scripts were written to index the data and group all symmetry related intensities [31••]. One of the main assumptions used to handle MicroED data is that the maximum intensity within a symmetry related group would originate from a diffraction pattern where the Ewald sphere passes through the largest cross-section of that given reflection. Therefore, only the maximum intensity within each symmetry related group was kept and taken to represent the most accurate measurement of the full intensity. By using this assumption, along with a few others, the data sets are concatenated and reflection files are generated. The final reflection files are then processed using standard refinement programs for X-ray crystallography as described above.
Future directions for MicroED
MicroED is a relatively new method and has room for much optimization with future work focusing on sample preparation, data collection strategies and data processing and phasing. Specifically, one area of work that we are currently focusing on is moving the technique away from the still diffraction patterns towards the collection of data from crystals that are oscillated. The obvious benefit is that the data sets collected this way would contain the measurement of full intensities as opposed to the partial intensities that still diffraction patterns provide.
Another method of collecting full intensities, which could be implemented in MicroED, is beam precession. This method relies on slightly tilting the beam and rotating it during data collection. Precession has been shown to provide fuller, more accurate intensities and reduce the effects of dynamic scattering, leading to greatly improved data quality [38–40].
MicroED promises to advance the field of structural biology by offering a parallel path to structure determination that has so far been dominated by X-ray crystallography. While there is much work to be done improving and optimizing the technique, this is an exciting time for electron crystallographers and those who would like to use micro and nanocrystals. While there will always be a place for traditional X-ray crystallography, one day — with the help of MicroED, microfocus X-ray beam lines, and femtosecond crystallography — many protein structures may be solved from extremely small crystals which have traditionally been discarded as undesirable.
Acknowledgments
We thank Dan Shi and Matthew G. Iadanza (JFRC) helpful insights and discussion. This work is supported by the Howard Hughes Medical Institute.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bill RM, Henderson PJ, Iwata S, Kunji ER, Michel H, Neutze R, Newstead S, Poolman B, Tate CG, Vogel H. Overcoming barriers to membrane protein structure determination. Nat Biotechnol. 2011;29:335–340. doi: 10.1038/nbt.1833. [DOI] [PubMed] [Google Scholar]
- 3.Moukhametzianov R, Burghammer M, Edwards PC, Petitdemange S, Popov D, Fransen M, McMullan G, Schertler GF, Riekel C. Protein crystallography with a micrometre-sized synchrotron-radiation beam. Acta Crystallogr D Biol Crystallogr. 2008;64:158–166. doi: 10.1107/S090744490705812X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pebay-Peyroula E, Rummel G, Rosenbusch JP, Landau EM. X-ray structure of bacteriorhodopsin at 2.5 angstroms from microcrystals grown in lipidic cubic phases. Science. 1997;277:1676–1681. doi: 10.1126/science.277.5332.1676. [DOI] [PubMed] [Google Scholar]
- 5.Bowler MW, Guijarro M, Petitdemange S, Baker I, Svensson O, Burghammer M, Mueller-Dieckmann C, Gordon EJ, Flot D, McSweeney SM, et al. Diffraction cartography: applying microbeams to macromolecular crystallography sample evaluation data collection. Acta Crystallogr D Biol Crystallogr. 2010;66:855–864. doi: 10.1107/S0907444910019591. [DOI] [PubMed] [Google Scholar]
- 6.Rasmussen SG, Choi HJ, Rosenbaum DM, Kobilka TS, Thian FS, Edwards PC, Burghammer M, Ratnala VR, Sanishvili R, Fischetti RF, et al. Crystal structure of the human beta2 adrenergic G-protein-coupled receptor. Nature. 2007;450:383–387. doi: 10.1038/nature06325. [DOI] [PubMed] [Google Scholar]
- 7.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, Choi HJ, Kuhn P, Weis WI, Kobilka BK, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8••.Shi D, Nannenga BL, Iadanza MG, Gonen T. Three-dimensional electron crystallography of protein microcrystals. Elife. 2013:2. doi: 10.7554/eLife.01345. This paper lays the foundation for MicroED and describes the proof of concept principles for the determination of the structure of lysozyme to 2.9 Å by electron diffraction of 3-dimensional protein microcrystals. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garman EF, Nave C. Radiation damage in protein crystals examined under various conditions by different methods. J Synchrotron Radiat. 2009;16:129–132. doi: 10.1107/S0909049509005238. [DOI] [PubMed] [Google Scholar]
- 10.Nave C. Radiation-damage in protein crystallography. Radiat Phys Chem. 1995;45:483–490. [Google Scholar]
- 11.Kmetko J, Husseini NS, Naides M, Kalinin Y, Thorne RE. Quantifying X-ray radiation damage in protein crystals at cryogenic temperatures. Acta Crystallogr D Biol Crystallogr. 2006;62:1030–1038. doi: 10.1107/S0907444906023869. [DOI] [PubMed] [Google Scholar]
- 12.Meents A, Gutmann S, Wagner A, Schulze-Briese C. Origin and temperature dependence of radiation damage in biological samples at cryogenic temperatures. Proc Natl Acad Sci U S A. 2010;107:1094–1099. doi: 10.1073/pnas.0905481107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cusack S, Belrhali H, Bram A, Burghammer M, Perrakis A, Riekel C. Small is beautiful: protein micro-crystallography. Nat Struct Biol. 1998;5:634–637. doi: 10.1038/1325. [DOI] [PubMed] [Google Scholar]
- 14••.Chapman HN, Fromme P, Barty A, White TA, Kirian RA, Aquila A, Hunter MS, Schulz J, DePonte DP, Weierstall U, et al. Femtosecond X-ray protein nanocrystallography. Nature. 2011;470:73–77. doi: 10.1038/nature09750. The first proof of principle of structure determination from femtosecond X-ray crystallography where a hard-X-ray free-electron laser (XFEL) was used solve the 8.5 structure of photosystem I. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15•.Boutet S, Lomb L, Williams GJ, Barends TR, Aquila A, Doak RB, Weierstall U, DePonte DP, Steinbrener J, Shoeman RL, et al. High-resolution protein structure determination by serial femtosecond crystallography. Science. 2012;337:362–364. doi: 10.1126/science.1217737. First high resolution structure (lysozyme to 1.9 Å) using XFELs and femtosecond X-ray crystallography. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fromme P, Spence JC. Femtosecond nanocrystallography using X-ray lasers for membrane protein structure determination. Curr Opin Struct Biol. 2011;21:509–516. doi: 10.1016/j.sbi.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chapman HN, Barty A, Bogan MJ, Boutet S, Frank M, Hau-Riege SP, Marchesini S, Woods BW, Bajt S, Benner H, et al. Femtosecond diffractive imaging with a soft-X-ray free-electron laser. Nat Phys. 2006;2:839–843. [Google Scholar]
- 18.Barends TR, Foucar L, Botha S, Doak RB, Shoeman RL, Nass K, Koglin JE, Williams GJ, Boutet S, Messerschmidt M, et al. De novo protein crystal structure determination from X-ray free-electron laser data. Nature. 2013;10 doi: 10.1038/nature12773. http://dx.doi.org/10.1038/nature12773. [DOI] [PubMed] [Google Scholar]
- 19.Redecke L, Nass K, DePonte DP, White TA, Rehders D, Barty A, Stellato F, Liang M, Barends TR, Boutet S, et al. Natively inhibited Trypanosoma brucei cathepsin B structure determined by using an X-ray laser. Science. 2013;339:227–230. doi: 10.1126/science.1229663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20•.Henderson R. The potential and limitations of neutrons electrons and X-rays for atomic-resolution microscopy of unstained biological molecules. Q Rev Biophys. 1995;28:171–193. doi: 10.1017/s003358350000305x. Insightful overview of the benefits and shortcomings of neutrons, electrons and X-rays for structure determination. Comparisons between the effects of radiation damage between electrons and X-rays are particularly pertinent to this MicroED. [DOI] [PubMed] [Google Scholar]
- 21.Henderson R, Unwin PN. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. This study presents the first structure of a membrane protein which was solved by 2-dimensional electron crystallography. This paper helped to set the stage for the field of 2-dimensional crystallography which has solved the structures of a number of membrane proteins within a lipid membrane. [DOI] [PubMed] [Google Scholar]
- 22.Unwin PN, Henderson R. Molecular structure determination by electron microscopy of unstained crystalline specimens. J Mol Biol. 1975;94:425–440. doi: 10.1016/0022-2836(75)90212-0. [DOI] [PubMed] [Google Scholar]
- 23.Wisedchaisri G, Reichow SL, Gonen T. Advances in structural and functional analysis of membrane proteins by electron crystallography. Structure. 2011;19:1381–1393. doi: 10.1016/j.str.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid–protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–638. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shi D, Lewis MR, Young HS, Stokes DL. Three-dimensional crystals of Ca2+-ATPase from sarcoplasmic reticulum: merging electron diffraction tilt series and imaging the (h, k, 0) projection. J Mol Biol. 1998;284:1547–1564. doi: 10.1006/jmbi.1998.2283. [DOI] [PubMed] [Google Scholar]
- 26.Jiang LH, Georgieva D, Nederlof I, Liu ZF, Abrahams JP. Image processing and lattice determination for three-dimensional nanocrystals. Microsc Microanal. 2011;17:879–885. doi: 10.1017/S1431927611012244. [DOI] [PubMed] [Google Scholar]
- 27.Nederlof I, van Genderen E, Li YW, Abrahams JP. A Medipix quantum area detector allows rotation electron diffraction data collection from submicrometre three-dimensional protein crystals. Acta Crystallogr D Biol Crystallogr. 2013;69:1223–1230. doi: 10.1107/S0907444913009700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jiang LH, Georgieva D, Zandbergen HW, Abrahams JP. Unit-cell determination from randomly oriented electron-diffraction patterns. Acta Crystallogr D Biol Crystallogr. 2009;65:625–632. doi: 10.1107/S0907444909003163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nannenga BL, Iadanza MG, Vollmar BS, Gonen T. Overview of electron crystallography of membrane proteins: crystallization and screening strategies using negative stain electron microscopy. Curr Protoc Protein Sci. 2013 doi: 10.1002/0471140864.ps1715s72. Chapter 17: Unit17 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30••.Gonen T. The collection of high-resolution electron diffraction data. Methods Mol Biol. 2013;955:153–169. doi: 10.1007/978-1-62703-176-9_9. This work provides a thorough description of the steps necessary to collect diffraction data with a TEM. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31••.Iadanza MG, Gonen T. A suite of software for processing microcrystal electron diffraction (MicroED) data. Journal of Applied Crystallography. 2014 doi: 10.1107/S1600576714008073. (submitted for publication). This paper describes the programs and methodology which were initially used for processing MicroED data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 34.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grigorieff N, Henderson R. Diffuse scattering in electron diffraction data from protein crystals. Ultramicroscopy. 1995;60:295–309. [Google Scholar]
- 36.Grigorieff N, Ceska TA, Downing KH, Baldwin JM, Henderson R. Electron-crystallographic refinement of the structure of bacteriorhodopsin. J Mol Biol. 1996;259:393–421. doi: 10.1006/jmbi.1996.0328. [DOI] [PubMed] [Google Scholar]
- 37.Glaeser RM, Ceska TA. High-voltage electron-diffraction from bacteriorhodopsin (purple membrane) is measurably dynamical. Acta Crystallogr A. 1989;45:620–628. doi: 10.1107/s0108767389004599. [DOI] [PubMed] [Google Scholar]
- 38.Gemmi M, Nicolopoulos S. Structure solution with three-dimensional sets of processed electron diffraction intensities. Ultramicroscopy. 2007;107:483–494. doi: 10.1016/j.ultramic.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 39.Sarakinou E, Mugnaioli E, Lioutas CB, Vouroutzis N, Frangis N, Kolb U, Nikolopoulos S. Structure characterization of hard materials by precession electron diffraction and automatic diffraction tomography: 6H-SiC semiconductor and Ni1+xTe1 embedded nanodomains. Semicond Sci Technol. 2012;27 [Google Scholar]
- 40.Gjonnes J, Hansen V, Berg BS, Runde P, Cheng YF, Gjonnes K, Dorset DL, Gilmore CJ. Structure model for the phase AlmFe derived from three-dimensional electron diffraction intensitydata collected by a precession technique. Comparison with convergent-beam diffraction. Acta Crystallogr A. 1998;54:306–319. [Google Scholar]