

Abstract
The brain is enriched in arachidonic acid (ARA) and docosahexaenoic acid (DHA), long-chain polyunsaturated fatty acids (LCPUFAs) of the n-6 and n-3 series, respectively. Both are essential for optimal brain development and function. Dietary enrichment with DHA and other long-chain n-3 PUFA, such as eicosapentaenoic acid (EPA), has shown beneficial effects on learning and memory, neuroinflammatory processes, and synaptic plasticity and neurogenesis. ARA, DHA and EPA are precursors to a diverse repertoire of bioactive lipid mediators, including endocannabinoids. The endocannabinoid system comprises cannabinoid receptors, their endogenous ligands, the endocannabinoids, and their biosynthetic and degradation enzymes. Anandamide (AEA) and 2-arachidonoylglycerol (2-AG) are the most widely studied endocannabinoids and are both derived from phospholipid-bound ARA. The endocannabinoid system also has well-established roles in neuroinflammation, synaptic plasticity and neurogenesis, suggesting an overlap in the neuroprotective effects observed with these different classes of lipids. Indeed, growing evidence suggests a complex interplay between n-3 and n-6 LCPUFA and the endocannabinoid system. For example, long-term DHA and EPA supplementation reduces AEA and 2-AG levels, with reciprocal increases in levels of the analogous endocannabinoid-like DHA and EPA-derived molecules. This review summarises current evidence of this interplay and discusses the therapeutic potential for brain protection and repair.
Keywords: Endocannabinoid system, Neurogenesis, Neuroinflammation, Omega-3 fatty acids, Omega-6 fatty acids
Introduction
N-6 and n-3 long-chain polyunsaturated fatty acids (LCPUFA) are essential components of membrane phospholipids and also precursors to a large and ever expanding repertoire of bioactive lipid mediators. The brain is highly enriched in the n-6 PUFA, arachidonic acid (ARA), and the n-3 PUFA, docosahexaenoic acid (DHA), with both essential for optimum brain development and function [1]. Elevated dietary intake of DHA and eicosapentaenoic acid (EPA), another n-3 LCPUFA, has beneficial effects on learning and memory, decreases neuroinflammatory processes and enhances synaptic plasticity and neurogenesis [2]. Similarly, inverse relationships are typically observed between fish intake or blood DHA levels and age-related cognitive decline [3]. However, recent estimates indicate that worldwide many populations are currently consuming DHA and EPA at levels well below the recommendations issued by many international authorities [4–6].
The mode of action of the LCPUFA is still poorly understood and is further complicated by the diverse repertoire of bioactive lipid mediators that can be generated. For example, ARA is the precursor to a wide range of mediators, including the two major endocannabinoids in the brain [7]. The endocannabinoid system has similarly been shown to have important roles in neuroprotective and pro-neurogenic processes, such as attenuating chronic neuroinflammation, regulating pro-inflammatory cytokine release and enhancing synaptic plasticity and adult neurogenesis [8, 9], and importantly has shown therapeutic potential in brain ageing and neurodegenerative conditions [10].
Thus, there is considerable overlap in effects of n-3 PUFA and the endocannabinoid system; however, these different classes of lipid mediators have traditionally been viewed and researched separately. This view is now being challenged as there are a growing number of independent lines of evidence suggesting a complex interplay between them. For example, analogous series of ethanolamide endocannabinoid-like molecules derived from DHA and EPA have been identified, although their biological roles have yet to be established [11, 12]. Furthermore, in recent elegant work long-term dietary n-3 PUFA deficiency in mice abolished endocannabinoid-mediated neuronal functions across a range of different brain regions, showing for the first time how the endocannabinoid system can be regulated by manipulation of the dietary n-6:n-3 PUFA ratio [13–15]. This is a cause for concern as the Western diet typically has an n-6:n-3 PUFA ratio of around 15:1, whereas the ideal ratio is thought to be closer to 4:1 [16]. This unbalanced intake is reflected in low to very low tissue levels of DHA and EPA [17], and may also be involved in the aetiology of many diseases, such as cardiovascular disease, cancer, inflammatory and autoimmune diseases [16].
The aim of this review is to summarise current evidence of the interplay between n-3 and n-6 LCPUFA and the endocannabinoid system and discusses the potential role of modifying their levels through dietary manipulation of n-6 and n-3 PUFA intake with the aim of ameliorating neuroinflammation and enhancing brain protection and repair, particularly in ageing.
Metabolism of PUFA and Endocannabinoids
ARA and DHA are the two main PUFA in the brain [2]. These LCPUFA can be supplied either preformed from the diet or synthesised in the liver from their shorter chain precursors, linoleic acid (LA, 18:2n-6) and α-linolenic acid (ALA, 18:3n-3), respectively [18, 19]. However, the efficiency of conversion in humans is extremely limited [20], and due to the shared nature of the biosynthetic pathways, imbalances in the dietary intake of LA and ALA will result in reciprocal inhibition of the opposing pathway and further limit conversion [21]. Therefore, the most efficient way to increase tissue levels of LCPUFA is by intake of the preformed LCPUFA. The n-6 and n-3 PUFA biosynthetic pathways are shown in detail in Fig. 1.
Fig. 1.

N-6 and n-3 PUFA metabolism and lipid mediators produced from ARA, DHA and EPA. Synthesis of n-6 and n-3 LCPUFA begins with desaturation of LA and ALA to γ-linolenic acid (GLA, 18:3n-6) and stearidonic acid (18:4n-4), respectively, catalysed by Δ6 desaturase (FADS2 gene). GLA is elongated to dihomo-γ-linolenic acid (DGLA, 20:3n-6) and SDA to eicosatetraenoic acid (20:4n-3) (ELOVL1 gene). Δ5-Desaturase (FADS1 gene) converts DGLA to ARA (20:4n-6) and 20:4n-3 to EPA (timnodonic acid, 20:5n-3). Two cycles of elongation (elongase-2, ELOVL2 gene) convert ARA to adrenic acid (AdA, 22:4n-6) and then tetracosatetraenoic acid (24:4n-6), and EPA to docosapentaenoic acid (DPAn-3, clupanodonic acid, 22:5n-3) and then tetracosapentaenoic acid (24:5n-3). A second desaturation by Δ6 desaturase produces tetracosapentaenoic acid (24:5n-6) and tetracosahexaenoic acid (nisinic acid, 24:6n-3), respectively. These are translocated to the peroxisome for β-oxidation by acyl-coenzyme-A oxidase (ACOX1 gene) and d-bifunctional enzyme (HSD1784 gene) and peroxisomal thiolases to produce docosapentaenoic acid (DPAn-6, osbond acid, 22:5n-6) and DHA (cervonic acid, 22:6n-3), which are translocated back to the endoplasmic reticulum
Endogenous synthesis of LCPUFA is low within the brain compared with uptake from the unesterified plasma fatty acid pool [22, 23], suggesting brain levels are maintained via uptake from dietary and/or liver sources in blood. Although LCPUFAs appear to cross the blood-brain barrier via simple diffusion [24], active transporters have been identified that may play a role in regulating the specificity of LCPUFA concentrations [20]. Further multiple mechanisms including β-oxidation, decreased incorporation, elongation and lower phospholipid recycling have also been identified, which maintain the high ARA and DHA concentration in relation to other LCPUFAs [25, 26]. However, brain LCPUFA composition is responsive to dietary intake, such that a diet high in LA, with an LA:an ALA ratio of 10:1 typical of a Western diet decreases brain DHA accretion and increases adrenic acid (AdA, docosatetraenoic acid, 22:4n-6) and docosapentaenoic acid (DPAn-6, 22:5n-6) levels [27], whereas a diet with an LA:ALA ratio of 1:1, more similar to that encountered during our evolution [16], leads to higher brain DHA levels. Imbalances in intake not only compromise brain LCPUFA content, but may also impact on the production of a wide range of mediators derived from these LCPUFA, thereby potentially negatively influencing brain activity and function.
The fatty acid composition of neuronal membranes influences cellular function through direct effects on membrane biophysical properties, but also by providing a precursor pool for signalling molecules and lipid-derived mediators [1]. N-6 and n-3 LCPUFA are the precursors to a vast array of bioactive mediators involved in many cellular processes, particularly related to the inflammatory response [2]. Three main pathways are involved in the production of these oxylipin mediators: (1) cyclooxygenase (COX, also known as prostaglandin endoperoxide H synthase or PGHS) and subsequent synthases, (2) lipoxygenase (LOX) and (3) cytochrome P450 mixed function oxidase enzymes (CYP) [28]. These canonical pathways produce the classic mediators, with those produced from C20 PUFA, such as ARA and EPA, called eicosanoids, whereas those from C22 PUFA, such as DHA, are called docosanoids. Analogous series of oxylipins generated from LA, dihomo-γ-linolenic acid (DGLA), AdA and ALA and the n-3 docosapentaenoic acid (DPAn-3) have also been identified, but their roles are not well characterised in the literature and are therefore not the focus of this review. However, the interested reader is referred to an excellent review by Gabbs and colleagues [29].
COX catalyses the initial oxygenation of non-esterified fatty acids to produce prostaglandin H (PGH), a short-lived intermediate, which is further metabolised into prostanoids, such as other prostaglandin series (PGD, PGE, PGF), prostacyclins (PGI), thromboxanes (Tx), and lipoxins (Lx), hydroxy and hydroperoxy fatty acids [30]. Vertebrates have two principal isoforms of COX: COX-1 and COX-2 [31]. COX-1 is constitutively expressed, whereas although COX-2 is an inducible enzyme in most tissues, in the cortex, hippocampus and amygdala constitutive expression is observed [32, 33]. COX-2 is not only a key enzyme in the inflammatory and neuroinflammatory processes, but has important roles in the regulation of neural activity, such as learning and memory [34]. COX-2 oxygenates a wide range of fatty acids and fatty esters [35].
COX-2 was traditionally thought to be responsible for causing inflammation and neuroinflammation by converting ARA to PG and Tx; however, this simplified model has been reconsidered with a greater understanding of the delicate balance between positive and negative feedback loops [36]. For example, PGE2 and PGD2 are pro-inflammatory mediators responsible for the induction of inflammation, but at a later stage in the process are also responsible for class switching of eicosanoid production from PG and leukotrienes (Lt) to Lx [36]. It has consistently been shown that increasing dietary n-3 PUFA changes the lipid profile of membranes and alters the balance of n-6 and n-3 PUFA competing as substrates for COX, consequently altering the series of prostaglandins synthesised, which ultimately alters cellular responses to mitogenic and inflammatory stimuli [37–41]. This has been demonstrated in many cells throughout the body, including glial cells [42].
LOX catalyse the formation of hydroxyl fatty acids and their metabolites, such as Lt, Lx and the “specialised lipid mediators” (SPM) [29]. These included the resolvins (Rv), protectins (PD) and maresins (MaR) derived from n-3 LCPUFA [43]. LOX enzymes are traditionally classified based on the position of the hydroxyl and hydroperoxy fatty acids they produce from ARA, e.g. 5-LOX forms 5-hydroxy-eicosatetraenoic acid (5-HETE) and 5-hydroperoxy-eicosateraenoic acid (5-HpETE); however, this system has limitations as the position varies according to different chain lengths of the substrates and some LOX act at more than one position [29].
The SPMs are a rapidly expanding class of molecules involved in the active resolution of inflammation produced via COX and LOX catalysed pathways [43]. D-series resolvins (RvD), PD and MaR are from produced from DHA, whereas E-series resolvins (RvE) are from EPA [44]. A further series of RvD and MaR has recently been identified generated from DPAn-3, including RvD1n-3 DPA and MaR1n-3 DPA, which demonstrate similar anti-inflammatory and pro-resolving properties to those from DHA and EPA [45, 46]. The SPMs act via a series of cell-type specific receptors, for example, RvD1 binds GPR32 and lipoxin A4 receptor (ALx), and RvE1 binds the ChemR23 orphan receptor and leukotriene B4 receptor (BLT1) [47]. The best characterised SPM in terms of nervous system protection is (neuro)protectin D1 (NPD1, 10R-17S-dihydroxy-docosahexaenoic acid), which is biosynthesised in response to injury and may have therapeutic potential in a wide range of neurological conditions [48, 49]. In addition, acetylation of COX-2 by aspirin blocks PG biosynthesis, but COX-2 is still able to produce HETE from ARA, hydroxy-docosahexaenoic acid (HDoHE) from DHA and hydroxy-eicosapentaenoic acid (HEPE) from EPA, which can be transformed by leukocytes to aspirin-triggered forms of Lx, Rv and PD [50].
A further class of metabolites generated from n-3 PUFA by LOX is the electrophilic fatty acid oxo-derivatives (EFOX), with 7-oxo-DHA 7-oxo-DPA and 5-oxo-EPA produced from DHA, DPAn-3 and EPA, respectively [51, 52]. EFOXs display a wide range of anti-inflammatory actions, including acting as agonists nuclear receptors, such as the peroxisome proliferator-activated receptor (PPAR) and inhibiting cytokine production in activated macrophages [52]. Furthermore, consistent with the formation of aspirin-triggered SPM, acetylation of COX-2 by aspirin also significantly increases the formation of EFOX [2].
The third oxidative pathway involves CYP epoxygenases and ϖ-hydrolases, which metabolise PUFA to lipid mediators with many diverse biological functions at both the systemic and cellular levels [53, 54]. Regio- and stereoisomers of epoxy-eicosatetraenoic acids (EET) and HETE are produced from ARA, whereas those derived from EPA include epoxy-eicosatetraenoic acids (EETeTR) and hydroxy-eicosapentaenoic acids (HEPE) and epoxy-docosapentaenoic acids (EDP) and HDoHE from DHA [54]. EPA is the preferred substrate for most isoforms of CYP, with metabolism of DHA and ARA occurring at similar rates [54]. Expression of CYP isoforms occurs in multiple cell types across the brain, including astrocytes, neurons and endothelial cells [53].
In addition, n-6 and n-3 PUFAs are also precursors to endogenous ligands of the endocannabinoid receptors (endocannabinoids). The endocannabinoid system is made up of the cannabinoid receptors (CB1 and CB2 receptors), endocannabinoids and the enzymes required for endocannabinoid synthesis and degradation [55]. Two families of endocannabinoids have been identified, 2-acylglycerols and ethanolamides; however, not all congeners are ligands of the cannabinoid receptors [56]. The most abundant and best characterised endocannabinoids in the brain are the 2-acylglycerol, 2-arachidonoylglycerol (2-AG) and the ethanolamide, N-arachidonoylethanolamine (AEA, anandamide), which are both derived from ARA [7]. Further n-6 PUFA-derived endocannabinoids include dihomo-γ-linolenoyl ethanolamide, docosatetraenoyl ethanolamide, 2-arachidonyl glycerol ether (noladin ether), O-arachidonoylethanolamine (virodhamine) and N-arachidonoyldopamine; however, although these endocannabinoids can bind to cannabinoid receptors, their function is still unclear and will not therefore be discussed further in this review [57]. Analogous series of endocannabinoids have been identified from n-3 PUFA. Alpha-linolenoylethanolamide (ALEA) is produced from ALA and has been identified in human plasma, where levels were shown to be responsive to dietary ALA supplementation [58]. However, the best characterised n-3 PUFA-derived endocannabinoids are produced from DHA and EPA, with the 2-acylglycerols, 2-docosahexaenoylglycerol (2-DHG) and 2-eicosapentaenoylglycerol (EPG), and the ethanolamides, N-docosahexaenoylethanolamine (DHEA) and N-eicosapentaenoylethanolamine (EPEA), generated from DHA and EPA, respectively [12, 59]. This review will focus on the endocannabinoids derived from ARA, DHA and EPA.
AEA and 2-AG are produced from membrane-bound phospholipid ARA, with synthesis occurring at the post-synaptic terminal via increased levels of intracellular calcium with both made in response to demand and rapidly degraded to ARA or oxygenated to further bioactive mediators [60]. The major pathways for the biosynthesis and degradation of AEA and 2-AG are described below and summarised in Fig. 3. However, the exact nature of these pathways is still to be resolved because of the complexity of the endocannabinoid system and presence of multiple often redundant pathways [61].
Fig. 3.
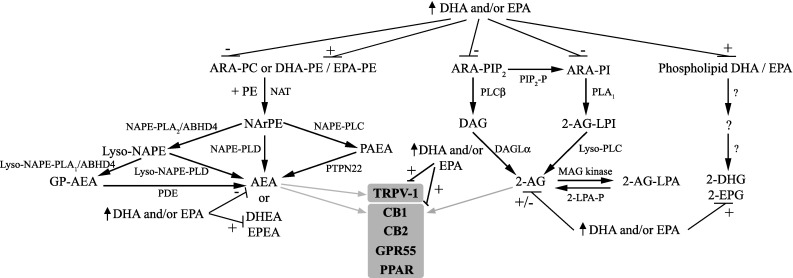
Interplay in the synthesis and actions of the 2-acylglycerols and ethanolamides derived from ARA, DHA and EPA. The major pathway for AEA production begins with N-acyltransferase (NAT) transferring ARA from phosphatidylcholine (ARA-PC) to phosphatidylethanolamine (PE) to generate N-arachidonoyl phosphatidylethanolamine (NArPE), which is followed by hydrolysis by N-acyl phosphatidylethanolamine-selective phospholipase D (NAPE-PLD) to produce AEA. Further pathways include NAPE deacylation by the α/β-hydrolase domain containing 4 (ABHD4) and either the glycerophosphoarachidonoylethanolamide produced (GP-NAPE) cleaved by phosphodiesterase (PDE) to produce AEA or lyso-NAPE is hydrolysed by lyso-NAPE-phospholipase D (PLD) directly to AEA. NAPE can also be hydrolysed by phospholipase C (NAPE-PLC) to generate phospho-anandamide (PAEA), which is dephosphorylated to AEA by phosphatases such as protein tyrosine phosphatase (PTPN22). DHEA and EPEA production from phospholipid bound DHA and EPA appears to share the same pathways. Synthesis of 2-AG occurs from phosphatidylinositol-bound ARA (ARA-PI) via phospholipase C-β (PLCβ) and production of an ARA-diacylglycerol (DAG), which is hydrolysed by diacylglycerol lipases-α to produce 2-AG. Further pathways include dephosphorylation of 2-AG-lysophosphatidic acid (2-AG-LPA) by LPA phosphatase (2-LPA-P) or via phospholipase A1 (PLA1) converting PI to 2-arachidonoyl-lyso PI (2-AG-LPI) and then to 2-AG by lyso phospholipase C (lyso-PLC). The pathways of 2-DPG and 2-EPG production are currently unknown. 2-AG and AEA act at CB1 and CB2 receptors, GPR55 and PPAR, with AEA additionally acting at TRPV-1 (shown in grey). Dietary DHA and EPA enrichment decreases phospholipid ARA and increases phospholipid DHA and EPA, and favours production of DHA and EPA-derived endocannabinoids, whereas acute DHA and EPA treatment in vitro increases 2-AG. DHA and EPA also regulate CB1, CB2 TRPV-1 and PPAR receptor activity and levels. For detailed explanations, refer to the text
AEA production occurs via a series of steps from the membrane phospholipid precursor, sn-1 ARA phosphatidylcholine [62]. A calcium-dependent N-acyltransferase (NAT) transfers ARA to the nitrogen atom of phosphatidylethanolamine (PE) to generate N-arachidonoyl phosphatidylethanolamine (NArPE), which is followed by hydrolysis by an N-acyl phosphatidylethanolamine-selective phospholipase D (NAPE-PLD) to produce AEA [63]. Further parallel pathways have been identified, whereby NAPE is deacylated by α/β-hydrolase domain containing 4 (ABHD4) and either the glycerophosphoarachidonoylethanolamide produced (GP-NAPE) cleaved by a metal-dependent phosphodiesterase (PDE) to produce AEA or lyso-NAPE is hydrolysed by lyso-NAPE-phospholipase D (PLD) directly to AEA. NAPE can also be hydrolysed by phospholipase C (NAPE-PLC) to generate phospho-anandamide (PAEA), which is dephosphorylated by phosphatases such as protein tyrosine phosphatase 22 (PTPN22) to AEA [63]. Studies with NAPE-PLD knock-out mice indicate that NAPE-PLD is the major pathway for NAPE hydrolysis; however, the formation of AEA in the brain readily occurs via NAPE-PLD-independent pathways [64, 65].
The major pathway for the synthesis of 2-AG in the brain occurs from phosphatidylinositol (PI)-bound ARA via phospholipase C-β (PLCβ), which produces sn-1-acyl-2-arachidonoylglycerol, an ARA-diacylglycerol (DAG) [66]. DAG is then hydrolysed into 2-AG by the action of diacylglycerol lipases-α or -β (DAGL-α or DAGL-β), with the removal of the acyl group [66]. DAGLα appears to be the main isoform for 2-AG formation in the brain, as basal and stimulus-induced 2-AG content of the brain is greatly reduced in DAGLα, but not DAGLβ knock-out mice. [67]. Further pathways for the synthesis of 2-AG include dephosphorylation of 2-AG-lysophosphatidic acid (2-AG-LPA) by an LPA phosphatase (2-LPA-P) or via the sequential action of PLA1 converting PI to 2-arachidonoyl-lyso PI (2-AG-LPI) and then to 2-AG by lyso phospholipase C (lyso-PLC) [66].
DHEA and EPEA appear to be produced by the same biosynthetic pathways as AEA [68], whereas the synthesis of 2-DHG and 2-EPG is not well characterised in the literature. However, it is likely they are produced via the same pathways as 2-AG, as chronic DHA and EPA supplementation reduces 2-AG and AEA levels across a range of tissues including the brain, with reciprocal increases in levels of DHEA and 2-DHG, and 2-EPG [12, 69–72]. These alterations suggest competition for shared biosynthetic pathways as DHA and EPA displace ARA from membrane phospholipids. Interestingly, recent work in our laboratory found that acute administration of DHA or EPA significantly increased 2-AG, although not AEA levels in neural stem cells [73]. This increase may be driven by competition for the inactivating enzymes, such as COX-2, although further work is needed to fully elucidate the underlying mechanisms.
AEA and 2-AG predominantly act at the guanine-nucleotide-binding protein (G protein)-coupled receptor (GPCR) cannabinoid receptors, CB1 and CB2 [74]. The CB1 receptor is widely expressed in the brain, where it is the most abundant GPCR, highly expressed in the cortex, hippocampus, cerebellum and basal ganglia [74]. CB2 receptors were initially identified in cells of the immune system [75], but more recently have additionally been described in glia and subsets of neurons in the brain [76]. In addition, AEA and 2-AG have also been shown to act at the orphan receptor, GPR55 [77], and peroxisome proliferator-activated receptors (PPAR) [78]. PPARs are nuclear acting transcription factors with three subtypes, α, β (δ) and γ, and are involved in many cellular processes; for example, PPARγ regulates genes involved in neuroinflammatory processes [79]. AEA is also a ligand for the transient receptor potential vanilloid receptor type 1 (TRPV-1), which is expressed in peripheral sensory neurons and in the central nervous (CNS) system, where they have a role in regulating synaptic function [80].
Endocannabinoids other than 2-AG and AEA either do not bind orthosterically with CB1 or CB2 receptors or bind with much lower affinity; however, they still exhibit cannabimimetic activities and potentiate the activity of 2-AG and AEA, in a phenomenon called the ‘entourage effect’ [56, 81]. However, evidence suggests that the relationship between 2-AG and AEA and their congeners is much more nuanced than this, and other endocannabinoids have been reported to either serve as functional antagonists [81] or act via non-endocannabinoid pathways. For example, DHEA activates protein kinase A (PKA)/cAMP response element binding protein (CREB) pathways [82].
Little is known about the process of endocannabinoid transport across cell membranes, although a putative endocannabinoid cell membrane transporter has been implicated in the control of AEA and 2-AG transport and metabolism [83]. The hydrolysis of AEA releases ARA and ethanolamine and is principally achieved by the fatty acid amide hydrolase (FAAH) enzyme [84], although further yet to be identified proteins are likely involved in the process [61]. DHEA is also a substrate for FAAH hydrolysis to release DHA and ethanolamine [68], whereas the process of EPEA hydrolysis has yet to be identified. Unlike the ethanolamides, a variety of enzymes are responsible for the degradation of 2-AG to ARA and glycerol, with three serine hydrolases accounting for approximately 99% of hydrolysis in the brain [85]. Approximately 85% of 2-AG hydrolysis occurs via monoacylglycerol lipase (MAGL), which is co-localised with CB1 receptors in axon terminals [85]. ABHD6 and ABHD12 account for approximately 4 and 9% of brain 2-AG hydrolase activity, respectively, with ABHD6 located in post-synaptic neurons and ABHD12 is highly expressed in microglia [85]. 2-AG hydrolysis may also be catalysed by FAAH [86]. The pathways(s) of 2-DPG and 2-EPG hydrolysis are currently unknown.
In addition to the direct signalling roles of 2-AG and AEA, both are important intermediates in lipid metabolism. They act as precursor pools for ARA for the subsequent production of eicosanoids [87] and are also converted to further classes of bioactive mediators. 2-AG and AEA are substrates for COX-2, producing prostamides and prostaglandin glycerol esters, LOX producing hydroperoxy derivatives (HPETE) and CYP enzymes, producing hydroxy-eicosatetraenoic ethanolamide molecules (HETE-EA) or epoxy-eicosatrienoic acids (EET) [30, 88, 89]. 2-AG can also be phosphorylated by acyl glycerol kinase(s) to produce lysophosphatidic acid (LPA) [66], another important bioactive lipid [90]. Interestingly, COX-2 metabolites of 2-AG and AEA have been shown to have opposing effects to those of 2-AG and AEA themselves, suggesting a fine balance in the control of synaptic transmission between these lipid mediators and their oxygenated products [91].
The oxidative metabolism of DHA and EPA-derived endocannabinoids is beginning to be elucidated, but there is much that is currently unknown. Lipidomic screening has identified oxygenated products of DHEA generated from LOX and includes 10,17-dihydroxy-docosahexaenoyl ethanolamide (10,17-diHDoHE) and hydroxy-16(17)-epoxy-docosapentaenoyl ethanolamide (HEDPEA) [68]. These molecules exhibit anti-inflammatory and organ-protective effects in a mouse reperfusion second organ injury [68].
The multiple lipid mediators derived from ARA, DHA and EPA are summarised in Fig. 2, where is can be seen that the lipidome of ARA is the best characterised; however, analogous repertoires of mediators are likely produced from DHA and EPA and potentially other PUFAs. Recent developments in lipidomic analyses have greatly increased interest in the discovery, identification and elucidation of the multiple mediators derived from PUFA and endocannabinoids, but much more work is needed to fully develop understanding of their biological activities and the effects of changing dietary intake and subsequent phospholipid PUFA composition on their formation. The remainder of this review will summarise current evidence of the interplay between n-3 and n-6 LCPUFA and endocannabinoids in neuroinflammation, neurogenesis and brain ageing.
Fig. 2.
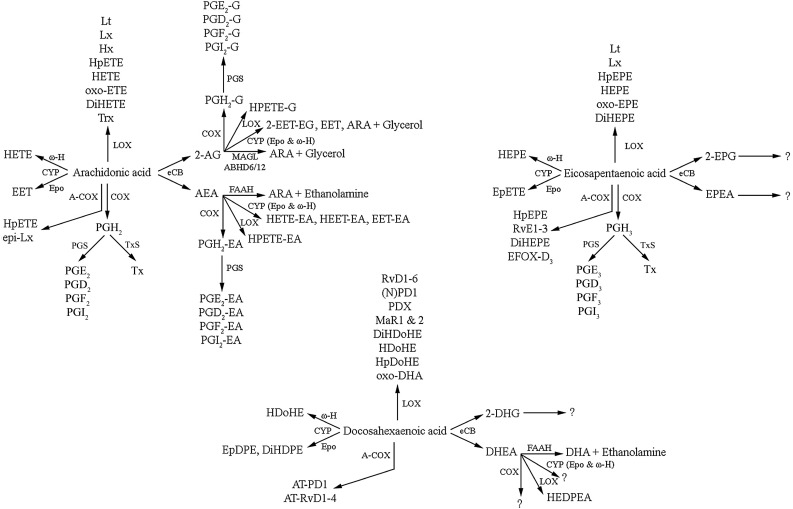
Main lipid mediators produced from ARA, DHA and EPA. ARA, DHA and EPA are precursors to multiple metabolites, including oxylipins produced by cyclooxygenase (COX) and acetylated COX-2 (A-COX), lipoxygenase (LOX) and cytochrome P450 (CYP) enzymes and the endocannabinoids (eCB). The major pathways in the synthesis of ARA, DHA and EPA-derived endocannabinoids are shown in Fig. 3. 2-AG 2-arachidonoylglycerol, 2-DHG 2-docosahexaenoylglycerol, 2-EET-EG 2-epoxy-eicosatrienoic acid glycerol, 2-EPG 2-eicosapentaenoylglycerol, ABHD6/12 α/β-Hydrolase domain containing 6 or 12, AEA N-arachidonoylethanolamide (anandamide), AT aspirin-triggered, DHEA N-docosahexanoylethanolamine (synaptamide), DiHDoHE dihydroxy-docosahexaenoic acid, DiHDPE dihydroxy-docosapentaenoic acid, DiHEPE dihydroxy-eicosapentaenoic acid, DiHETE dihydroxy-eicosatetraenoic acid, DiHETrE dihydroxy-eicosatrienoic acid, EDP epoxy-docosapentaenoic acids, EET epoxy-eicosatrienoic acid, EET-EA epoxy-eicosatrienoic acid ethanolamide, EETeTr epoxy-eicosatetraenoic acids, EFOX electrophilic fatty acid oxo-derivatives, EpDPE epoxy-docosapentaenoic acid, EPEA N-eicosapentaenoylethanolamine, EpETE epoxy-eicosapentaenoic acid, EpETrE epoxy-eicosatrienoic acid, Epo epoxygenase, FAAH fatty acid amide hydrolase, HDoHE hydroxy-docosahexaenoic acid, HEDPEA hydroxy-epoxy-docosapentaenoyl ethanolamide, HEET-EA hydroxyepoxy-eicosatrienoic acid ethanolamide, HEPE hydroxy-eicosapentaenoic acid, HETE hydroxy-eicosatetraenoic acid, HETE-EA hydroxy-eicosatetraenoic acid ethanolamide, HHTrE hydroxy-heptadecatrienoic acid, HpDoHE hydroperoxy-docosahexaenoic acid, HpEPE hydroperoxy-eicosapentaenoic acid, HpETE hydroperoxy-eicosatetraenoic acid, Hx hepoxilin, Lt leukotriene, Lx lipoxin, MAGL monoacylglycerol lipase, MaR maresin, (N)PD1 (neuro)protection D1, oxo-EET oxo-eicosatetraenoic acid, PGD prostaglandin D metabolite, PGE prostaglandin E metabolite, PGF prostaglandin F metabolite, PGI prostacyclin, PGS prostaglandin E, D or F or prostacyclin synthase, PD protectin, RvD resolvin D series, RvE resolvin E series, Tx thromboxane, TxS thromboxane synthase, Trx trioxilin, from DHA and hydroxy-eicosapentaenoic ϖ-hydrolase
Neuroinflammation
Neuroinflammation is the CNS process to restore damaged neurons and glia, with microglia and astrocytes the predominant effectors [92]. Activation of microglia initiates a rapid response involving migration, proliferation, and the release of cytokines and chemokines [93]. This is initially a protective response, but excess neuroinflammation may inhibit neuronal regeneration and if it becomes chronic play an important role in the pathogenesis of neurodegenerative diseases, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD), by secreting cytotoxic proteins and reactive oxygen species [94].
In the healthy brain microglia display a “resting” phenotype responsible for continuous immune monitoring and surveillance and also play a key role in regulating neuronal plasticity via processes including synaptic pruning and neurogenesis [95]. Pathological conditions such as damage to neural cells causes the local “resting” microglia to respond by “activation” and rapidly change their phenotype and redirect their activity [96]. Depending on the type and extent of stimulation the expression of specific genes is induced tailoring the microglial phenotype towards either the classically activated (M1) pro-inflammatory phenotype or the alternatively activated (M2) anti-inflammatory phenotype [96], although the further M2a and M2c phenotypes have been identified based on the stimulus of induction [97].
Work by our laboratory and others has shown the elevated levels of n-3 PUFA reduces microglial activation and subsequent production of pro-inflammatory cytokines in a wide variety of models of neuroinflammation, such as amyotrophic lateral sclerosis [98], spinal cord injury [99, 100], ischaemia [101] and brain ageing [102]. Recent work has begun to explore the mechanisms behind these effects. DHA down-regulates the cell-surface expression of cluster of differentiation 14 (CD14) and Toll-like receptor 4 (TLR4) in lipopolysaccharide (LPS)-stimulated microglial cells [103]. CD14 is a glycosylphosphatidylinositol-linked protein and transduces the signal by associating with other partners, especially TLR4 [104].
N-3 PUFA supplementation also inhibits microglial activation by inhibiting nuclear translocation and secretion of high-mobility group box 1 (HMGB1) and HMGB1-mediated activation of TLR4/NF-κβ signalling pathways in a model of traumatic brain injury [105]. HMGB1 is a central component of the late inflammatory response and the translocation and secretion of HMGB1 are important steps in HMGB1-induced inflammation [106]. After release, HMGB1 binds to transmembrane TLR4 and activates the TLR4/NF-κB signalling pathway, ultimately leading to neuroinflammation [107]. In this study n-3 PUFA supplementation inhibited the translocation of NF-κB p65 from the cytosol to the nucleus, reduced NF-κB p65 expression and inhibited the expression of the TLR4/NF-κB signalling pathway-associated proteins.
Taken together these results suggest n-3 PUFAs regulate microglial activation at several stages; however, these effects could be mediated by the n-PUFA themselves or their respective SPM. For example, both DHA and NPD1 block production of cytokines by microglial cells in a variety of retinal and brain injury models [108, 109]. RvD1 and MaR1 down-regulate in vitro microglia activation [110], RvD2 inhibits LPS-induced increase of TLR4 in microglia [111], and RvE1 alters the inflammatory response and decreases microglial activation in several in vivo models [112, 113].
During neuroinflammation there is a general up-regulation of the activity of the endocannabinoid system, with predominantly anti-inflammatory effects [114]. However, studies looking at the role of endocannabinoids in neuroinflammation tend to focus on the role of CB2 receptors, as CB2 receptors are more abundant than CB1 on microglia [115] and CB2 receptor expression is increased in microglia and astrocytes during neuroinflammation [74], where they attenuate the release of cytokines from activated microglia [8]. Furthermore, microglia from CB2 receptor knock-out mice show a decrease in phagocytic activity and CB2 receptor antagonists reduce motility of microglia in vitro [116]. Furthermore, microglia in brain tissue from patients with Alzheimer’s disease (AD), multiple sclerosis and amyotrophic lateral sclerosis express CB2 receptors [115]. However, recent work suggests a more complex story, with the endocannabinoid system responsive to the M2 phenotype [117]. CB1 and CB2 receptors are down-regulated in M1 microglia, whereas the M2a and M2c microglia show phenotypic changes in the endocannabinoid machinery, such that M2a favours 2-AG synthesis and M2c favours AEA. A recent study also highlighted the role of endocannabinoids in microglia-neuron signalling [118]. Endocannabinoids were secreted through microglial extracellular membrane vesicles and these extracellular vesicles carry AEA on their surface, which stimulates CB1 receptors on neurons and inhibits presynaptic transmission.
In addition to microglia, astrocytes respond to CNS damage and disease via the process of “reactive astrogliosis” [119]. In this process astrocytes respond to and also produce a wide range of cytokines and inflammatory mediators and interact with an array of cell types, thereby mediating crosstalk between neuroinflammatory and neural systems [120]. Astrocytes also have regulatory roles in PUFA metabolism and endocannabinoid signalling and promote endocannabinoid crosstalk with other lipid mediators. Astrocytes are able to synthesise ARA and DHA from LA and ALA, respectively [121], although astrocytic DHA synthesis is much lower than brain DHA uptake and utilisation rates, suggesting astrocyte synthesis does not provide a major contribution [20]. Astrocytes highly express MAGL and mice with specific astrocytic MAGL deletion exhibit moderately increased 2-AG and reduced ARA levels and reduced PGE2 and pro-inflammatory cytokine levels upon LPS administration, indicating an important role for astrocytes in endocannabinoid signalling in neuroinflammation [122]. Furthermore, using an inducible knock-out system the metabolism of 2-AG was shown to be co-ordinately regulated by neurons and astrocytes and involved transcellular shuttling of lipid substrates, such as ARA and eicosanoids [123]. This astrocyte-neuronal crosstalk may provide an integrated regulation of 2-AG metabolism and prevent excessive CB1 receptor activation.
Taken together, these studies show n-3 PUFA and their SPMs, and 2-AG and AEA play important roles in the regulation of the neuroinflammatory responses of microglia and astrocytes. However, with a greater understanding of the mechanisms by which these lipid mediators interact with each other and with microglia, astrocytes and surrounding neurons it may be possible to develop effective approaches to regulating neuroinflammation via manipulation of dietary n-6 and n-3 PUFA intake.
Learning, Memory and Synaptic Plasticity
N-3 PUFA supplementation benefits many aspects of learning and memory, and although a number of putative targets have been identified, the exact mechanisms underlying these effects are still unresolved [1]. A study by Pan and co-workers suggests that these positive effects may be dependent on modulation of the endocannabinoid system [124]. The spatial memory of rats treated with DHA significantly improved at lower doses (150 or 300 mg/kg/day), whereas at a higher level of intake (600 mg/kg/day) it was impaired. These in vivo dose-dependent effects were highly correlated with similar in vitro dose-dependent up-regulation of CB1 and TRPV-1 receptors in cultured hippocampal neurons. The authors concluded that CB1 and TRPV-1 may therefore be involved in positive effects of DHA supplementation on spatial memory, although further work is needed to confirm this.
Synaptic plasticity is a widespread CNS phenomenon that occurs at both excitatory and inhibitory synapses, where changes in synaptic efficacy and strength are induced in response to various stimuli, and this potentiation or depression is thought to underlie phenomena such as learning and memory [125]. The endocannabinoid system positively modulates many aspects of synaptic plasticity [126], and a recent elegant series of studies by Layė and co-workers shows the essential role of n-3 PUFA in these effects [13, 14, 127]. In the first of these studies, long-term n-3 PUFA deficiency prevented endocannabinoid-mediated long-term synaptic depression (LTD) in the prefrontal cortex and nucleus accumbens [13]. Cannabinoid receptors couple to G protein type Gi/o and activate signalling pathways [74], and in this study CB1 receptors were uncoupled from their G(i/o) proteins. In the follow-up studies, similar effects on other measures of endocannabinoid-dependent plasticity were also found in other brain regions, including the hypothalamus [14] and hippocampus [127]. In the hippocampus, loss of N-methyl-d-aspartate (NMDA) glutamate receptor-dependent LTP induced by n-3 PUFA deficiency was shown to be due to the ablation of endocannabinoid-mediated inhibitory LTD (iLTD) [127]. In the hippocampus LTP is gated by the process of heterosynaptic iLTD, which is dependent on the activation of CB1 receptors [80]. Overall, the role of n-PUFA regulation of the endocannabinoid system in learning, memory and synaptic plasticity appears more complex than simply the modulation of endocannabinoid levels, but also critically depends on modulating receptor function.
Neurogenesis
Neurogenesis in the adult brain from precursor neural stem cells has been identified consistently in two regions, the subgranular layer of the hippocampal dentate gyrus and the subventricular zone (SVZ), where it has been reported in all mammals studied, including humans [128]. The hippocampus is essential for learning and memory formation and consolidation and also important in regulating aspects of emotion, fear, anxiety and stress [129]. However, the hippocampus is particularly vulnerable to neuroinflammation, ageing and neurodegeneration [129]; indeed ageing is the greatest negative regulator of hippocampal neurogenesis [130]. It is therefore interesting to note that hippocampal neurogenesis has been shown to increase following ischaemia [131], stroke [132] and seizures [133], where the increases may be considered an attempt by the brain at self-repair. Enhancing hippocampal neurogenesis may therefore offer a novel therapeutic approach in the treatment of brain ageing and neurodegeneration.
DHA and EPA treatment has consistently been shown to increase adult hippocampal neurogenesis across a range of animal models [134], also in neural stem cells, where DHA appears to promote neuronal differentiation [73]. Similarly, the endocannabinoid system is essential for adult neurogenesis in both the hippocampus [135, 136] and SVZ [137], although studies into the pro-neurogeneic effects of endocannabinoids in the dentate gyrus have produced conflicting results. For example, adult rats treated with the AEA analogue methanandamide have significantly decreased hippocampal neurogenesis, which is increased by CB1 antagonists [136]. However, chronic treatment with a synthetic endocannabinoid agonist increases adult hippocampal neurogenesis in rats [138], and CB1 receptor knock-out mice show significant reductions in neurogenesis in the dentate gyrus and SVZ [135]. Pharmacological blockade of DAGL and CB2 with specific antagonists inhibits the proliferation of neural stem cells and the proliferation of progenitor cells in young animals [137]. A similar response is seen with a FAAH inhibitor [139]. Overall, the effects of the endocannabinoid system on neurogenesis appear to be a fine balance of receptor activation.
Work in our laboratory is the first to explore the role of the endocannabinoid system in the pro-neurogeneic effects of DHA and EPA [73]. In this study, addition of DHA or EPA to neural stem cells induces opposing effects on cell fate, which are directed by different signalling pathways. Although both DHA and EPA significantly increase 2-AG levels, only EPA utilises endocannabinoid signalling pathways to increase proliferation. EPA increases proliferation via CB1/2 receptors, which activate the p38 mitogen-activated protein kinase (p38 MAPK) signalling pathway. DHA was found to decrease cell proliferation, consistent with induction of differentiation. It may be hypothesised that although 2-AG is increased by DHA, the effects may be mitigated and cell fate directed towards differentiation via alternative pathways, such as through conversion to DHEA [82]. Rashid and co-workers show that DHEA induces differentiation of neural stem cells via protein kinase A (PKA)/cAMP response element binding protein (CREB). It may therefore be that DHA and EPA direct cell fate via alternative pathways determined by the levels and types of mediators produced.
In addition, our study also identified a previously unrecognised role of the immune system in the effects of DHA and EPA [73]. DHA and EPA treatment of neural stem cells from interleukin-1β (IL-1β) knock-out mice induced effects quite distinct from the wild-type cells, whereby proliferation was increased by DHA and reduced by EPA. As p38 MAPK was not activated by DHA, this suggests alterative non-endocannabinoid pathways were behind the increases in proliferation.
The Ageing Brain
Normal brain ageing is characterised by many detrimental changes, such as mitochondrial dysfunction and alterations in energy metabolism [140], damage to DNA [141], increased microglial activation [142] and increased oxidative stress [143]. The ageing brain is also prone to development of neurodegenerative diseases, such as AD and PD, but with the protracted pre-symptomatic stages it is hard to identify what are normal age-related changes and what are effects of undetected neurodegeneration [144].
Many epidemiological studies suggest positive associations between an elevated dietary intake of n-3 PUFA and the maintenance of cognitive function in old age [3]. However, the results of randomised controlled trials in this area have been mixed, although positive study outcomes with higher doses of DHA in particular in asymptomatic participants or those with very mild memory deficits suggest supplementation is most effective in the pre-symptomatic stage, prior to the onset of mild cognitive impairment or dementia [145–147].
Studies in both rodents and humans show that the endocannabinoid system is susceptible to age-related deficits [74]. For example, CB1 receptor levels decrease, along with the activity NAPE-PLD and DAGL [74]. Furthermore, decreases in DAGL, coupled with elevated MAGL, leads to specific decreases in 2-AG levels in the hippocampus of ageing mice [148]. Using mouse genetic CB1 receptor knock-out models, it is possible to mimic the effects of these age-related changes [74]. CB1 receptor deletion leads to an age-dependent acceleration of cognitive decline with accelerated hippocampal neuronal loss and increases aspects of neuroinflammation, such as reactive astrogliosis and microglial activation.
These studies suggest that the age-related decline of specific components of the endocannabinoid system accelerates key aspects of brain ageing; therefore, through the restoration or reversal of the age-related effects it may be able to decrease this decline. In addition to modulating the levels of 2-AG and AEA, expressions of CB1 receptors, TRPV-1 and PPARγ have all been shown to be responsive to n-3 PUFA treatment [79, 124], suggesting that n-3 PUFA may be able to mitigate or reverse some of these age-related losses. Furthermore, these positive effects on the endocannabinoid system may potentially contribute to some of the protective effects of n-3 PUFA observed in studies in ageing. However, much more research is required to develop our understanding of the mechanisms underlying these effects and the consequences for the endocannabinoid system to maximise the therapeutic potential of n-3 PUFA in brain protection and repair.
Conclusions
Due to their fundamental nature, ARA, DHA, EPA and their mediators and the endocannabinoid system have wide-ranging effects across the CNS and recent evidence strongly indicates a complex interplay between them. The levels of phospholipid-bound ARA determine the levels of 2-AG and AEA, which in addition to their own biological activities act as reservoirs of ARA for subsequent eicosanoid production. Importantly, brain LCPUFA levels are responsive to dietary intake, and the n-6:n-3 PUFA ratio of the current Western diet may lead to increased neuroinflammation and also overstimulation of the endocannabinoid system.
Neuroinflammation is a key feature of brain ageing and neurodegeneration and the development of new therapeutic approaches is necessary. Epidemiological studies consistently show beneficial effects of an elevated intake of DHA and EPA; however, these observations have so far failed to lead to new treatments. Trials typically provide n-3 PUFA in the form of fish oils, mixed DHA and EPA preparations or separate DHA and EPA, with limited consideration of the background levels of n-6 PUFA. It is hoped that a greater understanding of the relationship among ARA, DHA, EPA and the endocannabinoid system will lead to advances in developing their therapeutic potential and ultimately lead to the development of more targeted treatment options for brain protection and repair.
Abbreviations
- 2-AG
2-Arachidonoylglycerol
- 2-AG-LPA
2-Arachidonoylglycerol-lysophosphatidic acid
- 2-AG-LPI
2-Arachidonoyl-lysophosphatidylinositol
- 2-DHG
2-Docosahexaenoylglycerol
- 2-EET-EG
2-Epoxy-eicosatrienoic acid glycerol
- 2-EPG
2-Eicosapentaenoylglycerol
- A-COX
Acetylated COX-2
- ABHD4
α/β-Hydrolase domain containing 4
- ABHD6
α/β-Hydrolase domain containing 6
- ABHD12
α/β-Hydrolase domain containing 12
- AdA
Adrenic acid
- AEA
N-arachidonoylethanolamide (anandamide)
- ARA
Arachidonic acid
- AT
Aspirin-triggered
- COX-2
Cyclooxygenase-2
- CYP
Cytochrome P450 monooxygenase
- DAGL
Diacylglycerol lipase
- DGLA
Dihomo-γ-linolenic acid
- DHA
Docosahexaenoic acid
- DHEA
N-docosahexaenoylethanolamine (synaptamide)
- DiHDoHE
Dihydroxy-docosahexaenoic acid
- DiHDPE
Dihydroxy-docosapentaenoic acid
- DiHEPE
Dihydroxy-eicosapentaenoic acid
- DiHETE
Dihydroxy-eicosatetraenoic acid
- DiHETrE
Dihydroxy-eicosatrienoic acid
- DPA
Docosapentaenoic acid
- eCB
Endocannabinoid
- EDP
Epoxy-docosapentaenoic acid
- EET
Epoxy-eicosatrienoic acid
- EET-EA
Epoxy-eicosatrienoic acid ethanolamide
- EETeTr
Epoxy-eicosatetraenoic acids
- EFOX
Electrophilic fatty acid oxo-derivative
- EPA
Eicosapentaenoic acid
- EpDPE
Epoxy-docosapentaenoic acid
- EPEA
N-eicosapentaenoylethanolamine
- EpETE
Epoxy-eicosapentaenoic acid
- EpETrE
Epoxy-eicosatrienoic acid
- Epo
Epoxygenase
- FAAH
Fatty acid amide hydrolase
- GP-NAPE
Glycerophosphoarachidonoylethanolamide
- HDoHE
Hydroxy-docosahexaenoic acid
- HEDPEA
Hydroxy-epoxy-docosapentaenoylethanolamide
- HEET-EA
Hydroxy-epoxy-eicosatrienoic acid ethanolamide
- HEPE
Hydroxy-eicosapentaenoic acid
- HETE
Hydroxy-eicosatetraenoic acid
- HETE-EA
Hydroxy-eicosatetraenoic acid ethanolamide
- HHTrE
Hydroxy-heptadecatrienoic acid
- HpDoHE
Hydroperoxy-docosahexaenoic acid
- HpEPE
Hydroperoxy-eicosapentaenoic acid
- HpETE
Hydroperoxy-eicosatetraenoic acid
- Hx
Hepoxilin
- LCPUFA
Long-chain polyunsaturated fatty acid
- LOX
Lipoxygenase
- Lt
Leukotriene
- LTD
Long-term depression
- LTP
Long-term potentiation
- Lx
Lipoxin
- MAGL
Monoacylglycerol lipase
- MaR
Maresin
- (N)PD1
(Neuro)protection D1
- NAPE-PLD
N-acyl phosphatidylethanolamine-selective phospholipase D
- NArPE
N-arachidonoyl phosphatidylethanolamine
- NAT
N-acyltransferase
- oxo-EET
Oxo-eicosatetraenoic acid
- PAEA
Phospho-anandamide
- PD
Protectin
- PDE
Phosphodiesterase
- PE
Phosphatidylethanolamine
- PGD
Prostaglandin D metabolite
- PGE
Prostaglandin E metabolite
- PGF
Prostaglandin F metabolite
- PGI
Prostacyclin
- PGS
Prostaglandin E, D or F or prostacyclin synthase
- PI
Phosphatidylinositol
- PLA1
Phospholipase A1
- PLC
Phospholipase C
- PLD
Phospholipase D
- PPAR
Peroxisome proliferator-activated receptor
- PTPN22
Protein tyrosine phosphatase 22
- RvD
Resolvin D series
- RvE
Resolvin E series
- Trx
Trioxilin
- Tx
Thromboxane
- TXS
Thromboxane synthase
- SDA
Stearidonic acid
- SVZ
Subventricular zone
- TRPV-1
Transient receptor potential vanilloid receptor type 1
- ϖ-H
ϖ-hydrolase
Compliance with Ethical Standards
Conflict of interest
The author declares no conflict of interest.
References
- 1.Dyall SC, Michael-Titus AT. Neurological benefits of omega-3 fatty acids. Neuromol Med. 2008;10:219–235. doi: 10.1007/s12017-008-8036-z. [DOI] [PubMed] [Google Scholar]
- 2.Dyall SC. Long-chain omega-3 fatty acids and the brain: a review of the independent and shared effects of EPA, DPA and DHA. Front Ageing Neurosci. 2015;7:52. doi: 10.3389/fnagi.2015.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cederholm T, Salem N, Jr, Palmblad J. Omega-3 fatty acids in the prevention of cognitive decline in humans. Adv Nutr. 2013;4:672–676. doi: 10.3945/an.113.004556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Forsyth S, Gautier S, Salem N., Jr Global estimates of dietary intake of docosahexaenoic acid and arachidonic acid in developing and developed countries. Ann Nutr Metab. 2016;68:258–267. doi: 10.1159/000446855. [DOI] [PubMed] [Google Scholar]
- 5.Micha R, Khatibzadeh S, Shi P, Fahimi S, Lim S, Andrews KG, Engell RE, Powles J, Ezzati M, Mozaffarian D. Global, regional, and national consumption levels of dietary fats and oils in 1990 and 2010: a systematic analysis including 266 country-specific nutrition surveys. BMJ. 2014;348:g2272. doi: 10.1136/bmj.g2272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flock MR, Harris WS, Kris-Etherton PM. Long-chain omega-3 fatty acids: time to establish a dietary reference intake. Nutr Rev. 2015;71:692–707. doi: 10.1111/nure.12071. [DOI] [PubMed] [Google Scholar]
- 7.Katona I, Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. 2012;35:529–558. doi: 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cassano T, Calcagnini S, Pace L, De Marco F, Romano A, Gaetani S. Cannabinoid receptor 2 signaling in neurodegenerative disorders: from pathogenesis to a promising therapeutic target. Front Neurosci. 2017;11:30. doi: 10.3389/fnins.2017.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Molina-Holgado E, Molina-Holgado F. Mending the broken brain: neuroimmune interactions in neurogenesis. J Neurochem. 2010;114:1277–1290. doi: 10.1111/j.1471-4159.2010.06849.x. [DOI] [PubMed] [Google Scholar]
- 10.Bonnet AE, Marchalant Y. Potential therapeutical contributions of the endocannabinoid system towards aging and Alzheimer’s disease. Aging Dis. 2015;6:400–405. doi: 10.14336/AD.2015.0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berger A, Crozier G, Bisogno T, Cavaliere P, Innis SM, Di Marzo V. Anandamide and diet: inclusion of dietary arachidonate and docosahexaenoate leads to increased brain levels of the corresponding N-acylethanolamines in piglets. Proc Natl Acad Sci USA. 2001;98:6402–6406. doi: 10.1073/pnas.101119098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wood JT, Williams JS, Pandarinathan L, Janero DR, Lammi-Keefe CJ, Makriyannis A. Dietary docosahexaenoic acid supplementation alters select physiological endocannabinoid-system metabolites in brain and plasma. J Lipid Res. 2010;51:1416–1423. doi: 10.1194/jlr.M002436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lafourcade M, Larrieu T, Mato S, Duffaud A, Sepers M, Matias I, De Smedt-Peyrusse V, Labrousse VF, Bretillon L, Matute C, Rodriguez-Puertas R, Laye S, Manzoni OJ. Nutritional omega-3 deficiency abolishes endocannabinoid-mediated neuronal functions. Nat Neurosci. 2011;14:345–350. doi: 10.1038/nn.2736. [DOI] [PubMed] [Google Scholar]
- 14.Larrieu T, Madore C, Joffre C, Laye S. Nutritional n-3 polyunsaturated fatty acids deficiency alters cannabinoid receptor signaling pathway in the brain and associated anxiety-like behavior in mice. J Physiol Biochem. 2012;68:671–681. doi: 10.1007/s13105-012-0179-6. [DOI] [PubMed] [Google Scholar]
- 15.Thomazeau A, Bosch-Bouju C, Manzoni O, Laye S. Nutritional n-3 PUFA deficiency abolishes endocannabinoid gating of hippocampal long-term potentiation. Cereb Cortex. 2016;27(4):2571–2579. doi: 10.1093/cercor/bhw052. [DOI] [PubMed] [Google Scholar]
- 16.Simopoulos AP. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed Pharmacother. 2002;56:365–379. doi: 10.1016/S0753-3322(02)00253-6. [DOI] [PubMed] [Google Scholar]
- 17.Stark KD, Van Elswyk ME, Higgins MR, Weatherford CA, Salem N., Jr Global survey of the omega-3 fatty acids, docosahexaenoic acid and eicosapentaenoic acid in the blood stream of healthy adults. Prog Lipid Res. 2016;63:132–152. doi: 10.1016/j.plipres.2016.05.001. [DOI] [PubMed] [Google Scholar]
- 18.Gregory MK, Gibson RA, Cook-Johnson RJ, Cleland LG, James MJ. Elongase reactions as control points in long-chain polyunsaturated fatty acid synthesis. PLoS One. 2011;6:e29662. doi: 10.1371/journal.pone.0029662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sprecher H. Metabolism of highly unsaturated n-3 and n-6 fatty acids. Biochem Biophys Acta. 2000;1486:219–231. doi: 10.1016/s1388-1981(00)00077-9. [DOI] [PubMed] [Google Scholar]
- 20.Domenichiello AF, Kitson AP, Bazinet RP. Is docosahexaenoic acid synthesis from alpha-linolenic acid sufficient to supply the adult brain? Prog Lipid Res. 2015;59:54–66. doi: 10.1016/j.plipres.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 21.Lands B. Historical perspectives on the impact of n-3 and n-6 nutrients on health. Prog Lipid Res. 2014;55:17–29. doi: 10.1016/j.plipres.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 22.DeMar JC, Jr, Lee H-J, Ma K, Chang L, Bell JM, Rapoport SI, Bazinet RP. Brain elongation of linoleic acid is a negligible source of the arachidonate in brain phospholipids of adult rats. Biochem Biophys Acta. 2006;1761:1050–1059. doi: 10.1016/j.bbalip.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 23.Demar JC, Jr, Ma K, Chang L, Bell JM, Rapoport SI. alpha-Linolenic acid does not contribute appreciably to docosahexaenoic acid within brain phospholipids of adult rats fed a diet enriched in docosahexaenoic acid. J Neurochem. 2005;94:1063–1076. doi: 10.1111/j.1471-4159.2005.03258.x. [DOI] [PubMed] [Google Scholar]
- 24.Ouellet M, Emond V, Chen CT, Julien C, Bourasset F, Oddo S, LaFerla F, Bazinet RP, Calon F. Diffusion of docosahexaenoic and eicosapentaenoic acids through the blood-brain barrier: an in situ cerebral perfusion study. Neurochem Int. 2009;55:476–482. doi: 10.1016/j.neuint.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 25.Chen CT, Domenichiello AF, Trepanier MO, Liu Z, Masoodi M, Bazinet RP. The low levels of eicosapentaenoic acid in rat brain phospholipids are maintained via multiple redundant mechanisms. J Lipid Res. 2013;54:2410–2422. doi: 10.1194/jlr.M038505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaur G, Molero JC, Weisinger HS, Sinclair AJ. Orally administered [14C]DPA and [14C]DHA are metabolised differently to [14C]EPA in rats. Br J Nutr. 2013;109:441–448. doi: 10.1017/S0007114512001419. [DOI] [PubMed] [Google Scholar]
- 27.Novak EM, Dyer RA, Innis SM. High dietary omega-6 fatty acids contribute to reduced docosahexaenoic acid in the developing brain and inhibit secondary neurite growth. Brain Res. 2008;1237:136–145. doi: 10.1016/j.brainres.2008.07.107. [DOI] [PubMed] [Google Scholar]
- 28.Phillis JW, Horrocks LA, Farooqui AA. Cyclooxygenases, lipoxygenases, and epoxygenases in CNS: their role and involvement in neurological disorders. Brain Res Brain Res Rev. 2006;52:201–243. doi: 10.1016/j.brainresrev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 29.Gabbs M, Leng S, Devassy JG, Monirujjaman M, Aukema HM. Advances in our understanding of oxylipins derived from dietary PUFAs. Adv Nutr. 2015;6:513–540. doi: 10.3945/an.114.007732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alhouayek M, Muccioli GG. COX-2-derived endocannabinoid metabolites as novel inflammatory mediators. Trends Pharmacol Sci. 2014;35:284–292. doi: 10.1016/j.tips.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 31.Kulmacz RJ, van der Donk WA, Tsai A-L. Comparison of the properties of prostaglandin H synthase-1 and -2. Prog Lipid Res. 2003;42:377–404. doi: 10.1016/S0163-7827(03)00023-7. [DOI] [PubMed] [Google Scholar]
- 32.Breder CD, Dewitt D, Kraig RP. Characterization of inducible cyclooxygenase in rat brain. J Comp Neurol. 1995;355:296–315. doi: 10.1002/cne.903550208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–386. doi: 10.1016/0896-6273(93)90192-T. [DOI] [PubMed] [Google Scholar]
- 34.Yang H, Chen C. Cyclooxygenase-2 in synaptic signaling. Curr Pharm Des. 2008;14:1443–1451. doi: 10.2174/138161208784480144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vecchio AJ, Simmons DM, Malkowski MG. Structural basis of fatty acid substrate binding to cyclooxygenase-2. J Biol Chem. 2010;285:22152–22163. doi: 10.1074/jbc.M110.119867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buckley CD, Gilroy DW, Serhan CN. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity. 2014;40:315–327. doi: 10.1016/j.immuni.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mantzioris E, Cleland LG, Gibson RA, Neumann MA, Demasi M, James MJ. Biochemical effects of a diet containing foods enriched with n-3 fatty acids. Am J Clin Nutr. 2000;72:42–48. doi: 10.1093/ajcn/72.1.42. [DOI] [PubMed] [Google Scholar]
- 38.Yerram NR, Moore SA, Spector AA. Eicosapentaenoic acid metabolism in brain microvessel endothelium: effect on prostaglandin formation. J Lipid Res. 1989;30:1747–1757. [PubMed] [Google Scholar]
- 39.Bagga D, Wang L, Farias-Eisner R, Glaspy JA, Reddy ST. Differential effects of prostaglandin derived from w-6 and w-3 polyunsaturated fatty acids on COX-2 expression and IL-6 secretion. Proc Natl Acad Sci USA. 2003;100:1751–1756. doi: 10.1073/pnas.0334211100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luostarinen R, Wallin R, Saldeen T. Dietary (n-3) fatty acids increase superoxide dismutase activity and decrease thromboxane production in the rats heart. Nutr Res. 1997;17:163–175. doi: 10.1016/S0271-5317(96)00242-4. [DOI] [Google Scholar]
- 41.Broughton KS, Wade JW. Total fat and (n-3):(n-6) fat ratios influence eicosanoid production in mice. J Nutr. 2002;132:88–94. doi: 10.1093/jn/132.1.88. [DOI] [PubMed] [Google Scholar]
- 42.Petroni A, Salami M, Blasevich M, Papini N, Galli C. Inhibition by n-3 fatty acids of arachidonic acid metabolism in a primary culture of astroglial cells. Neurochem Res. 1994;19:1187–1193. doi: 10.1007/BF00965154. [DOI] [PubMed] [Google Scholar]
- 43.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Serhan CN, Dalli J, Colas RA, Winkler JW, Chiang N. Protectins and maresins: new pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim Biophys Acta. 2014;1851(4):397–413. doi: 10.1016/j.bbalip.2014.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Aursnes M, Tungen JE, Vik A, Colas R, Cheng CY, Dalli J, Serhan CN, Hansen TV. Total synthesis of the lipid mediator PD1n-3 DPA: configurational assignments and anti-inflammatory and pro-resolving actions. J Nat Prod. 2014;77:910–916. doi: 10.1021/np4009865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tungen JE, Aursnes M, Dalli J, Arnardottir H, Serhan CN, Hansen TV. Total synthesis of the anti-inflammatory and pro-resolving lipid mediator MaR1n-3 DPA utilizing an sp(3)–sp(3) Negishi cross-coupling reaction. Chemistry. 2014;20:14575–14578. doi: 10.1002/chem.201404721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serhan CN, Chiang N. Resolution phase lipid mediators of inflammation: agonists of resolution. Curr Opin Pharmacol. 2013;13(4):632–640. doi: 10.1016/j.coph.2013.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bazan NG. The docosanoid neuroprotectin D1 induces homeostatic regulation of neuroinflammation and cell survival. Prostaglandins Leukot Essent Fatty Acids. 2013;88:127–129. doi: 10.1016/j.plefa.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bazan NG, Musto AE, Knott EJ. Endogenous signaling by omega-3 docosahexaenoic acid-derived mediators sustains homeostatic synaptic and circuitry integrity. Mol Neurobiol. 2011;44:216–222. doi: 10.1007/s12035-011-8200-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Serhan CN, Chiang N, Dalli J. The resolution code of acute inflammation: novel pro-resolving lipid mediators in resolution. Semin Immunol. 2015;27:200–215. doi: 10.1016/j.smim.2015.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cipollina C, Salvatore SR, Muldoon MF, Freeman BA, Schopfer FJ. Generation and dietary modulation of anti-inflammatory electrophilic omega-3 fatty acid derivatives. PLoS One. 2014;9:e94836. doi: 10.1371/journal.pone.0094836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Groeger AL, Cipollina C, Cole MP, Woodcock SR, Bonacci G, Rudolph TK, Rudolph V, Freeman BA, Schopfer FJ. Cyclooxygenase-2 generates anti-inflammatory mediators from omega-3 fatty acids. Nat Chem Biol. 2010;6:433–441. doi: 10.1038/nchembio.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davis CM, Liu X, Alkayed NJ (2017) Cytochrome P450 eicosanoids in cerebrovascular function and disease. Pharmacol Ther. pii: S0163–7258(17)30123–7. doi:10.1016/j.pharmthera.2017.05.004 [DOI] [PMC free article] [PubMed]
- 54.Arnold C, Konkel A, Fischer R, Schunck WH. Cytochrome P450-dependent metabolism of omega-6 and omega-3 long-chain polyunsaturated fatty acids. Pharmacol Rep. 2010;62:536–547. doi: 10.1016/S1734-1140(10)70311-X. [DOI] [PubMed] [Google Scholar]
- 55.De Petrocellis L, Di Marzo V. An introduction to the endocannabinoid system: from the early to the latest concepts. Best Pract Res Clin Endocrinol Metab. 2009;23:1–15. doi: 10.1016/j.beem.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 56.Cascio MG. PUFA-derived endocannabinoids: an overview. Proc Nutr Soc. 2013;72:451–459. doi: 10.1017/S0029665113003418. [DOI] [PubMed] [Google Scholar]
- 57.Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- 58.Jones PJ, Lin L, Gillingham LG, Yang H, Omar JM. Modulation of plasma N-acylethanolamine levels and physiological parameters by dietary fatty acid composition in humans. J Lipid Res. 2014;55:2655–2664. doi: 10.1194/jlr.P051235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramsden CE, Zamora D, Makriyannis A, Wood JT, Mann JD, Faurot KR, MacIntosh BA, Majchrzak-Hong SF, Gross JR, Courville AB, Davis JM, Hibbeln JR. Diet-induced changes in n-3- and n-6-derived endocannabinoids and reductions in headache pain and psychological distress. J Pain. 2015;16:707–716. doi: 10.1016/j.jpain.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fonseca BM, Costa MA, Almada M, Correia-da-Silva G, Teixeira NA. Endogenous cannabinoids revisited: a biochemistry perspective. Prostaglandins Other Lipid Mediat. 2013;102–103:13–30. doi: 10.1016/j.prostaglandins.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 61.Piomelli D. More surprises lying ahead. The endocannabinoids keep us guessing. Neuropharmacology. 2014;76(Pt B):228–234. doi: 10.1016/j.neuropharm.2013.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz J-C, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- 63.Liu J, Wang L, Harvey-White J, Huang BX, Kim HY, Luquet S, Palmiter RD, Krystal G, Rai R, Mahadevan A, Razdan RK, Kunos G. Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology. 2008;54:1–7. doi: 10.1016/j.neuropharm.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tsuboi K, Okamoto Y, Ikematsu N, Inoue M, Shimizu Y, Uyama T, Wang J, Deutsch DG, Burns MP, Ulloa NM, Tokumura A, Ueda N. Enzymatic formation of N-acylethanolamines from N-acylethanolamine plasmalogen through N-acylphosphatidylethanolamine-hydrolyzing phospholipase D-dependent and -independent pathways. Biochim Biophys Acta. 2011;1811:565–577. doi: 10.1016/j.bbalip.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 65.Leung D, Saghatelian A, Simon GM, Cravatt BF. Inactivation of N-acyl phosphatidylethanolamine phospholipase D reveals multiple mechanisms for the biosynthesis of endocannabinoids. Biochemistry. 2006;45:4720–4726. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Murataeva N, Straiker A, Mackie K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br J Pharmacol. 2014;171:1379–1391. doi: 10.1111/bph.12411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron. 2010;65:320–327. doi: 10.1016/j.neuron.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 68.Yang R, Fredman G, Krishnamoorthy S, Agrawal N, Irimia D, Piomelli D, Serhan CN. Decoding functional metabolomics with docosahexaenoyl ethanolamide (DHEA) identifies novel bioactive signals. J Biol Chem. 2011;286:31532–31541. doi: 10.1074/jbc.M111.237990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Artmann A, Petersen G, Hellgren LI, Boberg J, Skonberg C, Nellemann C, Hansen SH, Hansen HS. Influence of dietary fatty acids on endocannabinoid and N-acylethanolamine levels in rat brain, liver and small intestine. Biochim Biophys Acta. 2008;1781:200–212. doi: 10.1016/j.bbalip.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 70.Batetta B, Griinari M, Carta G, Murru E, Ligresti A, Cordeddu L, Giordano E, Sanna F, Bisogno T, Uda S, Collu M, Bruheim I, Di Marzo V, Banni S. Endocannabinoids may mediate the ability of (n-3) fatty acids to reduce ectopic fat and inflammatory mediators in obese Zucker rats. J Nutr. 2009;139:1495–1501. doi: 10.3945/jn.109.104844. [DOI] [PubMed] [Google Scholar]
- 71.Matias I, Carta G, Murru E, Petrosino S, Banni S, Di Marzo V. Effect of polyunsaturated fatty acids on endocannabinoid and N-acyl-ethanolamine levels in mouse adipocytes. Biochim Biophys Acta. 2008;1781:52–60. doi: 10.1016/j.bbalip.2007.11.001. [DOI] [PubMed] [Google Scholar]
- 72.Watanabe S, Doshi M, Hamazaki T. n-3 Polyunsaturated fatty acid (PUFA) deficiency elevates and n-3 PUFA enrichment reduces brain 2-arachidonoylglycerol level in mice. Prostaglandins Leukot Essent Fatty Acids. 2003;69:51–59. doi: 10.1016/S0952-3278(03)00056-5. [DOI] [PubMed] [Google Scholar]
- 73.Dyall SC, Mandhair HK, Fincham RE, Kerr DM, Roche M, Molina-Holgado F. Distinctive effects of eicosapentaenoic and docosahexaenoic acids in regulating neural stem cell fate are mediated via endocannabinoid signalling pathways. Neuropharmacology. 2016;107:387–395. doi: 10.1016/j.neuropharm.2016.03.055. [DOI] [PubMed] [Google Scholar]
- 74.Di Marzo V, Stella N, Zimmer A. Endocannabinoid signalling and the deteriorating brain. Nat Rev Neurosci. 2015;16:30–42. doi: 10.1038/nrn3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Patel KD, Davison JS, Pittman QJ, Sharkey KA. Cannabinoid CB(2) receptors in health and disease. Curr Med Chem. 2010;17:1393–1410. doi: 10.2174/092986710790980041. [DOI] [PubMed] [Google Scholar]
- 76.Onaivi ES, Ishiguro H, Gu S, Liu QR. CNS effects of CB2 cannabinoid receptors: beyond neuro-immuno-cannabinoid activity. J Psychopharmacol. 2012;26:92–103. doi: 10.1177/0269881111400652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ryberg E, Larsson N, Sjogren S, Hjorth S, Hermansson NO, Leonova J, Elebring T, Nilsson K, Drmota T, Greasley PJ. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.O’Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dyall SC, Michael GJ, Michael-Titus AT. Omega-3 fatty acids reverse age-related decreases in nuclear receptors and increase neurogenesis in old rats. J Neurosci Res. 2010;88:2091–2102. doi: 10.1002/jnr.22390. [DOI] [PubMed] [Google Scholar]
- 80.Castillo PE, Younts TJ, Chavez AE, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76:70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Murataeva N, Dhopeshwarkar A, Yin D, Mitjavila J, Bradshaw H, Straiker A, Mackie K. Where’s my entourage? The curious case of 2-oleoylglycerol, 2-linolenoylglycerol, and 2-palmitoylglycerol. Pharmacol Res. 2016;110:173–180. doi: 10.1016/j.phrs.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rashid MA, Katakura M, Kharebava G, Kevala K, Kim HY. N-Docosahexaenoylethanolamine is a potent neurogenic factor for neural stem cell differentiation. J Neurochem. 2013;125:869–884. doi: 10.1111/jnc.12255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chicca A, Marazzi J, Nicolussi S, Gertsch J. Evidence for bidirectional endocannabinoid transport across cell membranes. J Biol Chem. 2012;287:34660–34682. doi: 10.1074/jbc.M112.373241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Placzek EA, Okamoto Y, Ueda N, Barker EL. Membrane microdomains and metabolic pathways that define anandamide and 2-arachidonyl glycerol biosynthesis and breakdown. Neuropharmacology. 2008;55:1095–1104. doi: 10.1016/j.neuropharm.2008.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Savinainen JR, Saario SM, Laitinen JT. The serine hydrolases MAGL, ABHD6 and ABHD12 as guardians of 2-arachidonoylglycerol signalling through cannabinoid receptors. Acta Physiol (Oxf) 2012;204:267–276. doi: 10.1111/j.1748-1716.2011.02280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Galve-Roperh I, Chiurchiu V, Diaz-Alonso J, Bari M, Guzman M, Maccarrone M. Cannabinoid receptor signaling in progenitor/stem cell proliferation and differentiation. Prog Lipid Res. 2013;52:633–650. doi: 10.1016/j.plipres.2013.05.004. [DOI] [PubMed] [Google Scholar]
- 87.Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MC, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science. 2011;334:809–813. doi: 10.1126/science.1209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zelasko S, Arnold WR, Das A. Endocannabinoid metabolism by cytochrome P450 monooxygenases. Prostaglandins Other Lipid Mediat. 2015;116–117:112–123. doi: 10.1016/j.prostaglandins.2014.11.002. [DOI] [PubMed] [Google Scholar]
- 89.Urquhart P, Nicolaou A, Woodward DF. Endocannabinoids and their oxygenation by cyclo-oxygenases, lipoxygenases and other oxygenases. Biochim Biophys Acta. 2015;1851:366–376. doi: 10.1016/j.bbalip.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 90.Yung YC, Stoddard NC, Mirendil H, Chun J. Lysophosphatidic acid signaling in the nervous system. Neuron. 2015;85:669–682. doi: 10.1016/j.neuron.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang H, Zhang J, Andreasson K, Chen C. COX-2 oxidative metabolism of endocannabinoids augments hippocampal synaptic plasticity. Mol Cell Neurosci. 2008;37:682–695. doi: 10.1016/j.mcn.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kempuraj D, Thangavel R, Natteru PA, Selvakumar GP, Saeed D, Zahoor H, Zaheer S, Iyer SS, Zaheer A. Neuroinflammation induces neurodegeneration. J Neurol Neurosurg Spine. 2017;1(1):1003. [PMC free article] [PubMed] [Google Scholar]
- 93.Farooqui AA, Horrocks LA, Farooqui T. Modulation of inflammation in brain: a matter of fat. J Neurochem. 2007;101:577–599. doi: 10.1111/j.1471-4159.2006.04371.x. [DOI] [PubMed] [Google Scholar]
- 94.Kim SU, de Vellis J. Microglia in health and disease. J Neurosci Res. 2005;81:302–313. doi: 10.1002/jnr.20562. [DOI] [PubMed] [Google Scholar]
- 95.Tremblay ME, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J Neurosci. 2011;31:16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Garden GA, Moller T. Microglia biology in health and disease. J Neuroimmune Pharmacol. 2006;1:127–137. doi: 10.1007/s11481-006-9015-5. [DOI] [PubMed] [Google Scholar]
- 97.Chhor V, Le Charpentier T, Lebon S, Ore MV, Celador IL, Josserand J, Degos V, Jacotot E, Hagberg H, Savman K, Mallard C, Gressens P, Fleiss B. Characterization of phenotype markers and neuronotoxic potential of polarised primary microglia in vitro. Brain Behav Immun. 2013;32:70–85. doi: 10.1016/j.bbi.2013.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yip PK, Pizzasegola C, Gladman S, Biggio ML, Marino M, Jayasinghe M, Ullah F, Dyall SC, Malaspina A, Bendotti C, Michael-Titus A. The omega-3 fatty acid eicosapentaenoic acid accelerates disease progression in a model of amyotrophic lateral sclerosis. PLoS One. 2013;8:e61626. doi: 10.1371/journal.pone.0061626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lim SN, Gladman SJ, Dyall SC, Patel U, Virani N, Kang JX, Priestley JV, Michael-Titus AT. Transgenic mice with high endogenous omega-3 fatty acids are protected from spinal cord injury. Neurobiol Dis. 2013;51:104–112. doi: 10.1016/j.nbd.2012.10.021. [DOI] [PubMed] [Google Scholar]
- 100.Huang WL, King VR, Curran OE, Dyall SC, Ward RE, Lal N, Priestley JV, Michael-Titus AT. A combination of intravenous and dietary docosahexaenoic acid significantly improves outcome after spinal cord injury. Brain. 2007;130:3004–3019. doi: 10.1093/brain/awm223. [DOI] [PubMed] [Google Scholar]
- 101.Belayev L, Khoutorova L, Atkins KD, Eady TN, Hong S, Lu Y, Obenaus A, Bazan NG. Docosahexaenoic Acid therapy of experimental ischemic stroke. Transl Stroke Res. 2011;2:33–41. doi: 10.1007/s12975-010-0046-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hopperton KE, Trepanier MO, Giuliano V, Bazinet RP. Brain omega-3 polyunsaturated fatty acids modulate microglia cell number and morphology in response to intracerebroventricular amyloid-beta 1-40 in mice. J Neuroinflammation. 2016;13:257. doi: 10.1186/s12974-016-0721-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.De Smedt-Peyrusse V, Sargueil F, Moranis A, Harizi H, Mongrand S, Laye S. Docosahexaenoic acid prevents lipopolysaccharide-induced cytokine production in microglial cells by inhibiting lipopolysaccharide receptor presentation but not its membrane subdomain localization. J Neurochem. 2008;105:296–307. doi: 10.1111/j.1471-4159.2007.05129.x. [DOI] [PubMed] [Google Scholar]
- 104.Lien E, Means TK, Heine H, Yoshimura A, Kusumoto S, Fukase K, Fenton MJ, Oikawa M, Qureshi N, Monks B, Finberg RW, Ingalls RR, Golenbock DT. Toll-like receptor 4 imparts ligand-specific recognition of bacterial lipopolysaccharide. J Clin Invest. 2000;105:497–504. doi: 10.1172/JCI8541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chen X, Wu S, Chen C, Xie B, Fang Z, Hu W, Chen J, Fu H, He H. Omega-3 polyunsaturated fatty acid supplementation attenuates microglial-induced inflammation by inhibiting the HMGB1/TLR4/NF-kappaB pathway following experimental traumatic brain injury. J Neuroinflamm. 2017;14:143. doi: 10.1186/s12974-017-0917-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lu B, Nakamura T, Inouye K, Li J, Tang Y, Lundback P, Valdes-Ferrer SI, Olofsson PS, Kalb T, Roth J, Zou Y, Erlandsson-Harris H, Yang H, Ting JP, Wang H, Andersson U, Antoine DJ, Chavan SS, Hotamisligil GS, Tracey KJ. Novel role of PKR in inflammasome activation and HMGB1 release. Nature. 2012;488:670–674. doi: 10.1038/nature11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Takizawa T, Shibata M, Kayama Y, Shimizu T, Toriumi H, Ebine T, Unekawa M, Koh A, Yoshimura A, Suzuki N. High-mobility group box 1 is an important mediator of microglial activation induced by cortical spreading depression. J Cereb Blood Flow Metab. 2017;37:890–901. doi: 10.1177/0271678X16647398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Bazan NG. Neuroprotectin D1-mediated anti-inflammatory and survival signaling in stroke, retinal degenerations, and Alzheimer’s disease. J Lipid Res. 2009;50(Suppl):S400–S405. doi: 10.1194/jlr.R800068-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bazan NG, Calandria JM, Gordon WC. Docosahexaenoic acid and its derivative neuroprotectin D1 display neuroprotective properties in the retina, brain and central nervous system. Nestle Nutr Inst Workshop Ser. 2013;77:121–131. doi: 10.1159/000351395. [DOI] [PubMed] [Google Scholar]
- 110.Zhu M, Wang X, Hjorth E, Colas RA, Schroeder L, Granholm AC, Serhan CN, Schultzberg M. Pro-resolving lipid mediators improve neuronal survival and increase Abeta42 phagocytosis. Mol Neurobiol. 2015;53:2733–2749. doi: 10.1007/s12035-015-9544-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tian Y, Zhang Y, Zhang R, Qiao S, Fan J. Resolvin D2 recovers neural injury by suppressing inflammatory mediators expression in lipopolysaccharide-induced Parkinson’s disease rat model. Biochem Biophys Res Commun. 2015;460:799–805. doi: 10.1016/j.bbrc.2015.03.109. [DOI] [PubMed] [Google Scholar]
- 112.Harrison JL, Rowe RK, Ellis TW, Yee NS, O’Hara BF, Adelson PD, Lifshitz J. Resolvins AT-D1 and E1 differentially impact functional outcome, post-traumatic sleep, and microglial activation following diffuse brain injury in the mouse. Brain Behav Immun. 2015;47:131–140. doi: 10.1016/j.bbi.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xu ZZ, Berta T, Ji RR. Resolvin E1 inhibits neuropathic pain and spinal cord microglial activation following peripheral nerve injury. J Neuroimmune Pharmacol. 2013;8:37–41. doi: 10.1007/s11481-012-9394-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bilkei-Gorzo A. The endocannabinoid system in normal and pathological brain ageing. Philos Trans R Soc Lond B Biol Sci. 2012;367:3326–3341. doi: 10.1098/rstb.2011.0388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stella N. Endocannabinoid signaling in microglial cells. Neuropharmacology. 2009;56(Suppl 1):244–253. doi: 10.1016/j.neuropharm.2008.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nadjar A, Leyrolle Q, Joffre C, Laye S. Bioactive lipids as new class of microglial modulators: when nutrition meets neuroimmunology. Prog Neuropsychopharmacol Biol Psychiatry. 2016;79(Pt A):19–26. doi: 10.1016/j.pnpbp.2016.07.004. [DOI] [PubMed] [Google Scholar]
- 117.Mecha M, Feliu A, Carrillo-Salinas FJ, Rueda-Zubiaurre A, Ortega-Gutierrez S, de Sola RG, Guaza C. Endocannabinoids drive the acquisition of an alternative phenotype in microglia. Brain Behav Immun. 2015;49:233–245. doi: 10.1016/j.bbi.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 118.Gabrielli M, Battista N, Riganti L, Prada I, Antonucci F, Cantone L, Matteoli M, Maccarrone M, Verderio C. Active endocannabinoids are secreted on extracellular membrane vesicles. EMBO Rep. 2015;16:213–220. doi: 10.15252/embr.201439668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sofroniew MV. Astrogliosis. Cold Spring Harb Perspect Biol. 2014;7:a020420. doi: 10.1101/cshperspect.a020420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sofroniew MV. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist. 2014;20:160–172. doi: 10.1177/1073858413504466. [DOI] [PubMed] [Google Scholar]
- 121.Moore SA, Yoder E, Murphy S, Dutton GR, Spector AA. Astrocytes, not neurons, produce docosahexaenoic acid (22:6 omega-3) and arachidonic acid (20:4 omega-6) J Neurochem. 1991;56:518–524. doi: 10.1111/j.1471-4159.1991.tb08180.x. [DOI] [PubMed] [Google Scholar]
- 122.Grabner GF, Eichmann TO, Wagner B, Gao Y, Farzi A, Taschler U, Radner FP, Schweiger M, Lass A, Holzer P, Zinser E, Tschop MH, Yi CX, Zimmermann R. Deletion of monoglyceride lipase in astrocytes attenuates lipopolysaccharide-induced neuroinflammation. J Biol Chem. 2016;291:913–923. doi: 10.1074/jbc.M115.683615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Viader A, Blankman JL, Zhong P, Liu X, Schlosburg JE, Joslyn CM, Liu QS, Tomarchio AJ, Lichtman AH, Selley DE, Sim-Selley LJ, Cravatt BF. Metabolic interplay between astrocytes and neurons regulates endocannabinoid action. Cell Rep. 2015;12:798–808. doi: 10.1016/j.celrep.2015.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pan JP, Zhang HQ, Wei W, Guo YF, Na X, Cao XH, Liu LJ. Some subtypes of endocannabinoid/endovanilloid receptors mediate docosahexaenoic acid-induced enhanced spatial memory in rats. Brain Res. 2011;1412:18–27. doi: 10.1016/j.brainres.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 125.Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- 126.Xu JY, Chen C. Endocannabinoids in synaptic plasticity and neuroprotection. Neuroscientist. 2015;21:152–168. doi: 10.1177/1073858414524632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Thomazeau A, Bosch-Bouju C, Manzoni O, Laye S. Nutritional n-3 PUFA deficiency abolishes endocannabinoid gating of hippocampal long-term potentiation. Cereb Cortex. 2017;27:2571–2579. doi: 10.1093/cercor/bhw052. [DOI] [PubMed] [Google Scholar]
- 128.Ehninger D, Kempermann G. Neurogenesis in the adult hippocampus. Cell Tissue Res. 2008;331:243–250. doi: 10.1007/s00441-007-0478-3. [DOI] [PubMed] [Google Scholar]
- 129.Bartsch T, Wulff P. The hippocampus in aging and disease: from plasticity to vulnerability. Neuroscience. 2015;309:1–16. doi: 10.1016/j.neuroscience.2015.07.084. [DOI] [PubMed] [Google Scholar]
- 130.Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Takagi Y, Nozaki K, Takahashi J, Yodoi J, Ishikawa M, Hashimoto N. Proliferation of neuronal precursor cells in the dentate gyrus is accelerated after transient forebrain ischemia in mice. Brain Res. 1999;831:283–287. doi: 10.1016/S0006-8993(99)01411-0. [DOI] [PubMed] [Google Scholar]
- 132.Darsalia V, Heldmann U, Lindvall O, Kokaia Z. Stroke-induced neurogenesis in aged brain. Stroke. 2005;36:1790–1795. doi: 10.1161/01.STR.0000173151.36031.be. [DOI] [PubMed] [Google Scholar]
- 133.Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dyall SC. The role of omega-3 fatty acids in adult hippocampal neurogenesis. Oléagineux Corps Gras Lipides. 2011;18:242–245. doi: 10.1051/ocl.2011.0392. [DOI] [Google Scholar]
- 135.Jin K, Xie L, Kim SH, Parmentier-Batteur S, Sun Y, Mao XO, Childs J, Greenberg DA. Defective adult neurogenesis in CB1 cannabinoid receptor knockout mice. Mol Pharmacol. 2004;66:204–208. doi: 10.1124/mol.66.2.204. [DOI] [PubMed] [Google Scholar]
- 136.Rueda D, Navarro B, Martinez-Serrano A, Guzman M, Galve-Roperh I. The endocannabinoid anandamide inhibits neuronal progenitor cell differentiation through attenuation of the Rap1/B-Raf/ERK pathway. J Biol Chem. 2002;277:46645–46650. doi: 10.1074/jbc.M206590200. [DOI] [PubMed] [Google Scholar]
- 137.Goncalves MB, Suetterlin P, Yip P, Molina-Holgado F, Walker DJ, Oudin MJ, Zentar MP, Pollard S, Yanez-Munoz RJ, Williams G, Walsh FS, Pangalos MN, Doherty P. A diacylglycerol lipase-CB2 cannabinoid pathway regulates adult subventricular zone neurogenesis in an age-dependent manner. Mol Cell Neurosci. 2008;38:526–536. doi: 10.1016/j.mcn.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 138.Jiang W, Zhang Y, Xiao L, Van Cleemput J, Ji SP, Bai G, Zhang X. Cannabinoids promote embryonic and adult hippocampus neurogenesis and produce anxiolytic- and antidepressant-like effects. J Clin Invest. 2005;115:3104–3116. doi: 10.1172/JCI25509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Aguado T, Monory K, Palazuelos J, Stella N, Cravatt B, Lutz B, Marsicano G, Kokaia Z, Guzman M, Galve-Roperh I. The endocannabinoid system drives neural progenitor proliferation. FASEB J. 2005;19:1704–1706. doi: 10.1096/fj.05-3995fje. [DOI] [PubMed] [Google Scholar]
- 140.Ames BN. Delaying the mitochondrial decay of aging. Ann N Y Acad Sci. 2004;1019:406–411. doi: 10.1196/annals.1297.073. [DOI] [PubMed] [Google Scholar]
- 141.Canugovi C, Misiak M, Ferrarelli LK, Croteau DL, Bohr VA. The role of DNA repair in brain related disease pathology. DNA Repair (Amst) 2013;12:578–587. doi: 10.1016/j.dnarep.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Marchalant Y, Cerbai F, Brothers HM, Wenk GL. Cannabinoid receptor stimulation is anti-inflammatory and improves memory in old rats. Neurobiol Aging. 2008;29:1894–1901. doi: 10.1016/j.neurobiolaging.2007.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Perluigi M, Swomley AM, Butterfield DA. Redox proteomics and the dynamic molecular landscape of the aging brain. Ageing Res Rev. 2014;13:75–89. doi: 10.1016/j.arr.2013.12.005. [DOI] [PubMed] [Google Scholar]
- 144.Fjell AM, McEvoy L, Holland D, Dale AM, Walhovd KB. What is normal in normal aging? Effects of aging, amyloid and Alzheimer’s disease on the cerebral cortex and the hippocampus. Prog Neurobiol. 2014;117:20–40. doi: 10.1016/j.pneurobio.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Quinn JF, Raman R, Thomas RG, Yurko-Mauro K, Nelson EB, Van Dyck C, Galvin JE, Emond J, Jack CR, Jr, Weiner M, Shinto L, Aisen PS. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: a randomized trial. JAMA. 2010;304:1903–1911. doi: 10.1001/jama.2010.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Strike SC, Carlisle A, Gibson EL, Dyall SC. A high omega-3 fatty acid multinutrient supplement benefits cognition and mobility in older women: a randomized, double-blind, placebo-controlled pilot study. J Gerontol A Biol Sci Med Sci. 2016;71:236–242. doi: 10.1093/gerona/glv109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yurko-Mauro K, McCarthy D, Rom D, Nelson EB, Ryan AS, Blackwell A, Salem N, Jr, Stedman M. Beneficial effects of docosahexaenoic acid on cognition in age-related cognitive decline. Alzheimers Dement. 2010;6:456–464. doi: 10.1016/j.jalz.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 148.Piyanova A, Lomazzo E, Bindila L, Lerner R, Albayram O, Ruhl T, Lutz B, Zimmer A, Bilkei-Gorzo A. Age-related changes in the endocannabinoid system in the mouse hippocampus. Mech Ageing Dev. 2015;150:55–64. doi: 10.1016/j.mad.2015.08.005. [DOI] [PubMed] [Google Scholar]