

Summary
Enteroviruses reorganize cellular endomembranes into replication organelles (ROs) for genome replication. Although enterovirus replication depends on phosphatidylinositol 4-kinase type IIIβ (PI4KB), its role, and that of its product, phosphatidylinositol 4-phosphate (PI4P), is only partially understood. Exploiting a mutant coxsackievirus resistant to PI4KB inhibition, we show that PI4KB activity has distinct functions both in proteolytic processing of the viral polyprotein and in RO biogenesis. The escape mutation rectifies a proteolytic processing defect imposed by PI4KB inhibition, pointing to a possible escape mechanism. Remarkably, under PI4KB inhibition, the mutant virus could replicate its genome in the absence of ROs, using instead the Golgi apparatus. This impaired RO biogenesis provided an opportunity to investigate the proposed role of ROs in shielding enteroviral RNA from cellular sensors. Neither accelerated sensing of viral RNA nor enhanced innate immune responses was observed. Together, our findings challenge the notion that ROs are indispensable for enterovirus genome replication and immune evasion.
Keywords: enterovirus, replication organelles, PI4P, phosphatidylinositol 4-kinase type IIIβ, polyprotein processing, innate immunity
Graphical Abstract
Highlights
-
•
PI4KB activity expedites the formation of coxsackievirus replication organelles (ROs)
-
•
PI4KB inhibition impairs polyprotein processing, which is rescued by a 3A mutation
-
•
Upon PI4KB inhibition, this mutant replicates at the Golgi in the absence of ROs
-
•
Innate immune responses are not enhanced when RO biogenesis is delayed
Like other positive-strand RNA viruses, enteroviruses reorganize host endomembranes for genome replication. Melia et al. demonstrate a pivotal role of PI4KB activity both in the rapid biogenesis of coxsackievirus replication organelles and in polyprotein processing. The notion that membrane rearrangements are indispensable for genome replication and innate immune evasion is challenged.
Introduction
Positive-strand RNA (+RNA) viruses comprise many human pathogens, such as hepatitis C virus, Zika virus, dengue virus, severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) coronaviruses, and enteroviruses. Despite substantial genetic divergence across virus families, some features of replication are common to all +RNA viruses infecting eukaryotes. One of the most striking is the remodeling of host cell endomembranes into novel membranous compartments in the cytoplasm of the infected cell. These compartments serve as compositionally unique platforms upon which the components of the viral RNA (vRNA) synthesis machinery assemble, and whose micro-environments may facilitate efficient genome replication (Romero-Brey and Bartenschlager, 2014, Paul and Bartenschlager, 2013). In addition, they have been postulated to play a role in the evasion of the innate antiviral host responses by shielding vRNA products from cytosolic sensors such as MDA5 and RIG-I, which signal to activate the type I interferon (IFN-α/β) pathway, and protein kinase R (PKR), which activates an integral stress response (Schulz and Mossman, 2016, White and Lloyd, 2012).
Members of the Enterovirus genus, belonging to the Picornaviridae family, include poliovirus, coxsackie A and B viruses, several numbered enteroviruses (e.g., EV-D68, EV-A71), and rhinoviruses, which are causative agents of various human diseases. Enteroviruses, like other positive-sense RNA viruses, modify host-cell membranes to form structures with novel morphologies. These modified membranes serve as platforms for viral replication, which we will refer to as replication organelles (ROs). At earlier stages of coxsackievirus B3 (CVB3) or poliovirus (PV) infection, ROs emerge as single-membrane tubules that appear to form at the expense of the Golgi apparatus. These tubules are interspersed with double-membrane vesicles (DMVs), which are believed to arise as tubules deform and enwrap small volumes of cytosol. In this way, most tubules transform into DMVs over the course of infection, and DMVs may be further enwrapped to form multilamellar vesicles (Limpens et al., 2011, Belov et al., 2012). Each stage of virus replication, including the transformation of cellular membranes into ROs, is dependent upon the interplay between viral proteins and host factors. The small, membrane-anchored enterovirus 3A protein has a key role in generating ROs (Suhy et al., 2000) and is known to recruit host factors that are essential for genome replication. One of these factors is phosphatidylinositol 4-kinase type IIIβ (PI4KB) (Hsu et al., 2010, van der Schaar et al., 2013). In uninfected cells, PI4KB is a Golgi-resident enzyme that generates phosphatidylinositol 4-phosphate (PI4P), while during enterovirus infection the viral 3A protein recruits PI4KB to ROs, enriching them in PI4P (Hsu et al., 2010, Greninger et al., 2012).
The importance of PI4P in viral infections has been the subject of several recent investigations, both for the Picornaviridae, such as Aichivirus (genus Kobuviruses) and encephalomyocarditis virus (EMCV) (genus Cardiovirus), and for other +RNA viruses, such as hepatitis C virus (HCV) (Altan-Bonnet and Balla, 2012, Dorobantu et al., 2015). In uninfected cells, PI4P is involved in a multitude of functions, including signaling, membrane trafficking, regulation of Golgi apparatus organization, and lipid homeostasis (Clayton et al., 2013, De Matteis et al., 2013). Of particular relevance for viruses that utilize PI4P is the exchange of PI4P and cholesterol between the Golgi apparatus and endoplasmic reticulum (ER), which is mediated by the oxysterol-binding protein (OSBP). OSBP acts as a bridge between these two membrane compartments to generate a membrane contact site, using PI4P as a Golgi-based anchor and vesicle-associated membrane protein-associated protein A (VAP-A) as an ER anchor (Mesmin et al., 2013). During enterovirus infection, OSBP is diverted to create a novel type of membrane contact site between ROs and the ER. These sites mediate the exchange of PI4P for cholesterol, which is another essential lipid for enterovirus genome replication (Arita, 2014, Strating et al., 2015, Roulin et al., 2014).
Recruitment of PI4KB by the viral 3A protein is critical for enterovirus genome replication (Hsu et al., 2010, van der Schaar et al., 2013), but its exact role is unclear. The stage of genome replication in the enterovirus life cycle encompasses different processes, including proteolytic processing of the polyprotein, RO biogenesis, and RNA synthesis by the viral replication machinery. In this study, we used a CVB3 mutant resistant to PI4KB inhibition, CVB3 3A-H57Y (van der Schaar et al., 2012), to further dissect the role of PI4KB and the PI4P-rich environment it generates during genome replication. We show that PI4KB activity facilitates efficient proteolytic processing of the CVB3 polyprotein. The 3A-H57Y substitution compensates for the impairment of polyprotein processing caused by PI4KB inhibition, which may represent the escape mechanism of this mutant virus. Distinct from its effect on polyprotein processing, we found that PI4KB inhibition also delayed RO formation. Remarkably, CVB3 3A-H57Y could replicate its genome in the absence of detectable ROs when PI4KB was inhibited. Under these conditions, vRNA synthesis was observed instead at a cellular organelle, the Golgi apparatus, which challenges the notion that ROs are essential for the exponential phase of genome replication. Golgi disintegration and RO formation did eventually occur under PI4KB inhibition, which suggests that PI4KB activity is not fundamentally required for RO biogenesis, but expedites the process. The delay in RO formation under PI4KB inhibition was exploited to experimentally test the hypothesis that ROs shield vRNA from cytoplasmic sensors of the innate immune system. Our results suggest that in addition to being dispensable for vRNA synthesis, enterovirus ROs do not play a pivotal role in suppression of the innate antiviral response pathways.
Results
CVB3 3A-H57Y RO Formation Is Impaired under PI4KB Inhibition
First, we set out to study the morphology of the ROs induced by CVB3 3A-H57Y under conditions where PI4KB is not inhibited. Infected BGM cells were prepared for electron microscopy (EM) by high-pressure freezing and freeze substitution at different times post-infection and analyzed for the presence of membrane modifications. The first ROs induced by CVB3 3A-H57Y were detected at 5 hr post-infection (hpi). At this stage, single-membrane tubules were predominant, interspersed with DMVs (Figure 1 A). The emergence of ROs coincides with disintegration of the Golgi apparatus in wild-type (WT) infection (Limpens et al., 2011) and, concordantly, the Golgi apparatus was not observed in cell sections that contained ROs at any time point in this analysis. Later in infection, tubules had largely transformed into DMVs (Figure 1A, 6 hpi) and multilamellar structures (Figure 1A, 7 hpi). This progression closely reflects observations of WT CVB3 RO development with regard to both the specific morphologies induced and the time frame over which they develop (Limpens et al., 2011) (Figure S1A). Together, these results indicate that the 3A-H57Y substitution does not affect RO development or their general architecture.
Figure 1.

CVB3 3A-H57Y RO Formation Is Impaired under PI4KB Inhibition
BGM cells infected with CVB3 3A-H57Y (MOI 50).
(A) Cells fixed for EM analysis at early (5 hpi), intermediate (6 hpi), or late (7 hpi) stages of infection show the progression in RO development.
(B) ROs were not observed in EM cell sections (up to 8 hpi, n = 153) from cells treated with BF738735. An example of the Golgi apparatus in cells fixed at 7 hpi (left). Parallel immunofluorescence data (right) (dsRNA, green; nuclear stain, blue).
Scale bars represent 1 μm. See also Figures S1 and S2.
Next, we investigated the effect of PI4KB inhibition on RO development during CVB3 3A-H57Y infection. To determine suitable time points for EM analysis of cells infected in the presence of a PI4KB inhibitor, we first measured viral replication in the presence of BF738735, a potent and specific PI4KB inhibitor without overt cytotoxicity (also known as compound 1; van der Schaar et al., 2013, MacLeod et al., 2013). Unlike CVB3 WT (Figure S2A), CVB3 3A-H57Y was resistant to BF738735 (Figure S2B), although its replication was nevertheless delayed and impaired in the presence of BF738735 with regard to both vRNA and infectious progeny virus. This is in agreement with observations using other PI4KB inhibitors (e.g., PIK93; van der Schaar et al., 2012). To examine whether the impairment of replication upon PI4KB inhibition was the consequence of a decrease in the number of infected cells and/or a reduced level of vRNA replication, we collected cells infected with EGFP-CVB3 3A-H57Y at different time points post-infection and analyzed them with flow cytometry. PI4KB inhibition reduced the number of CVB3 3A-H57Y-infected cells (by ∼3-fold), delayed replication (by ∼1–2 hr), and limited viral protein production, as reflected by reduced EGFP levels (Figure S2C). These results indicate that the PI4KB inhibitor imposes a critical barrier to CVB3 3A-H57Y replication in a subpopulation of cells and reduces the efficiency of replication in those cells where infection is established.
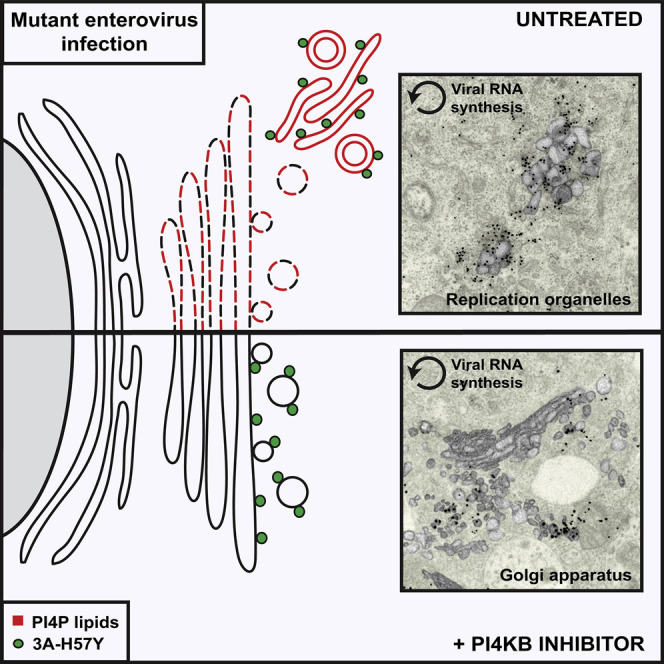
Based on the growth curve analysis, cells were fixed and processed for EM between 5 and 8 hpi, as this period encompasses the exponential phase of vRNA replication for CVB3 3A-H57Y under PI4KB inhibition. Remarkably, while ROs were detected at a frequency of ∼50% in EM cell sections from infections in the absence of PI4KB inhibitor (n = 43 cell sections assessed at 6 hpi), neither archetypal enterovirus ROs (i.e., clusters of single-membrane tubules and/or DMVs) nor cellular membranes with atypical morphologies were detected in cells infected under PI4KB inhibition (n = 153 cell sections), even at late time points (7–8 hpi, n = 47 cell sections, example shown in Figure 1B). Seemingly intact Golgi cisternae were apparent in these cells and detected at a frequency similar to that of mock-infected or BF738735-treated cells (Figures S1B and S1C, respectively). Corresponding immunofluorescence microscopy data showed a high percentage of cells positive for double-stranded RNA (dsRNA) labeling, confirming that the lack of membrane modifications in the EM analysis was not simply due to a low number of infected cells (Figure 1B, right). These data show that RO development is impaired by PI4KB inhibition and suggest that replication may be taking place instead at a cellular organelle.
CVB3 3A-H57Y Replicates Its Genome at the Golgi Apparatus under PI4KB Inhibition
As an initial indicator of the replication site under PI4KB inhibition, we performed immunofluorescence microscopy on infected cells (Figure 2 A). Cells were fixed at 5 hpi in the absence of inhibitor and at 6 hpi in the presence of inhibitor given the delay in replication. Similar to findings for WT virus (Hsu et al., 2010, van der Schaar et al., 2012), both 3A and dsRNA were detected throughout the cytoplasm in cells infected with CVB3 3A-H57Y in the absence of PI4KB inhibition (Figure 2A, left panels). Golgi disassembly could be readily visualized through the signal reduction and dispersion of the cis-Golgi marker GM130 and the trans-Golgi network marker TGN46, whereas the PI4KB signal overlapped with the 3A labeling. Upon PI4KB inhibitor treatment, however, 3A and dsRNA were primarily confined to the perinuclear region (Figure 2A, right panels). In wide-field images, the strength and distribution of GM130 and TGN46 signals were largely maintained and overlapped with the viral 3A signal. These results suggested that both the 3A-H57Y protein and dsRNA reside at the Golgi apparatus in the presence of the inhibitor. To confirm that these observations were due to specific inhibition of PI4KB, the localization of 3A-GFP was assessed using different PI4KB inhibitors. Similar to BF738735, enviroxime, GW5074, or PIK93 treatment resulted the accumulation of 3A specifically at the Golgi region, although confocal imaging revealed that 3A did not directly co-localize with the cis-Golgi marker GM130 (Figure 2B).
Figure 2.

The 3A Protein Localizes to the Golgi Apparatus under PI4KB Inhibition
BGM cells infected with CVB3 3A-H57Y (MOI 10).
(A) Untreated cells were fixed at 5 hpi (left panels), and drug-treated cells were fixed at 6 hpi (right panels).
(B) Cells were treated with different PI4KB inhibitors and fixed at 6 hpi.
Fixed cells were labeled with the indicated antibodies and a nuclear stain (blue). Scale bars represent 10 μm.
To unambiguously determine the subcellular location of the 3A-H57Y protein under PI4KB inhibitor treatment, correlative light and EM (CLEM) was performed. For this, we employed the split-GFP system (Cabantous et al., 2005) to label the 3A-H57Y protein, using an approach similar to that previously reported for WT CVB3 (van der Schaar et al., 2016b). To produce a split-GFP CVB3 3A-H57Y mutant, the final beta sheet of GFP (i.e., strand 11), further referred to as GFP(S11), was introduced into the 3A-H57Y protein to produce CVB3 3A-H57Y(S11). Upon CVB3 3A-H57Y(S11) infection of BGM cells stably expressing the remaining portion of GFP (GFPS1-10), the two fragments of GFP self-assemble to produce GFP-tagged 3A-H57Y. GFP-tagged 3A-H57Y was then visualized by confocal microscopy before chemical fixation and processing for EM. Overlays of 3A-H57Y-GFP signal with the corresponding EM cell sections fixed at 6 hpi revealed that the 3A protein localized specifically to the Golgi apparatus (Figure 3 A).
Figure 3.

Replication under PI4KB Inhibition Occurs at the Golgi Apparatus
(A) BGM(GFPS1-10) cells infected with CVB3 3A-H57Y(S11) (MOI 7) monitored by live-cell LM, fixed at 6 hpi, and processed for CLEM. Overlay of the 3A-GFP signal at the time of fixation with the corresponding EM image (left). Inset highlights one of two 3A-GFP foci present in the image shown, both of which correspond to Golgi membranes. Scale bars represent 2 μm.
(B and C) BGM cells infected with CVB3 3A-H57Y (MOI 50), metabolically labeled, fixed at 6 hpi (B) or 7 hpi (C), and processed for EM autoradiography to detect newly synthesized vRNA. The autoradiography signal is apparent as electron-dense grains. (B) ROs positive for ARG signal were readily observed in the absence of BF738735. (C) In cells treated with BF738735, ROs were not observed, and clusters of autoradiography signal were found exclusively at the Golgi apparatus. cis, Golgi apparatus cis face; trans, Golgi apparatus trans face; TGN, trans-Golgi network. Scale bars represent 1 μm.
The presence of the 3A-H57Y protein at the Golgi apparatus suggested that vRNA replication occurs at this compartment under PI4KB inhibition. To further investigate this, metabolic labeling was performed to detect newly synthesized vRNA in situ. Infected cells were pretreated with 10 μg/mL dactinomycin to inhibit cellular transcription and incubated with tritiated uridine for 45 min prior to fixation to label newly synthesized vRNA. As a control, mock-infected cells underwent the same treatment. After processing for EM, radiolabelled uridine was detected by autoradiography. In cells infected with CVB3 3A-H57Y without PI4KB inhibition, abundant autoradiography signal was found in regions containing typical ROs (Figure 3B). Although RO membranes in chemically fixed samples appeared somewhat distended in comparison to high-pressure frozen material (Figure 1), they were recognizable as clusters of single-membrane compartments (Figure 3B, black arrowhead) interspersed with DMVs (Figure 3B, white arrowhead) that closely resemble the ROs observed in chemically fixed PV-infected (Dales et al., 1965, Belov et al., 2012) or WT-CVB3-infected cells (van der Schaar et al., 2016b). In CVB3-3A-H57Y-infected cells treated with PI4KB inhibitor and fixed at 7 hpi, ROs were not observed (n = 148 cell sections), and abundant autoradiography signal was found at the Golgi apparatus (Figure 3C), with the vast majority of signal localizing to the trans side of the Golgi apparatus and trans-Golgi network rather than the Golgi cisternae. This demonstrates that under PI4KB inhibition, CVB3 3A-H57Y can replicate its genome in the absence of ROs on a seemingly intact Golgi apparatus.
Under PI4KB Inhibition, ROs Form Late in CVB3-3A-H57Y-Infected Cells
Our results thus far show that under PI4KB inhibition, CVB3 3A-H57Y replication is possible in association with an apparently intact cellular organelle. We next set out to establish whether PI4KB inhibition precludes Golgi disintegration and RO development entirely or simply delays the process. Confocal microscopy of live cells expressing mCherry-GM130 was performed to monitor the Golgi apparatus across the course of CVB3 3A-H57Y(S11) infection, either with or without BF738735 treatment (Figures 4 A–4C). In the absence of inhibitor, the process of Golgi fragmentation following CVB3 3A-H57Y(S11) infection was similar to that of WT split-GFP CVB3 infections (van der Schaar et al., 2016b). The onset of Golgi fragmentation occurred in conjunction with or even preceded 3A protein accumulation in the Golgi area, and Golgi disintegration was typically complete within 25 min (n = 23 cells) (Figures 4A and 4C, top graph). In the absence of PI4KB inhibition, the early 3A signal was often located at peripheral foci (Figure 4A arrowheads), and under PI4KB inhibition, the first 3A signal was often found in the Golgi region (Figure 4B, arrowheads). Remarkably, the Golgi apparatus of most CVB3-3A-H57Y(S11)-infected cells did disassemble during infections under PI4KB inhibition, but with markedly different dynamics. The onset of disintegration under these conditions did not coincide with 3A protein accumulation at the Golgi but suffered a relative average delay of 30 min (n = 48 cells). Additionally, the time needed for Golgi disintegration was highly variable under PI4KB inhibition. In a minority of infected cells treated with BF738735, Golgi disassembly was as rapid as in untreated cells. However, in most cells, the Golgi disintegration process was substantially prolonged (by up to 10 hr) under PI4KB inhibition (Figures 4B and 4C, bottom graph).
Figure 4.

Golgi Disintegration during CVB3 3A-H57Y Infection Is Prolonged under PI4KB Inhibition and Ultimately Results in RO Formation
(A–D) BGM(GFPS1-10) cells transduced with MLV mCherry-GM130, infected with CVB3 3A-H57Y(S11) (MOI 7), and imaged by live-cell confocal microscopy in the absence (A) or presence (B and D) of BF738735. Asterisks in (A) and (B) denote completion of Golgi disintegration, as evidenced by an entirely punctate mCherry-GM130 signal. Initial 3A-GFP signal was detected primarily at peripheral locations (A, arrowhead), and Golgi disintegration was typically complete within 25 min of its onset (C, top graph; n = 23 cells). In cells treated with BF738735, initial 3A-GFP signal was detected primarily at the Golgi apparatus (B, arrowhead). The Golgi disintegration duration was typically prolonged but highly variable (C, bottom graph; n = 48 cells). Cells were fixed for EM analysis at 9 hpi (D). The LM-EM overlay reveals single-membrane (black arrowhead) and double-membrane (white arrowhead) structures positive for 3A-GFP signal; scale bars represent 5 μm. See also Figure S3.
To determine whether Golgi disassembly under PI4KB inhibition was associated with the development of ROs, CLEM was performed on cells infected with split-GFP 3A-H57Y(S11) virus under BF738735 treatment and fixed at 9 hpi. While some cells retained seemingly intact Golgi membranes at this late stage in infection, in those cells lacking recognizable Golgi membranes, the 3A-GFP signal was found at structures resembling typical early ROs (Figure 4D; see Figure 3A for comparison). As verified by immunofluorescence microscopy, PI4P did not accumulate in cells infected under PI4KB inhibition (Figure S3), suggesting that the ROs that develop during CVB3 3A-H57Y infection under PI4KB inhibition do so independently of high PI4P levels.
PI4KB Activity Drives Rapid Golgi Disassembly and RO Formation
RO formation is likely associated with the amount of viral protein produced in the cell, which in turn depends upon vRNA replication levels. Therefore, rather than the absence of high PI4P levels, the delay in CVB3 3A-H57Y vRNA replication under PI4KB inhibition could be solely responsible for impeding RO formation. Previously, it was shown that replication-independent expression of PV non-structural proteins results in membranous structures reminiscent of ROs produced upon infection (Belov et al., 2008). Here, we engineered a replication-independent CVB3 expression system, which produces high amounts of viral proteins in the absence of vRNA replication. Hence, the inhibition of vRNA replication caused by PI4KB inhibition will not result in a delay in viral protein production in this setup. The system utilizes a CVB3 cDNA placed under the control of the bacteriophage T7 RNA polymerase and is rendered replication incompetent through modifications to the cloverleaf structure in the 5′ UTR of the viral genome that prevent replication via the CVB3 polymerase (Langereis et al., 2014). The 3A-H57Y substitution was introduced into this replication-incompetent CVB3 cDNA. Transfection of the CVB3 3A-H57Y cDNA plasmid in HuH-7/T7 cells, which stably express the T7 RNA polymerase, resulted in the expression of individual viral proteins whose amounts were not affected by the addition of PI4KB inhibitor (Figure S4). Immunofluorescence analysis of transfected cells showed that the first 3A signal could be detected as early as 3 hr post-transfection (hpt), but without significant effects on the Golgi apparatus. In the absence of PI4KB inhibitor, Golgi disintegration was observed from 4 hpt onward, as evidenced by the change in GM130 signal in transfected cells (Figure 5 A, top panels). Under PI4KB inhibition, Golgi disintegration was delayed by ∼1 hr (Figure 5A, middle and bottom panels). While Golgi disintegration is more rapid in the replication-independent system than during infection (see Figure 4), most likely because of the high protein expression levels generated with this system, these data nevertheless demonstrate that in cells with equal levels of viral proteins, PI4KB inhibition delays enterovirus-induced Golgi apparatus disassembly.
Figure 5.

Effect of PI4KB Inhibition on Enterovirus-Induced Golgi Disintegration and Proteolytic Polyprotein Processing in a Replication-Independent CVB3 System
HuH-7/T7 cells transfected with CVB3 3A-H57Y or WT CVB3 cDNA under the control of a T7 promoter, both rendered replication incompetent by altering the 5′ UTR of the genome. Where indicated, cells were treated with BF738735.
(A and B) Cells fixed at 4 hr or 5 hr post-transfection with CVB3 3A-H57Y cDNA (A) or CVB3 WT cDNA (B) and labeled for 3A (green) and GM130 (red) alongside a nuclear stain (blue). Representative 3A-positive cells are indicated (arrowheads). Scale bars represent 20 μm.
(C) Western blot analysis of lysates from cells at 16 hr post-transfection using antibodies against 3A or 3D. Actin was used as a loading control.
See also Figure S4.
To determine the contribution of the 3A-H57Y substitution to (delayed) Golgi disassembly, we examined whether the WT CVB3 cDNA could also induce Golgi disassembly under PI4KB inhibition in the replication-independent system. The results closely resembled those obtained for CVB3 3A-H57Y cDNA both with and without PI4KB inhibition (Figure 5B). This demonstrates that the 3A-H57Y substitution does not affect Golgi disintegration.
Altogether, these findings strongly suggest that enterovirus RO biogenesis is driven by PI4KB activity.
The 3A-H57Y Substitution Rectifies a Proteolytic Polyprotein Processing Impairment Induced by PI4KB Inhibition
Another important step in the enterovirus replication cycle that relies on membranes is the proteolytic processing of the polyprotein by the viral proteases (Molla et al., 1993). Proteolytic polyprotein processing is a co- and post-translational event mediated by the viral proteinases 2Apro, 3Cpro, and 3CDpro. These proteinases catalyze a cascade of cleavages in cis and in trans that liberate individual capsid proteins, as well as replication proteins and their precursors (e.g., 2BC, 3AB, and 3CD). Recent studies indicate that alterations to the lipid composition of membranes during PV infection impact polyprotein processing efficiency (Ilnytska et al., 2013, Ford Siltz et al., 2014, Arita, 2016). Given that WT virus replication is abolished under PI4KB inhibition, we used the replication-independent CVB3 expression system to study the effects of PI4KB inhibition on polyprotein processing. Cells were lysed at 16 hpt and processed for western blot analysis. PI4KB inhibition led to a relative accumulation of 3AB and a reduction in 3A for the WT polyprotein (Figure 5C, left). Remarkably, the 3A-H57Y substitution rectified this impaired polyprotein processing, as the relative levels of 3AB and 3A were not affected by PI4KB inhibition (Figure 5C, right). PI4KB inhibition did not affect levels of 3CD or 3D. Together, these results demonstrate that PI4KB activity is important for proteolytic processing at the 3A-3B junction, but not at the 3C-3D junction (or at the 2C-3A and 3B-3C junctions). The 3A-H57Y substitution restores processing to the level detected in the absence of inhibitor, which may point to a potential strategy of the mutant virus to escape PI4KB inhibition.
The Delay in RO Formation under PI4KB Inhibition Does Not Elicit a Strong Antiviral Response
One of the proposed advantages of ROs is that they may shield vRNA products against vRNA sensors present in the cytoplasm, such as MDA5, RIG-I, and PKR. Given that CVB3 3A-H57Y is able to replicate its genome at the Golgi apparatus under PI4KB inhibition, vRNA products may be more accessible and better detected in this situation, thereby triggering an antiviral response that might limit or delay replication. To investigate whether viral dsRNA is better sensed under PI4KB inhibition, we studied the activation status of the cytoplasmic RNA sensor PKR. Western blot analyses were performed on infected cell lysates to assess viral protein levels in parallel with dsRNA-activated phosphorylated PKR (p-PKR). The first appearance and the final levels of viral proteins were delayed and reduced respectively under PI4KB inhibition, which is in agreement with the observed reduction in the proportion of infected cells and the decrease in viral protein per cell (Figure S2C). Nevertheless, p-PKR emerged concomitantly with the accumulation of viral proteins in both the presence and absence of PI4KB inhibitor (Figure 6 A), suggesting that delayed RO formation under PI4KB inhibition does not lead to premature activation of PKR.
Figure 6.

Innate Antiviral Responses during CVB3 3A-H57Y Infection
Cells infected and treated with BF738735 where indicated.
(A) HeLa cells infected with CVB3 3A-H57Y (MOI 50) were lysed for western blot analysis at the indicated time points. Band sizes (in kilodaltons), full-length proteins (arrows), and putative cleavage products (arrowheads) are indicated.
(B) HeLa cells infected with CVB3 WT or CVB3 3A-H57Y (MOI 10). Total RNA was isolated from cells and subjected to qRT-PCR for interferon-β (IFN-β). Mengo Zn, whose L protein contains substitutions that render it unable to suppress IFN-β induction, was included as a positive control (left graph). vRNA levels were determined by qRT-PCR (right graph). Data were normalized to actin mRNA levels. Results are expressed as fold induction relative to quantities in mock-infected cells. Two independent experiments are shown, ranging from 2 to 8 hpi and 8 to 12 hpi.
(C and D) HeLa, HeLa PKR, and MAVS knockout (C) or HeLa AGO knockout (D) cells were infected with CVB3-Rluc 3A-H57Y (MOI 0.01) and lysed to determine intracellular luciferase levels. Values represent mean values of triplicates ± SEM.
See also Figures S5 and S6.
Another innate antiviral response that is activated upon sensing of vRNA products is the IFN-α/β pathway. Enteroviruses counteract the transcription of IFN-α/β genes by cleaving components in the signaling pathways that control their activation, such as MDA5 (Feng et al., 2014) and its downstream adaptor, MAVS (Feng et al., 2014, Mukherjee et al., 2011). The altered location of genome replication under PI4KB inhibition may influence the ability of the viral proteinases to cleave these components. However, we found that the appearance of MAVS degradation products in infected cell lysates coincided with the accumulation of viral proteins irrespective of the presence of a PI4KB inhibitor (Figure 6A). Furthermore, qPCR analysis showed that no substantial IFN-β response was triggered in infected cells in both conditions (Figure 6B). Together, these results indicate that the delayed formation of ROs during infection does not accelerate cellular sensing of viral dsRNA, nor does it affect the ability of the viral proteinases to cleave MAVS.
As a complementary approach to investigate whether the delay in replication in the presence of PI4KB inhibitor is related to activation of innate antiviral responses, the level of CVB3 3A-H57Y replication was determined in cells lacking sensors of these pathways. Knockout cells for PKR or MAVS, generated with CRISPR-Cas9 technology, were infected with CVB3 WT or 3A-H57Y that also encode Renilla luciferase as a sensitive measure to quantify the level of viral replication. In the absence of inhibitor, CVB3 3A-H57Y replicated to a similar extent in HeLa, HeLa PKR−/−, or HeLa MAVS−/− cells (Figures 6C and S6). In the presence of PI4KB inhibitor, CVB3 3A-H57Y replication was delayed not only in HeLa cells, but also in PKR−/− or MAVS−/− cells. Thus, replication under PI4KB inhibition was not significantly increased in PKR−/− or MAVS−/− cells compared to parental HeLa cells. Similar results were obtained in U2OS cells, another human cell line (Figures S5A and S6).
An alternative strategy employed by some eukaryotes, such as plants, worms, and insects, in response to viral infections is RNAi, although whether RNAi is an antiviral mechanism in mammalian cells remains controversial (Luna et al., 2016). In the RNAi pathway, the cytoplasmic sensor Dicer cleaves viral dsRNA into viral small interfering RNAs (siRNAs), which are loaded by Argonaute-2 (AGO2) into the RNA-induced silencing complex (RISC) in order to degrade vRNA (tenOever, 2016). Recently, it was reported that replication of the picornavirus EMCV was enhanced in mammalian somatic cells that lacked AGO2 (Li et al., 2016). To investigate whether the delay in CVB3 3A-H57Y replication in the presence of PI4KB inhibitor is due to triggering of antiviral RNAi, we studied its replication kinetics in HeLa and HEK293T cells that lacked AGO2. Similar to our findings in MAVS and PKR knockout cell lines, the delay in replication under PI4KB inhibition was not alleviated in cells lacking AGO2 (Figures 6D and S5B).
Altogether, these data suggest that the delay in CVB3 3A-H57Y replication under PI4KB inhibition is not caused by premature triggering of innate antiviral responses as result of impaired RO biogenesis.
Discussion
Like all other +RNA viruses infecting eukaryotes, enteroviruses modify host cell endomembranes to form structures with novel morphologies (ROs) dedicated to amplification of the viral genome. Enteroviruses hijack host factors and modulate lipid synthesis and homeostasis pathways to build ROs with a unique protein and lipid signature (van der Schaar et al., 2016a, Strating and van Kuppeveld, 2017). One of the lipid-modifying host factors previously shown to be essential for enterovirus genome replication is PI4KB (Hsu et al., 2010, van der Schaar et al., 2013). In this study, we clarify the role of PI4KB and the PI4P-enriched environment it generates in CVB3-infected cells by establishing its importance for proteolytic processing of the polyprotein and RO biogenesis. To do so, we exploited the CVB3 3A-H57Y mutant (van der Schaar et al., 2012), which is able to establish replication under PI4KB inhibition, albeit with a delay. CVB3 3A-H57Y generated ROs in a manner identical to WT CVB3 in the absence of the PI4KB inhibitor. Strikingly, under PI4KB inhibition, ROs were not observed during the peak hours of vRNA synthesis. Instead, vRNA synthesis was detected at the Golgi apparatus. Utilizing a replication-independent expression system, we ascertained that the impairment in RO formation was a consequence of PI4KB inhibition and not merely the result of delayed replication. Together, these findings demonstrate that PI4KB activity drives rapid RO formation.
In the absence of ROs, seemingly intact Golgi membranes are sufficient to facilitate vRNA replication of the mutant virus. Thus, the Golgi apparatus might also support initial genome replication during WT virus infection, prior to RO formation. The detection of CVB3 vRNA at the Golgi apparatus by fluorescence in situ hybridization early in infection (Hsu et al., 2010), while not directly proving RNA synthesis, would support this possibility. However, the Golgi apparatus is rapidly disassembled to form ROs during uninhibited WT CVB3 infections (Hsu et al., 2010, van der Schaar et al., 2016b), while during 3A-H57Y mutant infections, under PI4KB inhibition, this phase of replication at the Golgi apparatus was markedly prolonged, encompassing the exponential phase of genome replication. This illustrates a remarkable flexibility in the use of membranes as a platform for genome replication and contributes to the idea that +RNA-virus-induced membrane modification may be an adaptable process. Previous studies have reported that for some +RNA viruses, like tombusviruses and nodaviruses, replication can even be shifted to a different cellular organelle (Xu and Nagy, 2014). It is unknown whether these adoptive membranes are still remodeled to form ROs in all cases. Our findings also align with studies that show a relationship between RO formation and PI4P for viruses that depend on the activity of PI4KA (phosphatidylinositol 4-kinase type IIIα). When PI4KA is depleted or inhibited, HCV and EMCV were found to exhibit atypical RO development, as evidenced by clustering of ROs (Reghellin et al., 2014, Dorobantu et al., 2016) or altered RO morphology (Reiss et al., 2011), pointing to a central role for PI4P-producing kinases in RO biogenesis for these viruses as well.
PI4KB inhibition did not ultimately prevent the development of enterovirus ROs, which were readily detected at the late stages of CVB3 3A-H57Y infection. Immunofluorescence microscopy revealed that these late stages of infection were not associated with an accumulation of PI4P. Thus, despite the role of active PI4KB in facilitating rapid RO biogenesis in uninhibited infections, PI4P is not a strict requirement for their formation. What then is the role of PI4P in RO biogenesis? One possibility is that PI4P serves to recruit cholesterol, a lipid that significantly affects membrane structure and function. Cholesterol has been shown to accumulate at ROs (Ilnytska et al., 2013, Roulin et al., 2014, Strating et al., 2015, Arita, 2014) and may be involved in their formation. Recent evidence suggests that during WT enterovirus infection, cholesterol is trafficked to ROs via two PI4KB-dependent mechanisms. One of these is the PI4P-dependent exchange of cholesterol via OSBP (Albulescu et al., 2015, Strating et al., 2015). The second relies on the physical interaction between PI4KB and Rab11 on recycling endosomes, which can divert endosomal cholesterol to ROs (Ilnytska et al., 2013, Burke et al., 2014). PI4KB inhibitors limit PI4P accumulation and will thus limit OSBP-mediated cholesterol transfer. However, enzymatically inactive PI4KB may retain its ability to interact with Rab11 (de Graaf et al., 2004) and CVB3 3A-H57Y could in this way recruit sufficient cholesterol for (delayed) RO formation under PIK4B inhibition.
How can the acquisition of a single point mutation in the 3A protein allow enteroviruses to overcome their dependence on a critical host factor like PI4KB? Recent studies have indicated that the lipid composition of membranes is an important determinant for efficient proteolytic processing of the PV polyprotein and that PI4KB inhibition can perturb cleavage at specific junctions (Ilnytska et al., 2013, Ford Siltz et al., 2014, Arita, 2016). For WT CVB3 cDNA, we observed that 3AB accumulated and 3A levels were reduced under PI4KB inhibition, while levels of other viral proteins were unaffected. This suggests that PI4P facilitates processing specifically at the 3A-3B junction. Importantly, the 3A-H57Y substitution rendered polyprotein processing independent of high levels of PI4P lipids, as normal levels of precursor and products were detected under PI4KB inhibition. Restoring polyprotein processing efficiency may be a general resistance mechanism of enteroviruses to overcome PI4KB inhibition. The PV 3A-A70T substitution, which confers resistance to PI4KB inhibitors, in PV also restored a cleavage defect at the 3A-3B junction in the presence of PI4KB inhibitors (Arita, 2016). Notably, the H57Y substitution in CVB3 3A and A70T in PV 3A are respectively close to or present in the C-terminal hydrophobic domain. It is possible that these substitutions near or in the membrane anchor of 3A impose an altered conformation to 3AB that increases the accessibility of the 3AB cleavage site in the absence of high PI4P levels.
Why is the replication efficiency of CVB3 3A-H57Y lower under PI4KB inhibition? If enterovirus ROs enhance viral replication by expanding the membrane surface available for vRNA synthesis, it may be that impaired RO biogenesis in the absence of PI4KB activity directly limits replication. The formation of ROs might also contribute to host immune response evasion, leading to a higher replication efficiency. The disassembly of the secretory pathway, which occurs in conjunction with RO formation, has been suggested to limit cytokine secretion and downregulate major histocompatibility complex class I (MHC class I) surface expression (Dodd et al., 2001, Cornell et al., 2007), thereby curtailing activation of the adaptive immune system. ROs may serve as a barrier to activation of the innate immune system by physically segregating viral products and cellular sensors, such as RIG-I, MDA5, and PKR. This strategy is employed by HCV, which limits access of RIG-I and MDA5 to their ROs by diverting components of the nuclear transport machinery (Neufeldt et al., 2016). Some +RNA viruses, such as DENV (Welsch et al., 2009) or Zika virus (Cortese et al., 2017), induce negative membrane curvature to form invaginations in the boundary membranes of organelles, in which genome replication takes place. These invagination-type compartments have small pores toward the cytoplasm for selective import/export of material, which is proposed to restrict access of cellular RNA sensors. Enteroviruses, however, induce positive membrane curvature to generate ROs that protrude into the cytosol. Given that genome replication takes place on the cytosolic surface of membranes, enterovirus vRNA products may be more exposed and thus more vulnerable to cellular sensors. Early work with PV showed that “rosettes of virus-induced vesicles” isolated from infected cells provided protection against RNase-treatment in vitro (Bienz et al., 1992), but whether ROs in infected cells also protect vRNA from RNase treatment is unknown. In our study, we found no signs of premature sensing of viral dsRNA by PKR under PI4KB inhibition, a condition where RO formation was found to be delayed and replication occurred at a seemingly intact Golgi apparatus. Consistently, the delay in CVB3 3A-H57Y replication under PI4KB inhibition was not alleviated in PKR−/− cells. Furthermore, replication of CVB3 3A-H57Y was not enhanced in MAVS−/− cells, which are defective in signaling induced by MDA5 and RIG-I, or in AGO2−/− cells in which the Dicer-dependent RNAi pathway is rendered non-functional. Together, these findings challenge the idea that enterovirus ROs have a critical function in shielding viral dsRNA from cellular RNA sensors. Enteroviruses have acquired other mechanisms to suppress innate immune responses that rely on the cleavage of essential signaling pathway components (such as MDA5, MAVS, and the integral stress response protein G3BP1) by their proteinases (Lei et al., 2016). However, the possibility remains that ROs serve as a redundant protective measure against innate immune sensors, perhaps with a more prominent role in other cell types than those used here.
The pervasiveness of virus-induced membrane modifications across the Picornaviridae and other viral families suggests that they provide an inherent benefit to +RNA virus replication, perhaps by facilitating efficient vRNA synthesis or by coordinating different events in the viral life cycle. Through further investigations using drug-resistant viruses, replication-independent viral systems or other novel approaches, the intrinsic benefits of these morphologically diverse replication compartments will be unveiled.
Experimental Procedures
Replication-Independent System
HuH-7/T7 cells were seeded in 24-well plates containing glass coverslips. The next day, the cells were lipofectamine transfected with CVB3 cDNA rendered replication deficient through modifications to the cloverleaf structure in the 5′ UTR of the viral genome (Langereis et al., 2014). 1 hr later, the medium was replaced with fresh medium. At indicated hours post-transfection, cells were washed and either fixed for immunofluorescence or lysed for western blot analysis.
Live-Cell Imaging
BGM(GFPS1-10) cells were grown in glass-bottom 4-chamber 35-mm dishes (CELLview) to ∼35% confluency and transduced with MLV mCherry-GM130 particles described previously (van der Schaar et al., 2016b). Infection with CVB3 3A-H57Y(S11) was carried out 18–24 hr later. Prior to imaging, cells were washed with Fluorobrite medium (Thermo Fisher Scientific) supplemented with 8% fetal calf serum (FCS) and 25 mM HEPES. Cells were maintained in a live-cell imaging chamber at 37°C and 5% CO2. Imaging was carried out from ∼2.5 hpi using a Leica SP5 confocal microscope. Positions of interest (xyz) were marked and imaged sequentially at 5-min intervals.
High-Pressure Freezing and Freeze Substitution
BGM cells were grown on sapphire discs and infected with CVB3 or CVB3 3A-H57Y and refreshed at 1 hpi with medium supplemented with 25 mM HEPES buffer with or without 1 μm BF738735. Cells were high-pressure frozen using a Leica EM PACT2 at different time points post-infection. The instruments and procedures used for freeze substitution, epoxy resin infiltration, and polymerization were identical to those described previously (Limpens et al., 2011). Sections of 70 nm were then prepared for EM and post-stained with uranyl acetate and lead citrate.
CLEM Preparation
BGM (GFPS1-10) cells were cultured in gridded 8-well chamber μ-slides (Ibidi) ahead of infection with CVB3 3A-H57Y(S11). Just prior to imaging, cells were treated with 100 nM Mitotracker Deep Red FM for 30 min. Live-cell imaging was carried out to monitor the levels of 3A-H57Y-GFP. High-resolution (1,024 × 1,024) z stacks were collected of cells of interest just prior to fixation. To aid in the relocation of these cells for CLEM, tile scan overviews were taken and the positions of cells relative to nearby grid co-ordinates were recorded. Following imaging by live-cell light microscopy (LM), cells were prepared for EM as described previously (van der Schaar et al., 2016b). EM images were collected of cells previously identified by LM. The EM and LM data for each cell were overlaid using the Mitotracker Deep Red FM signal (LM images) and corresponding mitochondria (EM images) as a guide for image transformation.
Metabolic Labeling and Autoradiography
A subconfluent layer of BGM cells was grown in 35-mm dishes (Corning Life Sciences) and infected with CVB3 or CVB3 3A-H57Y at MOI 5. Cells were incubated with dactinomycin for 1 hr prior to a 45 min labeling with tritiated uridine ([5-3H], 1 mCi/mL) (Perkin Elmer). Cells were processed for EM as described previously (van der Schaar et al., 2016b). 50-nm sections of were collected, post-stained with lead citrate and uranyl acetate, and then prepared for autoradiography as described previously (Ginsel et al., 1979).
EM
Images were collected on an FEI Tecnai12 BioTWIN or TWIN electron microscope at 120 kV using an Eagle 4k slow-scan CCD camera (FEI) or OneView 4k high-frame-rate camera (Gatan), respectively. For the collection of larger EM datasets, meshes of overlapping areas across the grid were taken and then later stitched into a single composite image as described previously (Faas et al., 2012).
Author Contributions
Conceptualization, C.E.M., H.M.v.d.S., M.B., and F.J.M.v.K.; Investigation, C.E.M., H.M.v.d.S., H.L., R.W.A.L.L., Q.F., M.W., G.J.O., and M.B.; Writing – Original Draft, C.E.M., H.M.S., M.B., and F.J.M.v.K.; Writing – Review & Editing, C.E.M., H.M.v.d.S., R.P.v.R., A.J.K., E.J.S., M.B., and F.J.M.v.K.; Visualization, C.E.M. and H.M.S.; Supervision, H.M.S., E.J.S., A.J.K., R.P.v.R., M.B., and F.J.M.v.K.; Funding Acquisition, H.M.v.d.S., E.J.S., R.P.v.R., A.J.K., M.B., and F.J.M.v.K.
Acknowledgments
The authors would like to thank Huib Rabouw for excellent assistance with flow cytometry and the LUMC light microscopy facility. This work was supported by grants from the Netherlands Organisation for Scientific Research (NWO-VENI-863.12.005 to H.M.v.d.S., NWO-VICI-91812628 to F.J.M.v.K., ERASysApp project “SysVirDrug” ALW project 832.14.003 to F.J.M.v.K., NWO-MEERVOUD863.10.003 to M.B., and NWO-CW TOP 700.57.301 to E.J.S.) and the European Union (7th Framework: Marie Curie ITN “EUVIRNA” grant agreement 264286 to F.J.M.v.K., Horizon 2020: Marie Skłodowska-Curie ETN “ANTIVIRALS” grant agreement 642434 to F.J.M.v.K., and European Research Council consolidator grant CoG 615680 to R.P.v.R.). The funders had no role in study design, data collection, and interpretation or the decision to submit the work for publication.
Published: October 17, 2017
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and six figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.09.068.
Contributor Information
Montserrat Bárcena, Email: m.barcena@lumc.nl.
Frank J.M. van Kuppeveld, Email: f.j.m.vankuppeveld@uu.nl.
Supplemental Information
References
- Albulescu L., Wubbolts R., van Kuppeveld F.J., Strating J.R. Cholesterol shuttling is important for RNA replication of coxsackievirus B3 and encephalomyocarditis virus. Cell. Microbiol. 2015;17:1144–1156. doi: 10.1111/cmi.12425. [DOI] [PubMed] [Google Scholar]
- Altan-Bonnet N., Balla T. Phosphatidylinositol 4-kinases: hostages harnessed to build panviral replication platforms. Trends Biochem. Sci. 2012;37:293–302. doi: 10.1016/j.tibs.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arita M. Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure. Microbiol. Immunol. 2014;58:239–256. doi: 10.1111/1348-0421.12144. [DOI] [PubMed] [Google Scholar]
- Arita M. Mechanism of poliovirus resistance to host phosphatidylinositol-4 kinase III β inhibitor. ACS Infect. Dis. 2016;2:140–148. doi: 10.1021/acsinfecdis.5b00122. [DOI] [PubMed] [Google Scholar]
- Belov G.A., Feng Q., Nikovics K., Jackson C.L., Ehrenfeld E. A critical role of a cellular membrane traffic protein in poliovirus RNA replication. PLoS Pathog. 2008;4:e1000216. doi: 10.1371/journal.ppat.1000216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov G.A., Nair V., Hansen B.T., Hoyt F.H., Fischer E.R., Ehrenfeld E. Complex dynamic development of poliovirus membranous replication complexes. J. Virol. 2012;86:302–312. doi: 10.1128/JVI.05937-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienz K., Egger D., Pfister T., Troxler M. Structural and functional characterization of the poliovirus replication complex. J. Virol. 1992;66:2740–2747. doi: 10.1128/jvi.66.5.2740-2747.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke J.E., Inglis A.J., Perisic O., Masson G.R., McLaughlin S.H., Rutaganira F., Shokat K.M., Williams R.L. Structures of PI4KIIIβ complexes show simultaneous recruitment of Rab11 and its effectors. Science. 2014;344:1035–1038. doi: 10.1126/science.1253397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous S., Terwilliger T.C., Waldo G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005;23:102–107. doi: 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- Clayton E.L., Minogue S., Waugh M.G. Mammalian phosphatidylinositol 4-kinases as modulators of membrane trafficking and lipid signaling networks. Prog. Lipid Res. 2013;52:294–304. doi: 10.1016/j.plipres.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornell C.T., Kiosses W.B., Harkins S., Whitton J.L. Coxsackievirus B3 proteins directionally complement each other to downregulate surface major histocompatibility complex class I. J. Virol. 2007;81:6785–6797. doi: 10.1128/JVI.00198-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese M., Goellner S., Acosta E.G., Neufeldt C.J., Oleksiuk O., Lampe M., Haselmann U., Funaya C., Schieber N., Ronchi P. Ultrastructural characterization of Zika virus replication factories. Cell Rep. 2017;18:2113–2123. doi: 10.1016/j.celrep.2017.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dales S., Eggers H.J., Tamm I., Palade G.E. Electron microscopic study of the formation of Poliovirus. Virology. 1965;26:379–389. doi: 10.1016/0042-6822(65)90001-2. [DOI] [PubMed] [Google Scholar]
- de Graaf P., Zwart W.T., van Dijken R.A., Deneka M., Schulz T.K., Geijsen N., Coffer P.J., Gadella B.M., Verkleij A.J., van der Sluijs P., van Bergen en Henegouwen P.M. Phosphatidylinositol 4-kinasebeta is critical for functional association of rab11 with the Golgi complex. Mol. Biol. Cell. 2004;15:2038–2047. doi: 10.1091/mbc.E03-12-0862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Matteis M.A., Wilson C., D’Angelo G. Phosphatidylinositol-4-phosphate: the Golgi and beyond. BioEssays. 2013;35:612–622. doi: 10.1002/bies.201200180. [DOI] [PubMed] [Google Scholar]
- Dodd D.A., Giddings T.H., Jr., Kirkegaard K. Poliovirus 3A protein limits interleukin-6 (IL-6), IL-8, and beta interferon secretion during viral infection. J. Virol. 2001;75:8158–8165. doi: 10.1128/JVI.75.17.8158-8165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorobantu C.M., Albulescu L., Harak C., Feng Q., van Kampen M., Strating J.R.P.M., Gorbalenya A.E., Lohmann V., van der Schaar H.M., van Kuppeveld F.J.M. Modulation of the host lipid landscape to promote RNA virus replication: the picornavirus encephalomyocarditis virus converges on the pathway used by hepatitis C virus. PLoS Pathog. 2015;11:e1005185. doi: 10.1371/journal.ppat.1005185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorobantu C.M., Albulescu L., Lyoo H., van Kampen M., De Francesco R., Lohmann V., Harak C., van der Schaar H.M., Strating J.R., Gorbalenya A.E. Mutations in encephalomyocarditis virus 3A protein uncouple the dependency of genome replication on host factors phosphatidylinositol 4-kinase IIIalpha and oxysterol-binding protein. mSphere. 2016;1:e00068-16. doi: 10.1128/mSphere.00068-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faas F.G., Avramut M.C., van den Berg B.M., Mommaas A.M., Koster A.J., Ravelli R.B. Virtual nanoscopy: generation of ultra-large high resolution electron microscopy maps. J. Cell Biol. 2012;198:457–469. doi: 10.1083/jcb.201201140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q., Langereis M.A., Lork M., Nguyen M., Hato S.V., Lanke K., Emdad L., Bhoopathi P., Fisher P.B., Lloyd R.E., van Kuppeveld F.J. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014;88:3369–3378. doi: 10.1128/JVI.02712-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford Siltz L.A., Viktorova E.G., Zhang B., Kouiavskaia D., Dragunsky E., Chumakov K., Isaacs L., Belov G.A. New small-molecule inhibitors effectively blocking picornavirus replication. J. Virol. 2014;88:11091–11107. doi: 10.1128/JVI.01877-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsel L.A., Onderwater J.J., Daems W.T. Resolution of a gold latensification-elon ascorbic acid developer for Ilford L4 emulsion. Histochemistry. 1979;61:343–346. doi: 10.1007/BF00508456. [DOI] [PubMed] [Google Scholar]
- Greninger A.L., Knudsen G.M., Betegon M., Burlingame A.L., Derisi J.L. The 3A protein from multiple picornaviruses utilizes the golgi adaptor protein ACBD3 to recruit PI4KIIIβ. J. Virol. 2012;86:3605–3616. doi: 10.1128/JVI.06778-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu N.Y., Ilnytska O., Belov G., Santiana M., Chen Y.H., Takvorian P.M., Pau C., van der Schaar H., Kaushik-Basu N., Balla T. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell. 2010;141:799–811. doi: 10.1016/j.cell.2010.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilnytska O., Santiana M., Hsu N.Y., Du W.L., Chen Y.H., Viktorova E.G., Belov G., Brinker A., Storch J., Moore C. Enteroviruses harness the cellular endocytic machinery to remodel the host cell cholesterol landscape for effective viral replication. Cell Host Microbe. 2013;14:281–293. doi: 10.1016/j.chom.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langereis M.A., Feng Q., Nelissen F.H., Virgen-Slane R., van der Heden van Noort G.J., Maciejewski S., Filippov D.V., Semler B.L., van Delft F.L., van Kuppeveld F.J. Modification of picornavirus genomic RNA using ‘click’ chemistry shows that unlinking of the VPg peptide is dispensable for translation and replication of the incoming viral RNA. Nucleic Acids Res. 2014;42:2473–2482. doi: 10.1093/nar/gkt1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X., Xiao X., Wang J. Innate immunity evasion by enteroviruses: insights into virus-host interaction. Viruses. 2016;8:E22. doi: 10.3390/v8010022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Basavappa M., Lu J., Dong S., Cronkite D.A., Prior J.T., Reinecker H.C., Hertzog P., Han Y., Li W.X. Induction and suppression of antiviral RNA interference by influenza A virus in mammalian cells. Nat. Microbiol. 2016;2:16250. doi: 10.1038/nmicrobiol.2016.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limpens R.W.A.L., van der Schaar H.M., Kumar D., Koster A.J., Snijder E.J., van Kuppeveld F.J.M., Bárcena M. The transformation of enterovirus replication structures: a three-dimensional study of single- and double-membrane compartments. MBio. 2011;2:e00166-11. doi: 10.1128/mBio.00166-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna J.M., Wu X., Rice C.M. Present and not reporting for duty: dsRNAi in mammalian cells. EMBO J. 2016;35:2499–2501. doi: 10.15252/embj.201695933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod A.M., Mitchell D.R., Palmer N.J., Van de Poël H., Conrath K., Andrews M., Leyssen P., Neyts J. Identification of a series of compounds with potent antiviral activity for the treatment of enterovirus infections. ACS Med. Chem. Lett. 2013;4:585–589. doi: 10.1021/ml400095m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesmin B., Bigay J., Moser von Filseck J., Lacas-Gervais S., Drin G., Antonny B. A four-step cycle driven by PI(4)P hydrolysis directs sterol/PI(4)P exchange by the ER-Golgi tether OSBP. Cell. 2013;155:830–843. doi: 10.1016/j.cell.2013.09.056. [DOI] [PubMed] [Google Scholar]
- Molla A., Hellen C.U., Wimmer E. Inhibition of proteolytic activity of poliovirus and rhinovirus 2A proteinases by elastase-specific inhibitors. J. Virol. 1993;67:4688–4695. doi: 10.1128/jvi.67.8.4688-4695.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A., Morosky S.A., Delorme-Axford E., Dybdahl-Sissoko N., Oberste M.S., Wang T., Coyne C.B. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011;7:e1001311. doi: 10.1371/journal.ppat.1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeldt C.J., Joyce M.A., Van Buuren N., Levin A., Kirkegaard K., Gale M., Jr., Tyrrell D.L., Wozniak R.W. The hepatitis C Virus-induced membranous web and associated nuclear transport Machinery limit access of pattern recognition receptors to viral replication sites. PLoS Pathog. 2016;12:e1005428. doi: 10.1371/journal.ppat.1005428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul D., Bartenschlager R. Architecture and biogenesis of plus-strand RNA virus replication factories. World J. Virol. 2013;2:32–48. doi: 10.5501/wjv.v2.i2.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reghellin V., Donnici L., Fenu S., Berno V., Calabrese V., Pagani M., Abrignani S., Peri F., De Francesco R., Neddermann P. NS5A inhibitors impair NS5A-phosphatidylinositol 4-kinase IIIα complex formation and cause a decrease of phosphatidylinositol 4-phosphate and cholesterol levels in hepatitis C virus-associated membranes. Antimicrob. Agents Chemother. 2014;58:7128–7140. doi: 10.1128/AAC.03293-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss S., Rebhan I., Backes P., Romero-Brey I., Erfle H., Matula P., Kaderali L., Poenisch M., Blankenburg H., Hiet M.S. Recruitment and activation of a lipid kinase by hepatitis C virus NS5A is essential for integrity of the membranous replication compartment. Cell Host Microbe. 2011;9:32–45. doi: 10.1016/j.chom.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero-Brey I., Bartenschlager R. Membranous replication factories induced by plus-strand RNA viruses. Viruses. 2014;6:2826–2857. doi: 10.3390/v6072826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulin P.S., Lötzerich M., Torta F., Tanner L.B., van Kuppeveld F.J., Wenk M.R., Greber U.F. Rhinovirus uses a phosphatidylinositol 4-phosphate/cholesterol counter-current for the formation of replication compartments at the ER-Golgi interface. Cell Host Microbe. 2014;16:677–690. doi: 10.1016/j.chom.2014.10.003. [DOI] [PubMed] [Google Scholar]
- Schulz K.S., Mossman K.L. Viral evasion strategies in type I IFN signaling: a summary of recent developments. Front. Immunol. 2016;7:498. doi: 10.3389/fimmu.2016.00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strating J.R., van Kuppeveld F.J. Viral rewiring of cellular lipid metabolism to create membranous replication compartments. Curr. Opin. Cell Biol. 2017;47:24–33. doi: 10.1016/j.ceb.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strating J.R.P.M., van der Linden L., Albulescu L., Bigay J., Arita M., Delang L., Leyssen P., van der Schaar H.M., Lanke K.H.W., Thibaut H.J. Itraconazole inhibits enterovirus replication by targeting the oxysterol-binding protein. Cell Rep. 2015;10:600–615. doi: 10.1016/j.celrep.2014.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhy D.A., Giddings T.H., Jr., Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J. Virol. 2000;74:8953–8965. doi: 10.1128/jvi.74.19.8953-8965.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- tenOever B.R. The evolution of antiviral defense systems. Cell Host Microbe. 2016;19:142–149. doi: 10.1016/j.chom.2016.01.006. [DOI] [PubMed] [Google Scholar]
- van der Schaar H.M., van der Linden L., Lanke K.H., Strating J.R., Pürstinger G., de Vries E., de Haan C.A., Neyts J., van Kuppeveld F.J. Coxsackievirus mutants that can bypass host factor PI4KIIIβ and the need for high levels of PI4P lipids for replication. Cell Res. 2012;22:1576–1592. doi: 10.1038/cr.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Schaar H.M., Leyssen P., Thibaut H.J., de Palma A., van der Linden L., Lanke K.H., Lacroix C., Verbeken E., Conrath K., Macleod A.M. A novel, broad-spectrum inhibitor of enterovirus replication that targets host cell factor phosphatidylinositol 4-kinase IIIβ. Antimicrob. Agents Chemother. 2013;57:4971–4981. doi: 10.1128/AAC.01175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Schaar H.M., Dorobantu C.M., Albulescu L., Strating J.R., van Kuppeveld F.J. Fat(al) attraction: picornaviruses usurp lipid transfer at membrane contact sites to create replication organelles. Trends Microbiol. 2016;24:535–546. doi: 10.1016/j.tim.2016.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Schaar H.M., Melia C.E., van Bruggen J.A., Strating J.R., van Geenen M.E., Koster A.J., Barcena M., van Kuppeveld F.J. Illuminating the sites of enterovirus replication in living cells by using a split-GFP-tagged viral protein. mSphere. 2016;1 doi: 10.1128/mSphere.00104-16. e00104-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsch S., Miller S., Romero-Brey I., Merz A., Bleck C.K., Walther P., Fuller S.D., Antony C., Krijnse-Locker J., Bartenschlager R. Composition and three-dimensional architecture of the dengue virus replication and assembly sites. Cell Host Microbe. 2009;5:365–375. doi: 10.1016/j.chom.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J.P., Lloyd R.E. Regulation of stress granules in virus systems. Trends Microbiol. 2012;20:175–183. doi: 10.1016/j.tim.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K., Nagy P.D. Expanding use of multi-origin subcellular membranes by positive-strand RNA viruses during replication. Curr. Opin. Virol. 2014;9:119–126. doi: 10.1016/j.coviro.2014.09.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.