

Abstract
We present the first complete genome sequence of Odocoileus hemionus deer adenovirus 1 (OdAdV-1). This virus can cause sporadic haemorrhagic disease in cervids, although epizootics with high mortality have occurred in California. OdAdV-1 has been placed in the genus Atadenovirus, based on partial hexon, pVIII and fibre genes. Ten field isolates recovered from naturally infected mule deer (Odocoileus hemionus), white-tailed deer (Odocoileus virginiana) and moose (Alces alces) from Wyoming, black-tailed deer (Odocoileus hemionus columbianus) from California, and Rocky Mountain elk (Cervus elaphus nelsoni) from Colorado and Washington state were sequenced. The genome lengths ranged from 30 620 to 30 699 bp, contained the predicted proteins and gene organization typical of members of genus Atadenovirus, and had a high percentage of A/T nucleotides (66.7 %). Phylogenic analysis found that the closest ancestry was with ruminant atadenoviruses, while a divergence of the hexon, polymerase and penton base proteins of more than 15 % supports classification as a new species. Genetic global comparison between the 10 isolates found an overall 99 % identity, but greater divergence was found between those recovered from moose and elk as compared to deer, and a single variable region contained most of these differences. Our findings demonstrate that OdAdV-1 is highly conserved between 10 isolates recovered from multiple related cervid species, but genotypic differences, largely localized to a variable region, define two strains. We propose that the virus type name be changed to cervid adenovirus 1, with the species name Cervid atadenovirus A. Sequence data were used to develop molecular assays for improved detection and genotyping.
Keywords: cervid, adenovirus, Atadenovirus, moose, elk, deer
Abbreviations
A.a., moose; BAdV, bovine adenovirus; C.e., elk; EBM-2, endothelial cell basal medium-2; ITR, inverted terminal repeat; ML, maximum likelihood; OAdV, ovine adenovirus; OdAdV-1, Odocoileus hemionus deer adenovirus; O.h., mule deer; O.h.c, black-tailed deer; ORF, open reading frame; O.v, white-tailed deer; SNP, single nucleotide polymorphism.
Introduction
Adenoviruses have worldwide distribution and infect vertebrates of all types. They are typically well adapted to specific host species and infection is frequently subclinical, except for in young or immunocompromised individuals [1]. Exceptions occur, such as turkey haemorrhagic disease and canine infectious hepatitis [2]. Exceptions also occur for host-specific infections, as recently shown for a primate adenovirus (titi monkey adenovirus) with the potential to infect humans [3]. Adenoviruses of humans have been found to be extremely hardy and may remain in the environment for long periods in sheltered areas [4, 5], providing a source of infection for susceptible individuals.
Members of the family Adenoviridae have been divided into five genera [6]. The genus Atadenovirus was recently added in 2002 to include adenoviruses that were previously assigned to the genus Mastadenovirus, but varied significantly based on genomic size (20 % smaller), smaller structure, genes and gene arrangement, and codon usage biased to A/T-rich triplets – hence the genus name Atadenovirus [7, 8]. More recently, numerous diverse genus members have been described from reptile hosts that lack the A/T bias [9–12]. In general, a low GC content is associated with a recent host-switching event, suggesting that the original genus ancestor was from a squamate host [13, 14]. Atadenoviruses lack homologous genes for EIA, E3, V, IX and viral-associated RNA [7, 15], and there is an especially A/T-rich noncoding region of unknown function near the 3′ end of the genome of ruminant atadenoviruses [16]. This new genus contains seven species (Virus Taxonomy: 2016 Release. International Committee on Taxonomy of Viruses), with viruses that infect a diverse range of animals, including avian, ruminant, reptile and marsupial hosts [9, 17–20]. A complete or partial genome sequence has been determined for the genus members, including ovine adenovirus 7 (species Ovine atadenovirus D, OAdV-D; type species of the genus) [16], bovine adenovirus 4 [8] (within species Bovine atadenovirus D, BAdV-D), bovine adenovirus 6, duck adenovirus 1 [21], lizard adenovirus 2 [9], possum adenovirus 1 [17], psittacine adenovirus 3 [19] and snake adenovirus 1 [12, 14].
Odocoileus hemionus deer adenovirus 1 (OdAdV-1) is a member of the genus Atadenovirus that can cause systemic vasculitis with the potential for high fatality rates, and has been partially sequenced [22, 23]. Sporadic epizootics of adenovirus haemorrhagic disease in mule deer (Odocoileus hemionus, O.h.) and black-tailed deer (Odocoileus hemionus columbianus, O.h.c., a subspecies of mule deer) have been reported in California [24, 25]. White-tailed deer (Odocoileus virginiana, O.v.) and moose (Alces alces, A.a.) have also been found to be susceptible [26, 27]. Experimental challenge studies have induced fatal disease in young fawns, but mild or undetected infections in yearlings [28–30]. Lesions include pulmonary edema and haemorrhagic enteropathy, and may be similar to the haemorrhagic disease caused by the orbiviruses: bluetongue virus and epizootic haemorrhagic disease virus. Naturally occurring cases of adenovirus infection in wildlife are typically discovered as mortalities, but cases of mild disease with recovery are not likely to be observed. Serological testing to determine prevalence has proved challenging due to cross-reactions with related ruminant viruses [31], demonstrating the need for improved antibody detection assays. Little is known of the epidemiology and pathogenesis of OdAdV-1, or whether genetic variation in the virus contributes to variable outcomes of localized sporadic disease or large epizootics.
The purpose of our study was to sequence the complete genome of OdAdV-1, analyse the phylogenic relationship with other members of the genus Atadenoviruses, and determine the genetic variability between 10 isolates recovered from related host species in multiple states. Adenovirus recovered from naturally infected animals was identified by traditional methods of virus isolation, electron microscope imaging and PCR. Whole-genome sequences of field isolates collected from mule deer, white-tailed deer, black-tailed deer, elk and moose over 17 years were compared to each other by multisequence alignment and protein-coding regions were identified. The phylogenic relationship between isolates was examined using the whole-genome nucleotide sequence, and the relationship to other related adenoviruses was assessed based on amino acid sequences coding for the penton base, polymerase and hexon proteins. Sequence data were used to develop sensitive and specific assays for diagnosis and genotyping.
Results
Genome size and organization
The genomes of 10 OdAdV-1 isolates were fully sequenced and analysed. The genetic length varied between 30 620 and 30 699 bp, with 40 bp inverted terminal repeats (ITR) and an overall A/T content of 66.7 %. The gene arrangement and A/T-rich content are typical of the genus Atadenovirus from ruminant hosts. The genome arrangement of the predicted proteins is presented schematically in Fig. 1(a). Genus-specific findings, from 5′ to 3′, include: a p32K homologue in the EI genomic region, no E3 homologue in the region between pVIII and fibre genes, lack of homologues for V and IX, the presence of special E4 genes (E4.1) and a set of right-hand genes (RH2, 4 and 5) [9, 32]. Also present is a 1300 bp especially A/T-rich (72.6 %) noncoding region unique to the genus located between RH5 and E4.1 that has been described in ruminant atadenoviruses [16]. An unexpected finding was an apparent complete duplicate of the E1B gene located between the left-hand ITR and p32K, which is shown as E1B.1 in Fig. 1(a). This open reading frame (ORF) codes for a 379 aa protein that has 50 % identity with the second E1B gene (381 aa protein, E1B.2 in Fig. 1(a)). The E1B.2 gene is in the expected location and is >99 % identical to the current OdAdV-1 sequence in GenBank (AAF13267). These two E1B proteins have 46–52 % identity to E1B proteins of other ruminant atadenoviruses based on blastp search results. The whole-genome sequences of seven of the isolates have been submitted to GenBank and assigned the accession numbers KY468402- KY468407 and KY748210 (Table 1).
Fig. 1.

The genome organization of OdAdV-1 is depicted schematically (a). Potential ORFs were identified using blastx search to identify homologous proteins in GenBank. The genome sequences between the 10 isolates were highly identical (99 %), but had a variable region within the A/T-rich noncoding region (NCR). Multiple sequence alignment of the 10 isolates from the variable region is shown (b). This region contained two deletions totalling 54 nt (22 nt+32 nt, O.h. and O.v.) or three deletions totaling 75 nt (O.h.c.) compared to moose and elk (A.a., C.e.).
Table 1. Ten Odocoileus hemionus deer adenovirus isolates were sequenced as part of the study. Isolates were recovered from diagnostic cases received from 1999 to 2016.
Host species included Rocky Mountain elk (Cervus elaphus nelsoni, C.e.), moose (Alces alces, A.a.), mule deer (Odocoileus hemionus, O.h.), black-tailed deer (Odocoileus hemionus columbianus, O.h.c.) and white-tailed deer (Odocoileus virginiana O.v.).
| Isolate | Host | Year | Location | Age/diagnosis | GenBank accession |
|---|---|---|---|---|---|
| 99A.a | A.a. | 1999 | Wyoming | Calf/adenovirus mortality | KY468407 |
| 10A.a. | A.a. | 2010 | Wyoming | Calf/adenovirus mortality | KY468404 |
| 13C.e. | C.e. | 2013 | Washington | Juv/apparent health | KY468405 |
| 16C.e. | C.e. | 2016 | Colorado | Adult/adenovirus related mortality | KY468406 |
| 02O.h. | O.h. | 2002 | Wyoming | Yearling/adenovirus mortality | KY468402 |
| 14O.h. | O.h. | 2014 | Wyoming | Fawn/adenovirus mortality | |
| 15O.h.1 | O.h. | 2015 | Wyoming | 2 weeks/adenovirus mortality | |
| 15O.h.2 | O.h. | 2015 | Wyoming | 4 months/adenovirus mortality | |
| 06O.v. | O.v. | 2006 | Wyoming | 1.5 years/adenovirus mortality | KY468403 |
| CAO.h.c. | O.h.c. | 1998 | California | Adenovirus mortality | KY748210 |
Two strains based on a single variable region
The multiple sequence alignment of the 10 isolates found the genetic sequence is highly conserved, with greater than 99 % sequence identity. However, a variable region is present within the A/T-rich noncoding region between the RH5 and E4.1 genes (Fig. 1(a)). This region was longest in the isolates from elk and moose (N=4). The isolates from mule deer and white-tailed deer (N=5) had two deletions totalling 54 nt (22 plus 32 nt), and the California black-tailed deer isolate had an additional 21 nt deletion, as shown in the multiple sequence alignment (Fig. 1(b)). Largely based on this region, the genome length varied and resulted in two distinct strains. However, even outside of this region, over 100 single nucleotide polymorphisms (SNP) segregated in the same pattern, with 12–29 SNPs between members of the same strain, and 186 SNPs between members of the opposite strain. This difference resulted in an overall percentage identity of 99.9 % within a strain and 99.7 % between strains. The phylogenic tree based on whole-genome sequences of the ten isolates demonstrates two distinct clades, Fig. 2(a).
Fig. 2.

Maximum-likelihood phylogenic trees depicting the relationship between the 10 OdAdV-1 isolates in this study based on whole-genome sequences (a). Maximum-likelihood trees estimating the relationship between OdAdV-1 and the other atadenoviruses based on full amino acid sequences of the penton base (b), hexon (c) and polymerase proteins (d). Trees were constructed using PhyML in SeaView version 3.2 with a BioNJ starting tree and bootstrap values calculated from 1000 pseudo-replicates. A single OdAdV-1 sequence (99 A.a.) was used in (b)–(d) due to isolate identity in these proteins. Atadenoviruses: ovine adenovirus 7 (OAdVT, type species), duck adenovirus 1 (DAdV), psittacine adenovirus 3 (PsAdV), possum adenovirus 1 (PossAdV), lizard adenovirus 2 (LzAdV), snake adenovirus 1 (SnAdV), and bovine adenovirus 4 and 6 (BAdV-4 and 6). Representative ruminant viruses from the genus Mastadenovirus were included as outliers, bovine adenoviruses 1 and 3 (BAdV-1 and 3). Bootstrap values and branch lengths (b)–(d) are given. The accession numbers for the protein reference sequences retrieved from GenBank are listed in Table 2.
Phylogeny inference
The partial OdAdV-1 sequences currently in GenBank (AF198354, AF198355, AF198356 and AF361168) are 99 % identical to the nucleotide sequences of our isolates. Maximum-likelihood (ML) phylogenic estimation comparing OdAdV-1 with other genus members based on full-length predicted amino acid sequences for the penton base, hexon and polymerase proteins found that the closest relationship was with other ruminant atadenoviruses, OAdV-7, BAdV-4 and BAdV-6 (Fig. 2(b,d)). The phylogenic relationship was also analysed using partial amino acid sequences of the hexon and pol proteins so that BAdV-7 could be included, and this resulted in the same topology and similar branch lengths compared to the other ruminant atadenoviruses (data not shown). The most conserved genes were the structural proteins, hexon and penton base, with greater divergence being evident in the amino acid sequence for the polymerase protein, as has been found previously in phylogenic analysis for the genus [19].
Diagnostic and genotyping PCR
A PCR was developed to aid in the sensitive and specific detection of OdAdV-1 DNA in diagnostic material. Our use of a previously published assay [28] was found to give false positive reactions when applied to samples from pronghorn (Antilocapra americana), with Sanger sequencing of the amplicon finding pronghorn satellite DNA sequences. The new assay was able to detect OdAdV-1 in all of the species in the study, as well as pronghorn, with amplicons verified by Sanger sequencing using forward and reverse primers (Fig. 3(a)). To test for primer specificity, DNA from samples that were positive for bovine adenoviruses 1 and 4, canine adenovirus 2, ovine adenovirus (type not determined), bluetongue virus, epizootic haemorrhagic disease virus, pestivirus (bovine and pronghorn), chlamydophila sp. and cervid herpesvirus were not detected by the assay. A genotyping PCR was also designed using primers flanking the second and third deletions found in the variable region. This assay produced bands of 680, 701, or 733 bp, depending on the strain (Fig. 3(b)). This assay was used on DNA from an additional eight diagnostic cases positive for OdAdV-1 with amplicons run on a gel for size comparison and sequenced to verify the results. Of these eight, virus DNA from four additional black-tailed deer from a California epizootic contained the same changes as the isolate from the California black-tailed deer in our study. Virus DNA extracted from mule deer and pronghorn tissues had the deer strain sequence, and samples from two other mule deer had the longer sequence found in moose and elk virus from our study (Fig. 3(b)).
Fig. 3.
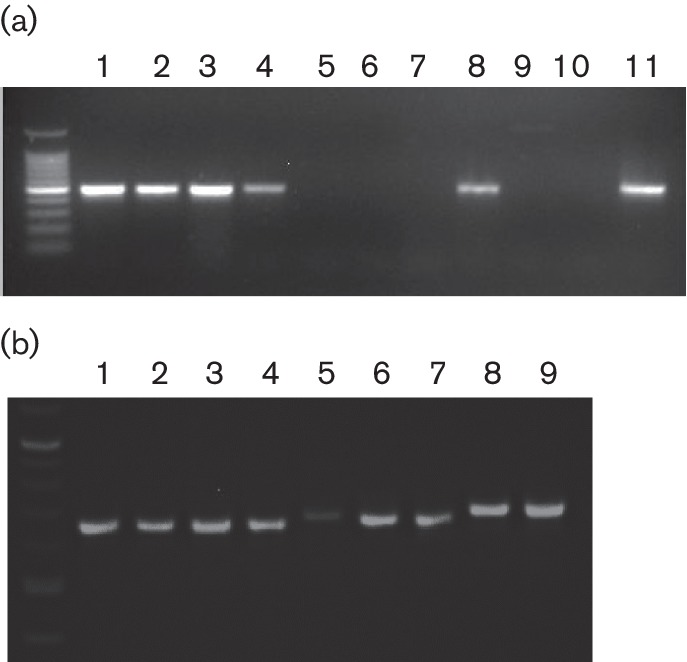
Primers specific for OdAdV-1 were designed for a diagnostic PCR and a genotyping PCR. Assays were run using DNA extracted from additional diagnostic cases that were positive for OdAdV-1 or other viral agents to test for specificity. (a) The diagnostic PCR amplified a 502 bp amplicon, a 100 bp molecular weight marker, and OdAdV-1 from mule deer (lanes 1, 3), elk (lane 2), pronghorn (lanes 4, 8) and moose (lane 11). No amplification product was produced with samples positive for bovine, canine, or ovine adenovirus of undetermined type (lanes 5–7), chlamydophila from a mule deer (lane 9), or the no template control (lane 10). (b) The genotyping PCR was designed to span part of the variable region and produced amplicons of 680, 701, or 733 bp, depending on the presence or absence of deletions: lanes 1–4, California black-tailed deer (680 bp due to 53 bp deletion); lanes 5–8, pronghorn, white-tailed deer, mule deer, (701 bp due to 32 bp deletion); and lane 8–9, mule deer with the moose/elk variation (733 bp with no deletions). Traditional Sanger sequencing using forward and reverse primers confirmed the expected sequence changes.
Discussion
The whole genome sequences in this report have 99 % identity with the partial OdAdV-1 sequences in GenBank, confirming that it is the same virus that was previously reported. The complete genome sequence contains homologous genes and a gene arrangement that is typical of the genus Atadenovirus, which confirms the previous classification in this genus [22, 23]. Of interest is an apparent duplication of the E1B gene in the region between the left-hand ITR and the p32 gene. The evolutionary and functional significance of this duplication remains to be determined. The exceptionally low GC content (33.3 %) of the genome may indicate that this virus is the result of a relatively recent host-switching event [13]. Phylogenic analysis showed that the closest ancestry was with other ruminant atadenoviruses, based on penton base, hexon and polymerase protein amino acid sequences (Fig. 2 (b–d)). The species demarcation criteria in the family Adenoviridae is 5–15 % divergence in the DNA polymerase amino acid sequence [6]. The divergence of greater than 15 % for the hexon and penton base proteins, and greater than 30 % for the polymerase protein, of OdAdV-1 compared to other genus members indicates this is a new species within the genus. This is supported by challenge studies where cross-infection of bovine adenoviruses and OdAdV-1 failed to produce infections in the opposite host [33]. The nearly identical isolates recovered from multiple related host species within the family Cervidae suggests that the species name should be Cervid atadenovirus A, and that the type name should be cervid adenovirus 1.
A distinct variable region was identified in the genus-unique A/T-rich noncoding region (Fig. 1(b)). Based largely on differences in this region, our isolates separated into two distinct clades (Fig. 2(a)). The function of this region is currently unknown [16]. These different clades segregated with host species, with virus from elk and moose being distinct from those from deer. However, the number of samples in this study is not sufficient to support a host-specific variation hypothesis. Indeed, using the genotype PCR and sequencing the amplicons from additional diagnostic cases, OdAdV-1-positive DNA from two mule deer mortalities in Wyoming in 2015 was found to have the elk/moose variation (Fig. 3(b)). These two cases were part of a cluster of mule deer adenovirus haemorrhagic disease mortalities in a region with large populations of moose and elk, providing potential for close contact. It is also of interest that the isolate recovered from a black-tailed deer from a large California epizootic [25] had an additional 21 bp deletion in this same region (Fig. 1(b)). The genotyping PCR found the same deletion in an additional four California cases from epizootic events (Fig. 3(b)). Large epizootics of adenovirus haemorrhagic disease resulting in thousands of mortalities have not been observed outside of California and Oregon, suggesting that the additional deletion might be significant. Sequencing of additional cases from California unrelated to large outbreaks would be useful in determining whether this additional deletion is likely to be related to pathogenesis, and this would need to be confirmed with challenge studies.
Most of the isolates (9/10) were recovered from animals succumbing to adenovirus haemorrhagic disease. However, the prevalence and distribution of OdAdV-1 and the potential for mild disease, chronic infections, and shedding are important unanswered questions. Studies have been limited due to the challenges of few diagnostic tools and the difficulty of sampling populations of wild animals. The isolate that came from an apparently healthy elk (Table 1, 13 C.e.) indicates that, at least transiently, OdAdV-1 can be detected in some healthy animals. Pathogenicity studies based on experimental infections have produced serious disease in fawns, but mild or inapparent disease in yearlings or adult animals [28–30]. Naturally occurring cases of adenovirus haemorrhagic disease are also more frequent in neonates and the young, but can occur animals of all ages. Factors affecting immune competence, such as stress and nutrition status, are likely to play a role in disease outcome. Adenovirus can survive for long periods outside of a host, and this, combined with the presence of susceptible animals, likely accounts for many cases of disease.
Our whole-genome sequence data and global comparison between 10 isolates identified a variable region distinguishing distinct genetic strains of OdAdV-1. The contribution of genetic variation to the epidemiology of adenovirus haemorrhagic disease remains to be tested. Further studies will be needed to determine the distribution and host preference of these strains, and whether the genetic variation contributes to variable pathogenesis. Complete genetic information and improved assays for diagnosis and genetic variation testing will be valuable tools for such pathogenicity and epidemiology studies.
Methods
Animals
Seven of the 10 isolates in the study were recovered from naturally occurring wildlife adenovirus haemorrhagic disease mortalities investigated by the Wyoming Game and Fish Department and submitted to the Wyoming State Veterinary Laboratory for diagnostic testing (Creekmore et al., manuscript in preparation). Of the remaining three isolates in the study, one was recovered from elk tissues received from the Colorado Division of Parks and Wildlife from an animal with ruminal ulcers as well as other multisystem lesions [34], one was recovered from an apparently healthy elk (unpublished finding) from the state of Washington that was a negative control from an unrelated study [35] and one was recovered from a black-tailed deer from the 1993/94 California epizootic [25]. These cases are listed in Table 1. These adenoviruses were identified by PCR, virus isolation, EM imaging and in some cases the detection of typical intranuclear bodies by histopathology.
Isolation and culture of mule deer umbilical vein endothelial cells
Umbilical cords from third-trimester twin mule deer foetuses were harvested aseptically and placed into 199E medium (Cellgro, Manassas, VA, USA) supplemented with 5 % ultimate grade FBS (Seradigm, Providence, UT, USA) and 2× penicillin/streptomycin (Lonza, Walkersville, MD, USA; 200 units ml−1 and 200 µg ml−1). The umbilical veins were dissected out and rinsed with PBS (Cellgro). One end of each vein was clamped and the veins filled with 1 mg ml−1 collagenase (Sigma-Aldrich, St Louis, MO, USA) in PBS, then the the other end was clamped and incubated for 5 min while resting in PBS. The veins were rinsed with collagenase, and then the digest was repeated, with incubation for 1 h. The veins were flushed with 199E into a centrifuge tube and the cells collected by centrifugation at 250 g for 8 min. The cells were resuspended in endothelial cell basal medium-2 (EBM-2; Lonza) supplemented with endothelial cell growth factors (EGM-2 single quots; Lonza) and 15 % FBS and incubated in cell culture-treated flasks at 37 °C, 5 % CO2 until confluent. Cells were processed using 0.05 % Trypsin-EDTA (Gibco, Grand Island, NY, USA) in PBS, and frozen at a low passage number (5–6) using dimethylsulfoxide (11 %, Sigma-Aldrich) in FBS. The white-tailed deer umbilical vein endothelial cells used in some cases were similarly prepared. Cultured cells were confirmed to be free of adenovirus, pestivirus and Mycoplasma species by PCR.
Virus isolation and purification
Tissue pools (a combination of two or more: lung, liver, kidney and spleen) from diagnostic cases submitted to the Wyoming State Veterinary Laboratory were homogenized in 1 : 5 vol of Bovarnick’s media (0.218 M sucrose, 3.8 mM KH2PO4, 7.2 mM K2HPO4, 4.9 mM glutamic acid; Sigma-Aldrich) and applied to 60-70 confluent mule deer umbilical vein endothelial cells or white-tailed deer umbilical vein endothelial cells drained of media. After 1 h adherence, inoculum was removed and cells were refed with complete media: EBM-2, 4 % FBS and 1× penicillin/streptomycin (Lonza; 100 units ml−1 and 100 µg ml−1) at 37 °C, 5 % CO2. Cultures were observed daily for cytopathic effect (CPE). Wells with no CPE after 7 days were frozen and then thawed, after which the supernatant was passed onto new cells for two blind passages. For sequencing, virus from the second to fifth passage was grown in 150 cm2 flasks to >/=90 % cytopathic effect, frozen, thawed and clarified by centrifugation at 5000 g for 20 min prior to centrifugation through a 20 % sucrose cushion at 107 000 g for 12 h.
DNA extraction
Pellets were resuspended in 400 ul PBS and the DNA extracted using filter columns (Qiagen Blood and Tissue Kit, Valencia, CA, USA) according to the manufacturer’s directions. The DNA concentration was estimated by fluorimetric measurement (Qubit, ThermoFisher Scientific, Waltham, MA, USA).
Sequencing and analyses
Shotgun libraries were prepared using the Hyper Library Construction kit (Kapa Biosystems, Wilmington, MA, USA) and sequenced on Illumina MiSeq with a Nano V2 kit (High-Throughput Sequencing and Genotyping Unit, Roy J. Carver Biotechnology Center/WM Keck Center, University of Illinois at Urbana-Champaign). Sequences were assembled de novo using the Ray Assembler program (Ray version 2.3.1, publicly available software) [36] and for mapping assembly MIRA version 4 was used [37]. In all cases, assembly of the next-generation data produced a single contiguous sequence of the expected size. These were examined using Tablet [38] for quality, SNP confirmation and depth of coverage. The average coverage depth for the 10 isolates was >2000 (214–11 480). PCR and traditional Sanger sequencing covering over 10 % of the genome (3115 bp) further checked the accuracy of the sequences. ORFs were identified and annotated using SeqBuilder, Lasergene 13 (DNASTAR, Madison, WI, USA), and the predicted transcription units were examined for homologous proteins in GenBank via Basic Local Alignment Search Tool (blast), using the blastx and blastp queries [39].
Phylogenic analysis
The phylogenic relationship between the isolates in our study was estimated, and it was also determined for OdAdV-1 and other atadenoviruses and representative bovine viruses from the genus Mastadenovirus as outliers. Sequences were aligned using BioNJ [40] and ML phylogenic trees (PhyML version 3.1) were constructed using programs in the Seaview version 4.6.1 software package [41, 42]. Distance was calculated with Kimura’s two-parameter model [43] and compared to the ML-generated tree branch length using programs in Seaview. The phylogenic tree comparing the 10 isolates to each other was based on whole-genome nucleotide sequences. Phylogenic trees comparing OdAdV-1 to other adenoviruses were based on the predicted complete amino acid sequences for the penton base (450 aa), hexon (911 aa) and polymerase (1076 aa) proteins. The accession numbers for the reference sequences retrieved from GenBank are listed in Table 2.
Table 2. The reference sequences used in phylogenic tree construction were retrieved from GenBank for full coding sequences for the penton base, hexon and polymerase proteins.
All seven species within the genus Atadenovirus are represented, and two ruminant mastadenoviruses are included as outliers.
| Adenovirus type (species) | Penton base aa | Hexon aa | Polymerase aa |
|---|---|---|---|
| Bovine adenovirus 4 (D) | NP_077393 | NP_077397 | NP_077389 |
| Bovine adenovirus 6 | YP_007347002 | YP_007347006 | YP_007346998 |
| Ovine adenovirus 7T (D) | NP_659519 | NP_659523 | NP_659515 |
| Possum adenovirus 1 (A) | AF249332 | AAL73247 | Not available |
| Duck adenovirus 1 (A) | NP_044706 | NP_044710 | AP_000080 |
| Psittacine adenovirus 3 (A) | YP_009112720 | YP_009112724 | YP_009112716 |
| Lizard adenovirus 2 (A) | YP_009051658 | YP_009051622 | YP_009051654 |
| Snake adenovirus 1 (A) | YP_001552251 | YP_001552255 | YP_001552247 |
| Bovine adenovirus 1 (A) | YP_094036 | YP_094039 | YP_094032 |
| Bovine adenovirus 3 (B) | AP_000028 | NP_ 046323 | AP_000026 |
T, type species of the genus Atadenovirus.
PCR assays
Whole-genome sequence data were used to develop molecular assays for diagnosis, genotyping and validation of next-generation sequencing results. The primers were in silico tested for specificity using National Center for Biotechnology Information (NCBI) Primer-blast [44], and the diagnostic primers were in vitro tested for cross-reactions with common bovine and cervid viruses. Sanger sequencing of the PCR amplicons using forward and reverse primers (GENEWIZ, South Plainfield, NJ, USA) was used to verify the amplicons. The primer pairs listed in Table 3 were used with Platinum PCR Supermix (Life Technologies, Carlsbad, CA, USA). A reaction volume of 25 ml contained 1 ul each 10 uM F and R primers, 3 ul DNA template and 20 ul Supermix. The thermocycler conditions were 94 °C 2 min, (94 °C 15 s, 62 °C 1 min, 72 °C 30 s) × 40, 72 °C 2 min.
Table 3. Whole-genome sequence data were used to design PCR assays for diagnosis, genotyping and validation of assembly data by Sanger sequencing.
| Primer | Sequence (5′– 3′) | Position* | Purpose, amplicon size (bp) |
|---|---|---|---|
| Ad Aa F | CCGCATTCAGGCCACTTCCATT | 29 915–29 936 | Diagnosis, 502 bp |
| Ad Aa R | TGCTGCGTCTGAACACTAATA | 30 396–30 416 | |
| Ad 6 L F | TTACCCAAAATTCCCTATCCA | 27 769–27 789 | Genotyping, 680, 701 and 733 |
| Ad 6 L R | TTGCGTGTTACTAATGCTGTTCG | 28 477–28 501 | |
| Ad13/14 F1 | GAACTTCCCAGACCATTTACC | 3 093–3 113 | Validation, 505 |
| Ad13/14 R1 | AGGCATTTCCGTTTTACTCTG | 3 577–3 597 | |
| Ad 13/14 F2 | GTGGCCTAGTAATTTTCGTCTGC | 4 107–4 129 | Validation, 653 |
| Ad 13/14 R2 | TTCCTCCTAGTTTGATTCCTGTGT | 4 736–4 759 | |
| Ad 13/14 F3 | AAGCGCTACACTGCAATAACAATC | 14 737–14 760 | Validation, 776 |
| Ad 13/14 R3 | GACCAGCTGAAACCACCTCTC | 15 492–15 512 |
*Based on the nucleotide sequence of OdAdV-1 99A.a. (GenBank KY468407.)
Nucleotide sequence accession numbers
Sequence data were submitted to GenBank with accession numbers KY468402- KY468407 and KY748210.
Funding information
This material is based upon work that is supported by the National Institute of Food and Agriculture, US Department of Agriculture, under award numbers WYO-568–16 and WYO-564–16. Support was also received from the W. Richard and Barbara Andrau Powell Wildlife/Livestock Disease Training Fund.
Acknowledgements
We thank Alan Greene and Noah C. Hull for assistance in the comparison of phylogenic analysis methods, and Drs Holly Ernest and Brant Schumaker for advice on manuscript content and submission.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical statement
No experimental animals were used in this study. Samples used were recovered from animals that died due to natural infection in their natural habitat, except for one case that was part of a previously published study (citation provided).
References
- 1.Echavarría M. Adenoviruses in immunocompromised hosts. Clin Microbiol Rev. 2008;21:704–715. doi: 10.1128/CMR.00052-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.MacLachlan NJ, Dubovi EJ, Fenner F. Fenner's Veterinary Virology. 4th ed. Amsterdam, Boston: Elsevier/AP; 2011. pp. 203–212. [Google Scholar]
- 3.Chen EC, Yagi S, Kelly KR, Mendoza SP, Tarara RP, et al. Cross-species transmission of a novel adenovirus associated with a fulminant pneumonia outbreak in a new world monkey colony. PLoS Pathog. 2011;7:e1002155. doi: 10.1371/journal.ppat.1002155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gordon YJ, Gordon RY, Romanowski E, Araullo-Cruz TP. Prolonged recovery of desiccated adenoviral serotypes 5, 8, and 19 from plastic and metal surfaces in vitro. Ophthalmology. 1993;100:1835–1839. doi: 10.1016/S0161-6420(93)31389-8. [DOI] [PubMed] [Google Scholar]
- 5.Mahl MC, Sadler C. Virus survival on inanimate surfaces. Can J Microbiol. 1975;21:819–823. doi: 10.1139/m75-121. [DOI] [PubMed] [Google Scholar]
- 6.Harrach B, Benkő M, Both GW, Brown M, Davison AJ, et al. Family Adenoviridae. In: Andrew MQ, King MJA, Carstesn EricB, Lefkowitz ElliotJ, editors. Virus Taxonomy: IXth Report of the International Committee on Taxonomy of Viruses IX. San Diego: Elsevier; 2011. pp. 125–141. (editors) [Google Scholar]
- 7.Benkö M, Harrach B. A proposal for a new (third) genus within the family Adenoviridae. Arch Virol. 1998;143:829–837. doi: 10.1007/s007050050335. [DOI] [PubMed] [Google Scholar]
- 8.Dán A, Ruzsics Z, Russell WC, Benkö M, Harrach B. Analysis of the hexon gene sequence of bovine adenovirus type 4 provides further support for a new adenovirus genus (Atadenovirus) J Gen Virol. 1998;79:1453–1460. doi: 10.1099/0022-1317-79-6-1453. [DOI] [PubMed] [Google Scholar]
- 9.Pénzes JJ, Menéndez-Conejero R, Condezo GN, Ball I, Papp T, et al. Molecular characterization of a lizard adenovirus reveals the first atadenovirus with two fiber genes and the first adenovirus with either one short or three long fibers per penton. J Virol. 2014;88:11304–11314. doi: 10.1128/JVI.00306-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ball I, Behncke H, Schmidt V, Geflügel FT, Papp T, et al. Partial characterization of new adenoviruses found in lizards. J Zoo Wildl Med. 2014;45:287–297. doi: 10.1638/2013-0143.1. [DOI] [PubMed] [Google Scholar]
- 11.Ball I, Ofner S, Funk RS, Griffin C, Riedel U, et al. Prevalence of neutralising antibodies against adenoviruses in lizards and snakes. Vet J. 2014;202:176–181. doi: 10.1016/j.tvjl.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 12.Farkas SL, Benko M, Elo P, Ursu K, Dán A, et al. Genomic and phylogenetic analyses of an adenovirus isolated from a corn snake (Elaphe guttata) imply a common origin with members of the proposed new genus Atadenovirus. J Gen Virol. 2002;83:2403–2410. doi: 10.1099/0022-1317-83-10-2403. [DOI] [PubMed] [Google Scholar]
- 13.Wellehan JF, Johnson AJ, Harrach B, Benkö M, Pessier AP, et al. Detection and analysis of six lizard adenoviruses by consensus primer PCR provides further evidence of a reptilian origin for the atadenoviruses. J Virol. 2004;78:13366–13369. doi: 10.1128/JVI.78.23.13366-13369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farkas SL, Harrach B, Benko M. Completion of the genome analysis of snake adenovirus type 1, a representative of the reptilian lineage within the novel genus Atadenovirus. Virus Res. 2008;132:132–139. doi: 10.1016/j.virusres.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 15.Venktesh A, Watt F, Xu ZZ, Both GW. Ovine adenovirus (OAV287) lacks a virus-associated RNA gene. J Gen Virol. 1998;79:509–516. doi: 10.1099/0022-1317-79-3-509. [DOI] [PubMed] [Google Scholar]
- 16.Vrati S, Brookes DE, Strike P, Khatri A, Boyle DB, et al. Unique genome arrangement of an ovine adenovirus: identification of new proteins and proteinase cleavage sites. Virology. 1996;220:186–199. doi: 10.1006/viro.1996.0299. [DOI] [PubMed] [Google Scholar]
- 17.Thomson D, Meers J, Harrach B. Molecular confirmation of an adenovirus in brushtail possums (Trichosurus vulpecula) Virus Res. 2002;83:189–195. doi: 10.1016/S0168-1702(01)00437-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nguyen TH, Vidovszky MZ, Ballmann MZ, Sanz-Gaitero M, Singh AK, et al. Crystal structure of the fibre head domain of bovine adenovirus 4, a ruminant atadenovirus. Virol J. 2015;12:81. doi: 10.1186/s12985-015-0309-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.To KK, Tse H, Chan WM, Choi GK, Zhang AJ, et al. A novel psittacine adenovirus identified during an outbreak of avian chlamydiosis and human psittacosis: zoonosis associated with virus-bacterium coinfection in birds. PLoS Negl Trop Dis. 2014;8:e3318. doi: 10.1371/journal.pntd.0003318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harrach B, Meehan BM, Benkö M, Adair BM, Todd D. Close phylogenetic relationship between egg drop syndrome virus, bovine adenovirus serotype 7, and ovine adenovirus strain 287. Virology. 1997;229:302–306. doi: 10.1006/viro.1996.8390. [DOI] [PubMed] [Google Scholar]
- 21.Fu G, Chen H, Huang Y, Cheng L, Fu Q, et al. Full genome sequence of egg drop syndrome virus strain FJ12025 isolated from muscovy duckling. Genome Announc. 2013;1:e00623-13. doi: 10.1128/genomeA.00623-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zakhartchouk A, Bout A, Woods LW, Lehmkuhl HD, Havenga MJ. Odocoileus hemionus deer adenovirus is related to the members of Atadenovirus genus. Arch Virol. 2002;147:841–847. doi: 10.1007/s007050200031. [DOI] [PubMed] [Google Scholar]
- 23.Lehmkuhl HD, Hobbs LA, Woods LW. Characterization of a new adenovirus isolated from black-tailed deer in California. Arch Virol. 2001;146:1187–1196. doi: 10.1007/s007050170114. [DOI] [PubMed] [Google Scholar]
- 24.Boyce WM, Woods LW, Keel MK, Maclachlan NJ, Porter CO, et al. An epizootic of adenovirus-induced hemorrhagic disease in captive black-tailed deer (Odocoileus hemionus) J Zoo Wildl Med. 2000;31:370–373. doi: 10.1638/1042-7260(2000)031[0370:AEOAIH]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 25.Woods LW, Swift PK, Barr BC, Horzinek MC, Nordhausen RW, et al. Systemic adenovirus infection associated with high mortality in mule deer (Odocoileus hemionus) in California. Vet Pathol. 1996;33:125–132. doi: 10.1177/030098589603300201. [DOI] [PubMed] [Google Scholar]
- 26.Sorden SD, Woods LW, Lehmkuhl HD. Fatal pulmonary edema in white-tailed deer (Odocoileus virginianus) associated with adenovirus infection. J Vet Diagn Invest. 2000;12:378–380. doi: 10.1177/104063870001200416. [DOI] [PubMed] [Google Scholar]
- 27.Shilton CM, Smith DA, Woods LW, Crawshaw GJ, Lehmkuhl HD. Adenoviral infection in captive moose (Alces alces) in Canada. J Zoo Wildl Med. 2002;33:73–79. doi: 10.1638/1042-7260(2002)033[0073:AIICMA]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 28.Woods LW, Hanley RS, Chiu PH, Burd M, Nordhausen RW, et al. Experimental adenovirus hemorrhagic disease in yearling black-tailed deer. J Wildl Dis. 1997;33:801–811. doi: 10.7589/0090-3558-33.4.801. [DOI] [PubMed] [Google Scholar]
- 29.Woods LW, Hanley RS, Chiu PH, Lehmkuhl HD, Nordhausen RW, et al. Lesions and transmission of experimental adenovirus hemorrhagic disease in black-tailed deer fawns. Vet Pathol. 1999;36:100–110. doi: 10.1354/vp.36-2-100. [DOI] [PubMed] [Google Scholar]
- 30.Woods LW, Lehmkuhl HD, Swift PK, Chiu PH, Hanley RS, et al. Experimental adenovirus hemorrhagic disease in white-tailed deer fawns. J Wildl Dis. 2001;37:153–158. doi: 10.7589/0090-3558-37.1.153. [DOI] [PubMed] [Google Scholar]
- 31.Lehmkuhl HD, Hobbs LA. Serologic and hexon phylogenetic analysis of ruminant adenoviruses. Arch Virol. 2008;153:891–897. doi: 10.1007/s00705-008-0063-4. [DOI] [PubMed] [Google Scholar]
- 32.Davison AJ, Benko M, Harrach B. Genetic content and evolution of adenoviruses. J Gen Virol. 2003;84:2895–2908. doi: 10.1099/vir.0.19497-0. [DOI] [PubMed] [Google Scholar]
- 33.Woods LW, Lehmkuhl HD, Hobbs LA, Parker JC, Manzer M. Evaluation of the pathogenic potential of cervid adenovirus in calves. J Vet Diagn Invest. 2008;20:33–37. doi: 10.1177/104063870802000106. [DOI] [PubMed] [Google Scholar]
- 34.Fox KA, Atwater L, Hoon-Hanks L, Miller M. A mortality event in elk (Cervus elaphus nelsoni) calves associated with malnutrition, pasteurellosis, and deer adenovirus in Colorado, USA. J Wildl Dis. 2017;53:674–676. doi: 10.7589/2016-07-167. [DOI] [PubMed] [Google Scholar]
- 35.Clegg SR, Mansfield KG, Newbrook K, Sullivan LE, Blowey RW, et al. Isolation of digital dermatitis treponemes from hoof lesions in Wild North American Elk (Cervus elaphus) in Washington State, USA. J Clin Microbiol. 2015;53:88–94. doi: 10.1128/JCM.02276-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boisvert S, Raymond F, Godzaridis E, Laviolette F, Corbeil J. Ray Meta: scalable de novo metagenome assembly and profiling. Genome Biol. 2012;13:R122. doi: 10.1186/gb-2012-13-12-r122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WE, et al. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14:1147–1159. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Milne I, Stephen G, Bayer M, Cock PJ, Pritchard L, et al. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform. 2013;14:193–202. doi: 10.1093/bib/bbs012. [DOI] [PubMed] [Google Scholar]
- 39.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 40.Gascuel O. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol Biol Evol. 1997;14:685–695. doi: 10.1093/oxfordjournals.molbev.a025808. [DOI] [PubMed] [Google Scholar]
- 41.Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol. 2010;27:221–224. doi: 10.1093/molbev/msp259. [DOI] [PubMed] [Google Scholar]
- 42.Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59:307–321. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 43.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 44.Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, et al. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]