

Abstract
Naturally occurring systems have the ability to self‐regulate, which plays a key role in their structural and functional adaptation. The autonomous behavior in living systems is biocatalytically controlled by the continuous consumption of energy to remain in a non‐equilibrium condition. In this work, we show the construction of a self‐regulated “breathing” microgel that uses chemical fuels to keep the system in the out‐of‐equilibrium state. The enzyme urease is utilized to program a feedback‐induced pH change, which in turn tunes the size switch and fluorescence intensity of the microgel. A continuous supply of chemical fuels to the system allows the process to be reversible. This microgel with tunable autonomous properties provides insights into the design of artificial systems and dynamic soft materials.
Keywords: breathing microgels, enzyme catalysis, feedback mechanism, non-equilibrium processes, self-regulation
The design and construction of stimuli‐responsive self‐assemblies has been the subject of intense research because of their exciting potential in the fields of materials science and nanotechnology.1 For these self‐assemblies, the produced switchable features are governed by external signals, generated by pH, light, temperature, enzymes, redox agents, gas or electrochemistry, leading to different amplified physical or chemical outputs.2 In general, such property changes are energetically downhill processes, which lead to thermodynamically stable states. In other words, a counter‐trigger is required to revert the one‐way transition of the synthetic responsive systems, which do not possess self‐regulating behavior. However, a hallmark of natural self‐assembly processes is the fact that they are energetically uphill and exhibit out‐of‐equilibrium behavior in their response to an external signal, making them self‐regulated and programmed in the time domain.3 This requires a continuous supply of energy to maintain, for example, biological adaptability in living cells, including cell division, organelle motility, and signal transduction.4 It is this principle that has inspired scientists over the past decades to design life‐like assemblies as a simplified way to mimic natural dynamic behavior and to create access to a variety of out‐of‐equilibrium molecular structures.5 These non‐equilibrium or dissipative self‐assemblies are dependent on the consumption of energy sources, which can activate the building blocks, and thereby temporarily form diverse structural conformations. In this regard, higher levels of self‐regulated systems can be obtained by the introduction of a chemical fuel, giving rise to the chemical modification of the building blocks, which are simultaneously deactivated by the external triggers.5b Autonomous cycles can be achieved by these two continuous opposing activation and deactivation processes.
However, how to precisely control the kinetics of competing activation and deactivation processes is the key towards the development and construction of time‐programmed systems. The principle underlying the main strategies to kinetically temporal programming systems is that activation has to be faster than deactivation to trigger the system, but in the long term deactivation needs to take over to reverse the process.6 By controlling the reaction rates of activation and deactivation, Ulijn and co‐workers have demonstrated the formation of supramolecular peptide nanofibers through non‐equilibrium biocatalytic assembly.5f In another example, Walther and co‐workers reported a facile approach to program the time domain of pH‐responsive self‐assemblies by installation of a base or an alkaline buffer promoter and a dormant deactivator, a lactone or an ester.7 They extended this generic concept to a transient acidic profile by the coupling of a fast acidic activator to the biocatalytically time‐controlled generation of a deactivator, alkaline ammonia, from urea by urease.8 The transient state can be time‐programmed by simply modulating the enzyme concentration and this platform was utilized to show biocatalytic feedback‐driven transient hydrogels (sol‐gel‐sol) with programmed lifetimes. More recently, they demonstrated a pH‐responsive block copolymer photonic gel with autonomous transient memories, remotely controlled signal propagation, and sensing by the propagation of pH‐waves through the urease‐based enzymatic switch.9
In non‐biological systems, shape or conformational changes under non‐equilibrium conditions have rarely been reported. In one recent example using enzymes, a synthetic supramolecular system with fuel‐driven temporal and tunable helical conformational changes could be achieved.5d With the long‐term goal of developing functional artificial systems with precisely controlled adaptive dynamic behavior (for example, “breathing” and shape transformation), herein we report a hybrid microgel with a pH‐induced temporal size control or “breathing” feature, which is mediated by an enzymatic reaction.
The pH‐responsive part of the microgel is formed by poly(N,N‐diethylaminoethyl methacrylate) (PDEAEMA), whereas the enzyme involved is urease. Upon exposure to an acidic buffer containing urea, first protonation of PDEAEMA leads to the swelling of the microgel. This swelling is subsequently counteracted by the urease‐mediated conversion of urea to ammonia, which leads to an increase in the local pH, deprotonation of PDEAEMA, and shrinkage of the hydrogel. Addition of the acidic buffer allows repetition of this cycle (Scheme 1).
Scheme 1.
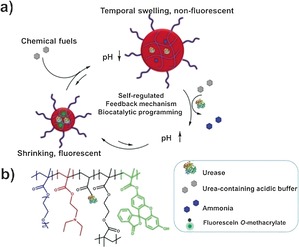
a) The self‐regulated “breathing” hybrid microgel with automatic fluorescence on/off switch mediated by the urease‐based enzymatic reaction. b) Chemical composition of the hybrid microgel with covalently‐immobilized urease.
The microgel was synthesized by free radical emulsion polymerization of poly(oligoethylene glycol methacrylate) (POEGMA), DEAEMA, and ethylene glycol dimethacrylate (EGDMA). POEGMA was used to ensure the solubility in water of the microgel particles irrespective of the protonation state of PDEAEMA; EGDMA was used for crosslinking (Scheme S1 in the Supporting Information). Furthermore, during polymerization, fluorescein moieties were introduced into the microgel, the pH‐dependent fluorescence of which was used as a measure of the local pH. Finally, urease was chemically attached to the microgel through the reaction between amino groups from the enzyme and N‐acryloxysuccinimide groups incorporated in the microgel. Urease was chosen first as it is able to change the pH upon substrate conversion. Furthermore, the bell‐shaped pH‐activity curve of urease with low enzyme activity at low and high pH provides a positive feedback, which allows a dormancy period before the subsequent pH elevation because of the generation of base.10 As a result, the microgel is able to first swell before it automatically shrinks, recovering its original contraction state. The transient swelling of the microgel is accompanied by the transient fluorescence off‐switch in the out‐of‐equilibrium low‐pH state. Upon return to a high pH, the fluorescence is switched back on again.
For the hybrid microgels, their size and morphology were confirmed by transmission electron microscopy (TEM) and scanning electron microscopy (SEM). The microgel exhibited a uniform round shape, and the introduction of the enzyme did not have an effect on the formation of the microgel (Supporting Information, Figures S1 and S2). To confirm the pH‐responsiveness of the microgel, dynamic light scattering (DLS) was first conducted to monitor changes in the particle diameter at different pH values. When the pH was decreased from 9 to 3.5 by adding acid to the microgel solution, DLS measurements indicated a remarkable size increase from 420 nm to 820 nm (Supporting Information, Figure S3). This significant size change was consistent with the result obtained from TEM images (Figure 1 a,b). The average microgel radius is plotted versus pH in the Supporting Information, Figure S4. Substantial swelling occurred from pH 6 to 4.5, which is viewed as a result of the protonation process of the tertiary amine groups from PDEAEMA, the pK a of which is around 7. Thereafter, a slight swelling was observed from pH 4.5 to 3.5. The reversibility of the pH‐induced size switch was also examined. Figure 1 c depicts multiple‐run reversibility experiments of the diameter responses of the microgel to pH variation. The results indicate that the size of the microgel alternately increases and decreases during a cycle of low pH and high pH. Thus, these results show adequate evidence that the swelling/collapse of the microgel can be reproducibly tuned in response to different pH values, which provides the platform for the development of a self‐regulated “breathing” out‐of‐equilibrium system.
Figure 1.
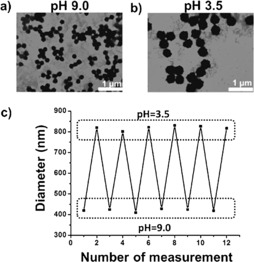
TEM images of the hybrid microgel at a) pH 9 and b) pH 3.5. c) DLS data showing multiple‐run reversibility of the microgel diameter between pH 9 and pH 3.5.
Contrary to other reports in which free urease was utilized as a pH regulator in bulk solution, in this case urease was conjugated to the microgel by covalent bonding in order to prevent the release of the enzyme and to ensure that the pH increase was achieved inside the microgel particles. The concentration of urease in the hybrid microgel was determined by measuring the concentration of free urease in the supernatant by using UV absorption at 280 nm (Supporting Information, Figure S5), which was subtracted from the amount of urease added during the conjugation reaction. The stability of the urease in the microgel was also studied, and it retained near 90 % activity after one month (Supporting Information, Figure S6), demonstrating that the hybrid microgel could be used for a long time.
At the start of the experiment the microgel particles were brought to pH 9, to bring them into a collapsed state and to keep urease activity low. Then, concentrated urea‐containing acidic buffer (citric acid/sodium citrate) was injected into the microgel solution. As expected, the pH of the microgel solution rapidly decreased to 3.5, at which the activity of urease is low. However, the activity was still sufficient to raise the pH of the system after a defined time period, from the conversion of urea into CO2 and NH3. This led to an increase in urease activity, which reached the maximum at intermediate pH values (from 5 to 8). At higher pH values the enzymatic reaction rate was self‐diminished, and thus the final pH levelled off at almost the same value as the original. The duration of the dormancy period could be adjusted by the variation of the chemical fuel concentration. As shown in Figure 2 a, higher urea concentrations resulted in faster recovery under otherwise constant conditions. In the absence of urea, the pH remained constant with respect to time. This relationship between the reaction rate and substrate concentration follows Michaelis–Menten kinetics, which rationalizes an asymptotic increase of the enzymatic reaction rate with substrate concentration.8, 11 The reversible fast pH decrease and slow pH increase was expected to occur by the repeated addition of chemical fuel (Figure 2 b).
Figure 2.
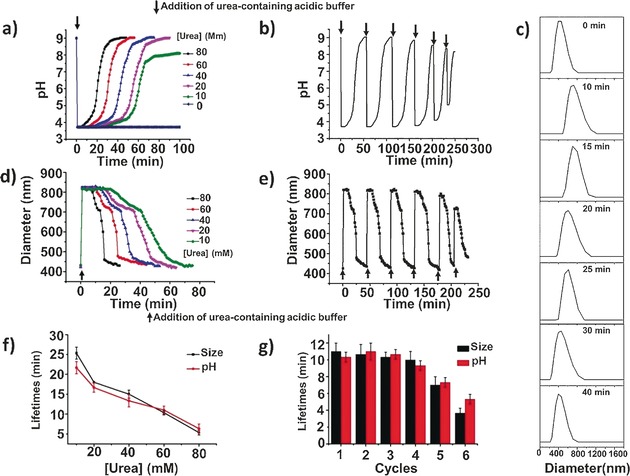
a) Influence of the substrate concentration on the pH changes of the microgel solution. b) Reversible pH changes of the microgel in time following repeated additions of chemical fuels (60 mm urea). c) Size distribution changes of the microgel after the addition of chemical fuel. d) Influence of the substrate concentration on the size changes of the microgel. e) Reversible diameter changes of the microgel in time following repeated additions of chemical fuels (60 mm urea). f) Average transient periods of pH and size as a function of the urea concentration. g) Average transient periods of pH and size within six cycles (60 mm urea). Experimental conditions: 0.6 mg mL−1 urease, 9 mm CA/Na3C (9:1) buffer.
As a control experiment, urea‐containing acidic buffer was also added into the microgel with the enzyme in the bulk solution instead of chemically conjugated with the microgel (Supporting Information, Figure S7). In this case, the enzyme‐regulated pH changes were also obtained except that it took less time to return to the starting level; after four cycles the reversibility was strongly affected compared to the hybrid microgel, which showed reversible behavior for at least six cycles. These results can be interpreted as that the diffusion of the substrate into the microgel is a non‐negligible process. Furthermore, the incorporation of the enzyme inside the microgel can have a stabilizing effect on urease activity, leading to good reversibility.
Next, we investigated if the pH changes can yield a hybrid microgel with switchable sizes. For this purpose, samples were taken out after the injection of chemical fuels to perform fast DLS measurements. Size distribution analysis reveals a fast size increase with an induction period and subsequent slow size recovery (Figure 2 c). As seen in Figure 2 d, in the first section, a size jump from 420 nm to 820 nm was observed. This was reversed gradually over a different time period, which can be changed by varying concentrations of the chemical fuel. Increasing the concentration of urea led to a fast size recovery with a short time delay because the rate of formation of ammonia is greatly enhanced by the enzymatic reaction. The generated swollen microgel is only the transient state, and the system is able to revert back to the initial contraction state; the microgel with an enzyme‐induced “breathing” feature thus follows a self‐regulated mechanism, corresponding to the dynamic pH changes of the system. More interestingly, this “breathing” cycle could be performed at least six times with the same sample, adding new batches of urea‐containing acidic buffer (Figure 2 e). More importantly, the urease activity assays show that the urease in the microgel maintained around 90 % activity after six cycles, demonstrating the high durability of the system (Supporting Information, Figure S8).
The transient period is highly dependent upon the rate of production of ammonia and increases with decreasing concentrations of chemical fuels. Figure 2 f clearly shows that the lifetimes of the temporal “breathing” state of the microgel can be regulated from 5 to 25 min by altering the urea concentration from 80 to 10 mm. Average induction periods within six cycles are depicted in Figure 2 g. Clearly, pH and size are almost synchronously changed and they have close transient time scales. It needs to be pointed out that the efficiency of the enzymatic reaction in adjusting the “breathing” behavior of the microgel decreased after six cycles. One possible reason is that the repeated addition of chemical fuels leads to the accumulation of waste products in the closed reaction system.
Finally, the dynamic behavior of the microgels was monitored by using fluorescence spectroscopy. At high pH, fluorescein exhibits strong fluorescence because of the ring‐opening of the lactone moiety, while gradual fluorescence quenching is observed when the pH decreases, which is attributed to ring‐closing of the lactone (Supporting Information, Figure S9). After formation, the microgel showed strong fluorescence with an emission maximum at 515 nm (Supporting Information, Figure S10), indicating that the formation of the microgel did not affect the fluorescence of the fluorescent dye. Kinetic fluorescence data demonstrated a rapid decay in fluorescence intensity upon the addition of urea‐containing acidic buffer, and a transient plateau was observed. Similar to the results obtained for the size switch in time, a gradual signal increase occurred, reaching a maximum with a rate that depended on the urea concentration (Figure 3 a). With an increase in the urea concentration, the ammonia production rate increased, and the lifetimes of the transient state decreased (Figure 3 b). Further addition of chemical fuels gave rise to a similar profile, and several cycles could be completed with controllable lifetimes (Figure 3 c,d).
Figure 3.
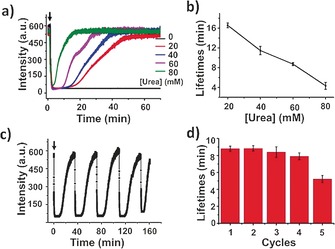
a) Influence of the substrate concentration on the changes in fluorescence of the microgel. b) Average transient periods of intensity as a function of the urea concentration. c) Reversible fluorescence changes in time following repeated additions of chemical fuels (60 mm urea). d) Average transient periods of intensity within five cycles (60 mm urea). Experimental conditions: 0.6 mg mL−1 urease, 9 mm CA/Na3C (9:1) buffer.
Laser scanning confocal microscopy studies allow for the direct visualization of fluorescence changes. At high pH, the microgel showed a strong fluorescent signal, while at low pH, quenched fluorescence was observed (Supporting Information, Figure S11), which is in accordance with the fluorescence data. The fluorescent objects disappeared immediately after addition of urea‐containing acidic buffer, after which they gradually reappeared in number (Supporting Information, Figure S12). These results clearly show the functioning of a “breathing” microgel accompanied by an on/off switching of a fluorescence signal with a lifetime that can be controlled by regulating the enzymatic reaction.
In conclusion, we have developed a self‐regulated, biocatalytically controlled and time programmed “breathing” hybrid microgel that requires a chemical fuel to maintain the non‐equilibrium state. The size switch and fluorescence on/off adjustment of the microgel is driven by urease‐catalyzed base generation in combination with urea‐containing acidic buffer that acts as the chemical fuel. Importantly, the microgel could be refueled several times upon the repeated addition of substrate. This hybrid microgel offers a step towards the construction of artificial systems and autonomous soft materials, and it is expected to be useful in adaptive biomedical applications. Additionally, limited cycle performance because of the accumulation of products is still an issue, and new approaches such as the introduction of microfluidics will be developed in future work.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to acknowledge the Dutch Science Foundation (NWO, VICI grant), the ERC Advanced grant Artisym 694120 and the Dutch Ministry of Education, Culture and Science (Gravitation program 024.001.035) for funding.
H. Che, B. C. Buddingh', J. C. M. van Hest, Angew. Chem. Int. Ed. 2017, 56, 12581.
References
- 1.
- 1a. Stoffelen C., Voskuhl J., Jonkheijm P., Huskens J., Angew. Chem. Int. Ed. 2014, 53, 3400–3404; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3468–3472; [Google Scholar]
- 1b. Chen L., Morris K., Laybourn A., Elias D., Hicks M. R., Rodger A., Serpell L., Adams D. J., Langmuir 2010, 26, 5232–5242; [DOI] [PubMed] [Google Scholar]
- 1c. Boal A. K., Ilhan F., DeRouchey J. E., Thurn-Albrecht T., Russell T. P., Rotello V. M., Nature 2000, 404, 746–748; [DOI] [PubMed] [Google Scholar]
- 1d. Davis J. T., Spada G. P., Chem. Soc. Rev. 2007, 36, 296–313; [DOI] [PubMed] [Google Scholar]
- 1e. Hirschberg J. K., Brunsveld L., Ramzi A., Vekemans J. A., Sijbesma R. P., Meijer E., Nature 2000, 407, 167–170. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Gohy J.-F., Zhao Y., Chem. Soc. Rev. 2013, 42, 7117–7129; [DOI] [PubMed] [Google Scholar]
- 2b. Hu J., Zhang G., Liu S., Chem. Soc. Rev. 2012, 41, 5933–5949; [DOI] [PubMed] [Google Scholar]
- 2c. Che H., van Hest J. C., J. Mater. Chem. B 2016, 4, 4632–4647; [DOI] [PubMed] [Google Scholar]
- 2d. Yan Q., Zhao Y., Chem. Commun. 2014, 50, 11631–11641; [DOI] [PubMed] [Google Scholar]
- 2e. De las Heras Alarcón C., Pennadam S., Alexander C., Chem. Soc. Rev. 2005, 34, 276–285; [DOI] [PubMed] [Google Scholar]
- 2f. Smith A. E., Xu X., McCormick C. L., Prog. Polym. Sci. 2010, 35, 45–93. [Google Scholar]
- 3.
- 3a. Mattia E., Otto S., Nat. Nanotechnol. 2015, 10, 111–119; [DOI] [PubMed] [Google Scholar]
- 3b. Karsenti E., Nat. Rev. Mol. Cell Biol. 2008, 9, 255–262. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Whitesides G. M., Ismagilov R. F., Science 1999, 284, 89–92; [DOI] [PubMed] [Google Scholar]
- 4b. Desai A., Mitchison T. J., Annu. Rev. Cell Dev. Biol. 1997, 13, 83–117; [DOI] [PubMed] [Google Scholar]
- 4c. Fletcher S. P., Dumur F., Pollard M. M., Feringa B. L., Science 2005, 310, 80–82. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Pappas C. G., Sasselli I. R., Ulijn R. V., Angew. Chem. Int. Ed. 2015, 54, 8119–8123; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8237–8241; [Google Scholar]
- 5b. Boekhoven J., Hendriksen W. E., Koper G. J., Eelkema R., van Esch J. H., Science 2015, 349, 1075–1079; [DOI] [PubMed] [Google Scholar]
- 5c. Leira-Iglesias J., Sorrenti A., Sato A., Dunne P. A., Hermans T. M., Chem. Commun. 2016, 52, 9009–9012; [DOI] [PubMed] [Google Scholar]
- 5d. Dhiman S., Jain A., George S. J., Angew. Chem. Int. Ed. 2017, 56, 1329–1333; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1349–1353; [Google Scholar]
- 5e. Krishna Kumar R., Harniman R. L., Patil A. J., Mann S., Chem. Sci. 2016, 7, 5879–5887; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5f. Debnath S., Roy S., Ulijn R. V., J. Am. Chem. Soc. 2013, 135, 16789–16792; [DOI] [PubMed] [Google Scholar]
- 5g. Postma S. G., Vialshin I. N., Gerritsen C. Y., Bao M., Huck W. T., Angew. Chem. Int. Ed. 2017, 56, 1794–1798; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1820–1824; [Google Scholar]
- 5h. Maiti S., Fortunati I., Ferrante C., Scrimin P., Prins L. J., Nat. Chem. 2016, 8, 725–731; [DOI] [PubMed] [Google Scholar]
- 5i. Boekhoven J., Brizard A. M., Kowlgi K. N., Koper G. J., Eelkema R., van Esch J. H., Angew. Chem. Int. Ed. 2010, 49, 4825–4828; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 4935–4938. [Google Scholar]
- 6.
- 6a. Heinen L., Walther A., Soft Matter 2015, 11, 7857–7866; [DOI] [PubMed] [Google Scholar]
- 6b.R. Merindol, A. Walther, Chem. Soc. Rev 2017, DOI: https://doi.org/10.1039/C6CS00738D.
- 7. Heuser T., Steppert A.-K., Molano Lopez C., Zhu B., Walther A., Nano Lett. 2015, 15, 2213–2219. [DOI] [PubMed] [Google Scholar]
- 8. Heuser T., Weyandt E., Walther A., Angew. Chem. Int. Ed. 2015, 54, 13258–13262; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13456–13460. [Google Scholar]
- 9. Heuser T., Merindol R., Loescher S., Klaus A., Walther A., Adv. Mater. 2017, 29, 1606842–1606848. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Hu G., Pojman J. A., Scott S. K., Wrobel M. M., Taylor A. F., J. Phys. Chem. B 2010, 114, 14059–14063; [DOI] [PubMed] [Google Scholar]
- 10b. Muzika F., Bansagi T., Schreiber I., Schreiberova L., Taylor A., Chem. Commun. 2014, 50, 11107–11109. [DOI] [PubMed] [Google Scholar]
- 11. Krajewska B., J. Mol. Catal. B 2009, 59, 9–21. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary