

Summary
There is increasing interest in the heritable bacteria of invertebrate vectors of disease as they present novel targets for control initiatives. Previous studies on biting midges (Culicoides spp.), known to transmit several RNA viruses of veterinary importance, have revealed infections with the endosymbiotic bacteria, Wolbachia and Cardinium. However, rickettsial symbionts in these vectors are underexplored. Here, we present the genome of a previously uncharacterized Rickettsia endosymbiont from Culicoides newsteadi (RiCNE). This genome presents unique features potentially associated with host invasion and adaptation, including genes for the complete non‐oxidative phase of the pentose phosphate pathway, and others predicted to mediate lipopolysaccharides and cell wall modification. Screening of 414 Culicoides individuals from 29 Palearctic or Afrotropical species revealed that Rickettsia represent a widespread but previously overlooked association, reaching high frequencies in midge populations and present in 38% of the species tested. Sequence typing clusters the Rickettsia within the Torix group of the genus, a group known to infect several aquatic and hematophagous taxa. FISH analysis indicated the presence of Rickettsia bacteria in ovary tissue, indicating their maternal inheritance. Given the importance of biting midges as vectors, a key area of future research is to establish the impact of this endosymbiont on vector competence.
Introduction
Heritable bacteria represent an important component of the biology of many arthropods. Carried by over half of all species (Weinert et al., 2015), many vertically transmitted microbes contribute to host function. This contribution is most commonly through specific services, such as nutrient provisioning or protection (Oliver et al., 2003; Douglas, 2009; Jaenike et al., 2010). Conversely, their maternal‐inheritance has led symbionts to favour production of daughters by their host, leading to the evolution of systems biasing offspring sex ratio towards females (reproductive parasitisms) (Hurst and Frost, 2015). The strength of symbiont impact on individual biology, combined with the high frequency with which arthropod species are infected with symbionts, has led to intense study. This study has the complementary motivations of understanding the dynamics and ecological impact of symbionts (Ferrari and Vavre, 2011) and applying this knowledge to modify the biological properties of target species (Iturbe‐Ormaetxe et al., 2011).
Particular attention has been focused on symbiont/host interactions in vector species. Through the induction of cytoplasmic incompatibility, the endosymbiont Wolbachia prevents the formation of viable progeny between infected males and uninfected females in various dipterans including Drosophila spp. and Aedes spp. (Werren et al., 2008). With respect to the latter, not only can this incompatibility lead to vector population suppression but, through unknown mechanisms, a strong RNA virus resistance phenotype (Moreira et al., 2009; Bian et al., 2010; Blagrove et al., 2012; Van den Hurk et al., 2012). Furthermore, experimental evidences show that both Wolbachia and another proteobacteria, Wigglesworthia, can act as obligate (required) symbionts, provisioning blood sucking vector hosts with B vitamins that are lacking in a blood‐diet (reviewed in Rio et al., 2016). This provisioning has evolved independently in bed bugs (Cimex lectularius) (Nikoh et al., 2014) and tsetse flies (Glossina sp.) (Akman et al., 2002; Snyder et al., 2010; Rio et al., 2012). Additional genomic surveys suggest that other proteobacterial symbionts including Coxiella are involved in metabolic homeostasis (Zhong et al., 2007; Manzano‐Marin et al., 2015; Smith et al., 2015). As such, these symbioses can have profound effects on the biology, ecology and evolutionary dynamics of vector–pathogen interactions.
Rickettsia (class: Alphaproteobacteria; order: Rickettsiales) symbionts are obligate intracellular bacteria most notable for containing species pathogenic to vertebrates, such as Rickettsia prowazekii, the causative agent of louse‐borne Typhus fever, Rickettsia rickettsii (Rocky Mountain spotted fever) and Rickettsia conorii (Boutonneuse or Mediterranean spotted fever). Despite this, vertebrate disease‐causing Rickettsia are atypical of the genus as a whole (Perlman et al., 2006; Weinert et al., 2009a) and many Rickettsia are maintained without infectious transfer. Members are known to induce a variety of reproductive manipulations, including male killing in ladybird beetles (Adalia bipunctata) (Werren et al., 1994; Hurst et al., 1999; Majerus et al., 1999) and parthenogenesis induction in parasitoid wasps (Pnigalio soemius; Neochrysocharis formosa) (Hagimori et al., 2006; Giorgini et al., 2010). Rickettsia symbiont infection can also be protective, enhancing resistance of aphids (Acyrthosiphon pisum) to fungal attack and whiteflies (Bemisia tabaci) to bacterial challenge (Łukasik et al., 2013; Hendry et al., 2014). Of significance to the study of vectors, Rickettsia are also known to increase the competence of Bemisia whiteflies for the transmission of tomato leaf curl virus (Kliot et al., 2014). Members of the genus can also be insect‐vectored plant pathogens in their own right, for example, underlying papaya bunchy top disease (Luis‐Pantoja et al., 2015). As such, symbiosis with Rickettsia is biologically important at the individual and population levels and both as vectored disease agents in themselves and as a symbiont facilitating the spread of other diseases.
In this work, we uncovered and examined a symbiotic association between Rickettsia and Culicoides biting midges which has been previously overlooked. Worldwide, biting midges of the genus Culicoides (Diptera: Ceratopogonidae) are known to transmit more than 50 arboviruses as well as some nematode and protozoan parasites. Midge‐vectored pathogens that threaten livestock and wildlife include bluetongue virus (BTV), Schmallenberg virus, African horse sickness virus, epizootic hemorrhagic disease virus, equine encephalosis virus and Akabane virus (Mellor et al., 2000). In South America, Culicoides midges spread Oropouche virus to humans. Previous studies of Culicoides symbionts have screened extensively for Cardinium and Wolbachia infections (Nakamura et al., 2009; Morag et al., 2012; Lewis et al., 2014; Mee et al., 2015) but failed to report presence of Rickettsia. However, a 16S metagenomic screening project in Culicoides sonorensis gut samples revealed amplicons allied to Rickettsia (Campbell et al., 2004), albeit with no phylogenetic or population‐based information. Complementary to this, when we performed a shallow whole‐genome sequencing of the Cardinium‐uninfected midge Culicoides newsteadii N5, we recovered a near complete genome of an uncharacterized, divergent Rickettsia species related to the Torix (also known as Limoniae) group of Rickettsia.
In this study, we first report on the genomic properties of the Rickettsia endosymbiont of C. newsteadi (RiCNE), which represents the first Rickettsia genome from the Torix group. We then examine the distribution and prevalence of Rickettsia in a wide‐range of Culicoides species from both Palearctic and Afrotropical regions and resolve the relationship of the Culicoides Rickettsia based on five gene sequences. We conclude that Rickettsia infection is common in Culicoides and raise the hypothesis that Torix group Rickettsia may be a dominant taxon in invertebrates with aquatic stages. Our genome data provide no support for a symbiont role in vitamin homeostasis but reveal unique features potentially related to the ecological attributes of this Rickettsia group.
Results
Serendipitous discovery of a Rickettsia symbiont during the shallow sequence of its Culicoides midge host
Culicoides newsteadi N5 is morphologically and genetically similar to C. punctatus, which has been previously reported to be infected with Cardinium symbiotic bacteria (Lewis et al., 2014). During a shallow illumina whole genome sequencing of C. newsteadi N5, we identified the presence of several contigs with homology to Rickettsia bacteria (Supporting Information Fig. S1).
General features and genetic repertoire of the RiCNE draft genome
The final assembly of the RiCNE draft genome consists of 193 scaffolds > 500 bp (N50 = 12.7 kb, largest scaffold = 71.2 kb) comprising a total size of 1,456,176 bp with an average GC content of 33% and an average depth of coverage 76× (Fig. 1B). Genome annotation identified 1352 protein coding sequences (CDSs) with an average length of 858 bp, a full set of rRNA genes (one each of 16S, 5S and 23S) and 35 tRNA genes accounting for a coding density of circa 80%. The proportion of missing BUSCO marker genes in RiCNE draft assembly fell well within the range of the previously completely sequenced Rickettsia genomes [BUSCO score = C: 93.2% (S: 93.2%, D: 0%), F: 0%, M: 6.8%, n: 148] (Supporting Information Fig. S2). These results suggest that the RiCNE draft assembly represents a nearly complete genome. From the 1352 predicted CDSs, 962 (∼ 71%) CDSs were annotated with putative functions, while 390 (∼ 29%) CDSs were annotated as hypothetical proteins. Additional searches for Pfam domains revealed that 122 of the hypothetical proteins had putative functional domains (Supporting Information Table S1).
Figure 1.
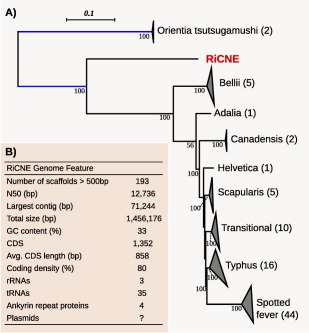
The Rickettsia endosymbiont of C. newsteadi N5 (RiCNE).
A. Phylogenomic placement of RiCNE was inferred using maximum likelihood (RaxML, model: Lag + G + I) from the concatenated protein alignments of 189 single copy ortholog genes. Support values are based on 100 rapid bootstrap replicates. Major Rickettsia groups have been collapsed for visualisation purposes and names are according to (Murray et al., 2016). Numbers in parenthesis represent the number of genomes used for the analysis (Table S2). The blue branches have been reduced 50% for visualisation purposes. The full phylogenetic tree is shown in Supporting Information Fig. S4.
B. RiCNE draft genome features. [Color figure can be viewed at wileyonlinelibrary.com]
Phylogeny
The phylogenetic relationships of RiCNE relative to other Rickettsiaceae were initially estimated from a set of 189 single copy panorthologs identified among 84 complete or draft Rickettsia genomes and its sister genus Orientia (Supporting Information Table S2). Maximum‐likelihood phylogeny placed RiCNE as a sister lineage of all other Rickettsia with strong support (bootstrap support = 100%) (Fig. 1A). Additionally, we performed a phylogenetic analysis using the conserved 16S rRNA which allowed us to include representative sequences from the Hydra and Torix groups of Rickettsia (Weinert et al., 2009a). Our analyses clearly positioned RiCNE sequence within the Torix group (Fig. 2) previously identified in leeches (Kikuchi et al., 2002), amoebae (Dyková et al., 2003) and several arthropod orders including Araneae, Diptera, Coleoptera, Psocoptera, Hemiptera and Hymenoptera (Goodacre et al., 2006; Perotti et al., 2006; Reeves et al., 2008; Küchler et al., 2009; Zouache et al., 2009; Machtelinckx et al., 2012; Weinert et al., 2015). The RiCNE genome represents the first to be sequenced from this group.
Figure 2.
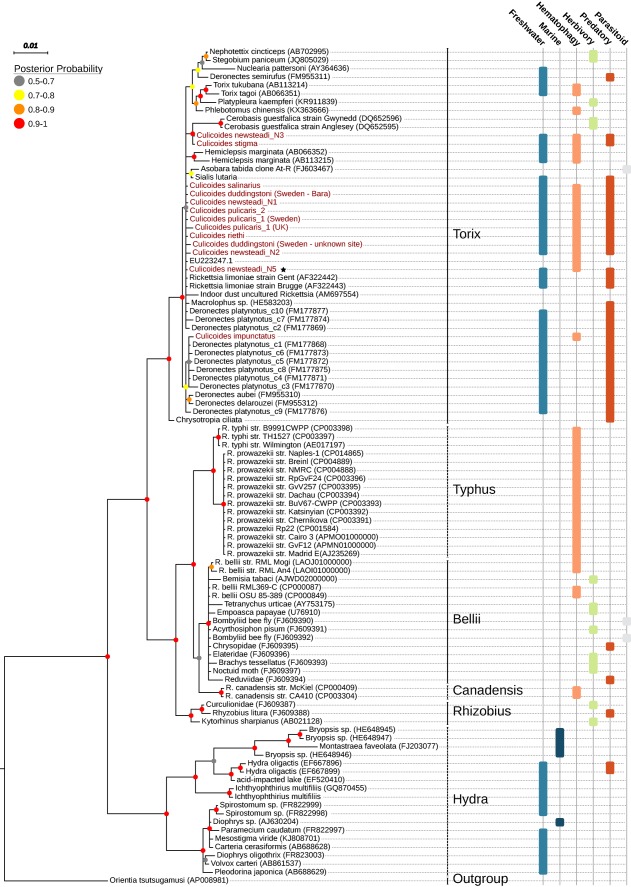
Phylogenetic placement of the Rickettsia symbionts of Culicoides midges based on the 16S rRNA gene.
Previously characterized Rickettsia groups including the basal group of Hydra are also presented. Host names are used in the absence of official Rickettsia species name. Tree topology and posterior probabilities (shown as color‐coded circles on each node) were inferred using Bayesian analysis in MrBayes under GTR + G + I (for details see the ‘Experimental procedures’ section). Sequence accession numbers are shown in brackets. A black star depicts the midges Rickettsia, which draft genome is presented in this study. The habitats and lifestyles of the host (or specific life stages of the host) are given to the right of the phylogeny. [Color figure can be viewed at wileyonlinelibrary.com]
Genome content
We compared the content of the RiCNE genome with other Rickettsiaceae (Supporting Information Table S2) to identify unique features potentially related to the biology of RiCNE and the Torix group Rickettsia. Overall, RiCNE presents typical features and genetic repertoire of a Rickettsia genome including the presence of a P‐like type IV secretion system (P‐T4SS) which is highly conserved among Rickettsiales (Supporting Information Fig. S3 and Table S3). The vir genes on the RiCNE genome are arranged into three major clusters (scaffold 1: virB3, virB4 and virB6; scaffold 4: virB8‐B11 and virD4; scaffold 10: two in tandem paralogs of the virB2 gene and a virB4 paralog) with additional paralogs of the virB8 and virB9 on scaffold 47. This scattered arrangement of the vir genes is typical of Rickettsia genomes (Gillespie et al., 2009). Additionally, the RiCNE genome encodes a tra conjugative DNA‐transfer element, which has been previously reported in several Rickettsia genomes (Ogata et al., 2006; Weinert et al., 2009b). RiCNE tra cluster is split into two scaffolds (scaffolds 5 and 34). The first unit contains the ‘F‐like’ T4SS (tra) genes including traE, traK, traB, traC, traW, traU, trbC, traN, traF, traH and traG_N (Supporting Information Fig. S3 and Table S3). The second unit contains the ‘Ti‐like’ genes traATi and traDTi previously identified in the Ti plasmid of Agrobacterium tumefaciens (Wood et al., 2001). Although we could not identify a traV homolog (a core glycoprotein, component of the pilus assembly structure), a hypothetical protein encoded by a gene located between the traB and traC homologs presented low similarities with TraV homologs from the Rickettsia endosymbiont of Ixodes scapularis and may represent a functional equivalent. Notably, the two scaffolds containing the conjugation genes were consistently represented at 2–3 times higher than average coverage (Supporting Information Fig. S3). However, this does not exceed the even higher coverage associated with repetitive loci such as insertion elements. This suggests that the conjugation system genes are likely encoded as multiple copies on the chromosome, as previously reported for other Rickettsia and Orientia (Cho et al., 2007; Gillespie et al., 2012). However, the presence of low‐copy‐number plasmids cannot be ruled out.
A shared feature among Rickettsia is the presence of several gene families potentially involved in environmental adaptation. These include multiple paralogous genes encoding the bifunctional (p)ppGpp synthase/hydrolase SpoT/RelA, a key component of the bacterial stringent response, several genes related to toxin‐antitoxin systems (see below) as well as genes encoding multidrug/efflux transporters. In the RiCNE genome, we identified 18 CDSs with homology to spoT paralogs shared with other Rickettsia genomes. Five of them were found at the ends of the scaffolds and may represent incomplete fragments, while another two truncated CDSs occurred in tandem and may represent a pseudogene.
A total of 187 of the 1352 predicted CDSs (∼ 14%) were unique to the RiCNE genome. Of these 187 CDSs, 43 CDSs were predicted to form hypothetical proteins of less than 70 amino acids and may therefore represent annotation artefacts or pseudogenised gene fragments. Forty of the remaining 144 RiCNE‐specific CDSs could be ascribed a putative function, either by significant matches in the NR database or by predicted Pfam domains (Supporting Information Table S4). Amongst these were genes putatively associated with host invasion and host–microbe interactions. These include a homolog of a putative exopolysaccharide synthesis (exoD) gene (RiCNE_02810), two paralogs of a putative lipid A 3‐O‐deacylase (pagL) gene (RiCNE_02710, RiCNE_13110), as well as a gene coding for a carbonic anhydrase (RiCNE_13200) and a gene coding for a leucine‐rich repeat protein (RiCNE_13500) (Supporting Information Table S5). Moreover, we identified four genes encoding cell wall biogenesis and modification proteins including UDP‐galactopyranose mutase (RiCNE_08860), N‐acetylmuramoyl‐l‐alanine amidase (RiCNE_08880), a putative Glycosyl transferases (RiCNE_08940) and a putative d‐alanyl‐d‐alanine carboxypeptidase (RiCNE_06020). Multiple genes coding for toxin–antitoxin systems (13 toxins and 9 antitoxins) were also detected. Of these, two CDSs encoding for a toxin (RiCNE_11100) and an antitoxin (RiCNE_07550) were specific to RiCNE. Finally, among the 14 multidrug/efflux transporters identified, two (RiCNE_09880 and RiCNE_13240) are specific to RiCNE.
The metabolic and biosynthetic potential of RiCNE
Overall, the metabolic capacities of the RiCNE genome are similar to other Rickettsia genomes. Like other Rickettsia, it is missing several central aspects of metabolism such as the glycolysis and gluconeogenesis pathways (Fig. 3). Likewise, pathways for nucleotide and amino acid biosynthesis are absent or defective. Instead, we identified genes encoding for putative transporters including five ATP/ADP translocase homologs, two amino acid permeases and several putative transporters belonging to major facilitator super‐family (MFS), suggesting that RiCNE likely relies on the exploitation of host resources.
Figure 3.
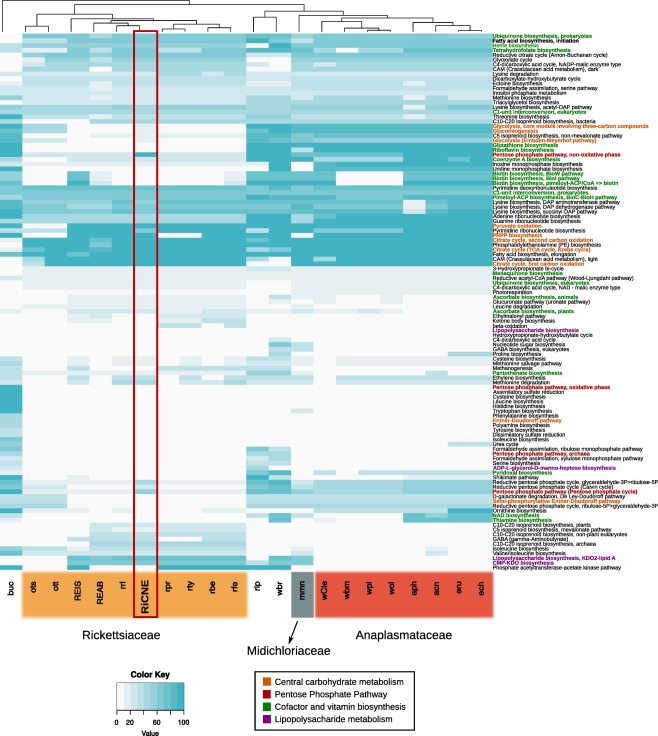
Assessment of metabolic potential of RiCNE genome (highlighted) and comparison to other members of the order Rickettsialles as well as representative known nutritional mutualists (primary symbionts) Buchnera, Riesia and Wigglesworthia.
Color gradient represents the module completion ratio (MCR) values calculated by MAPLE‐2.1.0. buc: Buchnera aphidicola APS, ots: Orientia tsutsugamushi Boryong, ott: Orientia tsutsugamushi Ikeda, REIS: Rickettsia endosymbiont of Ixodes scapularis, REAB: Rickettsia endosymbiont of Adalia bipunctata, rri: Rickettsia rickettsii Sheila Smith, RiCNE: Rickettsia endosymbiont of Culicoides newsteadi, rpr: Rickettsia prowazekii Madrid E, rty: Rickettsia typhi Wilmington, rbe: Rickettsia bellii RML369‐C, rfe: Rickettsia felis, rip: Candidatus Riesia pediculicola, wbr: Wigglesworthia glossinidia brevipalpis, mmm: Candidatus Midichloria mitochondrii, wcl: Wolbachia wCle, wbm: Wolbachia wBm, wpi: Wolbachia wPip, wol: Wolbachia wMel, aph: Anaplasma phagocytophilum HZ, acn: Anaplasma centrale, eru: Ehrlichia ruminantium Welgevonden and ech: Ehrlichia chaffeensis Arkansas. [Color figure can be viewed at wileyonlinelibrary.com]
A marked difference between RiCNE genome and all other sequenced Rickettsiaceae is that RiCNE encodes the complete set of proteins involved in the non‐oxidative phase of the pentose phosphate pathway (PPP), including transketolase, transaldolase, ribulose‐phosphate 3‐epimerase and a ribose 5‐phosphate isomerase B (RiCNE_05410, RiCNE_04320, RiCNE_00410 and RiCNE_09330, respectively) (Fig. 3). The oxidative phase is completely absent, as for other Rickettsia. Only one gene of the PPP (coding for the ribose 5‐phosphate isomerase B) has been detected in most other sequenced Rickettsiaceae including Orientia tsutsugamusi. To better understand the evolution of the non‐oxidative PPP branch in Rickettsia, we search the unpublished genome of the Rickettsia endosymbiont of Ichthyophirius multifilis for the presence of the same four key proteins. This rickettsial endosymbiont is affiliated to the basal Hydra group of Rickettsia (Weinert et al., 2009a,b) (Fig. 2) commonly found among diverse ciliates and recently provided with the unique genus name Megaira (Schrallhammer et al., 2013). This genome was obtained by sequencing its ciliate host (Sun et al., 2009) and was kindly provided by Prof. R.S. Coyne, Dr T. Doak and Dr H. Suzuki. Notably, all four proteins were encoded in this Rickettsia endosymbiont genome displaying moderate amino‐acid sequence similarity with the RiCNE homologs (rpe: 58.6%, tal: 58.1%, tkt: 52.2% and rpiB: 63.6%). We additionally conducted protein similarity searches against the NR database (NCBI) using the three sequences that did not have any homologs among the arthropod‐associated Rickettsia. The best BLAST hits for all three sequences fell within the α‐proteobacteria (RiCNE_05410 shared ∼ 50% amino acid identity with Ehrlichia homologs, RiCNE_04320 shared ∼ 59% identity with Sulfitobacter sp. EhC04 and RiCNE_00410 shared ∼ 58% identity with an uncultured α‐proteobacterium). Additional phylogenetic analyses of the individual PPP protein sequences clearly cluster the RiCNE sequences within the alpha‐proteobacteria and the Rickettsiales, and partial PPPs were detected in other members of the Rickettsiales including Wolbachia and Midichloria (Fig. 4). Finally, we also noticed that RiCNE_05410 gene contains an in‐frame insertion of a Rickettsia Palindromic Element (RPE) between the positions 1560 and 1666.
Figure 4.
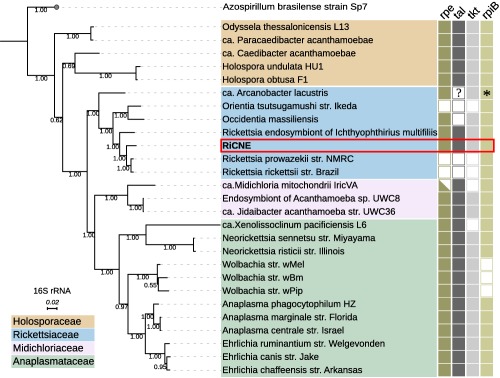
Loss of the non‐oxidative stage of the pentose phosphate pathway (PPP) among Rickettsiales.
Presence and absence of the four key enzymes (rpe: ribulose‐phosphate 3‐epimerase, tla: transaldolase, tkt: transketolase and rpi: ribose 5‐phosphate isomerase) are shown across the 16S rRNA phylogeny of representative members from the Rickettsiales as filled and empty rectangles, respectively. Completeness of the pathway in RiCNE is highlighted. Half‐filled rectangle indicates a truncated rpe gene homologue. The question mark indicates missing data rather than loss of tla homolog from ca. Arcanobacter lacustris since its genome is incomplete (completeness = 48%; Martijn et al., 2015). The asterisk indicates a possible gene fusion event in ca. Arcanobacter lacustris between the rpiB gene and the upstream gene coding for a NAD(P)H: quinone oxireductase. 16S rRNA tree constructions were performed with MrBayes under GTR + G + I. A phylogenetic analysis based on the individual protein sequences is presented in Supporting Information Fig. S5. [Color figure can be viewed at wileyonlinelibrary.com]
Inspection of predicted biosynthetic pathways for cofactors and B vitamin synthesis systems revealed no major differences from the rest of the Rickettsiaceae (Fig. 3). RiCNE features a reduced set of genes required for folate (vitamin B9) biosynthesis, with the gene for dihydrofolate synthase (folC) absent. The pathways required for the biosynthesis of biotin (vitamin B7), riboflavin (vitamin B2), thiamin (vitamin B1), pyridoxine (vitamin B6), nicotinate (vitamin B3) and pantothenate (vitamin B5) are completely absent. Moreover, the cofactor biosynthetic capacity of RiCNE appears to be limited, with only partial pathways for heme and ubiquinone biosynthesis.
Prevalence of Rickettsia in biting midges
Screening of field collected midge specimens revealed Rickettsia infections in 155 of 414 (37%) individuals and 11 of 29 (38%) Culicoides species sampled (Table 1 and Supporting Information Table S6). Rickettsia‐positive species of biting midge were recorded across Culicoides subgenera. Infection was identified across the subgenera Beltranmyia (1/3 species), Culicoides (7/11 species), Monoculicoides (2/2) and Oecacta (1/4 species) [as determined by Borkent (2016)]. There was no apparent host sex bias in the presence of Rickettsia for either Culicoides pulicaris haplotype 1 (UK) (Fisher's two‐tailed test; p = 1) or Culicoides impunctatus (Fisher's two‐tailed test; p = 0.36), the only infected species with both host sexes available to compare.
Table 1.
omp conventional PCR assay results for Rickettsia‐positive Culicoides sp. under study, given by subgenus, species, location, date and sex.
| Proportion of Rickettsia‐positive samples (n) [95% confidence interval] | |||||
|---|---|---|---|---|---|
| Subgenus | Culicoides species | Location | Year of collection | Females | Males |
| Beltranmyia | C. salinarius | Unknown site, Sweden | 2009 | 1 (2) [0.2–1] | |
| Culicoides | C. impunctatus | Torsås, Sweden | 2008 | 0.3 (20) [0.13–0.54] | |
| Bala, UK | 2012 | 0.81 (17) [0.5–0.92] | 0.5 (14) [0.27–0.73] | ||
| Kielder, UK | 2016 | 0.75 (16) [0.47–0.92] | 0.86 (7) [0.42–0.99] | ||
| C. newsteadi N1 a , * | Site 2, Corsica | 2015 | 0.5 (2) [0.1–0.91] | ||
| C. newsteadi N2 a , * | Site 2, Corsica | 2015 | 1 (2) [0.2–1] | ||
| C. newsteadi N3 b , * | Unknown site, Sweden | 2008–2010 | 1 (6) [0.52–1] | ||
| C. newsteadi N5 b , * | Wirral, UK | 2015 | 1 (13) [0.72–1] | ||
| C. pulicaris * (haplotype 1) | Canterbury, UK | 2014 | 1 (2) [0.2–1] | ||
| Hereford, UK | 2014 | 1 (1) [0.05–1] | |||
| Luton, UK | 2014 | 1 (1) [0.05–1] | |||
| Unknown site, Sweden | 2008–2010 | 1 (6) [0.52–1] | |||
| Wirral, UK | 2015 | 1 (32) [0.87–1] | |||
| Wolverhampton, UK | 2013 | 1 (11) [0.68–1] | 1 (4) [0.4–1] | ||
| Worcester, UK | 2014 | 1 (6) [0.52–1] | |||
| C. pulicaris * (haplotype 2) | Site 2, Corsica | 2015 | 1 (13) [0.72–1] | ||
| Monoculicoides | C. riethi | Ljungbyholm, Sweden | 2010 | 1 (1) [0.05–1] | |
| C. stigma | Unknown site, Sweden | 2008 | 1 (3) [0.31–1] | ||
| Oecacta | C. duddingstoni | Bara, Sweden | 2008 | 1 (4) [0.4–1] | |
| Unknown site, Sweden | 2008–2010 | 1 (3) [0.31–1] | |||
Rickettsia was found at fixation in all individuals in 16 of the 20 positive populations screened, being at low or intermediate prevalence in the remaining 4 (1 C. newsteadi N1 population and 3 C. impunctatus populations). Where multiple samples of particular species were tested, there was no significant difference in the fraction infected (C. impunctatus populations from Bala vs. Kielder in the UK, N1 = 31, N2 = 23, Fisher's two‐tailed test; p = 0.37). Mitochondrial DNA barcoding of infected (KY765353) and uninfected (KY765354) individuals of C. impunctatus confirmed these individuals shared a barcode, consistent with infection showing within‐species polymorphism.
Rickettsia diversity in Culicoides
The level of 16S rRNA divergence within the Culicoides Rickettsia was low (0.9% segregating sites, Pi = 0.002) (Supporting Information Table S7), such that the strains would all be considered as belonging to a single species in classic bacteriological nomenclature (Stackebrandt and Goebel, 1994). To resolve patterns of relatedness more fully, we obtained the sequence of three further housekeeping loci as well as the omp gene, for each of the specimens. Housekeeping gene PCR amplification was successful for 13 typings; The C. pulicaris strain (I) from the UK failed to amplify with the COX primers after more than one attempt. An exclusive allele was designated to this locus, because non‐amplification implies the genotype of this strain is unique at the priming site, as failure to amplify occurred on a background of successful amplification for other loci in these specimens. The number of alleles per locus ranged from 6 to 10, with a total of 11 unique allelic profiles found (Supporting Information Table S8). All gene sequences, including the non‐housekeeping gene omp, maintained an intact coding frame, consistent with their presence in a symbiont genome, rather than a nuclear insertion of a Rickettsia gene. The most polymorphic housekeeping locus was atpA, with 9.9% variable sites and the highest level of nucleotide diversity per site (Pi = 0.046) (Supporting Information Table S7). This gene exhibited evidence of intragenic recombination suggested by atypical pairwise divergence in closely related isolates (Supporting Information Table S9), as well as detection by RDPv4 (Martin et al., 2015) (p < 0.001, determined by MaxChi) (Supporting Information Table S7). All genes showed average K a/K s of less than 1 (Supporting Information Table S7), indicating that the genes were subject to purifying selection, conforming to the general requirements for reliable indicators of genetic relatedness between bacterial isolates. Predictably, as an antigenic protein with less intense purifying selection and potential episodes of positive selection, omp had a greater average K a/K s than the other loci, although no signs of positive selection were observed at the gene‐level.
Whilst there was evidence that the strains found within Culicoides were closely related, it is not clear if they are monophyletic. Some loci demonstrated 100% sequence identity with Rickettsia strains from other taxa. These included the partial gltA sequences of C. impunctatus, which was identical to the Rickettsia symbionts of the beetle Deronectes platynotus (Dytiscidae; FM177878) (Küchler et al., 2009), the Dipteran fly Chrysotimus flaviventris (Dolichopodidae; JQ925578) (Martin et al., 2013) and the spider Pityohyphantes phrygianus (Linyphiidae; DQ 231491) (Goodacre et al., 2006), and the partial 16S sequences of clonal complex 2 strains (C. duddingstoni (Bara, Sweden), C. pulicaris haplotype 1 (Sweden), C. newsteadi N1, C. pulicaris (haplotype 2), which were identical to the 16S sequence of the Rickettsia in the cranefly Limonia chorea (Limoniidae; AF322443). Furthermore, a coxA 995 bp region of the Hemipteran bug Macrolophus sp. Rickettsia 1 (Miridae; HE583223) (Machtelinckx et al., 2012) was > 99% similar to all Culicoides’ strains except for C. impunctatus and C. salinarius. Moreover, enforcing the monophyly of Culicoides Rickettsia on the 16S phylogeny (Fig. 2) did not result in a significantly worse tree (SH‐test, p > 0.05). Similar results were obtained when a phylogenetic analysis was conducted using the available Rickettsia gltA sequences (data not shown). Thus, it is unclear (largely due to lack of multi locus data from other taxa) whether the Culicoides Rickettsia represents a monophyletic group.
We next examined the relationship of the Rickettsia strains from different host species using allelic profiles across loci (Fig. 5). Most allelic profiles obtained from different host populations (11/13) were unique. Furthermore, of these 11 unique allelic profiles, 4 (H, I, J and K) allelic profiles shared no alleles with other strains. Allelic profiles that were shared by more than one host species were designated as central strains (CSs), whereas isolates that varied at one locus to these CSs were termed single locus variants (SLVs). Together the CSs and SLVs form clonal complexes, as they are presumed to be closely related. Two clonal complexes were identified in this study (Fig. 5); the central strain A from C. stigma and C. newsteadi N3 formed clonal complex 1 with the SLV strain from C. riethi (B), whereas the central strain C from C. newsteadi N1 and C. duddingstoni (Bara, Sweden) formed clonal complex 2 with the SLV strains from C. pulicaris haplotype 1 (Sweden) (D) and C. pulicaris haplotype 2 (E).
Figure 5.
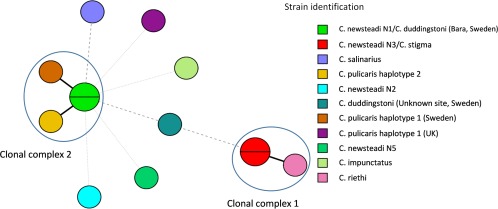
Minimum spanning tree using Unweighted Pair Group Method with Arithmetic Mean (UPGMA) cluster analysis of isolates.
Allelic profiles that are shared by more than one host species are designated as central strains (CSs). Strains differing at one locus (SLVs) are connected by a solid line, strains sharing one or more loci are connected by a dashed line and unique strains sharing no allele identity are connected by a faded line. [Color figure can be viewed at wileyonlinelibrary.com]
Visualisation of Rickettsia in C. impunctatus’ ovaries
Fluorescent in situ hybridization (FISH) of C. impunctatus’ dissected ovaries, using a Rickettsia‐specific probe, showed strong positive signals within the ovarioles (Fig. 6A). The strongest signal was localized inside the developing oocytes. In addition, hybridization signals were detected inside nurse and follicle cells. No signal was detected in the Rickettsia‐uninfected controls used (Fig. 6B), suggesting the specificity of the detection.
Figure 6.
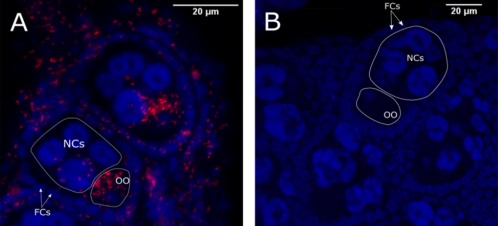
Rickettsia localization in midge ovaries via FISH.
The combined z‐stack optical sections of infected C. impunctatus (A) and Rickettsia free C. nubeculosus (B) ovarioles stained with DAPI (blue) and an ATTO633‐labeled Rickettsia‐specific probe (red). FCs: follicle cells, NCs: nurse cells and OO: oocyte. [Color figure can be viewed at wileyonlinelibrary.com]
Discussion
In this study, we serendipitously recovered the genome of a Rickettsia bacterium (RiCNE) from the WGS sequencing of C. newsteadi N5, the 16S rRNA sequence of which paralleled a Rickettsia identified in a screen of the midgut microbiota of C. sonorensis (Campbell et al., 2004). Phylogenetic analyses placed the RiCNE isolate within the Torix group, a sister lineage of the arthropod‐associated Rickettsia. We report the draft genome sequence for RiCNE, which represents the first sequenced genome of a Rickettsia belonging to the Torix group. Furthermore, we show Torix group Rickettsia are common in biting midges and, thus, represent a previously unrecognized component of the biology of this important vector group.
The draft genome of RiCNE provides valuable insights into the potential role of Rickettsia in midges and can further our understanding on the evolution of Rickettsia lifestyle and pathogenicity. The RiCNE draft genome shares many features with the previously sequenced Rickettsia, associated with genome reduction in the obligately intracellular genus. The genome size (∼ 1.5 Mb), the number of the protein‐coding genes (1352) and the coding density (80%) fell well within the range reported for Rickettsia (Merhej and Raoult, 2011; Gillespie et al., 2012).
Analysis of the metabolic potential of RiCNE shows a reduced biosynthetic and catabolic capacity typical of other Rickettsia, including absent or deficient pathways for glycolysis, nucleotide metabolism and amino‐acid biosynthesis. The blood feeding lifestyle of midges led us to particularly investigate the capacity for B vitamin synthesis, as recorded for Wigglesworthia symbionts in tsetse flies and Wolbachia in Cimex bedbugs (Snyder et al., 2010; Nikoh et al., 2014). However, with the exception of a reduced pathway for folate biosynthesis (also found in other Rickettsia), RiCNE lacks known pathways for the biosynthesis of cofactors and B‐vitamins.
A striking difference between the RiCNE genome and other arthropod‐associated Rickettsia is the presence of the complete set of genes encoding for the non‐oxidative branch of the PPP in RiCNE. The PPP is a major component of central metabolism in prokaryotes and eukaryotes (Stincone et al., 2015). The PPP is associated with both regulatory processes and biochemical functions, including carbon and redox homeostasis, response to oxidative stress and provision of precursors for nucleotide and amino acid biosynthesis. Notably, some parasites rely on the PPP to overcome the oxidative stress suffered during host invasion (Maugeri et al., 2003; Husain et al., 2012). Additionally, the non‐oxidative branch of the PPP in bacteria plays an essential role in the biosynthesis of lipopolysaccharides (LPS) by providing intermediates for the production of LPS precursors (Tzeng et al., 2002; Taylor et al., 2008). The biological role of the non‐oxidative PPP in RiCNE is unclear. Its presence in the Rickettsia endosymbiont of I. multifilis (Hydra group – ‘Megaira’) and its partial presence in Occidentia massiliensis, a sister species to Orientia isolated from a soft tick (Mediannikov et al., 2014), suggest that the non‐oxidative branch of the PPP has been independently lost in Rickettsia and Orientia lineages upon their transition to an arthropod host. Its absence from all other arthropod‐associated Rickettsiaceae may suggests specific functions to the lifestyle of Torix and Hydra group Rickettsia or to specific host microhabitats used by these symbionts (Fuchs et al., 2012). Alternatively, this can be suggestive of a relatively recent Rickettsia host shift to the midge host from a yet unknown ciliate host. Among the Rickettsiales, complete non‐oxidative PPP but absent oxidative PPP (as found in RiCNE) have been noted within the genera Anaplasma, Ehrlichia and Neorickettsia and the newly discovered member of the Midichloriaceae ‘Candidatus Jidaibacter acanthamoeba’, but in contrast, these pathways are incomplete in the genera Wolbachia and Midichloria (Fig. 4A). Our phylogenetic analysis suggests that the ancestor of the Rickettsiales had at least a partial PPP with a complete non‐oxidative phase, which was subsequently lost from certain lineages including most of the Rickettsiaceae. Further work should establish the degree to which the pathway is present in other Torix group Rickettsia, and the reasons for its loss more widely in the genus.
Rickettsiae have a complex surface structure, encoded by the presence of many of genes involved in LPS and peptidoglycan biosynthesis (Fuxelius et al., 2007). LPS are major components of the outer membrane in several Gram‐negative bacteria and constitute strong elicitors of the immune response both in insects and mammals (Raetz and Whitfield, 2002). Moreover, the capacity of intracellular, Gram‐negative, bacteria to modify their LPS components is essential for host immune evasion and host adaptation, influencing both pathogenicity and symbiosis (Li et al., 2012). Aside from the potential role of the PPP above in LPS biosynthesis, we found additional RiCNE‐specific genes associated with LPS and cell wall modification. Of note are the two paralogs of the lipid A 3‐O‐deacylase (pagL), a gene reported to be essential for establishing symbiosis in the nitrogen‐fixing endosymbiont Rhizobium etli (Brown et al., 2013). Recently the role of lipid A 3‐O‐deacylase in LPS remodelling and outer membrane vesicles (OMV) formation in bacteria has been reported (Elhenawy et al., 2016). Interestingly, OMVs have been reported to play essential roles in pathogenicity and symbiosis in several Gram‐negative bacteria. These roles include the delivery of virulence factors, modulation of host immune system, gut microbiota establishment and homoeostasis as well as horizontal DNA transfer (Ellis and Kuehn, 2010; Velimirov and Hagemann, 2011). Another example of a system associated with cell wall modification is the putative N‐acetylmuramoyl‐l‐alanine amidase (AmiD) gene encoding for a periplasmic lipoprotein involved in peptidoglycan recycling (Uehara and Park, 2007). It is noteworthy that aphids appear to have acquired horizontally an AmiD homologue, presumably from a rickettsial bacterium. This gene is highly upregulated specifically in the aphid bacteriocytes (the specialized host cells hosting its Buchnera symbiont), suggesting a potential role in bacteriocyte homeostasis and host–symbiont interaction (Nikoh et al., 2010).
Our second finding was that Torix group Rickettsia was found commonly across biting midges. Previous work on Culicoides, using conventional PCR to establish the presence of the heritable symbiont Cardinium, revealed interspecies infection rates ranging from 16% to 29% (Nakamura et al., 2009; Lewis et al., 2014; Mee et al., 2015). Thus, our PCR screen suggests that Rickettsia is the most common known symbiont of Culicoides, being present in 11 of 29 species tested (38%) and in 100% of specimens examined in 9 of the Rickettsia positive species. Hence, this Rickettsia clade represents an important associate found widely in Culicoides midges. It is noteworthy that our assessment of incidence is conservative, being based on a conventional PCR assay which will likely report false negatives for low titre infections.
The Torix group of Rickettsia has been recorded previously in an array of invertebrate species. Many of these species share ecological characteristics including an aquatic phase and predatory larval stages (e.g., midges, diving beetles, leeches and crane flies) (see Fig. 2). Others are notable for hematophagy (e.g., biting midges, leeches and sandflies). Moreover, no secondary associations with vertebrate hosts or pathogenicity have been associated so far with this Rickettsia group. Given the scarcity of available multilocus sequence data within Torix group, it is unclear whether the midge Rickettsia forms a monophyletic assemblage. More sequence data from other Torix Rickettsia will be needed to increase the phylogenetic resolution and determine the degree of relatedness among Torix Rickettsia strains. Nevertheless, our results support the hypothesis that Torix Rickettsia is a dominant taxon among invertebrates with aquatic life stages (Fig. 2).
The impact of the Rickettsia on host biology is uncertain. Rickettsia infections are known to be associated with a variety of reproductive manipulations of their host (reproductive parasitisms), including male‐killing in ladybird beetles (Werren et al., 1994) and parthenogenesis induction in parasitoids (Hagimori et al., 2006; Giorgini et al., 2010). However, equal likelihood of male and female midges being infected indicates sex ratio distortion is unlikely to be a phenotype for the Rickettsia in midges. Further to this, Rickettsia represents an obligate symbiont in book lice (Liposcelis bostrychophila) required for egg production (Perotti et al., 2006). However, the sporadic distribution of Rickettsia across subgenera suggests a lack of co‐speciation making it unlikely that the host requires symbiont presence for its function. Overall, the data suggests the Torix group Rickettsia identified in this study may have some facultative benefit to their host. Indeed, Rickettsia from this clade has been linked with a fitness benefit (increased body size) in leeches (Kikuchi and Fukatsu, 2005).
The strong tropism of Rickettsia bacteria for the midge oocytes unambiguously supports a vertical transmission route commonly seen in endosymbionts. This result also gives an indication of the likely routes driving Rickettsia to fixation within most of the populations in this study. There are a few routes that drive infection to fixation; combined horizontal and vertical transmission (Perlman et al., 2006), cytoplasmic incompatibility or non‐frequency‐dependent benefits combined with high fidelity maternal transmission and finally combined paternal and maternal transmission. The latter of these was described for the first time in Torix Rickettisa infecting leaf hoppers (Nephotettix cincticeps) (Watanabe et al., 2014). A peculiarity of note is the detection of coexisting infected and uninfected individuals in C. impunctatus populations, a scenario contrary to the more common fixed infections observed in this study. However, low titre infections cannot be ruled out (Mee et al., 2015). Alternative explanations for this difference are that the strain in C. impunctatus has a different role in its host in comparison to the other isolates at fixation in midges. In fact, the divergence of the omp gene in C. impunctatus (Supporting Information Fig. S6), one of the surface antigen coding genes, suggests possible differences in host specificity. Rickettsia surface antigens have previously been identified to be evolving under positive selection and may have key roles in host adherence and infiltration (Blanc et al., 2005). A major research effort for the future lies in identifying the impact of Rickettsia on host biology.
In conclusion, we have identified a common but neglected association between Rickettsia and biting midges and have described its unique genetic properties. Given the importance of biting midges as vectors, two key areas of future research are to establish the impact of Rickettsia presence on vector competence and on vector dispersal. Symbionts may reduce vector competence (as for Wolbachia in Aedes aegypti), increase it (as for Rickettsia in Bemisia tabaci) or have no impact. Rickettsia infections are also known to affect host dispersal tendencies, with Torix Rickettsia‐infected spiders showing lower motivation for dispersal (Goodacre et al., 2009). Symbiont impact on either of these characteristics would significantly alter the local and spatial spread of vector‐borne infections, thus pressingly deserve attention.
Experimental procedures
Genome sequencing, assembly and annotation
Genomic DNA from C. newsteadi N5 was extracted from single individuals using the QIAGEN DNAeasy™ Blood & Tissue Kit following the protocol for purification of total DNA from Insects. Equal concentrations of DNA from three individuals was pooled and used to construct a 500 bp paired‐end library (Illumina TruSeq Nano) that was sequenced on 1/3 lane of a HiSeq2500 platform at the Centre for Genomic Research (CGR), University of Liverpool, with 2 × 125 bp paired reads.
Quality assessment and filtering of the Illumina reads were performed using FastQC (Andrews, 2016) and FastX‐Toolkit (Gordon, 2010). A preliminary assembly was performed using SPAdes version 3.7.0 (Nurk et al., 2013) with k‐mer sizes 21, 33, 55 and 77 under ‘careful’ mode and a coverage cutoff of 5. Identification and filtering of putative symbiont contigs was performed by visualizing the data in taxon‐annotated GC‐coverage plots using Blobtools (Kumar et al., 2013; Laetsch, 2016) and TBLASTX searches against a local Rickettsia‐genomic database . Rickettsia contigs were extracted and any host contamination was removed by BLASTX searches against the non‐redundant protein database (NR). Rickettsia‐specific reads were retrieved using Bowtie2 (Langmead and Salzberg, 2012) and samtools (Li et al., 2009) and re‐assembled de novo with SPAdes assembler (k‐mer sizes: 21, 33, 55 and 77, ‘careful’ mode). The final assembly produced 224 contigs ≥ 500 bp, which were subjected to a final decontamination step removing only four contigs which had strong similarities to Enterobacteriaceae and lower than average coverage. Assembly errors were assessed using REAPR software (Hunt et al., 2013) (Supporting Information) and a final scaffolding was performed with SSPACE (Boetzer et al., 2011) with the following parameters, k = 5, a = 0.5 and n = 15.
The draft genome of the Rickettsia symbiont from C. newsteadi (RiCNE) was annotated using Prokka software v.1.12 (Seemann, 2014) (Supporting Information), and completeness was assessed using BUSCO v.2 based on 148 single‐copy universal bacterial markers (Simão et al., 2015). COG functional categories were assigned using the eggNOG 4.5 database (Huerta‐Cepas et al., 2016), and Pfam domains were predicted using InterProScan 5 (Jones et al., 2014). We evaluated the metabolic potential of RiCNE genome using the Metabolic and Physiological Potential Evaluator (MAPLE‐2.1.0) based on the calculation of the KEGG‐defined module completion ratio (MCR) (Takami et al., 2016). These results were compared with other Rickettsia from major Rickettsia groups (Belli, Adalia, Scapularis, Transitional, Typhus and Spotted Fever) as well as other members of the order Rickettsiales including the genera Orientia, Wolbachia, Anaplasma, Ehrlichia and Midichloria. Three known nutritional mutualists (Wigglesworthia, Buchnera and Riesia) were also included in the analyses.
Ortholog identification and phylogenomic analyses
Identification of orthologous gene clusters (Orthogoups) was performed using OrthoFinder method (Emms and Kelly, 2015) on a dataset of 84 publicly available Rickettsia genomes as well as two Orientia tsutsugamushi strains (outgroup) (Supporting Information Table S2). In order to avoid inconsistencies arising from different annotation practices, all Rickettsia genomes used were re‐annotated using Prokka software as described above. A set of 189 single‐copy core orthogroups were selected (Supporting Information Table S10) and automatically aligned with MAFFT v7 (Katoh and Standley, 2013) using default settings. For phylogenetic analyses, a super‐matrix was generated by concatenating the protein alignments of the 189 single‐copy core genes and subsequently trimmed with trimAl version 1.4 (Capella‐Gutiérrez et al., 2009) using the ‘automated’ option. Phylogenetic relationships were reconstructed using maximum likelihood. The best protein model and substitution matrix was selected using ProtTest version 3.4.2 (Darriba et al., 2011) and maximum likelihood (ML) phylogeny were inferred with RAxML version 8.2.8 (Stamatakis, 2014) using 100 rapid bootstrap replicates under the PROTGAMMAILG model.
Culicoides collection identification and DNA extractions
Overall, 414 specimens of 29 Culicoides species were collected using light traps from May 2007 to July 2016 across sites spanning France, South Africa, Sweden and the UK (Table 1 and Supporting Information Table S6). Sampled species included both vectors and non‐vectors of BTV. All midge specimens were stored in 70% ethanol for preservation before being sexed and separated morphologically down to the species level using relevant keys (Downes and Kettle, 1952; Campbell and Pelham‐Clinton, 1960; Delécolle, 1985; Meiswinkel, 1994). Morphological identification was confirmed by sequencing a fragment of the mitochondrial cytochrome c oxidase subunit 1 (COI) barcode (Pagès et al., 2009; Ander et al., 2013; Nielsen and Kristensen, 2015). DNA extractions were prepared based on the protocol of Ander et al. (2013) and details are presented in the Supporting Information. The COI gene fragment was amplified using different universal primer sets (Folmer et al., 1994; Dallas et al., 2003) and sequenced through the Sanger method by GATC Ltd. Samples that did not amplify were deemed to contain low quality DNA and were removed from further analysis.
PCR screening for Rickettsia and analysis of strain relatedness
Presence of Rickettsia was initially assessed by PCR assay using Rickettsia‐specific primers designed to amplify a 320‐bp region of the omp (17 kDa surface antigen precursor) gene (Supporting Information Table S11). Cycling conditions were as follows: initial denaturation at 95°C for 5 min, followed by 35 cycles of denaturation (94°C, 30 s), annealing (54°C, 30 s), extension (72°C, 120 s), and a final extension at 72°C for 7 min. Amplicons identified by gel electrophoresis were subsequently purified enzymatically (ExoSAP) and sequenced (GATC Biotech AG, Konstanz, Germany).
Based on previous studies (Fournier et al., 2003; Weinert et al., 2009a; Li et al., 2010; Machtelinckx et al., 2012; Santibáñez et al., 2013) that profile Rickettsia diversity, the 16S rRNA, gltA (Citrate synthase), coxA (cytochrome oxidase) and atpA (ATP synthase) genes were chosen as indicators of genetic relatedness between isolates. With the consideration that these housekeeping loci may be too conserved to resolve recently diverged strains, the omp gene was included to allow higher resolution in typing, alongside an inference of selection pressure due to the divergent nature of antigen genes compared to housekeeping genes.
Primers to amplify these loci (Supporting Information Table S11) were designed on conserved regions based on RiCNE and available complete gene sequences so that they could amplify across several Rickettsia groups but would not cross amplify any alpha‐proteobacteria outgroups. All PCR amplifications were performed as described above. Sanger sequencing through both strands allowed for the clarification of ambiguous base calls as well as giving greater sequence coverage at individual loci. Raw sequences were edited in UGENE (Okonechnikov et al., 2012) and alignments for each locus were generated in MEGA6 using the ClustalW algorithm (Tamura et al., 2013).
A profile of each locus was constructed by calculating GC content, selective pressure (K a/K s), nucleotide diversity per site (π) and the percentage of variable sites using DNAsp v5 (Librado and Rozas, 2009). As phylogenetic inferences can be complicated by recombination, the presence of intragenic recombination was investigated using the program RDP v4 (Martin et al., 2015). To this end, the MaxChi algorithm was utilised with the following criteria to assess a true recombination positive: a p value of < 0.01, sequences were considered linear with 1200 permutations being performed. Recombination events detected by the programme were visually inspected for congruency between the recombinant and putative parent strains to confirm a true positive. As an additional aid, genetic divergence at each locus was determined using pairwise divergence. The omp locus was also assessed for evidence of diversifying selection at the gene level via pairwise non‐synonymous/synonymous rate ratio analysis (K a/K s ratio).
Similar to multilocus sequence typing (MLST) convention, all unique genotypes were designated allele numbers (used as a unique identifier) which, when combined at all loci, produce an allelic profile (Maiden et al., 1998). Aside from identifying specific isolates, this multigenic approach also allows for clonal complexes to be identified (conventionally allelic profiles which are identical at three or more loci). Allelic profiles and complexes were designated based on Unweighted Pair Group Method with Arithmetic Mean (UPGMA) cluster analysis and visualised as a minimum spanning tree (MST) implemented by Bionumerics v7 (Applied Maths, Austin, TX, USA).
Phylogenetic analyses
The phylogenetic position of the midge Rickettsia within the Rickettsiaceae was first assessed using the 16S rRNA gene sequence. Briefly, sequences of 16S rRNA from selected Rickettsia genomes used for the phylogenomic analyses (Bellii, Canadensis and Typhus groups) were extracted and combined with sequences from the Hydra and Torix Rickettsia groups (Weinert et al., 2009a) obtained from GenBank. All sequences were aligned using SSU‐ALIGN software (Nawrocki, 2009) and unambiguously aligned columns were automatically selected by ssu‐mask program. A Bayesian phylogeny was estimated with MrBayes v3.2.6 (Ronquist et al., 2012) under the GTR + G + I model. Two independent runs were carried out for 1,000,000 generations with sampling every 100 generations using four Markov chains. The first 25% of the samples were discarded as burn‐in. Alternative phylogenetic hypotheses were tested using constrain tree searches and the Shimodaira–Hasegawa (SH) test as implemented in RAxML version 8.2.8. The ML phylogeny for the omp gene was estimated with RaxML version 8.2.8 (Stamatakis, 2014) using 100 rapid bootstrap replicates under the GTR + G model of nucleotide substitutions. Single protein phylogenies of the PPP were estimated with Bayesian analyses using a mixed model of amino‐acid substitutions (two runs of 1,500,000 generations with sampling every 100 generations using four Markov chains). Additional ML analyses were inferred with RAxML version 8.2.8 (Stamatakis, 2014) using 100 rapid bootstrap replicates under the PROTGAMMAAUTO model optimization setting. Finally, trees were drawn using the iTOL (Letunic and Bork, 2007) and EvolView (He et al., 2016) online tree annotation and visualization tools.
Fluorescent in situ hybridisation
Live nulliparous female C. impunctatus were collected from Kielder, UK and ovaries were dissected in 70% ethanol. Samples were fixed overnight in Carnoy's solution (chloroform:ethanol:glacial acetic acid, 6:3:1) and decolorized with 6% H2O2 in ethanol for 1 h. Hybridisation was performed overnight in hybridisation buffer (20 mM Tris‐HCl, pH 8.0, 0.9 M NaCl, 0.01% sodium dodecyl sulfate, 30% formamide) containing 10 pmol/ml of the Rickettsia specific probe [5′‐CCATCATCCCCTACTACA‐(ATTO 633)‐3′] adapted from Perotti et al. (2006). After hybridisation, the samples were thoroughly washed twice in hybridisation buffer (without the probe) and slide mounted in Vectashield with DAPI (Vector Laboratories) and viewed under a Zeiss LSM 880 BioAFM confocal microscope. The specificity of the detection and any autofluorescent properties of midge tissue was assessed using Rickettsia‐free midges (Culicoides nubeculosus; Pirbright Institute) as negative controls.
Nucleotide sequence accession numbers
Raw reads and the RiCNE draft genome assembly have been submitted to the DDBJ/EMBL/GenBank database under the BioProject accession number PRJNA376033 (WGS project: MWZE00000000). COI barcodes and sequences generated for individual Rickettsia loci in this study were deposited in GenBank under deposition numbers KY765346‐KY765408, KY777722‐KY777733 and KY778697‐KY778698.
Author contributions
Acquisition, analysis and interpretation of the data were undertaken by JP and SS, as well as drafting of the manuscript. GH and MB assisted in the conception and design of the study, in addition to critical revision of the manuscript. CG and MA aided in the collection and identification of midge specimens and the critical revision of the manuscript.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Taxon annotated GC‐coverage plots. (A) Primary genome assembly of Culicoides newsteadi N5. (B) Postfiltering against a local database containing all available (complete and draft) Rickettsia genomes.
Fig. S2. BUSCO completeness assessment results for RiCNE draft genome in relation to selected complete Rickettsia genomes. The results are based on the presence or absence of 148 single‐copy universal bacterial markers. BUSCO notation: complete (C), single‐copy (S), duplicated (D), fragmented (F) and missing (M).
Fig. S3. Circular representation of the RiCNE draft genome. For visualization purposes, the 193 scaffolds were concatenated into a pseudomolecule. Alternating gray and white strips indicate scaffold borders. Inwards, the first, second and third circles are color‐coded according to the eggNOG functional categories and represent: (a) the complete RiCNE protein‐coding genes (CDSs), (b) CDSs universally present in all Rickettsiacea genomes used in this study and (c) RiCNE unique CDSs respectively. The fourth circle shows the clusters of the T4SS (red) and conjugation (purple) genes. The first line plot represents the genome coverage. An orange line indicates the average coverage of the draft assembly (∼ 76×). The asterisks indicate the two scaffolds containing the conjugation genes represented by 2–3× higher than average coverage indicating possible multiple copies. The innermost line plot represents the GC% coverage calculated based on a 1 kb sliding window. The circular plot was generated with Circos v0.69 (Krzywinski et al., 2009).
Fig. S4. Cladogram of the complete core‐genome phylogeny. Phylogenomic placement of the Rickettsia endosymbiont of C. newsteadii were inferred using maximum likelihood (RaxML, model: Lag + G + I) from the concatenated protein alignments of 189 single copy ortholog genes. Support values are based on 100 rapid bootsrap replicates.
Fig. S5. Individual trees for the pentose phosphate pathway (PPP) proteins. Tree topology and posterior probabilities were inferred with MrBayes using a mixed model of amino acid substitution. The trees were midpoint rooted.
Fig. S6. Maximum likelihood phylogeny of the omp gene. The tree topology was estimated using RaxML and the GTR + G model of nucleotide substitutions. Support values are based on 1000 rapid bootstrap replicates. The tree was midpoint rooted.
Table S1. Functional annotation of RiCNE draft genome including Interproscan results against Pfam and eggNOG results.
Table S2. Publicly available Rickettsiaceae genomes used in this study for ortholog identification and phylogenomic analysis.
Table S3. Genes encoding for the P‐T4SS and the tra conjugative DNA‐transfer element in RiCNE genome.
TableS4. RiCNE unique genes.
Table S5. RiCNE unique genes putatively associated with host invasion and host–microbe interactions.
Table S6. omp conventional PCR assay results for Rickettsia‐negative Culicoides species under study, given by subgenus, species, location, date and sex. a Culicoides newsteadi haplotype N1 designated by Ander et al. (2013). b Culicoides newsteadi N6 previously undesignated.
Table S7. Genetic characteristics of housekeeping and omp alleles.
Table S8. Rickettsia strains recovered from Culicoides midges, with allelic profiles; strains sharing the same allelic profiles at all five loci were designated as a single strain. NA: non amplifiable.
Table S9. Pairwise divergence at individual loci between strains from clonal complex 2 showing remarkable divergence in the ATPase allele compared to the average of all strains, most likely as a result of a recombination event.
Table S10. Core genes used for phylogenomic analysis.
Table S11. Housekeeping and omp gene primer attributes.
Acknowledgements
We wish to thank three anonymous referees for constructive comments that improved the manuscript. The sequencing was carried out at the Centre for Genomic Research, University of Liverpool, United Kingdom. We would like to thank Dr. Michael Gerth for kindly providing the unpublished sequences of additional Torix group Rickettsia endosymbionts from the green lacewing Chrysotropia ciliata and the alderfly Sialis lutaria. We also thank Kenneth Sherlock, Georgette Kluiters, Steve Price, Lukasz Lukomski, the Direction générale de l' alimentation (DGAL) and the national veterinary services (France) for their support with the collection of midges samples. We acknowledge the Liverpool Centre for Cell Imaging (CCI) for provision of imaging equipment and technical assistance (BB/M012441/1). We are grateful to Jan Chirico for the identification of Culicoides from Sweden, Ignace Rakotoarivony (CIRAD) for the identification of Culicoides from France and Gert Venter and Karien Labuschagne for the identification of Culicoides from South Africa. We wish to thanks Prof. R.S. Coyne, Dr T. Doak and Dr H. Suzuki for permission to use unpublished genomic data for the Rickettsia endosymbiont of Ichthyophirius multifilis. Culicoides nubeculosus were provided via a Core Capability BBSRC Grant awarded to Simon Carpenter (The Pirbright Institute) and produced by Eric Denison (BBS/E/I/00001701). This work was supported by a Marie Curie Individual Fellowship (H2020‐MSCA‐IF‐2014) grant 657135 ‘MIDGESYM’ to Stefanos Siozios and a BBSRC DTP studentship to Jack Pilgrim. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, the Department of Health or Public Health England. The authors declare no conflict of interest.
References
- Akman, L. , Yamashita, A. , Watanabe, H. , Oshima, K. , Shiba, T. , Hattori, M. , and Aksoy, S. (2002) Genome sequence of the endocellular obligate symbiont of tsetse flies, Wigglesworthia glossinidia. Nat Genet 32: 402–407. [DOI] [PubMed] [Google Scholar]
- Ander, M. , Troell, K. , and Chirico, J. (2013) Barcoding of biting midges in the genus Culicoides: a tool for species determination. Med Vet Entomol 27: 323–331. [DOI] [PubMed] [Google Scholar]
- Andrews, S. (2016) FastQC Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Bian, G. , Xu, Y. , Lu, P. , Xie, Y. , and Xi, Z. (2010) The endosymbiotic bacterium Wolbachia induces resistance to dengue virus in Aedes aegypti . PLoS Pathog 6: e1000833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagrove, M.S.C. , Arias‐Goeta, C. , Failloux, A.‐B. , and Sinkins, S.P. (2012) Wolbachia strain wMel induces cytoplasmic incompatibility and blocks dengue transmission in Aedes albopictus . Proc Natl Acad Sci USA 109: 255–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc, G. , Ngwamidiba, M. , Ogata, H. , Fournier, P.‐E. , Claverie, J.‐M. , and Raoult, D. (2005) Molecular evolution of Rickettsia surface antigens: evidence of positive selection. Mol Biol Evol 22: 2073–2083. [DOI] [PubMed] [Google Scholar]
- Boetzer, M. , Henkel, C.V. , Jansen, H.J. , Butler, D. , and Pirovano, W. (2011) Scaffolding pre‐assembled contigs using SSPACE. Bioinformatics 27: 578–579. [DOI] [PubMed] [Google Scholar]
- Borkent, A. (2016) The subgeneric classification of species of Culicoides – thoughts and a warning. Available at: http://wwx.inhs.illinois.edu/files/7413/4219/9567/CulicoidesSubgenera.pdf.
- Brown, D.B. , Muszyński, A. , Salas, O. , Speed, K. , and Carlson, R.W. (2013) Elucidation of the 3‐O‐deacylase gene, pagL, required for the removal of primary β‐hydroxy fatty acid from the lipid A in the nitrogen‐fixing endosymbiont Rhizobium etli CE3. J Biol Chem 288: 12004–12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, C.L. , Mummey, D.L. , Schmidtmann, E.T. , and Wilson, W.C. (2004) Culture‐independent analysis of midgut microbiota in the arbovirus vector Culicoides sonorensis (Diptera: Ceratopogonidae). J Med Entomol 41: 340–348. [DOI] [PubMed] [Google Scholar]
- Campbell, J.A. , and Pelham‐Clinton, E.C. (1960) A taxonomic review of the British species of Culicoides Latreille (Diptera, Ceratopogonidæ). Proc R Soc Edinburgh 67: 181–302. [Google Scholar]
- Capella‐Gutiérrez, S. , Silla‐Martínez, J.M. , and Gabaldón, T. (2009) trimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics 25: 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho, N.‐H. , Kim, H.‐R. , Lee, J.‐H. , Kim, S.‐Y. , Kim, J. , Cha, S. , et al (2007) The Orientia tsutsugamushi genome reveals massive proliferation of conjugative type IV secretion system and host–cell interaction genes. Proc Natl Acad Sci USA 104: 7981–7986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallas, J.F. , Cruickshank, R.H. , Linton, Y.M. , Nolan, D.V. , Patakakis, M. , Braverman, Y. , et al (2003) Phylogenetic status and matrilineal structure of the biting midge, Culicoides imicola, in Portugal, Rhodes and Israel. Med Vet Entomol 17: 379–387. [DOI] [PubMed] [Google Scholar]
- Darriba, D. , Taboada, G.L. , Doallo, R. , and Posada, D. (2011) ProtTest 3: fast selection of best‐fit models of protein evolution. Bioinformatics 27: 1164–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delécolle, J.C. (1985) Nouvelle contribution à l’étude systématique et iconographique des espéces du genre Culicoides (Diptera: Ceratopogonidae) du Nord‐Est de la France. PhD Thesis. Université Louis Pasteur du Strasbourg.
- Douglas, A.E. (2009) The microbial dimension in insect nutritional ecology. Funct Ecol 23: 38–47. [Google Scholar]
- Downes, J.A. , and Kettle, D.S. (1952) Descriptions of three species of Culicoides Latreille (Diptera: Ceratopogonidae) new to science, together with notes on, and a revised key to the British species of the pulicaris and obsoletus groups. Proc R Entomol Soc Lond B 21: 3–78. [Google Scholar]
- Dyková, I. , Veverková, M. , Fiala, I. , Machácková, B. , and Pecková, H. (2003) Nuclearia pattersoni sp. n. (Filosea), a new species of amphizoic amoeba isolated from gills of roach (Rutilus rutilus), and its rickettsial endosymbiont. Folia Parasitol (Praha) 50: 161–170. [PubMed] [Google Scholar]
- Elhenawy, W. , Bording‐Jorgensen, M. , Valguarnera, E. , Haurat, M.F. , Wine, E. , and Feldman, M.F. (2016) LPS remodeling triggers formation of outer membrane vesicles in Salmonella . mBio 7: e00940–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, T.N. , and Kuehn, M.J. (2010) Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol Mol Biol Rev 74: 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emms, D.M. , and Kelly, S. (2015) OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol 16: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari, J. , and Vavre, F. (2011) Bacterial symbionts in insects or the story of communities affecting communities. Philos Trans R Soc B 366: 1389–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folmer, O. , Black, M. , Hoeh, W. , Lutz, R. , and Vrijenhoek, R. (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotechnol 3: 294–299. [PubMed] [Google Scholar]
- Fournier, P.‐E. , Dumler, J.S. , Greub, G. , Zhang, J. , Wu, Y. , and Raoult, D. (2003) Gene sequence‐based criteria for identification of new Rickettsia isolates and description of Rickettsia heilongjiangensis sp. nov. J Clin Microbiol 41: 5456–5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs, T.M. , Eisenreich, W. , Heesemann, J. , and Goebel, W. (2012) Metabolic adaptation of human pathogenic and related nonpathogenic bacteria to extra‐ and intracellular habitats. FEMS Microbiol Rev 36: 435–462. [DOI] [PubMed] [Google Scholar]
- Fuxelius, H.‐H. , Darby, A. , Min, C.‐K. , Cho, N.‐H. , and Andersson, S.G.E. (2007) The genomic and metabolic diversity of Rickettsia . Res Microbiol 158: 745–753. [DOI] [PubMed] [Google Scholar]
- Gillespie, J.J. , Ammerman, N.C. , Dreher‐Lesnick, S.M. , Rahman, M.S. , Worley, M.J. , Setubal, J.C. , et al (2009) An anomalous type IV secretion system in Rickettsia is evolutionarily conserved. PLoS One 4: e4833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie, J.J. , Joardar, V. , Williams, K.P. , Driscoll, T. , Hostetler, J.B. , Nordberg, E. , et al (2012) A Rickettsia genome overrun by mobile genetic elements provides insight into the acquisition of genes characteristic of an obligate intracellular lifestyle. J Bacteriol 194: 376–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giorgini, M. , Bernardo, U. , Monti, M.M. , Nappo, A.G. , and Gebiola, M. (2010) Rickettsia symbionts cause parthenogenetic reproduction in the parasitoid wasp Pnigalio soemius (Hymenoptera: Eulophidae). Appl Environ Microbiol 76: 2589–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodacre, S.L. , Martin, O.Y. , Thomas, C.F.G. , and Hewitt, G.M. (2006) Wolbachia and other endosymbiont infections in spiders. Mol Ecol 15: 517–527. [DOI] [PubMed] [Google Scholar]
- Goodacre, S.L. , Martin, O.Y. , Bonte, D. , Hutchings, L. , Woolley, C. , Ibrahim, K. , et al (2009) Microbial modification of host long‐distance dispersal capacity. BMC Biol 7: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon, A. (2010) FASTX‐Toolkit Available at: http://hannonlab.cshl.edu/fastx_toolkit/.
- Hagimori, T. , Abe, Y. , Date, S. , and Miura, K. (2006) The first finding of a Rickettsia bacterium associated with parthenogenesis induction among insects. Curr Microbiol 52: 97–101. [DOI] [PubMed] [Google Scholar]
- He, Z. , Zhang, H. , Gao, S. , Lercher, M.J. , Chen, W.‐H. , and Hu, S. (2016) Evolview v2: an online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Res 44: W236–W241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendry, T.A. , Hunter, M.S. , and Baltrus, D.A. (2014) The facultative symbiont Rickettsia protects an invasive whitefly against entomopathogenic Pseudomonas syringae strains. Appl Environ Microbiol 80: 7161–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huerta‐Cepas, J. , Szklarczyk, D. , Forslund, K. , Cook, H. , Heller, D. , Walter, M.C. , et al (2016) eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res 44: D286–D293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt, M. , Kikuchi, T. , Sanders, M. , Newbold, C. , Berriman, M. , and Otto, T.D. (2013) REAPR: a universal tool for genome assembly evaluation. Genome Biol 14: R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst, G.D.D. , and Frost, C.L. (2015) Reproductive parasitism: maternally inherited symbionts in a biparental world. Cold Spring Harb Perspect Biol 7: a017699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst, G.D.D. , von der Schulenburg, J.H.G. , Majerus, T.M.O. , Bertrand, D. , Zakharov, I.A. , Baungaard, J. , et al (1999) Invasion of one insect species, Adalia bipunctata, by two different male‐killing bacteria. Insect Mol Biol 8: 133–139. [DOI] [PubMed] [Google Scholar]
- Husain, A. , Sato, D. , Jeelani, G. , Soga, T. , and Nozaki, T. (2012) Dramatic increase in glycerol biosynthesis upon oxidative stress in the anaerobic protozoan parasite Entamoeba histolytica . PLoS Negl Trop Dis 6: e1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturbe‐Ormaetxe, I. , Walker, T. , O’ Neill, S.L. (2011) Wolbachia and the biological control of mosquito‐borne disease. EMBO Reports 12: 508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenike, J. , Unckless, R. , Cockburn, S.N. , Boelio, L.M. , and Perlman, S.J. (2010) Adaptation via symbiosis: recent spread of a Drosophila defensive symbiont. Science 329: 212–215. [DOI] [PubMed] [Google Scholar]
- Jones, P. , Binns, D. , Chang, H.‐Y. , Fraser, M. , Li, W. , McAnulla, C. , et al (2014) InterProScan 5: genome‐scale protein function classification. Bioinformatics 30: 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K. , and Standley, D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi, Y. , and Fukatsu, T. (2005) Rickettsia infection in natural leech populations. Microb Ecol 49: 265–271. [DOI] [PubMed] [Google Scholar]
- Kikuchi, Y. , Sameshima, S. , Kitade, O. , Kojima, J. , and Fukatsu, T. (2002) Novel clade of Rickettsia spp. from leeches. Appl Environ Microbiol 68: 999–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliot, A. , Cilia, M. , Czosnek, H. , and Ghanim, M. (2014) Implication of the bacterial endosymbiont Rickettsia spp. in interactions of the whitefly Bemisia tabaci with tomato yellow leaf curl virus. J Virol 88: 5652–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski, M. , Schein, J. , Birol, I. , Connors, J. , Gascoyne, R. , Horsman, D. , et al (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Küchler, S.M. , Kehl, S. , and Dettner, K. (2009) Characterization and localization of Rickettsia sp. in water beetles of genus Deronectes (Coleoptera: Dytiscidae). FEMS Microbiol Ecol 68: 201–211. [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Jones, M. , Koutsovoulos, G. , Clarke, M. , and Blaxter, M. (2013) Blobology: exploring raw genome data for contaminants, symbionts and parasites using taxon‐annotated GC‐coverage plots. Front Genet 4: 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laetsch, D.R. (2016) blobtools:blobtools v0.9.19.4 Available at: http://doi.org/10.5281/zenodo.61799.
- Langmead, B. , and Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. , and Bork, P. (2007) Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23: 127–128. [DOI] [PubMed] [Google Scholar]
- Lewis, S.E. , Rice, A. , Hurst, G.D.D. , and Baylis, M. (2014) First detection of endosymbiotic bacteria in biting midges Culicoides pulicaris and Culicoides punctatus, important Palaearctic vectors of bluetongue virus. Med Vet Entomol 28: 453–456. [DOI] [PubMed] [Google Scholar]
- Li, A.Y. , Adams, P.J. , Abdad, M.Y. , and Fenwick, S.G. (2010) High prevalence of Rickettsia gravesii sp. nov. in Amblyomma triguttatum collected from feral pigs. Vet Microbiol 146: 59–62. [DOI] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Powell, D.A. , Shaffer, S.A. , Rasko, D.A. , Pelletier, M.R. , Leszyk, J.D. , et al (2012) LPS remodeling is an evolved survival strategy for bacteria. Proc Natl Acad Sci USA 109: 8716–8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Librado, P. , and Rozas, J. (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452. [DOI] [PubMed] [Google Scholar]
- Luis‐Pantoja, M. , Ramos‐González, P.L. , Naranjo, M. , Hernández‐Rodríguez, L. , Rodríguez, J. , and Pérez‐López, E. (2015) Rickettsia‐related bacteria associated with papaya plants showing bunchy top disease in Cuba. J Gen Plant Pathol 81: 166–168. [Google Scholar]
- Łukasik, P. , Guo, H. , van Asch, M. , Ferrari, J. , and Godfray, H.C.J. (2013) Protection against a fungal pathogen conferred by the aphid facultative endosymbionts Rickettsia and Spiroplasma is expressed in multiple host genotypes and species and is not influenced by co‐infection with another symbiont. J Evol Biol 26: 2654–2661. [DOI] [PubMed] [Google Scholar]
- Machtelinckx, T. , Van Leeuwen, T. , Van De Wiele, T. , Boon, N. , De Vos, W.H. , Sanchez, J.‐A. , et al (2012) Microbial community of predatory bugs of the genus Macrolophus (Hemiptera: Miridae). BMC Microbiol 12: S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden, M.C.J. , Bygraves, J.A. , Feil, E. , Morelli, G. , Russell, J.E. , Urwin, R. , et al (1998) Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci USA 95: 3140–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majerus, T.M.O. , Von Der Schulenburg, J.H.G. , Majerus, M.E.N. , and Hurst, G.D.D. (1999) Molecular identification of a male‐killing agent in the ladybird Harmonia axyridis (Pallas) (Coleoptera: Coccinellidae). Insect Mol Biol 8: 551–555. [DOI] [PubMed] [Google Scholar]
- Manzano‐Marin, A. , Oceguera‐Figuerora, A. , Latorre, A. , Jiménez‐Garcia, L.F. , and Moya, A. (2015) Solving a bloody mess: B‐vitamin independent metabolic convergence among gammaproteobacterial obligate endosymbionts from blood‐feeding arthropods and the leech Haementeria officinalis . Genome Biol Evol 7: 2871–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, D.P. , Murrell, B. , Golden, M. , Khoosal, A. , and Muhire, B. (2015) RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol 1: 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, O.Y. , Puniamoorthy, N. , Gubler, A. , Wimmer, C. , and Bernasconi, M.V. (2013) Infections with Wolbachia, Spiroplasma, and Rickettsia in the Dolichopodidae and other Empidoidea. Infect Genet Evol 13: 317–330. [DOI] [PubMed] [Google Scholar]
- Maugeri, D.A. , Cazzulo, J.J. , Burchmore, R.J.S. , Barrett, M.P. , and Ogbunude, P.O.J. (2003) Pentose phosphate metabolism in Leishmania mexicana . Mol Biochem Parasitol 130: 117–125. [DOI] [PubMed] [Google Scholar]
- Mediannikov, O. , Nguyen, T.‐T. , Bell‐Sakyi, L. , Padmanabhan, R. , Fournier, P.‐E. , and Raoult, D. (2014) High quality draft genome sequence and description of Occidentia massiliensis gen. nov., sp. nov., a new member of the family Rickettsiaceae. Stand Genomic Sci 9: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mee, P.T. , Weeks, A.R. , Walker, P.J. , Hoffmann, A.A. , and Duchemin, J.‐B. (2015) Detection of low‐level Cardinium and Wolbachia infections in Culicoides . Appl Environ Microbiol 81: 6177–6188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiswinkel, R. (1994) Preliminary Wing Picture Atlas of Afrotropical Culicoides (119 Species), Pretoria, South Africa: ARC‐Onderstepoort Veterinary Institute. [Google Scholar]
- Mellor, P.S. , Boorman, J. , and Baylis, M. (2000) Culicoides biting midges: their role as arbovirus vectors. Annu Rev Entomol 45: 307–340. [DOI] [PubMed] [Google Scholar]
- Merhej, V. , and Raoult, D. (2011) Rickettsial evolution in the light of comparative genomics. Biol Rev 86: 379–405. [DOI] [PubMed] [Google Scholar]
- Morag, N. , Klement, E. , Saroya, Y. , Lensky, I. , and Gottlieb, Y. (2012) Prevalence of the symbiont Cardinium in Culicoides (Diptera: Ceratopogonidae) vector species is associated with land surface temperature. FASEB J 26: 4025–4034. [DOI] [PubMed] [Google Scholar]
- Moreira, L.A. , Iturbe‐Ormaetxe, I. , Jeffery, J.A. , Lu, G. , Pyke, A.T. , Hedges, L.M. , et al (2009) A Wolbachia symbiont in Aedes aegypti limits infection with dengue, chikungunya, and Plasmodium . Cell 139: 1268–1278. [DOI] [PubMed] [Google Scholar]
- Murray, G.G.R. , Weinert, L.A. , Rhule, E.L. , and Welch, J.J. (2016) The phylogeny of Rickettsia using different evolutionary signatures: how tree‐like is bacterial evolution? Syst Biol 65: 265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura, Y. , Kawai, S. , Yukuhiro, F. , Ito, S. , Gotoh, T. , Kisimoto, R. , et al (2009) Prevalence of Cardinium bacteria in planthoppers and spider mites and taxonomic revision of “Candidatus Cardinium hertigii” based on detection of a new Cardinium group from biting midges. Appl Environ Microbiol 75: 6757–6763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki, E.P. (2009), Structural RNA homology search and alignment using covariance models. PhD Thesis. Washington University in Saint Louis, School of Medicine.
- Nielsen, S.A. , and Kristensen, M. (2015) Delineation of Culicoides species by morphology and barcode exemplified by three new species of the subgenus Culicoides (Diptera: Ceratopogonidae) from Scandinavia. Parasit Vectors 8: 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoh, N. , McCutcheon, J.P. , Kudo, T. , Miyagishima, S. , Moran, N.A. , and Nakabachi, A. (2010) Bacterial genes in the aphid genome: absence of functional gene transfer from Buchnera to its host. PLoS Genet 6: e1000827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikoh, N. , Hosokawa, T. , Moriyama, M. , Oshima, K. , Hattori, M. , and Fukatsu, T. (2014) Evolutionary origin of insect‐Wolbachia nutritional mutualism. Proc Natl Acad Sci USA 111: 10257–10262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk, S. , Bankevich, A. , Antipov, D. , Gurevich, A. , Korobeynikov, A. , Lapidus, A. , et al (2013) Assembling genomes and mini‐metagenomes from highly chimeric reads In Deng M., Jiang R., Sun F., and Zhang X. (eds). Research in Computational Molecular Biology, Lecture Notes in Computer Science. Berlin/Heidelberg: Springer, pp. 158–170. [Google Scholar]
- Ogata, H. , La Scola, B. , Audic, S. , Renesto, P. , Blanc, G. , Robert, C. , et al (2006) Genome sequence of Rickettsia bellii illuminates the role of amoebae in gene exchanges between intracellular pathogens. PLoS Genet 2: e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okonechnikov, K. , Golosova, O. , and Fursov, M. (2012) Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28: 1166–1167. [DOI] [PubMed] [Google Scholar]
- Oliver, K.M. , Russell, J.A. , Moran, N.A. , and Hunter, M.S. (2003) Facultative bacterial symbionts in aphids confer resistance to parasitic wasps. Proc Natl Acad Sci USA 100: 1803–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagès, N. , Muñoz‐Muñoz, F. , Talavera, S. , Sarto, V. , Lorca, C. , and Núñez, J.I. (2009) Identification of cryptic species of Culicoides (Diptera: Ceratopogonidae) in the subgenus Culicoides and development of species‐specific PCR assays based on barcode regions. Vet Parasitol 165: 298–310. [DOI] [PubMed] [Google Scholar]
- Perlman, S.J. , Hunter, M.S. , and Zchori‐Fein, E. (2006) The emerging diversity of Rickettsia . Proc R Soc Lond B: Biol Sci 273: 2097–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perotti, M.A. , Clarke, H.K. , Turner, B.D. , and Braig, H.R. (2006) Rickettsia as obligate and mycetomic bacteria. FASEB J 20: 2372–2374. [DOI] [PubMed] [Google Scholar]
- Raetz, C.R. , and Whitfield, C. (2002) Lipopolysaccharide endotoxins. Annu Rev Biochem 71: 635–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves, W.K. , Kato, C.Y. , and Gilchriest, T. (2008) Pathogen screening and bionomics of Lutzomyia apache (Diptera: Psychodidae) in Wyoming, USA. J Am Mosq Control Assoc 24: 444–447. [DOI] [PubMed] [Google Scholar]
- Rio, R.V.M. , Symula, R.E. , Wang, J. , Lohs, C. , Wu, Y. , Snyder, A.K. , et al (2012) Insight into the transmission biology and species‐specific functional capabilities of tsetse (Diptera: Glossinidae) obligate symbiont Wigglesworthia . mBio 3: e00240–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rio, R.V.M. , Attardo, G.M. , and Weiss, B.L. (2016) Grandeur alliances: symbiont metabolic integration and obligate arthropod hematophagy. Trends Parasitol 32: 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist, F. , Teslenko, M. , van der Mark, P. , Ayres, D.L. , Darling, A. , Höhna, S. , et al (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61: 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santibáñez, S. , Portillo, A. , Santibáñez, P. , Palomar, A.M. , and Oteo, J.A. (2013) Usefulness of rickettsial PCR assays for the molecular diagnosis of human rickettsioses. Enferm Infecc Microbiol Clin 31: 283–288. [DOI] [PubMed] [Google Scholar]
- Schrallhammer, M. , Ferrantini, F. , Vannini, C. , Galati, S. , Schweikert, M. , Görtz, H.‐D. , et al (2013) “Candidatus Megaira polyxenophila” gen. nov., sp. nov.: considerations on evolutionary history, host range and shift of early divergent Rickettsiae. PLoS One 8: e72581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann, T. (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069. [DOI] [PubMed] [Google Scholar]
- Simão, F.A. , Waterhouse, R.M. , Ioannidis, P. , Kriventseva, E.V. , and Zdobnov, E.M. (2015) BUSCO: assessing genome assembly and annotation completeness with single‐copy orthologs. Bioinformatics 31: 3210–3212. [DOI] [PubMed] [Google Scholar]
- Smith, T.A. , Driscoll, T. , Gillespie, J.J. , and Raghavan, R. (2015) A Coxiella‐like endosymbiont is a potential vitamin source for the lone star tick. Genome Biol Evol 7: 831–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder, A.K. , Deberry, J.W. , Runyen‐Janecky, L. , and Rio, R.V.M. (2010) Nutrient provisioning facilitates homeostasis between tsetse fly (Diptera: Glossinidae) symbionts. Proc R Soc Lond B: Biol Sci 277: 2389–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stackebrandt, E. , and Goebel, B.M. (1994) Taxonomic note: a place for DNA–DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol 44: 846–849. [Google Scholar]
- Stamatakis, A. (2014) RAxML version 8: a tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics 30: 1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stincone, A. , Prigione, A. , Cramer, T. , Wamelink, M.M.C. , Campbell, K. , Cheung, E. , et al (2015) The return of metabolism: biochemistry and physiology of the pentose phosphate pathway. Biol Rev 90: 927–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, H.Y. , Noe, J. , Barber, J. , Coyne, R.S. , Cassidy‐Hanley, D. , Clark, T.G. , et al (2009) Endosymbiotic bacteria in the parasitic ciliate Ichthyophthirius multifiliis . Appl Environ Microbiol 75: 7445–7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takami, H. , Taniguchi, T. , Arai, W. , Takemoto, K. , Moriya, Y. , and Goto, S. (2016) An automated system for evaluation of the potential functionome: MAPLE version 2.1.0. DNA Res 23: 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura, K. , Stecher, G. , Peterson, D. , Filipski, A. , and Kumar, S. (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30: 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, P.L. , Blakely, K.M. , Leon, G.P. , , de Walker, J.R. , McArthur, F. , Evdokimova, E. , et al (2008) Structure and function of sedoheptulose‐7‐phosphate isomerase, a critical enzyme for lipopolysaccharide biosynthesis and a target for antibiotic adjuvants. J Biol Chem 283: 2835–2845. [DOI] [PubMed] [Google Scholar]
- Tzeng, Y.‐L. , Datta, A. , Strole, C. , Kolli, V.S.K. , Birck, M.R. , Taylor, W.P. , et al (2002) KpsF is the arabinose‐5‐phosphate isomerase required for 3‐deoxy‐d‐manno‐octulosonic acid biosynthesis and for both lipooligosaccharide assembly and capsular polysaccharide expression in Neisseria meningitidis . J Biol Chem 277: 24103–24113. [DOI] [PubMed] [Google Scholar]
- Uehara, T. , and Park, J.T. (2007) An anhydro‐N‐acetylmuramyl‐l‐alanine amidase with broad specificity tethered to the outer membrane of Escherichia coli . J Bacteriol 189: 5634–5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Hurk, A.F. , Hall‐Mendelin, S. , Pyke, A.T. , Frentiu, F.D. , McElroy, K. , Day, A. , et al (2012) Impact of Wolbachia on infection with chikungunya and yellow fever viruses in the mosquito vector Aedes aegypti . PLoS Negl Trop Dis 6: e1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velimirov, B. , and Hagemann, S. (2011) Mobilizable bacterial DNA packaged into membrane vesicles induces serial transduction. Mob Genet Elem 1: 80–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, K. , Yukuhiro, F. , Matsuura, Y. , Fukatsu, T. , and Noda, H. (2014) Intrasperm vertical symbiont transmission. Proc Natl Acad Sci USA 111: 7433–7437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert, L.A. , Werren, J.H. , Aebi, A. , Stone, G.N. , and Jiggins, F.M. (2009a) Evolution and diversity of Rickettsia bacteria. BMC Biol 7: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert, L.A. , Welch, J.J. , and Jiggins, F.M. (2009b) Conjugation genes are common throughout the genus Rickettsia and are transmitted horizontally. Proc R Soc Lond B: Biol Sci 276: 3619–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert, L.A. , Araujo‐Jnr, E.V. , Ahmed, M.Z. , and Welch, J.J. (2015) The incidence of bacterial endosymbionts in terrestrial arthropods. Proc R Soc Lond B: Biol Sci 282: 20150249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren, J.H. , Hurst, G.D. , Zhang, W. , Breeuwer, J.A. , Stouthamer, R. , and Majerus, M.E. (1994) Rickettsial relative associated with male killing in the ladybird beetle (Adalia bipunctata). J Bacteriol 176: 388–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren, J.H. , Baldo, L. , and Clark, M.E. (2008) Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol 6: 741–751. [DOI] [PubMed] [Google Scholar]
- Wood, D.W. , Setubal, J.C. , Kaul, R. , Monks, D.E. , Kitajima, J.P. , Okura, V.K. , et al (2001) The genome of the natural genetic engineer Agrobacterium tumefaciens C58. Science 294: 2317–2323. [DOI] [PubMed] [Google Scholar]
- Zouache, K. , Voronin, D. , Tran‐Van, V. , and Mavingui, P. (2009) Composition of bacterial communities associated with natural and laboratory populations of Asobara tabida infected with Wolbachia . Appl Environ Microbiol 75: 3755–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong, J. , Jasinskas, A. , and Barbour, A.G. (2007) Antibiotic treatment of the tick vector Amblyomma americanum reduced reproductive fitness. PLoS One 2: e405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's web‐site:
Fig. S1. Taxon annotated GC‐coverage plots. (A) Primary genome assembly of Culicoides newsteadi N5. (B) Postfiltering against a local database containing all available (complete and draft) Rickettsia genomes.
Fig. S2. BUSCO completeness assessment results for RiCNE draft genome in relation to selected complete Rickettsia genomes. The results are based on the presence or absence of 148 single‐copy universal bacterial markers. BUSCO notation: complete (C), single‐copy (S), duplicated (D), fragmented (F) and missing (M).
Fig. S3. Circular representation of the RiCNE draft genome. For visualization purposes, the 193 scaffolds were concatenated into a pseudomolecule. Alternating gray and white strips indicate scaffold borders. Inwards, the first, second and third circles are color‐coded according to the eggNOG functional categories and represent: (a) the complete RiCNE protein‐coding genes (CDSs), (b) CDSs universally present in all Rickettsiacea genomes used in this study and (c) RiCNE unique CDSs respectively. The fourth circle shows the clusters of the T4SS (red) and conjugation (purple) genes. The first line plot represents the genome coverage. An orange line indicates the average coverage of the draft assembly (∼ 76×). The asterisks indicate the two scaffolds containing the conjugation genes represented by 2–3× higher than average coverage indicating possible multiple copies. The innermost line plot represents the GC% coverage calculated based on a 1 kb sliding window. The circular plot was generated with Circos v0.69 (Krzywinski et al., 2009).
Fig. S4. Cladogram of the complete core‐genome phylogeny. Phylogenomic placement of the Rickettsia endosymbiont of C. newsteadii were inferred using maximum likelihood (RaxML, model: Lag + G + I) from the concatenated protein alignments of 189 single copy ortholog genes. Support values are based on 100 rapid bootsrap replicates.
Fig. S5. Individual trees for the pentose phosphate pathway (PPP) proteins. Tree topology and posterior probabilities were inferred with MrBayes using a mixed model of amino acid substitution. The trees were midpoint rooted.
Fig. S6. Maximum likelihood phylogeny of the omp gene. The tree topology was estimated using RaxML and the GTR + G model of nucleotide substitutions. Support values are based on 1000 rapid bootstrap replicates. The tree was midpoint rooted.
Table S1. Functional annotation of RiCNE draft genome including Interproscan results against Pfam and eggNOG results.
Table S2. Publicly available Rickettsiaceae genomes used in this study for ortholog identification and phylogenomic analysis.
Table S3. Genes encoding for the P‐T4SS and the tra conjugative DNA‐transfer element in RiCNE genome.
TableS4. RiCNE unique genes.
Table S5. RiCNE unique genes putatively associated with host invasion and host–microbe interactions.
Table S6. omp conventional PCR assay results for Rickettsia‐negative Culicoides species under study, given by subgenus, species, location, date and sex. a Culicoides newsteadi haplotype N1 designated by Ander et al. (2013). b Culicoides newsteadi N6 previously undesignated.
Table S7. Genetic characteristics of housekeeping and omp alleles.
Table S8. Rickettsia strains recovered from Culicoides midges, with allelic profiles; strains sharing the same allelic profiles at all five loci were designated as a single strain. NA: non amplifiable.
Table S9. Pairwise divergence at individual loci between strains from clonal complex 2 showing remarkable divergence in the ATPase allele compared to the average of all strains, most likely as a result of a recombination event.
Table S10. Core genes used for phylogenomic analysis.
Table S11. Housekeeping and omp gene primer attributes.