

Abstract
Aim: The mammalian lung expresses at least three aquaporin (AQP) water channels whose precise role in lung injury or inflammation is still controversial. Materials and Methods: Three murine models of lung inflammation and corresponding controls were used to evaluate the expression of Aqp1, Aqp4, Aqp5 and Aqp9: lipopolysaccharide (LPS)-induced lung injury; HCl-induced lung injury; and ventilation-induced lung injury (VILI). Results: All models yielded increased lung vascular permeability, and inflammatory cell infiltration in the broncho-alveolar lavage fluid; VILI additionally produced altered lung mechanics. Lung expression of Aqp4 decreased in the models that targeted primarily the alveolar epithelium, i.e. acid aspiration and mechanical ventilation, while Aqp5 expression decreased in the model that appeared to target both the capillary endothelium and alveolar epithelium, i.e. LPS. Conclusion: Participation of aquaporins in the acute inflammatory process depends on localization and the type of lung injury.
Keywords: Aquaporins, ARDS, inflammation
Acute respiratory distress syndrome (ARDS) is a distressing illness associated with increased morbidity and mortality. It is characterized by diffuse alveolar injury, intense activation of the coagulation system and increased capillary permeability. ARDS-inducing events (e.g. sepsis, trauma, aspiration and pneumonia) are quite common, yet only a percentage of patients develop the syndrome. This differential response has led to the investigation of potential factors that contribute to ARDS susceptibility and prognosis (1,2).
The discovery of the aquaporin water-channels unveiled the molecular mechanism of water transport across membranes (3). So far 13 mammalian aquaporins involved in water movement have been detected in the kidney, lung, eye and brain (4). Aquaporins are hence being considered a target for therapy in diseases caused by abnormalities in water homeostasis (5).
In lung injury, neutrophilic recruitment characterizes the inflammatory response, with severe injury to the alveolar endothelial and epithelial barriers (6). At least three aquaporin water channels are located in the mammalian lung: AQP1 in microvascular endothelia and some pneumocytes (7,8), AQP4 at the basolateral membrane of airway epithelium (9), and AQP5 at the type-I alveolar epithelial cell apical membranes (9); additionally, AQP9, an aquaglyceroporin that is also permeable to a wide variety of solutes, plays a role in membrane protrusion formation and cell migration (10). This distinct distribution in the lung suggests that aquaporins are central in water movement between the air space and the capillary compartments (3). Furthermore, results from our laboratory using gene expression profiling demonstrated that leukocyte aquaporin-1 is differentially expressed in sepsis and ARDS (11). Since it is evident that the lung vascular epithelium and endothelium are crucial in the lung inflammatory response (12) and AQPs facilitate water permeability between the alveolar compartment and vasculature (13), in the present study we aimed to address the potential role of AQP1, 4, 5 and 9 in the acute inflammatory process of three murine experimental models of lung injury, namely lipopolysaccharide (LPS)-induced and HCl-induced direct lung injury, and ventilation-induced lung injury (VILI).
We chose to study the aforementioned three models, which produce lung injury amongst which increased lung vascular permeability, inflammatory cell infiltration in the broncho-alveolar lavage fluid (BALF), and altered lung mechanics. The fact that alveolar oedema is a major injurious feature in these models makes them a good choice for the study of AQPs, molecules highly involved in osmotic water movement in the lung. It should be noted that as yet no single experimental animal model has been able to yield all the histopathological features of ARDS adequately (14), while the chosen models reflect common underlying causes of lung injury, that is sepsis – severe pneumonia, aspiration of harmful substances and injury occurring from mechanical ventilation.
Materials and Methods
Animals. All experimentation was approved by the Evangelismos Hospital Research Review Board - Ethics Committee (experimental study approval number 206), as well as by the Veterinary Service of the governmental prefecture of Attica, Greece (approval protocol number K/3792, K/2176, K/2654). The study was conducted in compliance with the European Union Directive 2010/63/EU on animal experimentation. Animals were monitored daily and were randomly assigned into groups. All surgery was performed under deep anesthesia achieved by intraperitoneal administration of ketamine/xylazine (100 mg/kg and 10 mg/kg, respectively). All efforts were made to minimize animal distress and suffering. Euthanasia was performed under deep anesthesia by exsanguination.
Eight- to 14-week-old male mice, with a mean±standard deviation weight at the time of experiments of 23.8±2.7 g, were bred and maintained on a C57BL/6 background in the animal facilities of the Hellenic Pasteur Institute (Athens, Greece) under specific pathogen-free conditions. Mice were then transported to the Animal Model Research Unit of Evangelismos Hospital where they were housed at 20-22˚C, with 55±5% humidity, and a 12-hour light-dark cycle; food and water was given ad libitum during the experimental period.
Experimental groups and treatments. Three murine models of acute lung inflammation and corresponding controls (a separate control group per each model) were used to evaluate the expression of aquaporins: i) LPS-induced lung injury, ii) HCl-induced lung injury, and iii) ventilation-induced lung injury (VILI).
To induce LPS-induced lung injury, mice were exposed to intratracheal (i.t.) instillation of 5 mg/kg LPS (Sigma-Aldrich, St Louis, MO, USA). Control mice received i.t. placebo (normal saline-NS) at equal volumes. The study interventions were instillation of LPS (n=9) or NS (n=9) to mice. The animals were sacrificed 24 h post treatment.
To induce HCl-induced lung injury, mice were exposed to i.t. instillation of 0.1 N HCl (n=9). Control mice received i.t. placebo (normal saline; NS) at equal volumes (n=9) to mice. The animals were sacrificed 24 h post treatment.
To induce VILI, mice were ventilated using high tidal volume (HVt) of 25 ml/kg and a respiratory rate of 50 breaths/min (n=9). Control mice were ventilated with low tidal volume (LVt) of 8 ml/kg and respiratory rate of 150 breaths/min (n=9), simulating normally breathing in adult (8-week-old) mice. Mice were sacrificed after 4 h of mechanical ventilation.
Following sacrifice, lung tissues were obtained and snap-frozen in liquid nitrogen. Total RNA and protein were isolated from all lung tissues. Aquaporin mRNA was quantified by real-time polymerase chain reaction (RT-PCR), and protein levels were measured by immunoblotting.
Intratracheal injection. Mice were anesthetized with ketamine/xylazine (100/5 mg/kg) intraperitoneally, the trachea was exposed via median incision and 50 μl of LPS, HCl, or NS were injected using a 500 μl syringe and 27-gauge needle. The incision was closed with continuous 3-0 silk suture.
Mechanical ventilation. Mice were anesthetized with ketamine/ xylazine (100/5 mg/kg) intraperitoneally, the trachea was exposed under sterile conditions, cannulated with a 22-gauge catheter and sutured. Mechanical ventilation was performed using a small animal ventilator (Flexivent, Scireq, Ontario, Canada) and ambient air. Positive end-expiratory pressure was set at 2 cm H2O and Vt was set at 8 ml/kg. After an initial 5 min run-in period, two deep inflations to total lung capacity were applied in order to standardize lung volume history, followed by a 3-minute default ventilation period at the end of which baseline measurements of lung function were obtained, followed by LVt or HVt ventilation according to experimental design. Respiratory mechanics were evaluated by measuring tissue elastance coefficient (H) via forced oscillation technique. Five successive measurements were obtained at 30 s intervals (15). Following these manoeuvres, a single quasi-static pressure-volume curve was transduced in order to measure the static compliance (Cst) of the respiratory system (15).
Tissue handling and sample collection. The inferior vena cava and abdominal aorta were transected and animals were allowed to exsanguinate. Broncho-alveolar lavage (BAL) was performed by injecting and slowly aspirating three 0.5 ml aliquots of phosphate-buffered saline (PBS) at ambient temperature into the tracheal cannula. BAL fluid return from the three consecutive instillations was pooled intro pre-weighed 1.5 ml tubes. The tubes were weighed again to obtain the weight (and volume) of the recovered BAL fluid. The chest was then opened and the pulmonary circulation was flushed free of blood by slowly injecting 10 ml of PBS into the right ventricle. The right lung was then dissected lobe-by-lobe, the tissue fragments were rinsed with PBS, blotted dry on tissue paper and snap-frozen in liquid nitrogen.
Total and differential cell counts in BAL fluid. BAL fluid was centrifuged and the supernatant was aspirated and stored at −80˚C. The pellet containing BAL cells was reconstituted in PBS at a volume equal to the recovered volume of BAL fluid, as described above. After obtaining the manual total cell count in BAL fluid with a hemocytometer, 50,000 cells were centrifuged on glass slides and stained with May-Gruenwald-Giemsa stain. Differential cell counts were obtained by measuring and characterizing 100 cells/slide.
RNA isolation from lung tissues. RNA extraction was performed using Trizol® reagent and PureLink RNA Mini kit (Invitrogen, Thermofisher Scientific, Waltham, MA, USA), following the manufacturer’s instructions. Total RNA concentration and quality were determined spectrophotometrically at 260 and 280 nm, while RNA integrity was evaluated with formaldehyde agarose gel electrophoresis. Total RNA was stored at −80˚C until used.
RT-PCR. One hundred nanogrammes of total RNA from each sample were reverse-transcribed into single-stranded cDNA in a 10 μl reaction mixture, using Primescript RT reagent kit from Takara (Takara Bio Inc., Shiga, Japan), following the manufacturer’s instructions. The success of the synthesis of the single-stranded cDNA was tested by its PCR amplification. PCR was performed using 5.0 μl of cDNA, 1.5 mM MgCl2, 400 μM dNTPs, 500 nM primers, 1.5 U of Dream-Taq DNA polymerase and 1x reaction buffer (Fermentas, Thermofisher Scientific), in a PTC-200 thermocycler (MJ Research Inc., Waltham, MA, USA). Equal amounts (10 μl) of all amplicons were electrophoresed on a 2.5% agarose gel, visualized following RedGel staining (Biotium Inc., Hayward, CA, USA) and photographed under ultraviolet light with a Kodak DC120 digital camera (Kodak, Rochester, NY, USA).
Quantitative real-time PCR. A highly sensitive quantitative real-time PCR method has been developed for the quantification of both glyceraldehyde 3-phosphate dehydrogenase (Gapdh) and Aqp1, -4, -5 and -9 mRNAs, with the use of SYBR® Green Dye detection systems. For the amplification of Gapdh (endogenous reference gene), as well as the different Aqp target gene mRNA sequences, gene-specific sets of primers were designed according to the information on the National Centre for Biotechnology Information Sequence database and Primer Express program (Applied Biosystems, Thermofisher Scientific). In order to avoid genomic DNA amplification, the primers were chosen to span at least two exons. The sequences of the primers used and the size of the amplicons in base pairs (bp) produced are listed in Table I.
Table I. Sequences of the primers used and the size of the amplicons produced.

Quantitative real-time PCR analysis was performed in 96-well plates on a PTC-200 thermocycler (MJ Research Inc.). The 25 μl reaction mixture contained 10 ng cDNA, 300 nM primers and 1xKAPA SYBR® Fast qPCR Master Mix Universal (Kapa Biosystems, Boston, MA, USA), in which a KAPA SYBR® DNA polymerase is included. The thermal protocol conditions consisted of 2 min polymerase activation step at 95˚C, 40 cycles of denaturation at 95˚C for 15 s, primer annealing at 53.8˚C for 15 s and extension at 72˚C for 10 s. All samples were amplified in triplicates and the average threshold cycle (CT) values were calculated for subsequent expression analysis. Following amplification, a dissociation curve was generated to distinguish the PCR products of interest from the non-specific ones or any primer-dimers, through their specific melting temperatures (Tm), recorded in the software. Using the comparative CT method 2−DDCT (16) and NS or LVt samples as a calibrator, the relative quantification of the expression analysis of all lung samples was carried out. Gapdh expression was used for the normalization of Aqp mRNA expression levels between samples from the different groups.
Lung tissue homogenate. Approximately 50 mg of lung tissue was homogenized in a triplicate volume of lysis buffer (250 mM sucrose, 1% Triton X-100, 1 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride, 10 mM Tris-HCl, pH 7.4, 150 mM NaCl and protease inhibitors using a hand-held homogenizer (Tissue-Tearor; Biospec Products, Bartlesville, OK, USA) and 3×30 s bursts. The homogenate was centrifuged in a 5804 R Eppendorf centrifuge (rotor A-4-44; Eppendorf AG, Hamburg, Germany) for 5 min at 4˚C and 1,000 × g. The supernatant, referred to as “total protein”, was collected and stored at −80˚C until used.
Total protein determination. Total protein content in BAL fluid and lung tissue homogenates was obtained using the bicinchoninic acid reaction, as described (17).
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). SDS-PAGE was performed on a Biorad Mini Protean II electrophoresis apparatus (Bio-Rad, Hercules, CA, USA), as described (18), using 12% polyacrylamide slab gels. Electrophoresis was carried out at 150 V for 1 h at room temperature.
Immunoblotting of lung tissue homogenates. Following electrophoresis, samples were transferred onto an Immobilon-P polyvinylidene difluoride membrane (0.45 μl pore size; Merck Millipore, Merck KGaA, Darmstadt, Germany). Western transfer was performed on a wet transfer apparatus (Bio-Rad). Immunological detection was performed according to the method of Batteiger et al. (19). Polyclonal antibodies against AQP1 and AQP9 were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA); AQP4, AQP5 and actin from Merck Millipore and GAPDH from Cell Signaling Technology, Inc. (Danvers, MA, USA). The final blots were visualized by enhanced chemiluminescence (PerkinElmer, Inc., Waltham, MA, USA) developed on Agfa Ortho CP-G Plus films (Agfa HealthCare NV, Mortsel, Belgium). The image was then analyzed by densitometry (Gel-pro analyzer 4.5; Media Cybernetics, Inc., Silver Spring, MD, USA), which evaluates the relative amount of protein staining and quantifies the results in terms of optical density. The density of each band was compared with the band expressed by the loading controls, actin, or in the VILI model by GAPDH, since HVt caused an obvious decrease in actin expression. The intensity ratio of the difference was used to compare protein expression following injury.
Statistical analysis. Data are presented as the mean±SEM. Two-group comparisons were performed by unpaired Student’s t-test or the non-parametric Mann–Whitney test for skewed data. Statistical analysis was performed using Graphpad Prism 6.01 for Windows (GraphPad Software, San Diego, CA, USA); differences with two-tailed p-values of less than 0.05 were considered significant.
Results
Respiratory function in VILI. Decreased lung compliance can provoke respiratory failure in ARDS. Using a high-stretch ventilation protocol we observed significant alterations in lung mechanics at 4 h (Figure 1A and B). Specifically, under HVt, the elastance coefficient H peaked at 82.27±21.24 cmH2O/ml, with LVt values at 29.68±1.36 cmH2O/ml (Figure 1A; p<0.05). To support the above, the reciprocal of elastance, quasi-static lung compliance, was measured by pressure volume curve and was reduced by approximately 35% in the HVt group compared to LVt ventilation (Figure 1B; p<0.01).
Figure 1. Respiratory mechanics, microvascular permeability and alveolar airspace inflammation evaluation in mice undergoing ventilation with low (LVt; 8 ml/kg, N=9) or high (HVt; 25 ml/kg, N=9) tidal volume for 4 h, VILI model. A, B: Lung mechanical changes. Lung tissue elastance coefficient (H) was evaluated via forced oscillation technique (A), while static compliance (Cst) was evaluated by a single quasi-static pressurevolume curve (B). C: Microvascular permeability. Total protein in bronchoalveolar lavage fluid samples obtained from mice 4 h after LVt or HVt ventilation (VILI model) was used to quantify microvascular barrier integrity in the pulmonary circulation. D: Alveolar airspace inflammation. Total cell count in bronchoalveolar lavage fluid collected as above served as a marker of lung infiltration by inflammatory cells. Data are presented as mean±SEM. Unpaired Student’s t-test was performed. Statistically significantly different from respective LVt control animals at *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.

Airspace protein leak and airspace inflammation. The degree of alveolo-capillary membrane permeability in lung injury/ARDS can be approximated by measuring total protein in BAL fluid, while cell counts in BAL fluid serve as a marker of the lungs’ inflammatory response to various lung injury-inducing molecules. Mice treated with HVt ventilation compared to mice treated with LVt ventilation showed significantly higher levels of total protein in BAL fluid (8.8-fold; Figure 1C; p<0.001), and furthermore there was an inflammatory response in mice treated with HVt ventilation compared to the animals given LVt ventilation, as reflected by higher cell counts in BAL fluid (1.8-fold rise; Figure 1D; p<0.0001).
Likewise, in LPS-challenged mice, a 2.6-fold increase in BAL fluid protein was noted compared to the respective controls (Figure 2A; p<0.01). Following LPS exposure in mice, we also noted a nearly 30-fold rise in cell counts in BAL fluid compared to corresponding NS-receiving controls (Figure 2B; p<0.0001).
Figure 2. Evaluation of microvascular permeability (A,C) and alveolar airspace inflammation (B, D) in the lipopolysaccharide (LPS) (A, B) and HCl (C, D) models. Total protein in bronchoalveolar lavage fluid samples obtained from mice 24 h after intratracheal instillation of normal saline (NS), lipolysaccharide (LPS) or HCl was used to quantify microvascular barrier integrity in the pulmonary circulation. The total cell count in bronchoalveolar lavage fluid collected as above served as a marker of lung infiltration by inflammatory cells. Data are presented as mean±SEM. Unpaired Student’s t-test (D) or Mann-Whitney test for skewed data (A, B, C) was performed, as appropriate. Statistically significantly different from respective NS control animals at **p<0.01, ***p<0.001, ****p<0.0001 (N=9).

In mice subjected to i.t. HCl, total protein in BAL fluid rose 2.4-fold compared to the respective NS-treated mice (Figure 2C; p<0.001), while cell counts were also significantly higher in HCl-treated mice compared to respective controls (4.5-fold rise; Figure 2D; p<0.0001).
The aforementioned rise was associated with a shift in cell type in BAL fluid, from primarily monocytic to overwhelmingly neutrophilic (data not shown).
Aquaporin mRNA expression. To evaluate changes in Aqp expression associated with the acute inflammatory process of the tested experimental lung injury models, RT-PCR was used to analyze mRNA expression 24 h following LPS or HCl instillation, and after 4 h of mechanical ventilation. Real-time PCR showed that in the LPS model, Aqp5 mRNA expression significantly decreased and Aqp9 mRNA expression increased compared to the respective control group (Figure 3A; p<0.05 respectively). Aqp1 and Aqp4 mRNA expression in the experimental group did not significantly change compared to the respective control group (Figure 3A).
Figure 3. Relative expression of aquaporin mRNA in the lungs of mice of the three experimental models. Relative expression of Aqp1, Aqp4, Aqp5 and Aqp9 was determined by real-time polymerase chain reaction. Normal saline (NS) or low tidal volume (LVt) comprised the calibrator. A: Lipopolysaccharide (LPS) model: Lungs were obtained from mice 24 h after intratracheal instillation of NS or 5 mg/kg LPS. B: HCl model: Lungs were obtained from mice 24 h after intratracheal instillation of normal saline (NS) or 0.1 N HCl. C: VILI model: Lungs were obtained from mice 4 h after LVt (8 ml/kg) or high tidal volume ventilation (25 ml/kg). The amount of the target gene normalized to that of the endogenous control gene (glyceraldehyde 3-phosphate dehydrogenase), and relatively to the corresponding calibrator, was calculated for each lung injury model separately using the formula 2–DDCT. Levels of each gene are expressed as the fold change relative to that of the corresponding control animals. Data are presented as mean±SEM. Closed bars, controls (NS or LVE); open bars, treatment. Mann-Whitney test for skewed data was performed. Statistically significantly different from respective NS or LVt control animals at *p<0.05 (N=9).
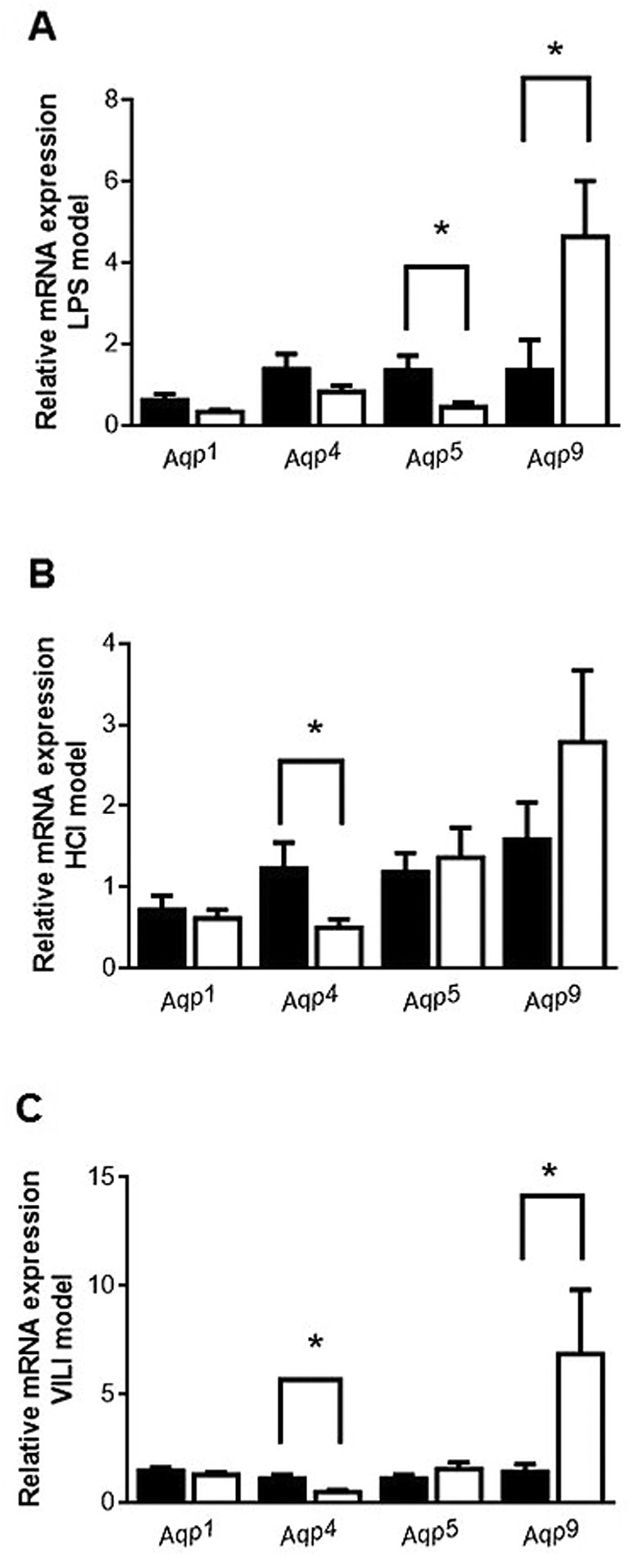
In the HCl-induced lung injury model, Aqp4 mRNA expression significantly decreased compared to the respective control group (Figure 3B; p<0.05), while Aqp1, Aqp5 and Aqp9 mRNA expression did not show any change compared to the respective control group (Figure 3B).
In the VILI model, Aqp4 mRNA expression was reduced in HVt mice compared to the mice given default ventilation, while Aqp9 mRNA was increased (Figure 3C; p<0.05, respectively). Aqp1 and Aqp5 mRNA showed no change in expression (Figure 3C).
Aquaporin protein expression. Immunoblotting was performed on lung homogenates of the experimental animals using antibodies raised against AQP1, AQP4, AQP5 and AQP9 and reference antibodies against actin and GAPDH (for VILI).
The results of the immunoblotting experiments showed that in the LPS model, only protein expression of AQP5 significantly decreased in the animals given LPS compared to the control animals, whereas the protein levels of the other three aquaporins remained unaltered (Figure 4A; p<0.01).
Figure 4. Expression of aquaporin protein in the lungs of the three experimental models. A: Lipopolysaccharide (LPS) model: Lungs were obtained from mice 24 h after intratracheal instillation of NS or 5 mg/kg LPS. B: HCl model: Lungs were obtained from mice 24 h after intratracheal instillation of normal saline (NS) or 0.1 N HCl. C: VILI model: Lungs were obtained from mice 4 h after low (8 ml/kg) or high tidal volume ventilation (25 ml/kg). Change in protein expression of AQP1, -4, -5 and -9 was determined in crude protein samples isolated from lungs, as analyzed by electrophoresis and immunoblotting. The density of each band was compared with the band expressed by the loading controls (actin, or glyceraldehyde 3-phosphate dehydrogenase in the VILI model). The intensity ratio of the difference was used to compare protein expression results following injury. Closed bars, controls (NS or low tidal volume ventilation); open bars, treatment. Data are presented as mean±SEM. Mann-Whitney test for skewed data was performed. Statistically significantly different from respective NS or low tidal volume control animals at *p<0.05, **p<0.01 (N=9).
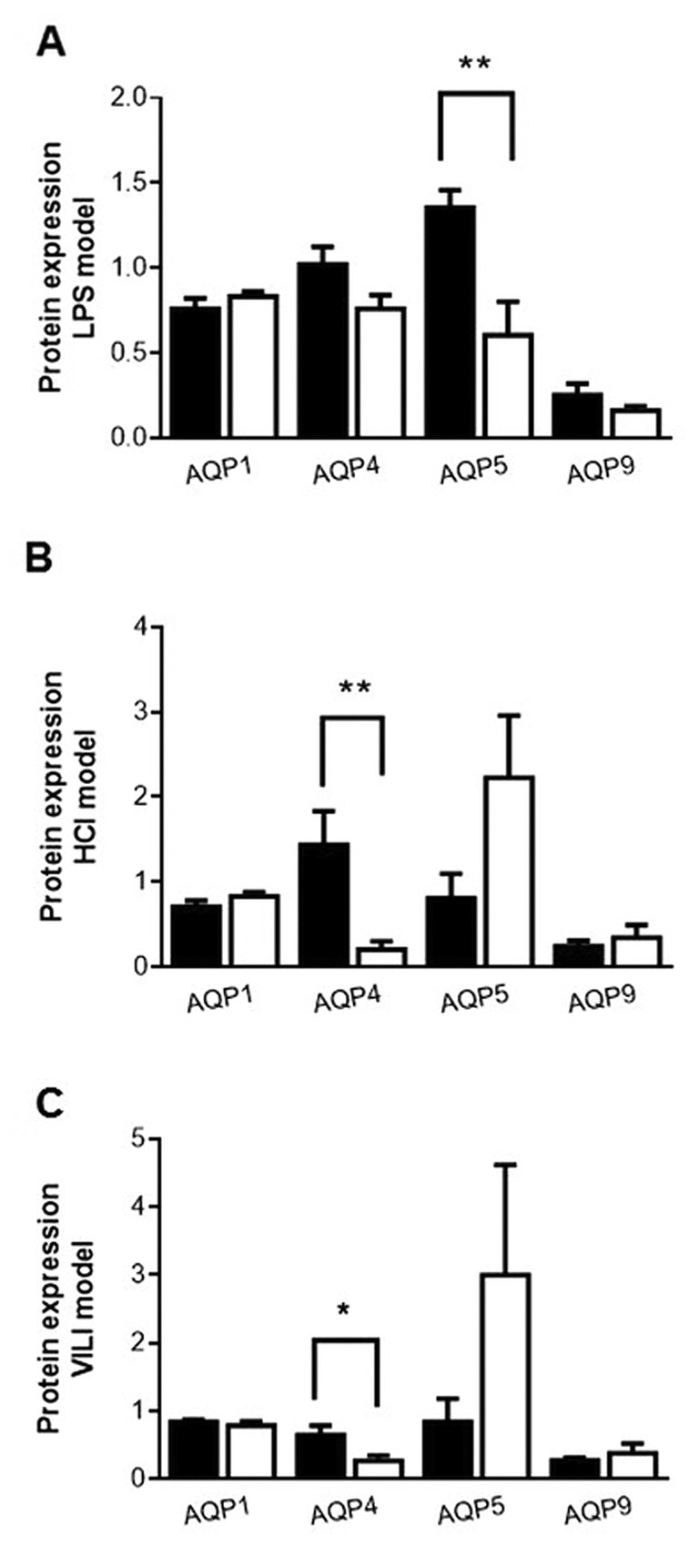
In the HCl-induced model, in agreement with the real-time PCR results, only the levels of AQP4 protein decreased in the HCl-treated animals compared to the respective control group (Figure 4B; p<0.01). Levels of AQP1, AQP5 and AQP9 protein expression remained unaltered (Figure 4B).
In the VILI model, AQP4 protein expression was reduced in the HVt animals (Figure 4C; p<0.05), in accordance with the mRNA expression results, and levels of AQP1 and AQP5 remained unaltered; however, AQP9 protein expression did not show any change in expression, despite its differential mRNA expression (Figure 4C).
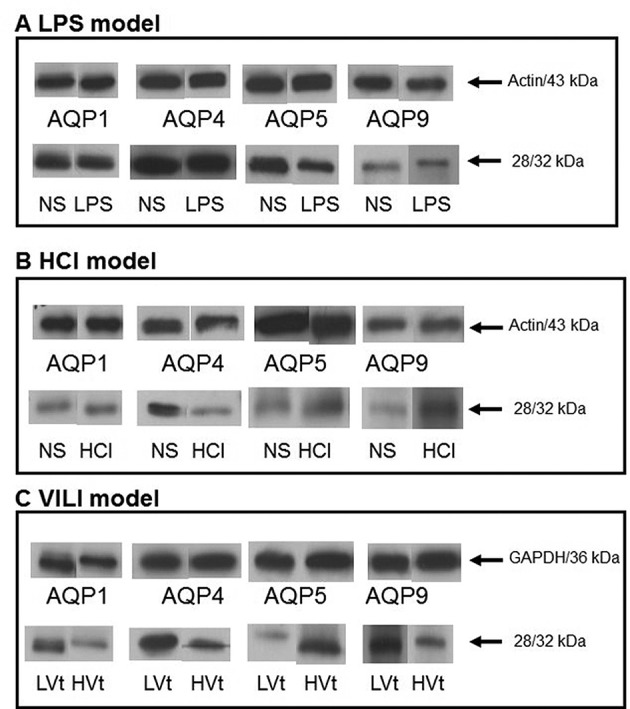
Figure 5 shows representative immunoblotting performed on lung homogenates of the experimental animals using antibodies raised against AQP1, AQP4, AQP5 and AQP9.
Figure 5. Representative protein expression of aquaporins AQP1, -4, -5 and -9. Lungs were obtained from mice 24 h after intratracheal instillation of normal saline (NS) or 5 mg/kg lipopolysaccharide (LPS) (LPS model) (A), or 0.1 N HCl (HCl model) (B), or 4-h after low (8 ml/kg; LVt) or high (25 ml/kg; HVt) tidal volume ventilation (VILI model) (C). The samples were analysed by sodium dodecyl sulfate– polyacrylamide gel electrophoresis followed by immunoblotting using actin (or glyceraldehyde 3-phosphate dehydrogenase in VILI) as loading controls. MW: AQP1=28 kDa; AQP4=32 kDa; AQP5=28 kDa; AQP9=32 kDa.
Discussion
The aim of this study was to test whether different aquaporins expressed in different lung compartments have a role in the acute lung inflammatory process observed in response to i.t. instillation of LPS and HCl, and mechanical ventilation. Our results indicate that aquaporin involvement is not consistent but depends both on localization and the lung injury model chosen to reflect the common underlying causes of lung injury and ARDS.
More specifically, our results showed that Aqp1 expression remained unaltered in all three models of lung injury tested, Aqp4 significantly decreased in HCl-induced lung injury and VILI, whereas Aqp5 significantly decreased in the LPS model. As far as Aqp9 is concerned, its mRNA expression increased in the LPS-induced and VILI models; nevertheless its protein expression remained unaltered in all three models.
At present, studies on the function of aquaporins in lung injury are mainly focused on AQP1 and AQP5, with limited research on AQP4 and AQP9. The main conclusion so far has been that, despite differences in expression in the various studies, AQP1 and AQP5 provide the principal route for osmotically driven water transport between airspace and capillary compartments (20-27). However, Song et al. used mice deficient in AQP1, AQP4 and AQP5 to test the hypothesis that aquaporins are important in neonatal lung fluid balance, adult lung fluid clearance and formation of lung oedema after acute lung injury (24). They demonstrated that despite their established role in epithelial and endothelial osmotic water permeability, the major lung aquaporins have little importance in active alveolar fluid clearance in the neonatal and adult lung, or in the accumulation of fluid in the injured lung.
In view of these rather inconsistent findings, and the fact that leukocyte AQP1 expression has been found increased in patients with severe sepsis and ARDS by gene-expression profiling analysis (11), we sought to investigate the expression of three water-selective aquaporins, namely AQP1, AQP4, AQP5, and of the aquaglyceroporin AQP9 in three murine models of direct acute lung injury, mimicking human ARDS, which target the capillary or alveolar epithelium. Aquaporin expression was evaluated 24-h following LPS-induced injury (28), 24-h following a previously employed model of acid aspiration lung injury (29,30), and after 4-h of mechanical ventilation-induced injury (31). All three models of induced lung injury produced increased lung vascular permeability (endothelial cell injury), inflammatory cell infiltration (mainly neutrophilic) in the BAL fluid, and in addition, altered lung mechanics in VILI.
Our results showed that in all three lung injury models both Aqp1 mRNA and protein expression levels were similar to those in control animals. Up to now, studies have shown rather controversial results on this subject. More specifically, Aqp1 expression has been shown to be either reduced or increased in various lung injury models (20-23,26,32,33); nevertheless studies on knock-out animals have not been able to confirm the role of Aqp1 in lung inflammation and oedema (25,34,35). Hence, our results for Aqp1 agree with the suggestion that alveolar fluid clearance and oedema accumulation in response to acute lung injury do not appear to require high AQP1-mediated lung water permeability.
Aqp4 and Aqp5 exhibited a lung-injury target-specific regulation of gene and protein expression. Decreased expression of Aqp4, which is found at the basolateral membrane of airway epithelium, was observed in the acid aspiration and VILI models, in which injury primarily occurs at the alveolar epithelium. Several studies have shown that acid aspiration causes impairment in the alveolar epithelial fluid transport function, resulting in changes in alveolar fluid clearance (36,37), whereas overstretching of alveolar walls results in endothelial and epithelial breaks and interstitial oedema (38). In the airways, although AQP3 and AQP4 facilitate osmotic water transport, their gene deletion does not impair airway hydration, regulation of airway surface liquid or fluid absorption (27). However, in various non-endothelial cell lines, Aqp4 knockout or knockdown has been found to inhibit proliferation and migration (39,40). Thus, the observed decreased expression of Aqp4 might impair lung regeneration, via reduced cell migration and wound healing.
On the other hand, Aqp5 expression decreased in the LPS model. Although this model has been suggested to primarily target the capillary endothelium (41), AQP5 is mainly localized in alveolar epithelia. This apparent discrepancy may be explained by a recent study by Woods et al. (42) who demonstrated that in LPS-induced lung injury, the endothelium response is less apparent than that of epithelial cells, with the clearest response exhibited by alveolar macrophages. On that note, AQP5 has been suggested to play a role in epithelial barrier maintenance and its deletion has been shown to aggravate lung injury by Pseudomonas aeruginosa (43). Cellular injury induced by LPS may be related to increased apoptosis (41), while AQP5 has been shown to play a role in promoting cell proliferation and inhibiting apoptosis in various cancer types (44-46). Thus the decreased expression of Aqp5 in our LPS model may partially contribute in such an apoptosis-related injurious mechanism.
Lung expression of Aqp9 has scarcely been reported; it appears to be mainly expressed and play an active role in neutrophil volume and migration (47). In our experiments, Aqp9 mRNA expression increased in the LPS and VILI models; LPS administration has been shown to be followed by large increases of neutrophils in the air spaces (48), whilst in addition to mechanical damage, increased permeability in VILI also requires the presence of neutrophils (49). AQP9 protein levels remained, however, unaltered, denoting the complexity of aquaporin regulation, which includes transcriptional, post-translational, protein-trafficking and channel-gating mechanisms that are frequently distinct for each family member. The observed Aqp9 expression could be lung tissue- and neutrophil-derived since tight neutrophil adherence to the lung vascular bed and alveoli has been shown post perfusion of the lungs (50). This unique subpopulation of neutrophils exhibits decreased apoptosis when compared to cells from control animals (51); such a mechanism might have contributed to the increased Aqp9 mRNA expression in our animals.
Our results show that Aqp1 expression remains unaltered in all tested lung injury models, Aqp4 decreases in the models that primarily target the alveolar epithelium, i.e. acid aspiration and mechanical ventilation, while Aqp5 decreases in the model that targets both the endothelium and epithelium, i.e. LPS. Increased Aqp9 expression is observed at a molecular level in the LPS and VILI models, however at the protein/functional level does not seem to be part of the inflammatory response.
Conflicts of Interest
The Authors declare that there is no conflict of interest in regard to this study.
Acknowledgements
This work was funded by the nonprofit institute THORAX Research Center for Intensive and Emergency Thoracic Medicine, Athens, Greece.
References
- 1.Garcia JG. Searching for candidate genes in acute lung injury: SNPs, Chips and PBEF. Trans Am Clin Climatol Assoc. 2005;116:205–219. [PMC free article] [PubMed] [Google Scholar]
- 2.Kamp R, Sun X, Garcia JG. Making genomics functional: deciphering the genetics of acute lung injury. Proc Am Thorac Soc. 2008;5:348–353. doi: 10.1513/pats.200709-152DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Verkman AS, Matthay MA, Song Y. Aquaporin water channels and lung physiology. Am J Physiol Lung Cell Mol Physiol. 2000;278:L867–879. doi: 10.1152/ajplung.2000.278.5.L867. [DOI] [PubMed] [Google Scholar]
- 4.Agre P, King LS, Yasui M, Guggino WB, Ottersen OP, Fujiyoshi Y, Engel A, Nielsen S. Aquaporin water channels–from atomic structure to clinical medicine. J Physiol. 2002;542:3–16. doi: 10.1113/jphysiol.2002.020818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yasui M. Molecular mechanisms and drug development in aquaporin water channel diseases: structure and function of aquaporins. J Pharmacol Sci. 2004;96:260–263. doi: 10.1254/jphs.fmj04004x4. [DOI] [PubMed] [Google Scholar]
- 6.Lai KN, Leung JC, Metz CN, Lai FM, Bucala R, Lan HY. Role for macrophage migration inhibitory factor in acute respiratory distress syndrome. J Pathol. 2003;199:496–508. doi: 10.1002/path.1291. [DOI] [PubMed] [Google Scholar]
- 7.Folkesson HG, Matthay MA, Hasegawa H, Kheradmand F, Verkman AS. Transcellular water transport in lung alveolar epithelium through mercury-sensitive water channels. Proc Natl Acad Sci USA. 1994;91:4970–4974. doi: 10.1073/pnas.91.11.4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nielsen S, DiGiovanni SR, Christensen EI, Knepper MA, Harris HW. Cellular and subcellular immunolocalization of vasopressin-regulated water channel in rat kidney. Proc Natl Acad Sci USA. 1993;90:11663–11667. doi: 10.1073/pnas.90.24.11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nielsen S, King LS, Christensen BM, Agre P. Aquaporins in complex tissues. II. Subcellular distribution in respiratory and glandular tissues of rat. Am J Physiol. 1997;273:C1549–1561. doi: 10.1152/ajpcell.1997.273.5.C1549. [DOI] [PubMed] [Google Scholar]
- 10.Loitto VM, Forslund T, Sundqvist T, Magnusson KE, Gustafsson M. Neutrophil leukocyte motility requires directed water influx. J Leukoc Biol. 2002;71:212–222. [PubMed] [Google Scholar]
- 11.Vassiliou AG, Maniatis NA, Orfanos SE, Mastora Z, Jahaj E, Paparountas T, Armaganidis A, Roussos C, Aidinis V, Kotanidou A. Induced expression and functional effects of aquaporin-1 in human leukocytes in sepsis. Crit Care. 2013;17:R199. doi: 10.1186/cc12893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ward PA, Hunninghake GW. Lung inflammation and fibrosis. Am J Respir Crit Care Med. 1998;157:S123–129. doi: 10.1164/ajrccm.157.4.nhlbi-10. [DOI] [PubMed] [Google Scholar]
- 13.Borok Z, Verkman AS. Lung edema clearance: 20 years of progress: invited review: role of aquaporin water channels in fluid transport in lung and airways. J Appl Physiol (1985) 2002;93:2199–2206. doi: 10.1152/japplphysiol.01171.2001. [DOI] [PubMed] [Google Scholar]
- 14.Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS and Kuebler WM. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44:725–738. doi: 10.1165/rcmb.2009-0210ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maniatis NA, Harokopos V, Thanassopoulou A, Oikonomou N, Mersinias V, Witke W, Orfanos SE, Armaganidis A, Roussos C, Kotanidou A, Aidinis V. A critical role for gelsolin in ventilator-induced lung injury. Am J Respir Cell Mol Biol. 2009;41:426–432. doi: 10.1165/rcmb.2008-0144OC. [DOI] [PubMed] [Google Scholar]
- 16.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(–Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 17.Lleu PL, Rebel G. Interference of Good’s buffers and other biological buffers with protein determination. Anal Biochem. 1991;192:215–218. doi: 10.1016/0003-2697(91)90210-k. [DOI] [PubMed] [Google Scholar]
- 18.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 19.Batteiger B, Newhall WJt, Jones RB. The use of Tween 20 as a blocking agent in the immunological detection of proteins transferred to nitrocellulose membranes. J Immunol Methods. 1982;55:297–307. doi: 10.1016/0022-1759(82)90089-8. [DOI] [PubMed] [Google Scholar]
- 20.Fabregat G, Garcia-de-la-Asuncion J, Sarria B, Cortijo J, De Andres J, Mata M, Pastor E and Belda FJ. Increased expression of AQP1 and AQP5 in rat lungs ventilated with low tidal volume is time dependent. PLoS One. 2014;9:e114247. doi: 10.1371/journal.pone.0114247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fabregat G, Garcia-de-la-Asuncion J, Sarria B, Mata M, Cortijo J, de Andres J, Gallego L and Belda FJ. Expression of aquaporins 1 and 5 in a model of ventilator-induced lung injury and its relation to tidal volume. Exp Physiol. 2016;101(11):1418–1431. doi: 10.1113/EP085729. [DOI] [PubMed] [Google Scholar]
- 22.Jiao G, Li E, Yu R. Decreased expression of AQP1 and AQP5 in acute injured lungs in rats. Chin Med J (Engl) 2002;115:963–967. [PubMed] [Google Scholar]
- 23.Rump K, Brendt P, Frey UH, Schafer ST, Siffert W, Peters J, Adamzik M. Aquaporin 1 and 5 expression evoked by the beta2 adrenoreceptor agonist terbutaline and lipopolysaccharide in mice and in the human monocytic cell line THP-1 is differentially regulated. Shock. 2013;40:430–436. doi: 10.1097/SHK.0000000000000035. [DOI] [PubMed] [Google Scholar]
- 24.Song Y, Fukuda N, Bai C, Ma T, Matthay MA, Verkman AS. Role of aquaporins in alveolar fluid clearance in neonatal and adult lung, and in oedema formation following acute lung injury: studies in transgenic aquaporin null mice. J Physiol 525 Pt. 2000;3:771–779. doi: 10.1111/j.1469-7793.2000.00771.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Su X, Song Y, Jiang J, Bai C. The role of aquaporin-1 (AQP1) expression in a murine model of lipopolysaccharide-induced acute lung injury. Respir Physiol Neurobiol. 2004;142:1–11. doi: 10.1016/j.resp.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 26.Towne JE, Harrod KS, Krane CM, Menon AG. Decreased expression of aquaporin (AQP)1 and AQP5 in mouse lung after acute viral infection. Am J Respir Cell Mol Biol. 2000;22:34–44. doi: 10.1165/ajrcmb.22.1.3818. [DOI] [PubMed] [Google Scholar]
- 27.Verkman AS. Role of aquaporins in lung liquid physiology. Respir Physiol Neurobiol. 2007;159:324–330. doi: 10.1016/j.resp.2007.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhao Y, Gorshkova IA, Berdyshev E, He D, Fu P, Ma W, Su Y, Usatyuk PV, Pendyala S, Oskouian B, Saba JD, Garcia JG, Natarajan V. Protection of LPS-induced murine acute lung injury by sphingosine-1-phosphate lyase suppression. Am J Respir Cell Mol Biol. 2011;45:426–435. doi: 10.1165/rcmb.2010-0422OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kardara M, Hatziantoniou S, Sfika A, Vassiliou AG, Mourelatou E, Muagkou C, Armaganidis A, Roussos C, Orfanos SE, Kotanidou A, Maniatis NA. Caveolar uptake and endothelial-protective effects of nanostructured lipid carriers in acid aspiration murine acute lung injury. Pharm Res. 2013;30:1836–1847. doi: 10.1007/s11095-013-1027-2. [DOI] [PubMed] [Google Scholar]
- 30.Maniatis NA, Sfika A, Nikitopoulou I, Vassiliou AG, Magkou C, Armaganidis A, Roussos C, Kollias G, Orfanos SE, Kotanidou A. Acid-induced acute lung injury in mice is associated with P44/42 and c-Jun N-terminal kinase activation and requires the function of tumor necrosis factor alpha receptor I. Shock. 2012;38:381–386. doi: 10.1097/SHK.0b013e3182690ea2. [DOI] [PubMed] [Google Scholar]
- 31.Manitsopoulos N, Orfanos SE, Kotanidou A, Nikitopoulou I, Siempos I, Magkou C, Dimopoulou I, Zakynthinos SG, Armaganidis A, Maniatis NA. Inhibition of HMGCoA reductase by simvastatin protects mice from injurious mechanical ventilation. Respir Res. 2015;16:24. doi: 10.1186/s12931-015-0173-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hasan B, Li FS, Siyit A, Tuyghun E, Luo JH, Upur H, Ablimit A. Expression of aquaporins in the lungs of mice with acute injury caused by LPS treatment. Respir Physiol Neurobiol. 2014;200:40–45. doi: 10.1016/j.resp.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 33.Jin LD, Wang LR, Wu LQ, Shan YL, Zhao XY, Xiong XQ, Zhou JH, Lin LN, Jin LL. Effects of COX-2 inhibitor on ventilator-induced lung injury in rats. Int Immunopharmacol. 2013;16:288–295. doi: 10.1016/j.intimp.2013.03.031. [DOI] [PubMed] [Google Scholar]
- 34.Song Y, Jayaraman S, Yang B, Matthay MA, Verkman AS. Role of aquaporin water channels in airway fluid transport, humidification, and surface liquid hydration. J Gen Physiol. 2001;117:573–582. doi: 10.1085/jgp.117.6.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bai C, Fukuda N, Song Y, Ma T, Matthay MA, Verkman AS. Lung fluid transport in aquaporin-1 and aquaporin-4 knockout mice. J Clin Invest. 1999;103:555–561. doi: 10.1172/JCI4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.McAuley DF, Frank JA, Fang X, Matthay MA. Clinically relevant concentrations of beta2-adrenergic agonists stimulate maximal cyclic adenosine monophosphate-dependent airspace fluid clearance and decrease pulmonary edema in experimental acid-induced lung injury. Crit Care Med. 2004;32:1470–1476. doi: 10.1097/01.ccm.0000129489.34416.0e. [DOI] [PubMed] [Google Scholar]
- 37.Modelska K, Pittet JF, Folkesson HG, Courtney Broaddus V, Matthay MA. Acid-induced lung injury. Protective effect of anti-interleukin-8 pretreatment on alveolar epithelial barrier function in rabbits. Am J Respir Crit Care Med. 1999;160:1450–1456. doi: 10.1164/ajrccm.160.5.9901096. [DOI] [PubMed] [Google Scholar]
- 38.Fu Z, Costello ML, Tsukimoto K, Prediletto R, Elliott AR, Mathieu-Costello O, West JB. High lung volume increases stress failure in pulmonary capillaries. J Appl Physiol. 1992;73:123–133. doi: 10.1152/jappl.1992.73.1.123. [DOI] [PubMed] [Google Scholar]
- 39.Kong H, Fan Y, Xie J, Ding J, Sha L, Shi X, Sun X, Hu G. AQP4 knockout impairs proliferation, migration and neuronal differentiation of adult neural stem cells. J Cell Sci. 2008;121:4029–4036. doi: 10.1242/jcs.035758. [DOI] [PubMed] [Google Scholar]
- 40.Saadoun S, Papadopoulos MC, Hara-Chikuma M, Verkman AS. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature. 2005;434:786–792. doi: 10.1038/nature03460. [DOI] [PubMed] [Google Scholar]
- 41.Matute-Bello G, Frevert CW and Martin TR. Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L379–399. doi: 10.1152/ajplung.00010.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Woods SJ, Waite AA, O’Dea KP, Halford P, Takata M, Wilson MR. Kinetic profiling of in vivo lung cellular inflammatory responses to mechanical ventilation. Am J Physiol Lung Cell Mol Physiol. 2015;308:L912–921. doi: 10.1152/ajplung.00048.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang ZQ, Song YL, Chen ZH, Shen Y, Bai CX. Deletion of aquaporin 5 aggravates acute lung injury induced by Pseudomonas aeruginosa. J Trauma. 2011;71:1305–1311. doi: 10.1097/TA.0b013e3182128528. [DOI] [PubMed] [Google Scholar]
- 44.Chae YK, Kang SK, Kim MS, Woo J, Lee J, Chang S, Kim DW, Kim M, Park S, Kim I, Keam B, Rhee J, Koo, NH, Park G, Kim SH, Jang SE, Kweon, IY, Sidransky D, C Moon. Human AQP5 plays a role in the progression of chronic myelogenous leukemia (CML) PLoS One. 2008;3:e2594. doi: 10.1371/journal.pone.0002594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chae YK, Woo J, Kim MJ, Kang SK, Kim MS, Lee J, Lee SK, Gong G, Kim YH, Soria JC, Jang SJ, Sidransky D, Moon C. Expression of aquaporin 5 (AQP5) promotes tumor invasion in human non small cell lung cancer. PLoS One. 2008;3:e2162. doi: 10.1371/journal.pone.0002162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Z, Chen Z, Song Y, Zhang P, Hu J, Bai C. Expression of aquaporin 5 increases proliferation and metastasis potential of lung cancer. J Pathol. 2010;221:210–220. doi: 10.1002/path.2702. [DOI] [PubMed] [Google Scholar]
- 47.Karlsson T, Glogauer M, Ellen RP, Loitto VM, Magnusson KE, Magalhaes MA. Aquaporin 9 phosphorylation mediates membrane localization and neutrophil polarization. J Leukoc Biol. 2011;90:963–973. doi: 10.1189/jlb.0910540. [DOI] [PubMed] [Google Scholar]
- 48.Wiggs BR, English D, Quinlan WM, Doyle NA, Hogg JC, Doerschuk CM. Contributions of capillary pathway size and neutrophil deformability to neutrophil transit through rabbit lungs. J Appl Physiol. 1994;77:463–470. doi: 10.1152/jappl.1994.77.1.463. [DOI] [PubMed] [Google Scholar]
- 49.Kawano T, Mori S, Cybulsky M, Burger R, Ballin A, Cutz E, Bryan AC. Effect of granulocyte depletion in a ventilated surfactant-depleted lung. J Appl Physiol. 1987;62:27–33. doi: 10.1152/jappl.1987.62.1.27. [DOI] [PubMed] [Google Scholar]
- 50.Lavnikova N, Prokhorova S, Lakhotia AV, Gordon R, Laskin DL. Distinct inflammatory responses of adherent vascular lung neutrophils to pulmonary irritants. J Inflamm. 1998;48:56–66. [PubMed] [Google Scholar]
- 51.Sunil VR, Connor AJ, Zhou P, Gordon MK, Laskin JD, Laskin DL. Activation of adherent vascular neutrophils in the lung during acute endotoxemia. Respir Res. 2002;3:21. doi: 10.1186/rr171. [DOI] [PMC free article] [PubMed] [Google Scholar]