

Abstract
Despite the fact that pharmacokinetic exposure of kinase inhibitors (KIs) is highly variable and clear relationships exist between exposure and treatment outcomes, fixed dosing is still standard practice. This review aims to summarize the available clinical pharmacokinetic and pharmacodynamic data into practical guidelines for individualized dosing of KIs through therapeutic drug monitoring (TDM). Additionally, we provide an overview of prospective TDM trials and discuss the future steps needed for further implementation of TDM of KIs.
Numerous KIs have become available for the treatment of solid tumors and have improved outcomes for a wide range of malignant diseases. In contrast to most classical cytotoxic drugs, these agents target specific molecular aberrations of cancer cells and are administered orally.
Many KIs show exposure–response and exposure–toxicity relationships. As pharmacokinetic (PK) exposure [e.g., area under the plasma concentration time curve (AUC) or plasma trough level (C min)] varies highly between patients, some patients may be at risk of treatment‐related toxicity due to high exposure, while others may experience suboptimal efficacy caused by low exposure.
Therefore, PK is a relevant and obvious biomarker that could be used to optimize treatment through TDM (Figure 1). For some anticancer drugs, TDM targets have already been recommended previously.1 Nonetheless, expansion and an update of these previous works is warranted given the rapid developments in oncology demonstrated by the large volume of new PK and pharmacodynamic (PD) data that has become available and the abundance of new agents in this class that have been approved in recent years.
Figure 1.
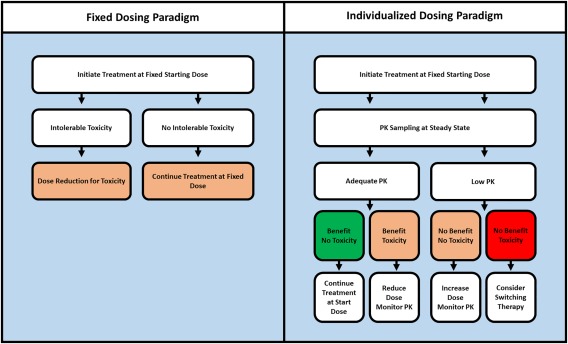
Current fixed dosing paradigm (left) vs. the proposed individualized or TDM dosing algorithm (right).
The purpose of this review is to integrate the available clinical PK and PD data into practical recommendations that can be used to personalize the treatment with KIs approved for the treatment of solid tumors, using TDM. An overview of the selected KIs used in the treatment of solid tumors and their pharmacokinetic properties (most relevant to TDM) are provided in Supplemental Table 1. A discussion of the available data for each KI is provided below. First, an overview of the available exposure–toxicity studies is given and exposure–response data are discussed. Concentrations for metabolites are taken into account if these have been shown to be pharmacologically active and contribute substantially to the anticancer effect. Then, based on these data, TDM recommendations are provided, focusing on the PK target. These TDM recommendations for each drug are summarized in Tables 1 and 2. Where evidence‐based target exposure is lacking, the average exposure of the approved efficacious dose will be provided as a proxy (also see Figure 2). Additionally, we provide a comprehensive general discussion on a broadly applicable PK‐guided dosing algorithm, a weighting of the evidence for TDM of each drug, the use of the mean exposure as proxy for a PK target, and an overview of previously conducted prospective TDM trials in oncology.
Table 1.
Overview of practical TDM recommendations for KIs approved by the FDA for the treatment of solid tumorsa
| Drug | TDM recommendation |
Proposed target (ng/mL) |
Mean/median exposure (Cmin in ng/mL) |
Outcome parameter associated with TDM target | References |
|---|---|---|---|---|---|
| Afatinib | Exploratory | 14.4 | |||
| Alectinib | Promising | Cmin ≥435 | 572 | Increased ORR | 2 |
| Axitinib | Promising | AUC ≥300b | 375b | Increased OS | 42 |
| Ceritinib | Exploratory | 871 | |||
| Cabozantinib | Exploratory | 1,380 | |||
| Cobimetinib | Exploratory | 127 | |||
| Crizotinib | Promising | Cmin ≥235 | 274 | Increased PFS | 6 |
| Dabrafenib | Exploratory | 96.1 | |||
| Erlotinib | Exploratory | 1,010 | |||
| Everolimus | Promising | Cmin ≥10.0 | 13.2 | Increased PFS | 95 |
| Gefitinib | Promising | Cmin ≥200 | 291 | Increased OS | 35 |
| Imatinib | Viable | Cmin ≥1,100 | 1,193 | Increased PFS | 100 |
| Lapatinib | Exploratory | 780 | |||
| Lenvatinib | Exploratory | 51.5 | |||
| Nintedanib | Exploratory | 13.1 | |||
| Osimertinib | Exploratory | 166 | |||
| Palbociclib | Exploratory | 61 | |||
| Pazopanib | Viable | Cmin ≥20,000 | 24,000 | Increased PFS | 57, 64 |
| Regorafenib | Exploratory | 1,400 | |||
| Sorafenib | Exploratory | 3,750 | |||
| Sunitinib | Viable |
Cmin ≥50 (inter), ≥37.5 (cont) |
51.6 (sum of parent & SU12662) | Increased OS | 76 |
| Trametinib | Promising | Cmin ≥10.6 | 12.1 | Increased PFS | 110 |
| Vandetanib | Exploratory | 795 | |||
| Vemurafenib | Promising | Cmin ≥42,000 | 39,000 | Increased PFS | 85, 89 |
AUC, area under the curve; Cmin, minimum plasma concentration/trough concentration; ORR, objective response rate; OS, overall survival; PFS, progression‐free survival.
The provided recommendation is considered promising if a pharmacokinetic TDM target is available or viable if a prospective TDM study has been conducted. Otherwise the recommendations should be considered exploratory.
For axitinib the AUC is provided in units of ng*h/mL.
Table 2.
Overview of practical TDM recommendations for KIs approved by the FDA for the treatment of hematological malignanciesa
| Drug | TDM recommendation |
Proposed target (ng/mL) |
Mean/median exposure (Cmin in ng/mL) |
Outcome parameter associated with TDM target | References |
|---|---|---|---|---|---|
| Bosutinib | Exploratory | 147 | |||
| Dasatinib | Exploratory | 2.61 | |||
| Nilotinib | Promising | Cmin ≥469 | 1,165 | Prolonged TTP | 14 |
| Idelalisib | Exploratory | 318 | |||
| Ibrutinib | Exploratory | 680b | |||
| Imatinib | Viable | Cmin ≥1,000 | 1,170 | Improved MMR, CCYR | 19 |
| Ponatinib | Exploratory | 34.2 |
CCYR, complete cytogenetic response; MMR, major molecular response; TTP, time to progression.
The provided recommendation is considered promising if a pharmacokinetic TDM target is available or viable if a prospective TDM study has been conducted. Otherwise the recommendations should be considered exploratory.
For Ibrutinib the AUC is provided in units of ng*h/mL.
Figure 2.
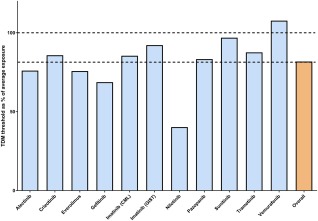
The TDM thresholds of selected KIs as percent of the mean/median exposure of the approved dose (blue bars). Overall, the thresholds were 81.7% of the mean exposure across all agents (orange bar), with a standard deviation of 17.4%. Dotted horizontal lines indicate 100% of mean exposure and 81% (the mean of the thresholds). This analysis suggests that across all kinase inhibitors, the target exposure matches with 81.7% of the population exposure and supports the view that targeting the population average could serve as a proxy, in the absence of a definitive TDM target.
PRACTICAL RECOMMENDATIONS FOR TDM OF KIs IN ONCOLOGY
Anaplastic lymphoma kinase (ALK) inhibitors
Alectinib
In previous studies, no relationships between alectinib exposure and Grade 3 toxicity were found.2 No relationship between best overall response and the combined average concentration of alectinib and its metabolite M4 was found (n = 49). However, in a population pharmacokinetic analysis, a higher than median steady‐state alectinib C min ≥435 ng/mL has been associated with greater reduction in tumor size (n = 46).2
Based on the available data, the best estimate for a cutoff for efficacy at this time is C min ≥435 ng/mL. Yet this preliminary finding should be confirmed in future studies.
Ceritinib
Higher ceritinib C min has been associated with an increase of Grade ≥3 adverse events (AEs) (P = 0.002), specifically with Grade ≥3 alanine transaminase (ALT) elevation, aspartate transaminase (AST) elevation, Grade ≥2 hyperglycemia and probability of dose reduction (all P < 0.01), but not with Grade ≥2 diarrhea (P = 0.11), Grade ≥3 gastrointestinal tract AEs (P = 0.86), or fatigue (P = 0.92).3 No significant exposure–response relationships were identified for the primary efficacy endpoint objective response rate (ORR) and secondary efficacy endpoint progression‐free survival (PFS) in the pivotal trial in nonsmall‐cell lung cancer (NSCLC),3 but a trend towards higher ORR with higher C min was reported.4
Based on the limited data, no specific threshold can be proposed yet. For now, ceritinib concentrations measured for TDM could be interpreted in relation to the mean C min of 871 ng/mL at the approved dose.3
Crizotinib
No relationships between exposure and toxicity have been reported for crizotinib, except for a suggested relationship with QTc prolongation.5 In two trials (n = 120 and 114), the ORR was 60% in the patients with a C min in the upper three quartiles (≥235 ng/mL) compared to 47% in the lowest quartile (<235 ng/mL).6 An increase in PFS with increasing C min was also found. A stepwise Cox proportional analysis pointed toward a higher hazard of disease progression in the lowest quartile compared to the higher quartiles with a hazard ratio of 3.2 (90% confidence interval (CI): 1.62–6.36).6 This threshold of >235 ng/mL is in accordance with the EC50 of 233 ng/mL found in preclinical models.7
Based on these data, it seems reasonable to use the threshold of C min ≥235 ng/mL for TDM of crizotinib.
Break point cluster region: Abelson (Bcr‐Abl) oncoprotein inhibitors
Bosutinib
Few exposure–response and exposure–toxicity data have been reported for bosutinib in chronic myelogenous leukemia (CML).8 PK‐PD analyses indicated weak relationships between the incidence (but not severity) of diarrhea and rash and PK described by an Emax model.9 The same study identified limited associations between AUC and C min for both complete cytogenetic response and complete hematological response and between AUC, maximum plasma concentration (Cmax), and C min with major molecular response. Moreover, C min was reported to be higher in responders than in nonresponders in the pivotal CML trial.10 Although the limited data point towards both exposure–response and exposure–toxicity relationships, no cutoff values have yet been proposed. Therefore, the most pragmatic PK target for TDM would be the median C min on the approved 500 mg once daily (q.d.) dose of 147 ng/mL.9, 11
Dasatinib
In a population PK‐PD analysis of the several clinical trials including the phase III study in CML (n = 981), the dasatinib trough concentration was significantly related to pleural effusion (P < 0.01).12
Moreover, the dasatinib weighted average steady‐state concentration was significantly associated with major cytogenetic response, with the odds of response increasing 2.11‐fold for every doubling of the average steady‐state concentration (P < 0.001).12
Another study in Japanese patients (n = 51) found that the time above the IC50 of phosphorylated CT10 regulator of kinase like (p‐CrkL) in CD43+ cells was related to early molecular response to dasatinib in CML.13
Given the solid relation of exposure (weighted average steady‐state concentration) with treatment response, TDM could be of value for dasatinib. However, using an average concentration for TDM is not feasible. Therefore, using the geometric mean C min of 2.61 ng/mL may serve as a more practical proxy.
Nilotinib
Large population PK‐PD analyses identified several exposure–response and exposure–safety relationships for nilotinib. Higher C min was associated with the occurrence of all‐grade elevations in total bilirubin and lipase levels and increases in QTcF changes.14, 15
Also, patients in the lowest C min quartile had significantly longer time to complete cytogenetic response or major molecular response and shorter time to progression compared with patients in the higher quartiles.14 For each of these analyses this Q1‐Q2 threshold varied from 469 to 553 ng/mL. Based on the above, nilotinib TDM could be employed with a target of C min ≥469 ng/mL.
Imatinib
Several relationships between imatinib concentrations and toxicity, including C min with thrombocytopenia16 and AUC (unbound) with absolute neutrophil count decrease, have been established.17 A trend towards higher incidences of hematological Grade 3/4 AEs for patients with patients with very high C min (>3,180 ng/mL) was reported.18
Multiple studies in CML patients point towards increased efficacy of imatinib in CML with higher exposure.
In a subanalysis of the IRIS trial (n = 351), significantly reduced incidences of major molecular and complete cytogenetic response and a trend towards reduced event‐free survival were observed in the lowest C min quartile.19 Another study in Japanese patients found (n = 254) found a significant correlation between C min ≥1,002 ng/ml and higher probability of achieving a major molecular response.20
An Israeli study (n = 191) also found a significantly higher C min in CML patients who achieved a complete cytogenetic response compared to those without (1,078 vs. 827 ng/mL, P = 0.045).21
A study in 353 CML patients found higher incidences of major molecular response and complete cytogenetic response rates for patients with an exposure >1,165 ng/mL.18 A subanalysis of an imatinib adherence study (n = 84) also found a statistically significant increased incidence of major molecular response (83.2 vs. 60.1%) for patients with C min >1,000 ng/mL.22
Several other studies have also found that patients with better treatment outcomes also had higher C min values.23, 24
Given the large number of studies reporting the importance of imatinib C min, a prospective TDM study was conducted in 56 CML patients.25 It set a PK target of 750–1,500 ng/mL. Due to low adherence to the dosing recommendations, this study did not meet its formal endpoint. Yet in patients who were dosed in accordance with the recommendation they experienced significantly fewer unfavorable events (28 vs. 77%, P = 0.03).25
The studies above all seem to support the use of a threshold of ≥1,000 ng/mL for efficacy for imatinib in CML. Moreover, the feasibility of imatinib dosing based on C min has been established in a prospective study.25 Future studies are needed to conclusively demonstrate the added benefit of personalized imatinib dosing in CML patients.
Ponatinib
For ponatinib, analyses of the dose intensity–safety relationship (defined as the average ponatinib dose of each subject while on study, which ranged from 0.34–45.2 mg) indicated a significant increase in Grade ≥3 safety events such as AST, ALT, and lipase increases, myelosuppression, hypertension, pancreatitis, rash, neutropenia, and thrombocytopenia, with increasing dose intensity.26 A statistically significant relationship between dose intensity and probability of major cytogenetic responses in CML patients has been described.26
Given the relationship between dose intensity and major cytogenetic response, targeting the geometric mean (CV%) C min of the approved 45 mg q.d. dose 34.2 (45.4) ng/mL (corresponding to 64.3 nM) seems a reasonable target.27
Epidermal growth factor receptor (EGFR) inhibitors
Afatinib
Diarrhea and rash are the most common AEs of afatinib. These toxicities have been correlated with AUC and Cmax (P < 0.0005).28 C min in patients experiencing Grade 3 diarrhea was higher (35.8 ng/mL) than those experiencing Grade 1–2 diarrhea (25.2–31.6 ng/mL). In patients experiencing Grade 3 rash, C min was 31.4 ng/mL vs. 26.8–27.6 ng/mL in those with only Grade 1–2 rash.29 A consistent relationship between exposure and response has not been found yet for afatinib.30
Awaiting future exposure–response analyses, TDM of afatinib could focus on targeting a steady‐state C min of the 40 mg q.d. dose of 14.4–27.4 ng/mL.30
Erlotinib
Erlotinib exposure has been significantly correlated with rash in several studies.31 However, there was significant overlap in the range of PK values with patients who had no rash. No correlation was found with diarrhea.31 Two clinical exposure–response studies have been reported. The first was conducted in head and neck squamous cell carcinoma (HNSCC) patients and found a trend toward increased overall survival (OS) for a C min >950 ng/mL (P = 0.09). The second found a relationship between the ratio of erlotinib and its O‐desmethyl metabolite and PFS and OS (both P < 0.01).32 This metabolite ratio was also associated with Grade 2 rash (P = 0.02). This study found no relationships between PFS or OS and erlotinib concentrations. It should be noted, however, that these results are based on a pooled analysis of NSCLC and pancreatic cancer patients (n = 63 and 33, respectively).
More studies are needed to elaborate the role of erlotinib and O‐desmethyl erlotinib concentrations, as no threshold for monitoring of the metabolic ratio is currently available.32 At the moment, the previously established preclinical threshold of >500 ng/mL still seems the most rational target for TDM.1, 33
Gefitinib
Gefitinib AUC0‐24 and C min were higher in patients experiencing diarrhea and hepatotoxicity.34, 35 Rash‐based dosing of gefitinib has been explored in head and neck squamous cell carcinoma, but even though this was found to be feasible, it did not result in increased antitumor activity, measured as response rate or PFS.36 This study did find higher gefitinib C min levels in patient with disease control compared to patient with progressive disease as best response, 1,117 ng/ml vs. 520 ng/ml (P = 0.01). In another study, OS was linked to gefitinib C min in NSCLC patients (n = 30). Patients with C min <200 ng/mL had an OS of 4.7 months compared to 14.6 months for patients ≥200 ng/mL (P = 0.007).35 The available data support TDM of gefitinib in NSCLC using a threshold C min of ≥200 ng/mL.
Lapatinib
No thorough exposure–response or exposure–toxicity studies have been reported for lapatinib, although one trial found that the majority of responders had a C min in the 300–600 ng/ml range.37 Future studies should focus on establishing exposure–response and exposure–toxicity relationships.
Meanwhile, lapatinib C min could be interpreted in reference to the mean C min of 780 ng/mL.1
Osimertinib
For osimertinib, a relationship was found between steady‐state AUC and the probability of rash (P = 0.0023) and diarrhea (P = 0.0041) in a population of NSCLC patients.38 However, no evidence of a relationship between exposure and tumor response, duration of response, or change in tumor size has been established.38, 39
In the absence of conclusive exposure–response analyses, C min could be compared to the geometric mean [coefficient of variation (CV)] of the approved 80 mg daily dose of 166 (48.7) ng/mL (corresponding to 332 nM).39
Vascular endothelial growth factor receptor (VEGFR) inhibitors
Axitinib
Exposure–safety analysis has demonstrated that axitinib AUC was significantly related to increased hypertension, proteinuria, fatigue, and diarrhea.40 Diastolic blood pressure (dBP) ≥90 mmHg has been associated with increased probability of response, PFS, and OS in RCC patients.41, 42 Based on these results, a randomized phase II trial to individualize axitinib dose based on dBP has been performed.43 In total, 122 RCC patients were randomized to either axitinib or placebo dose titration. The axitinib dose titration group showed an increased ORR compared to the placebo group (P = 0.019)43, but this did not result in improved OS (P = 0.162).44 One small study (n = 24) also found a relationship between axitinib C min >5 ng/mL and tumor response and the occurrence of hypertension, hyperthyroidism, and proteinuria.45 In renal cell carcinoma (RCC) patients, an AUC ≥300 ng*h/mL was significantly associated with increased PFS (13.8 vs. 7.4 months, P = 0.03) and OS (37.4 vs. 15.8 months, P < 0.01).42
The available data support using an AUC ≥300 ng*h/mL as a target for TDM.42 However, given that prospective studies using dBP are already available, an integrated approach using both PK and dBP to guide dosing may be the most appropriate strategy to optimize treatment, as has been advocated previously.46 Although more evidence is available to support the AUC target, the more practical C min target of >5 ng/mL could also be considered (as it requires only a single plasma sample).45
Cabozantinib
Steady‐state AUC derived from a population PK model of combined phase I, II, and III studies has been correlated with dose reductions and lower achieved dose intensity. These dose modifications, however, did not appear to impact PFS.47 Population pharmacodynamic modeling suggested that a concentration of only 59–78 ng/mL would already result in 50% of maximum effect in medullary thyroid cancer patients.48
As no PK thresholds for cabozantinib have been reported, future studies should first establish these before TDM of cabozantinib can move forward. Meanwhile, cabozantinib concentrations could be referenced relative to the mean C min in the medullary thyroid cancer phase III trial of 1,380 ng/mL (on 140 mg) or 1,125 ng/mL in renal cell carcinoma.49, 50
Lenvatinib
An increase in the incidence of Grade 3 or higher hypertension, Grade 3 or higher proteinuria, nausea, and vomiting with higher lenvatinib dose intensity has been observed.51 Analyses of the pivotal study in thyroid cancer indicated similar PFS across the full range of exposures (AUC0‐24 between 1,410 and 10,700 ng*h/mL).52 However, a model‐based PK/PD analysis indicated that lenvatinib AUC0‐24 was correlated with reduction in tumor size.52
As no exposure–response and exposure–toxicity thresholds are established yet for lenvatinib, TDM could target the mean C min of 51.5 ng/mL.51
Nintedanib
Nintedanib has only shown modest relationships between exposure and safety and efficacy.53 In exploratory analyses, higher nintedanib concentrations have been associated with hepatotoxicity, but not with gastrointestinal AEs. Exposure–response analyses are currently not available for clinical endpoints, except for a statistically significant association between nintedanib exposure and dynamic contrast‐enhanced magnetic resonance imaging (MRI) response54 and a decrease in soluble VEGFR levels with increasing C min in a phase I study (r = –0.46, n = 15).55
As no specific threshold for nintedanib has been proposed, TDM should focus on targeting the mean C min value of the approved dose (calculated for a 200 mg dose, based on the dose‐normalized C min value of 0.0654 ng/mL/mg) of 13.1 ng/mL.56
Pazopanib
Pazopanib exposure has been correlated with hypertension.57 This correlation was stronger for C min than for AUC0‐t (R2 of 0.91, P = 0.0075 and 0.25, respectively, P = 0.23). Relations were also found between C min and diarrhea, ALT‐elevations, hand‐foot syndrome, and stomatitis.58 The probability of Grade ≥3 ALT increased with a higher pazopanib concentration.59 However, a recent study suggested that pazopanib hepatotoxicity maybe related to genetic mutations in human leukocyte antigen (HLA) and, therefore, unrelated to PK.60 Analysis of data from 177 RCC patients showed an increased PFS in patients with C min ≥20.5 mg/L compared to patients with a C min below this threshold (52.0 vs. 19.6 weeks, P = 0.0038).57 This threshold seems to be in accordance with preclinical data showing optimal VEGFR2 inhibition by pazopanib in vivo at a concentration ≥17.5 mg/L.61 Plasma concentrations have also been correlated with radiographic response in a phase II study of patients with progressive, radioiodine‐refractory, metastatic differentiated thyroid cancer.62 Two trials have investigated individualized dosing of pazopanib in cancer patients. The first used pazopanib AUC0‐24h as a target (715–920 mg*h/L) and set a reduction in variability as the primary endpoint.63 AUC‐guided dosing did not significantly reduce interpatient variability, probably due to intrapatient variability or sampling time issues. Based on this trial, the authors concluded it may be more beneficial to target the C min threshold rather than an AUC window. The second study was a prospective study in 30 patients with advanced solid tumors, using a C min ≥20 mg/L as target.64 The dosing algorithm, based on dose adjustments after 2, 4, and 6 weeks, led to patients being treated at dosages ranging from 400–1,800 mg daily. C min in patients whose dose was successfully escalated above 800 mg (n = 10) increased significantly from 13.2 (38.0%) mg/L [mean (CV%)] to 22.9 mg/L (44.9%).
This study demonstrated the safety and feasibility of C min (≥20 mg/L) guided dosing for pazopanib and merits further investigation of pazopanib TDM, for instance, in a randomized clinical trial (RCT) to demonstrate the relevance of individualized over fixed dosing on a clinical endpoint such as PFS or OS.
Regorafenib
Regorafenib is metabolized by CYP3A4 into the active metabolites M2 (N‐oxide) and M5 (N‐oxide, N‐desmethyl), which at steady state form a major component of the total exposure.65 An exposure–dependent increase was seen for rash, total bilirubin, and median indirect bilirubin in gastrointestinal stromal tumor (GIST) patients, for parent and total (including M2 and M5) regorafenib exposure.66 No exposure–response relationships for efficacy have been reported for regorafenib hitherto. 65
More studies are needed to investigate exposure–response and –toxicity relationships of regorafenib. These should take into account M2 and M5, as these have been shown to be pharmacologically active and present at similar or higher concentrations than the parent compound. Currently, the most appropriate TDM target for regorafenib (parent compound only) is the mean C min of 1.4 mg/L.65
Sorafenib
In a study of patients with advanced solid tumors (n = 54), a cutoff at a cumulative AUC (calculated over day 0 to 30) of 3,161 mg*h/L was associated with the highest risk to develop any Grade ≥3 toxicity (P = 0.018).67 A patient series found that sorafenib C min was higher in patients who experienced Grade 3 AEs (n = 8) than those who did not (n = 14), 7.7 ± 3.6 mg/L vs. 4.4 ± 2.4 mg/L (P = 0.0083).68 Sorafenib steady‐state concentrations were found to be higher in patients with Grade ≥2 hand‐foot syndrome and hypertension than in those not experiencing these AEs (P = 0.0045 and 0.0453, respectively). Optimal cutoffs were 5.78 mg/L for hand‐foot syndrome and 4.78 mg/L for hypertension.69 In a small cohort of 25 hepatocellular carcinoma patients, the AUC‐ratio of sorafenib and its metabolites resulted in even better prediction of toxicity (P = 0.002).70 The same cohort found that not sorafenib AUC but that of its metabolite seemed significantly associated with dose reduction or discontinuation (P = 0.031) and increased PFS (P = 0.048).70 A study in Japanese patients (n = 91) found a trend toward increased OS in hepatocellular carcinoma patients at a sorafenib Cmax of ≥4.78 mg/L (12.0 vs. 6.5 months, P = 0.08).69
Future studies need to confirm the proposed exposure–response and –toxicity relations described in these small patients cohorts, taking into account the N‐oxide metabolite. Currently, the most appropriate target for sorafenib TDM is >3.75–4.30 mg/L (parent compound only), based on preclinical experiments and the mean exposure in humans, as was advocated previously.1
Sunitinib
Sunitinib is metabolized by CYP3A4 into its active metabolite N‐desethylsunitinib, also known as SU12662. TDM for sunitinib is generally performed using the sum of concentrations (total C min) of both sunitinib and SU12662.71 Dose‐limiting and Grade ≥3 toxicities of sunitinib have been associated with total C min ≥100 ng/mL.17, 72 Grade ≥2 mucositis and altered taste have also been related to higher total C min.73 A relationship was also found between sunitinib AUC and Grade ≥3 toxicity (P = 0.0005).74 Based on the above, an upper C min cutoff of <100 ng/mL could be considered.
In RCC, increasing AUC has been related to higher response rates, longer PFS, and OS.74, 75, 76, 77 A meta‐analysis found AUC of sunitinib combined with its active metabolite N‐desethylsunitinib to be significantly associated with PFS and OS in both GIST (n = 401) and RCC (n = 169), all P < 0.01.76 An increased OS was found for an AUC ≥1,973 ng*h/mL in another study in RCC patients (n = 55).74 C min correlated with AUC (r2 = 0.8–0.9), suggesting C min could be used for TDM as a substitute.76 A PK target of 50–100 ng/mL17 has been suggested for intermittent dosing in RCC (50 mg daily for 4 weeks in a 6‐week cycle) and based on PK linearity a target of ≥37.5 ng/mL was extrapolated for continuous dosing in GIST (37.5 mg daily continuously).1
A TDM‐feasibility trial has been conducted in cancer patients using C min ≥50 ng/mL as a PK target allowing for dose adjustments after 3 and 5 weeks of treatment.78 A third of the patients <50 ng/mL at the standard dose could be treated successfully at an increased dose and additional patients reached the target exposure. This study demonstrates the feasibility using C min ≥50 ng/mL (sunitinib + metabolite) as a TDM target. Future studies are now needed to confirm the efficacy of TDM over fixed dosing for sunitinib.
Vandetanib
Grade ≥2 diarrhea and fatigue have significantly been associated with steady‐state vandetanib C min (P = 0.03 and 0.02, respectively), but no relationship was found for hypertension or rash.79 Importantly, a substantial dose‐ and exposure‐related QTc prolongation has been observed.79 No clear relationship between PFS and exposure has been found in the pivotal trial in patients with thyroid cancer,79 although multiple studies have used IC50 values established in vitro (190 ng/mL) to support dose selection in early clinical trials.80
In the absence of studies that establish specific PK thresholds, current exploratory TDM efforts could focus on targeting the population mean exposure of 795 ng/mL.
Serine/threonine‐protein kinase B‐Raf (BRAF) inhibitors
Dabrafenib
Dabrafenib is metabolized into its carboxy, hydroxyl, and desmethyl metabolites.81 The hydroxyl metabolite showed similar IC50 values to dabrafenib in vitro. No relationships between AEs and exposure, except for pyrexia, have been reported.82 Pyrexia seemed to be related to Caverage dabrafenib and hydroxy‐dabrafenib C min, but not to desmethyldabrafenib C min.83 At the moment, no evident exposure–response relationships have been reported for dabrafenib and/or for any of its metabolites.84
In the absence of a validated target, current TDM efforts could target the median C min (sum of parent dabrafenib and its hydroxyl metabolite) of 99.6 ng/mL.84
Vemurafenib
In melanoma patients, vemurafenib concentrations were significantly higher in those patients who developed Grade ≥2 rash compared to those who did not (mean ± standard deviation (SD) of 61.7 ± 25.0 vs. 36.3 ± 17.9 mg/L, P < 0.0001).85 Another study found an exposure–dependent QTc prolongation for vemurafenib.86 Vemurafenib concentrations have also been related to treatment response. Responders had a mean concentration of 56.4 mg/L, while nonresponders had a mean of 38.8 mg/L (P = 0.013).87, 88 Moreover, melanoma patients in the lowest exposure quartile (<40.4 mg/L) had a PFS of 1.5 months compared to that of 4.5 months of patients in the higher three quartiles (P = 0.029).85 This effect was confirmed in an independent cohort after 12 months of follow‐up with a threshold of 42 mg/L (P = 0.005).89
The available data support the use of a threshold C min of >42 mg/L. A real‐world study, however, found that in routine care only half of patients had a C min <42 mg/L,90 demonstrating the opportunities for dose optimization.90
Mitogen‐activated protein kinase kinase (MEK) inhibitors
Cobimetinib
Exploratory exposure–toxicity analyses for safety identified a trend towards increased diarrhea with increasing cobimetinib and vemurafenib exposure.91 No significant exposure–response relationship has been established for cobimetinib on the primary endpoint of PFS in the pivotal registration trial.91
On the basis of the available data, no clear PK target can yet be identified for cobimetinib. Therefore, the currently most appropriate target would be the mean C min of the approved dose of 127 ng/mL.91
Trametinib
No exposure–toxicity relationships have been identified for trametinib. A population analysis was performed to explore the effect of trametinib C min and average concentration on ORR and PFS.92 The proportion of responders seemed to increase with increasing exposure and reached a plateau at a C min of 10 ng/mL. No relationship between exposure above or below the mean C min of 13.6 ng/mL and PFS has been identified in phase III trials. However, in an analysis of the phase II study, patients with C min above 10.6 ng/mL, had longer PFS than those below this C min value.92 Furthermore, the C min threshold of 10.6 ng/mL is supported by preclinical data pointing towards a target of 10.4 ng/mL based on efficacy in BRAF mutant melanoma cell lines.93
Given the above, the threshold of a C min ≥10.6 ng/mL seems the most appropriate target to be used for trametinib TDM.
Other kinase inhibitors used in oncology
Everolimus
In transplantation medicine, TDM is routinely applied for everolimus, using a window of 6–10 ng/mL or 3–8 ng/mL in combination therapy.94 No target for TDM has been validated in oncology. Higher C min has been associated with increased risk of high‐grade pulmonary and metabolic (such as hyperglycemia) AEs and stomatitis. However, this meta‐analysis of everolimus phase II trials (n = 945) found that a 2‐fold increase in everolimus C min was associated with improved tumor size reduction, regardless of cancer type.95 No specific target window has been proposed, but in RCC and pNET cutoffs at ≥10 and 30 ng/mL resulted in numerically higher PFS values than C min<10 ng/mL.95 A retrospective analysis of 45 RCC patients showed a trend toward increased PFS for patients with a C min ≥14.1 ng/mL of 13.3 vs. 3.9 months, P = 0.06.96
Based on experience in transplant and pediatric patients, everolimus TDM seems feasible.94, 97 Although exposure–response relations are seen for everolimus in oncology, no formal PK‐target has been established yet. Based on the available data, a cutoff for efficacy of C min ≥10 ng/mL seems a reasonable target for TDM of everolimus in oncology.
Ibrutinib
For ibrutinib no exposure–safety relationships were found.98 A phase I study indicated maximum Bruton's tyrosine kinase occupancy at doses of ≥2.5 mg/kg (corresponding to a 175 mg dose for average weight of 70 kg). This complete target inhibition was already seen at an AUC of 160 ng*h/mL.99
In the absence of clearly defined pharmacokinetic thresholds for clinical patient outcomes, ibrutinib TDM could target the mean ±SD AUC at the approved 560 mg q.d. dose of 953 ± 705 ng*h/mL for mantle cell lymphoma patients or 680 ± 517 ng*h/mL at 420 mg q.d. for patients with chronic lymphocytic leukemia (no C min data were reported).98
Imatinib
In addition to its use in CML, imatinib is also used as an inhibitor of the stem cell receptor KIT and platelet‐derived growth factor receptor (PDGFR) in GIST. In an analysis of 73 GIST patients randomized to either 400 or 600 mg q.d., an increase in time to disease progression was found for patients with a C min >1,100 ng/mL.100 Another study did not find a relationship between imatinib C min and treatment response, but did find a relationship between free (unbound to plasma proteins) imatinib concentration >20 ng/mL and complete response.101 Two real‐world studies suggest a relationship of imatinib C min and efficacy. The first found that responders had a median C min of 1271 ng/mL, while C min in nonresponders was 920 ng/mL (P = 0.23).102 The second did not find a significant relationship between a C min >1,100 ng/mL threshold of imatinib and PFS (P = 0.1107). However, a threshold of >760 ng/mL was associated with a significantly longer PFS (P = 0.0256).103
The available studies point towards different targets for imatinib TDM in GIST patients (≥760 and ≥1,100 ng/mL). The more pragmatic approach may be to use the C min >1,100 ng/mL threshold, as it is based on PFS data from an RCT 100 and seems to be confirmed by data from an independent observational cohort.102 Moreover, a retrospective cohort study of 68 GIST patients indicated the feasibility of dosing imatinib based on the 1,100 ng/mL threshold, with more patients reaching the prespecified target exposure.104
Idelalisib
No exposure–response or exposure–safety relationships have been identified for idelalisib in chronic lymphocytic leukemia or non‐Hodgkin's lymphoma, using either AUC or C min as pharmacokinetic parameters.105 However, dose selection was supported by the fact that the exposure achieved on the approved dose achieved an EC90 of 125 ng/mL for inhibition of PI3Kδ in vitro.105 In the absence of more conclusive data, TDM of idelalisib should, for now, target the median C min at the approved 150 mg q.d. dose of 318 ng/mL.105, 106
Palbociclib
A greater reduction in absolute neutrophil count appears to be associated with increased palbociclib exposure.107 No conclusive exposure–response relationship has been found in 81 patients treated at the 125 mg fixed dose.
Based on the limited exposure–response and –toxicity analyses, no specific PK target for palbociclib can be formulated. More thorough PK/PD analyses are needed. Until these come available palbociclib concentrations can be compared to the population mean (CV) C min of 61 (42) ng/mL.107
DISCUSSION
Currently, KIs are administered at a fixed starting dose which is only adjusted in case of intolerable toxicity (Figure 1, left). As many KIs show an exposure–response and exposure–toxicity relationship and exposure varies highly between patients, we propose that an individualized PK‐guided dosing or TDM algorithm should be explored for KIs (Figure 1, right).
Based on the PK targets discussed above, dose increments could be considered for patients with low exposure in the absence of significant toxicity. These dose increments could, for instance, follow the dose‐escalation schedule explored in the phase I dose‐escalation study of the respective drug. Yet if available, a prospectively validated and safe TDM‐dose algorithm would be preferred (Table 3).
Table 3.
Overview of prospective dose individualization trials of KIs
| Drug | n |
Patient population |
PK parameter |
Target | Dose change | PK‐guided dose escalations (↑) or reductions (↓)a | Endpoint | Reference |
|---|---|---|---|---|---|---|---|---|
| Everolimus | 28 |
Pediatric SEGA patients |
Cmin | 5–15 ng/mL | — | ↑ and ↓ | PD | 97 |
| Sunitinib | 37 | Advanced solid tumors | Cmin | ≥50 ng/mL | After 3 and 5 weeks | ↑ only | PK | 78 |
| Imatinib | 56 |
Chronic myelogenous leukemia patients |
Cmin | 750–1,500 ng/mL | — | ↑ and ↓ | PK | 25 |
| Pazopanib | 13 | Renal cell carcinoma patients | AUC | 715–920 mg*h/L | After 2 weeks | ↑ and ↓ | PK | 63 |
| Pazopanib | 30 | Advanced solid tumors | Cmin | ≥20 mg/L | After 2, 4, and 6 weeks | ↑ only | PK | 64 |
AUC, area under the curve; Cmin, minimum plasma concentration/trough concentration; PD, pharmacodynamic; PK, pharmacokinetic; SEGA, subendymal giant cell astrocytoma.
Per protocol, some trials had dosing algorithms which allowed for dose reductions (in the absence of toxicity) based on PK, while others only allowed for dose escalation based on PK. All allowed for dose reductions based on toxicity.
For patients with a high plasma concentration not experiencing toxicity, dose reductions could be considered. However, in contrast to, for example, TDM of aminoglycosides in infectious diseases, in oncology the main focus of TDM will probably be directed towards improving efficacy by increasing the dose in low‐exposure patients. Concerns for lasting side‐effects may in most cases be less relevant.
Nonetheless, monitoring of plasma concentrations may be useful in patients requiring dose reductions for toxicity. Here, it could be used to differentiate between patients who had toxicity due to high exposure (who might be successfully treated at the lower dose) and those who do not tolerate treatment despite an exposure below the efficacious concentration (red box, Figure 1). Taking together the considerations above, a proposal for a generic decision tree for PK‐guided dosing is provided in Figure 1.
Ideally, individualized dosing should be based on thorough exposure–response and exposure–toxicity analyses. A weighting of the robustness of the evidence has been provided for each of the proposed TDM recommendations in Tables 1 and 2 as either negative, exploratory, promising, viable, or standard of care.
None of the included drugs has been qualified as negative. Based on the mechanism of action of KIs and the clinical pharmacological properties, exposure–response relationships are to be expected for most of these drugs. A fully negative recommendation can only be provided if evidence from an adequately sized and powered study demonstrates that at the recommended dose no relationship between drug exposure and response exist.
For the drugs in the exploratory category (Tables 1, 2), no PK‐targets have been specified yet. Therefore, it is too early to recommend implementation of TDM for these drugs. Further PK sampling in clinical trials and routine patient care could help to identify exposure–response and exposure–toxicity relationships. TDM, however, could already be of value in specific patient populations, such as patients with hepatic impairment, patients not able to swallow medication, or patients having possible drug interactions and compliance issues. The mean population exposure could be used as a reference for interpretation of the exposure of these individual patients.1 An updated analysis of the relationship between available TDM targets and the average population exposure support this (Figure 2). Overall, the targets (n = 11) amounted to 81.7% of the population exposure, with a relatively small SD of 17.4%. Although this is no substitute for thorough exposure–response analyses, the data support the view that targeting the mean or median exposure will generally result in efficacious concentrations for KIs in oncology.
If an exposure–outcome relationship and a PK target have been established, TDM could be considered a promising strategy for treatment optimization. The agents for which a TDM target is available are therefore classified as promising in Tables 1 and 2. For these drugs, the feasibility of individualized dosing based on this target should preferably be demonstrated in a prospective clinical trial.
For KIs where feasibility studies have already been conducted (Table 3), TDM is classified as viable (Tables 1, 2). All but one of these studies used PK endpoints, aiming to establish the safety and feasibility of reaching the target exposure.25, 63, 64, 78, 108 One study used a PD endpoint, a one‐armed trial with the purpose to show efficacy in a rare pediatric tumor (subependymal giant cell astrocytoma).97
Currently, for none of the discussed agents is TDM performed as the standard of care. Before TDM can become standard for drugs in the viable category, the relevance of this dosing strategy over fixed dosing should, if feasible, be clinically validated in a prospective randomized trial. Such studies are scarce, but have been conducted previously for TDM of cytotoxic drugs such as paclitaxel,109 indicating the feasibility of conducting randomized individualized dosing trials in cancer patients. This type of trial should now be initiated to demonstrate an effect of TDM for targeted anticancer agents on relevant clinical endpoints in oncology.
CONCLUSION
For KIs with an exposure–response and/or exposure–toxicity relationship and high interpatient variability in exposure, a PK parameter such as C min is an obvious and relevant biomarker for dose individualization through TDM.
Several clinical trials demonstrate the safety and feasibility of TDM of KIs, such as imatinib, pazopanib, tamoxifen, everolimus, and sunitinib. Randomized clinical trials are now needed to confirm an effect of TDM over fixed dosing on relevant clinical efficacy endpoints such as PFS and OS, before TDM can become universally implemented as standard care of cancer patients treated with KIs.
Supporting information
Additional supporting information can be found in the online version of this article.
Supporting Information
CONFLICT OF INTEREST
The authors declared no conflict of interest.
References
- 1. Yu, H. et al Practical guidelines for therapeutic drug monitoring of anticancer tyrosine kinase inhibitors: focus on the pharmacokinetic targets. Clin. Pharmacokinet. 53, 305–325 (2014). [DOI] [PubMed] [Google Scholar]
- 2. Food and Drug Administration . Center for Drug Evaluation and Research Alectinib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/208434Orig1s000ClinPharmR.pdf> (2016).
- 3. Food and Drug Administration . Center for Drug Evaluation and Research Ceritinib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205755Orig1s000ClinPharmR.pdf> (2014).
- 4. Committee for Medicinal Products for Human Use (CHMP) European Medicines Agency Ceritinib European Public Assessment report. 44, (2015).
- 5. European Medicines Agency Committee for Medicinal Products For Human Use (CHMP) Crizotinib European Public Assessment Report. <http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002489/WC500134761.pdf> (2012).
- 6. Food and Drug Administration . Center for Drug Evaluation and Research Crizotinib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202570Orig1s000ClinPharmR.pdf> (2011).
- 7. Yamazaki, S. Translational pharmacokinetic‐pharmacodynamic modeling from nonclinical to clinical development: a case study of anticancer drug, crizotinib. AAPS J. 15, 354–366 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abbas, R. & Hsyu, P.‐H. Clinical pharmacokinetics and pharmacodynamics of bosutinib. Clin. Pharmacokinet. 55, 1191–1204 (2016). [DOI] [PubMed] [Google Scholar]
- 9. Hsyu, P.‐H. , Mould, D.R. , Upton, R.N. & Amantea, M. Pharmacokinetic‐pharmacodynamic relationship of bosutinib in patients with chronic phase chronic myeloid leukemia. Cancer Chemother. Pharmacol. 71, 209–218 (2013). [DOI] [PubMed] [Google Scholar]
- 10. Committee for Medicinal Products for Human Use (CHMP) European Medicines Evaluation Agency Bosutinib European Public Assessment Report. <http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002373/WC500141745.pdf> (2013).
- 11. Hsyu, P. , Mould, D.R. , Abbas, R. & Amantea, M. Population pharmacokinetic and pharmacodynamic analysis of bosutinib. Drug Metab. Pharmacokinet. 29, 4411–448 (2014). [DOI] [PubMed] [Google Scholar]
- 12. Wang, X. et al Differential effects of dosing regimen on the safety and efficacy of dasatinib: Retrospective exposure‐response analysis of a phase III study. Clin. Pharmacol. 5, 85–97 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ishida, Y. et al Pharmacokinetics and pharmacodynamics of dasatinib in the chronic phase of newly diagnosed chronic myeloid leukemia. Eur. J. Clin. Pharmacol. 72, 185–193 (2016). [DOI] [PubMed] [Google Scholar]
- 14. Giles, F.J. et al Nilotinib population pharmacokinetics and exposure‐response analysis in patients with imatinib‐resistant or ‐intolerant chronic myeloid leukemia. Eur. J. Clin. Pharmacol. 69, 813–823 (2013). [DOI] [PubMed] [Google Scholar]
- 15. Larson, R.A. et al Population pharmacokinetic and exposure‐response analysis of nilotinib in patients with newly diagnosed Ph+ chronic myeloid leukemia in chronic phase. Eur. J. Clin. Pharmacol. 68, 723–733 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Francis, J. , Dubashi, B. , Sundaram, R. , Pradhan, S.C. & Chandrasekaran, A. A study to explore the correlation of ABCB1, ABCG2, OCT1 genetic polymorphisms and trough level concentration with imatinib mesylate‐induced thrombocytopenia in chronic myeloid leukemia patients. Cancer Chemother. Pharmacol. 76, 1185–1189 (2015). [DOI] [PubMed] [Google Scholar]
- 17. Faivre, S. et al Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J. Clin. Oncol. 24, 25–35 (2006). [DOI] [PubMed] [Google Scholar]
- 18. Guilhot, F. et al Plasma exposure of imatinib and its correlation with clinical response in the Tyrosine Kinase Inhibitor OPtimization and Selectivity trial. Haematologica 97, 731–738 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Larson, R.A. et al Imatinib pharmacokinetics and its correlation with response and safety in chronic‐phase chronic myeloid leukemia: A subanalysis of the IRIS study. Blood 111, 4022–4028 (2008). [DOI] [PubMed] [Google Scholar]
- 20. Takahashi, N. et al Correlation between imatinib pharmacokinetics and clinical response in Japanese patients with chronic‐phase chronic myeloid leukemia. Clin. Pharmacol. Ther. 88, 809–813 (2010). [DOI] [PubMed] [Google Scholar]
- 21. Koren‐Michowitz, M. et al Imatinib plasma trough levels in chronic myeloid leukaemia: results of a multicentre study CSTI571AIL11TGLIVEC. Hematol. Oncol. 30, 200–205 (2012). [DOI] [PubMed] [Google Scholar]
- 22. Marin, D. et al Adherence is the critical factor for achieving molecular responses in patients with chronic myeloid leukemia who achieve complete cytogenetic responses on imatinib. J. Clin. Oncol. 28, 2381–2388 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Van Obbergh, F. et al The clinical relevance of imatinib plasma trough concentrations in chronic myeloid leukemia. A Belgian study. Clin. Biochem. 50, 452–454 (2017). [DOI] [PubMed] [Google Scholar]
- 24. Singh, N. , Kumar, L. , Meena, R. & Velpandian, T. Drug monitoring of imatinib levels in patients undergoing therapy for chronic myeloid leukaemia: comparing plasma levels of responders and non‐responders. Eur. J. Clin. Pharmacol. 65, 545–549 (2009). [DOI] [PubMed] [Google Scholar]
- 25. Gotta, V. et al Clinical usefulness of therapeutic concentration monitoring for imatinib dosage individualization: results from a randomized controlled trial. Cancer Chemother. Pharmacol. 74, 1307–1319 (2014). [DOI] [PubMed] [Google Scholar]
- 26. Food and Drug Administration . Center for Drug Evaluation and Research Ponatinib Clinical Pharmacology and Biopharmaceutics Review. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203469Orig1s000ClinPharmR.pdf>
- 27. Cortes, J.E. et al Ponatinib in refractory Philadelphia chromosome‐positive leukemias. N. Engl. J. Med. 367, 2075–2088 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wind, S. , Schmid, M. , Erhardt, J. , Goeldner, R.G. & Stopfer, P. Pharmacokinetics of afatinib, a selective irreversible ErbB family blocker, in patients with advanced solid tumours. Clin. Pharmacokinet. 52, 1101–1109 (2013). [DOI] [PubMed] [Google Scholar]
- 29. Wind, S. , Schnell, D. , Ebner, T. , Freiwald, M. & Stopfer, P. Clinical Pharmacokinetics and Pharmacodynamics of Afatinib. Clin. Pharmacokinet. [Epub ahead of print] (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Food and Drug Administration . Center for Drug Evaluation and Research Afatinib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/201292Orig1s000ClinPharmR.pdf> (2012).
- 31. Lu, J.‐F. et al Clinical pharmacokinetics of erlotinib in patients with solid tumors and exposure‐safety relationship in patients with non‐small cell lung cancer. Clin. Pharmacol. Ther. 80, 136–145 (2006). [DOI] [PubMed] [Google Scholar]
- 32. Steffens, M. et al Dosing to rash? — The role of erlotinib metabolic ratio from patient serum in the search of predictive biomarkers for EGFR inhibitor‐mediated skin rash. Eur. J. Cancer 55, 131–139 (2016). [DOI] [PubMed] [Google Scholar]
- 33. Hidalgo, M. et al Phase I and pharmacologic study of OSI‐774, an epidermal growth factor receptor tyrosine kinase inhibitor, in patients with advanced solid malignancies. J Clin Oncol 19, 3267–3279 (2001). [DOI] [PubMed] [Google Scholar]
- 34. Kobayashi, H. et al Relationship among gefitinib exposure, polymorphisms of its metabolizing enzymes and transporters, and side effects in Japanese patients with non‐small‐cell lung cancer. Clin. Lung Cancer 16, 274–281 (2015). [DOI] [PubMed] [Google Scholar]
- 35. Zhao, Y.‐Y. et al The relationship between drug exposure and clinical outcomes of non‐small cell lung cancer patients treated with gefitinib. Med. Oncol. 28, 697–702 (2011). [DOI] [PubMed] [Google Scholar]
- 36. Perez, C.A. et al Phase II study of gefitinib adaptive dose escalation to skin toxicity in recurrent or metastatic squamous cell carcinoma of the head and neck. Oral Oncol. 48, 887–892 (2012). [DOI] [PubMed] [Google Scholar]
- 37. Burris, H.A. et al Phase I safety, pharmacokinetics, and clinical activity study of lapatinib (GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J. Clin. Oncol. 23, 5305–5313 (2005). [DOI] [PubMed] [Google Scholar]
- 38. Food and Drug Administration . Center for Drug Evaluation and Research Osimertinib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/208065Orig1s000ClinPharmR.pdf> (2015).
- 39. Committee for Medicinal Products for Human Use (CHMP) European Medicines Agency Osimertinib European Public Assessment Report. <http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/004124/WC500202024.pdf> (2015).
- 40. Food and Drug Administration . Center for Drug Evaluation and Research Axitinib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202324Orig1s000ClinPharmR.pdf> (2012).
- 41. Rini, B.I. et al Diastolic blood pressure as a biomarker of axitinib efficacy in solid tumors. Clin. Cancer Res. 17, 3841–3849 (2011). [DOI] [PubMed] [Google Scholar]
- 42. Rini, B.I. et al Axitinib in metastatic renal cell carcinoma: results of a pharmacokinetic and pharmacodynamic analysis. J. Clin. Pharmacol. 53, 491–504 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rini, B.I. et al Axitinib with or without dose titration for first‐line metastatic renal‐cell carcinoma: a randomised double‐blind phase 2 trial. Lancet Oncol. 14, 1233–1242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rini, B.I. et al Overall survival analysis from a randomized phase II study of axitinib with or without dose titration in first‐line metastatic renal cell carcinoma. Clin. Genitourin. Cancer 1–5 (2016). [DOI] [PubMed] [Google Scholar]
- 45. Tsuchiya, N. et al Association of pharmacokinetics of axitinib with treatment outcome and adverse events in advanced renal cell carcinoma patients. In Genitourinary Cancers Symposium. J. Clin. Oncol. 33, 2015 (suppl 7; abstr 506) (2015). [Google Scholar]
- 46. Rini, B.I. et al Axitinib dose titration: analyses of exposure, blood pressure and clinical response from a randomized phase II study in metastatic renal cell carcinoma. Ann. Oncol. 26, 1372–1377 (2015). [DOI] [PubMed] [Google Scholar]
- 47. Miles, D. , Jumbe, N.L. , Lacy, S. & Nguyen, L. Population pharmacokinetic model of cabozantinib in patients with medullary thyroid carcinoma and its application to an exposure‐response analysis. Clin. Pharmacokinet. 55, 93–105 (2016). [DOI] [PubMed] [Google Scholar]
- 48. Miles, D.R. , Wada, D.R. , Jumbe, N.L. , Lacy, S.A. & Nguyen, L.T. Population pharmacokinetic/pharmacodynamic modeling of tumor growth kinetics in medullary thyroid cancer patients receiving cabozantinib. Anticancer Drugs 27, 328–341 (2016). [DOI] [PubMed] [Google Scholar]
- 49. Food and Drug Administration . Center for Drug Evaluation and Research Cabozantinib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203756Orig1s000ClinPharmR.pdf> (2012).
- 50. Singh, H. et al U.S. Food and Drug Administration Approval: cabozantinib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 23, 330–335 (2017). [DOI] [PubMed] [Google Scholar]
- 51. Food and Drug Administration . Center for Drug Evaluation and Research Lenvatinib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206947Orig1s000ClinPharmR.pdf> (2014).
- 52. Committee for Medicinal Products for Human Use (CHMP) European Medicines Agency Lenvatinib European Public Assessment report. <http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/003727/WC500188676.pdf> (2015).
- 53. Committee for Medicinal Products for Human Use (CHMP) European Medicines Agency Nintedanib European Public Assessment Report. <http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002569/WC500179972.pdf> (2014).
- 54. Committee for Medicinal Products for Human Use (CHMP) European Medicines Agency Nintedanib Summary of Product Characteristics. <http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Product_Information/human/002569/WC500179970.pdf> (2014).
- 55. Okamoto, I. et al Phase I safety, pharmacokinetic, and biomarker study of BIBF 1120, an oral triple tyrosine kinase inhibitor in patients with advanced solid tumors. Mol. Cancer Ther. 9, 2825–2833 (2010). [DOI] [PubMed] [Google Scholar]
- 56. Food and Drug Administration . Center for Drug Evaluation and Research Nintedanib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2014/205832Orig1s000ClinPharmR.pdf> (2009).
- 57. Suttle, A.B. et al Relationships between pazopanib exposure and clinical safety and efficacy in patients with advanced renal cell carcinoma. Br. J. Cancer 111, 1–8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lin, Y. et al Relationship between plasma pazopanib concentration and incidence of adverse events in renal cell carcinoma. In J. Clin. Oncol. 29 (suppl 7; abstr 345) (2011). [Google Scholar]
- 59. Food and Drug Administration . Center for Drug Evaluation and Research Pazopanib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2009/022465s000_ClinPharmR.pdf> (2008).
- 60. Xu, C.‐F. et al HLA‐B*57:01 confers susceptibility to pazopanib‐associated liver injury in patients with cancer. Clin. Cancer Res. 22, 1371–1377 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kumar, R. et al Pharmacokinetic‐pharmacodynamic correlation from mouse to human with pazopanib, a multikinase angiogenesis inhibitor with potent antitumor and antiangiogenic activity. Mol. Cancer Ther. 6, 2012–2021 (2007). [DOI] [PubMed] [Google Scholar]
- 62. Bible, K.C. et al Efficacy of pazopanib in progressive, radioiodine‐refractory, metastatic differentiated thyroid cancers: results of a phase 2 consortium study. Lancet Oncol. 11, 962–972 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. de Wit, D. et al Therapeutic drug monitoring to individualize the dosing of pazopanib: a pharmacokinetic feasibility study. Ther. Drug Monit. 37, 331–338 (2014). [DOI] [PubMed] [Google Scholar]
- 64. Verheijen, R.B. et al Individualized pazopanib dosing: a prospective feasibility study in cancer patients. Clin. Cancer Res. 22, 5738–5746 (2016). [DOI] [PubMed] [Google Scholar]
- 65. Food and Drug Administration . Center for Drug Evaluation and Research Regorafenib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203085Orig1s000ClinPharmR.pdf> (2012).
- 66. Committee for Medicinal Products for Human Use (CHMP) European Medicines Agency Regorafenib CHMP Extension of Indication Variation Assessment Report. <http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Assessment_Report_‐_Variation/human/000701/WC500215995.pdf> (2014).
- 67. Boudou‐Rouquette, P. et al Early sorafenib‐induced toxicity is associated with drug exposure and UGTIA9 genetic polymorphism in patients with solid tumors: a preliminary study. PLoS One 7, 1–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Blanchet, B. et al Validation of an HPLC‐UV method for sorafenib determination in human plasma and application to cancer patients in routine clinical practice. J. Pharm. Biomed. Anal. 49, 1109–1114 (2009). [DOI] [PubMed] [Google Scholar]
- 69. Fukudo, M. et al Exposure‐toxicity relationship of sorafenib in Japanese patients with renal cell carcinoma and hepatocellular carcinoma. Clin. Pharmacokinet. 53, 185–196 (2014). [DOI] [PubMed] [Google Scholar]
- 70. Shimada, M. et al Monitoring serum levels of sorafenib and its N‐oxide is essential for long‐term sorafenib treatment of patients with hepatocellular carcinoma. Tohoku J. Exp. Med. 237, 173–182 (2015). [DOI] [PubMed] [Google Scholar]
- 71. Yu, H. et al Integrated semi‐physiological pharmacokinetic model for both sunitinib and its active metabolite SU12662. Br. J. Clin. Pharmacol. 79, 809–819 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Noda, S. et al Assessment of sunitinib‐induced toxicities and clinical outcomes based on therapeutic drug monitoring of sunitinib for patients with renal cell carcinoma. Clin. Genitourin. Cancer 13, 350–358 (2015). [DOI] [PubMed] [Google Scholar]
- 73. Teo, Y.L. et al Association of drug exposure with toxicity and clinical response in metastatic renal cell carcinoma patients receiving an attenuated dosing regimen of sunitinib. Target. Oncol. 10, 429–437 (2015). [DOI] [PubMed] [Google Scholar]
- 74. Narjoz, C. et al Role of the lean body mass and of pharmacogenetic variants on the pharmacokinetics and pharmacodynamics of sunitinib in cancer patients. Invest. New Drugs 33, 257–268 (2015). [DOI] [PubMed] [Google Scholar]
- 75. Ravaud, A. & Bello, C.L. Exposure‐response relationships in patients with metastatic renal cell carcinoma receiving sunitinib: maintaining optimum efficacy in clinical practice. Anticancer Drugs 22, 377–383 (2011). [DOI] [PubMed] [Google Scholar]
- 76. Houk, B.E. et al Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta‐analysis. Cancer Chemother. Pharmacol. 66, 357–371 (2010). [DOI] [PubMed] [Google Scholar]
- 77. Houk, B.E. , Bello, C.L. , Kang, D. & Amantea, M. A population pharmacokinetic meta‐analysis of sunitinib malate (SU11248) and its primary metabolite (SU12662) in healthy volunteers and oncology patients. Clin. Cancer Res. 15, 2497–2506 (2009). [DOI] [PubMed] [Google Scholar]
- 78. Lankheet, N.A.G. et al Pharmacokinetically guided sunitinib dosing: a feasibility study in patients with advanced solid tumours. Br. J. Cancer 110, 2441–2449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Food and Drug Administration . Center for Drug Evaluation and Research Vandetanib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/022405Orig1s000ClinPharmR.pdf> (2010).
- 80. Holden, S.N. et al Clinical evaluation of ZD6474, an orally active inhibitor of VEGF and EGF receptor signaling, in patients with solid, malignant tumors. Ann. Oncol. 16, 1391–1397 (2005). [DOI] [PubMed] [Google Scholar]
- 81. Bershas, D.A. et al Metabolism and disposition of oral dabrafenib in cancer patients: proposed participation of aryl nitrogen in carbon‐carbon bond cleavage via decarboxylation following enzymatic oxidation. Drug Metab. Dispos. 41, 2215–2224 (2013). [DOI] [PubMed] [Google Scholar]
- 82. Committee for Medicinal Products for Human Use (CHMP) European Medicines Agency Dabrafenib European Public Assessment Report. <http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002604/WC500149673.pdf> (2013).
- 83. Menzies, A.M. et al Characteristics of pyrexia in BRAFV600E/K metastatic melanoma patients treated with combined dabrafenib and trametinib in a phase I/II clinical trial. Ann. Oncol. 26, 415–421 (2015). [DOI] [PubMed] [Google Scholar]
- 84. Food and Drug Administration . Center for Drug Evaluation and Research Dabrafenib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/202806Orig1s000ClinPharmR.pdf> (2013).
- 85. Kramkimel, N. et al Vemurafenib pharmacokinetics and its correlation with efficacy and safety in outpatients with advanced BRAF‐mutated melanoma. Target. Oncol. 11, 59–69 (2016). [DOI] [PubMed] [Google Scholar]
- 86. Committee for Medicinal Products for Human Use (CHMP) European Medicines Agency Vemurafenib European Public Assessment Report. <http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002409/WC500124400.pdf> (2011).
- 87. Funck‐Brentano, E. et al Plasma vemurafenib concentrations in advanced BRAFV600mut melanoma patients: impact on tumour response and tolerance. Ann. Oncol. 26, 1470–1475 (2015). [DOI] [PubMed] [Google Scholar]
- 88. Funck‐Brentano, E. et al Is there a plasma vemurafenib concentration which predicts outcome in advanced BRAFV600 melanoma patients? Ann. Oncol. 2–6 (2015). [Google Scholar]
- 89. Goldwirt, L. et al. Reply to ‘Plasma vemurafenib concentrations in advanced BRAFV600mut melanoma patients: impact on tumour response and tolerance’ by Funck‐Brentano et al . Ann. Oncol. 27, 363.1–364 (2016). [DOI] [PubMed] [Google Scholar]
- 90. Nijenhuis, C. M. et al Clinical Pharmacokinetics of vemurafenib in BRAF‐mutated melanoma patients. J. Clin. Pharmacol. (2016) doi:10.1002/jcph.788 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 91. Food and Drug Administration . Center for Drug Evaluation and Research Cobimetinib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/206192Orig1s000ClinPharmR.pdf> (2014).
- 92. Ouellet, D. et al Population pharmacokinetics and exposure‐response of trametinib, a MEK inhibitor, in patients with BRAF V600 mutation‐positive melanoma. Cancer Chemother. Pharmacol. 77, 807–817 (2016). [DOI] [PubMed] [Google Scholar]
- 93. Committee for Medicinal Products for Human Use (CHMP) European Medicines Agency Trametinib European Public Assessment Report. <http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_‐_Public_assessment_report/human/002643/WC500169708.pdf> (2014).
- 94. Shipkova, M. et al Therapeutic drug monitoring of everolimus: a consensus report. Ther. Drug Monit. 38, 143–169 (2016). [DOI] [PubMed] [Google Scholar]
- 95. Ravaud, A. et al Relationship between everolimus exposure and safety and efficacy: meta‐analysis of clinical trials in oncology. Eur. J. Cancer 50, 486–495 (2014). [DOI] [PubMed] [Google Scholar]
- 96. Thiery‐Vuillemin, A. et al Impact of everolimus blood concentration on its anti‐cancer activity in patients with metastatic renal cell carcinoma. Cancer Chemother. Pharmacol. 73, 999–1007 (2014). [DOI] [PubMed] [Google Scholar]
- 97. Krueger, D.A. et al Everolimus for subependymal giant‐cell astrocytomas in tuberous sclerosis. N Engl J Med 363, 1801–1811 (2010). [DOI] [PubMed] [Google Scholar]
- 98. Food and Drug Administration . Center for Drug Evaluation and Research Ibrutinib Clinical Pharmacology and Biopharmaceutics Review. <https://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/205552Orig1s000ClinPharmR.pdf> (2013).
- 99. Advani, R.H. et al Bruton tyrosine kinase inhibitor ibrutinib (PCI‐32765) has significant activity in patients with relapsed/refractory B‐cell malignancies. J. Clin. Oncol. 31, 88–94 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Demetri, G.D. et al Imatinib plasma levels are correlated with clinical benefit in patients with unresectable/metastatic gastrointestinal stromal tumors. J. Clin. Oncol. 27, 3141–3147 (2009). [DOI] [PubMed] [Google Scholar]
- 101. Widmer, N. et al Imatinib plasma levels: correlation with clinical benefit in GIST patients. Br. J. Cancer 102, 1198–1199 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Farag, S. et al Imatinib pharmacokinetics in a large observational cohort of gastrointestinal stromal tumour patients. Clin. Pharmacokinet. [Epub ahead of print] (2016). [DOI] [PubMed] [Google Scholar]
- 103. Bouchet, S. et al Relationship between imatinib trough concentration and outcomes in the treatment of advanced gastrointestinal stromal tumours in a real‐life setting. Eur. J. Cancer 57, 31–38 (2016). [DOI] [PubMed] [Google Scholar]
- 104. Lankheet, N.A.G. et al Optimizing the dose in cancer patients treated with imatinib, sunitinib and pazopanib. Br. J. Clin. Pharmacol. (2017) doi:10.1111/bcp.13327 [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ramanathan, S. , Jin, F. , Sharma, S. & Kearney, B.P. Clinical pharmacokinetic and pharmacodynamic profile of idelalisib. Clin. Pharmacokinet. 55, 33–45 (2016). [DOI] [PubMed] [Google Scholar]
- 106. Jin, F. , Robeson, M. , Zhou, H. , Hisoire, G. & Ramanathan, S. The pharmacokinetics and safety of idelalisib in subjects with severe renal impairment. Cancer Chemother. Pharmacol. 76, 1133–1141 (2015). [DOI] [PubMed] [Google Scholar]
- 107. Food and Drug Administration . Center for Drug Evaluation and Research Palbociclib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/207103Orig1s000ClinPharmR.pdf> (2014).
- 108. Fox, P. et al Dose escalation of tamoxifen in patients with low endoxifen level: evidence for therapeutic drug monitoring — the TADE Study. Clin. cancer Res. 22, 3164–3171 (2016). [DOI] [PubMed] [Google Scholar]
- 109. Joerger, M. et al Open‐label, randomised study of individualized, pharmacokinetically (PK)‐guided dosing of paclitaxel combined with carboplatin or cisplatin in patients with advanced non‐small cell lung cancer (NSCLC). Ann. Oncol. 1–22 (2016).doi:10.1093/annonc/mdw290. [DOI] [PubMed] [Google Scholar]
- 110. Food and Drug Administration . Center for Drug Evaluation and Research Trametinib Clinical Pharmacology and Biopharmaceutics Review. <http://www.accessdata.fda.gov/drugsatfda_docs/nda/2013/204114Orig1s000ClinPharmR.pdf> (2013).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional supporting information can be found in the online version of this article.
Supporting Information