

Abstract
Recent studies by Stoltz, Grubbs et al. have shown that triethylsilane and potassium tert‐butoxide react to form a highly attractive and versatile system that shows (reversible) silylation of arenes and heteroarenes as well as reductive cleavage of C−O bonds in aryl ethers and C−S bonds in aryl thioethers. Their extensive mechanistic studies indicate a complex network of reactions with a number of possible intermediates and mechanisms, but their reactions likely feature silyl radicals undergoing addition reactions and SH2 reactions. This paper focuses on the same system, but through computational and experimental studies, reports complementary facets of its chemistry based on a) single‐electron transfer (SET), and b) hydride delivery reactions to arenes.
Keywords: density-functional calculations, electron transfer, hydrides, reaction mechanisms, silicon
Recently, Stoltz, Grubbs et al.1 have discovered a simple and elegant system comprising Et3SiH (2) and KOtBu which achieves a number of remarkable reactions: 1) converting arenes and heteroarenes, and their alkylated counterparts, into silyl‐substituted products, often with excellent regiocontrol1a–1c (e.g. 1→3; Scheme 1); 2) achieving reductive C−S bond cleavage in aryl thioethers (e.g. 4→5) in a reaction which has potential importance in removing sulfur traces from hydrocarbon fuels;1d 3) triggering reductive C−O bond cleavage in aryl ethers (e.g. 6→7) in a reaction with potential applications to controlled lignin degradation.1a,1d A number of intermediates likely arise from reaction of these two reagents, and spectroscopic evidence has resulted in informed proposals being made for their structures. These reactions have proved puzzling, but a recent coordinated study by synthetic, mechanistic, and computational chemists has allowed significant advances to be made.1e,1f The conclusions are: 1) the combination of Et3SiH and KOtBu leads to triethylsilyl radicals which have a major role to play in the reductive cleavage of the C−O and C−S bonds,1d 2) triethylsilyl radicals are also likely to be involved in the silylation reactions, although nonradical routes to the silylation have also been considered in depth and may also play a central role.1e,1f The mechanistic details are not fully in place, for example, on how formation of the silyl radicals occurs, but rational working hypotheses have been advanced.1e
Scheme 1.
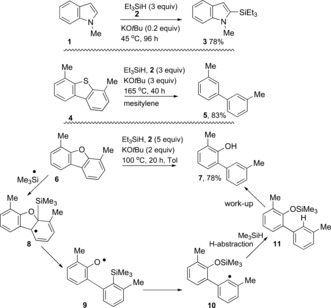
Selected transformations of the KOtBu/Et3SiH system.1
We had wondered if single‐electron transfer mechanisms were playing a significant role in some of these reactions, notably for the cleavage of C−O and C−S bonds. An early suggestion1a mentioned pentavalent silicates (e.g. 13 b; see Scheme 2) as reagents that were likely involved in the C−O cleavage, but the more recent computational studies on the substrates 4 and 6 instead support an alternative mechanism.1d In this regard, Scheme 1 shows ipso addition to the carbon atom of the C−O bond by triethylsilyl radicals, followed by C−O bond cleavage in conversion of 6 into 7.
Scheme 2.
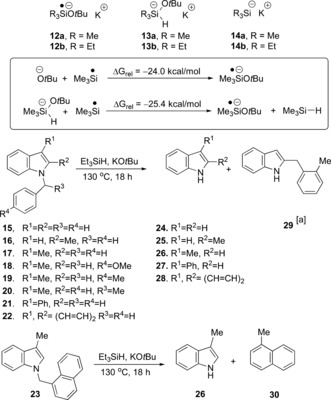
Indole‐based substrates as probes of electron‐transfer activity. [a] See the Supporting Information for a discussion of the mechanism of formation of this compound.
Our recent interest in reductive chemistry carried out by reactions involving KOtBu attracted us to this area.2 Studies mentioned above1e suggest that the reactive species produced could include the radical anion 12 b (Scheme 2) and the silicate anion 13 b.1a,1e Because of their subsequent importance in this paper, we mention here that the radical anions 12 may be formed in a number of ways, two of which are shown (inset) in Scheme 2 (see Figure 14 in Ref. 1e for an additional route). For these studies, we used the computationally less costly trimethylsilyl group instead of the triethylsilyl group.1d,1e To these, we add the triethylsilyl anion 14 b as another putative intermediate. At first sight, these compounds are potentially excellent electron donors, although, as will be seen below, computational chemistry is very helpful in eliminating species and mechanisms which are unlikely to contribute. In recent years, we have reported on many highly reducing organic electron donors that demonstrate remarkable behavior.3 We were therefore keen to test the KOtBu/Et3SiH system for evidence of single‐electron transfer (SET) activity and, if found, to calibrate the system's reactivity.
A literature search reveals that N‐benzylindole substrates are reductively cleaved to indoles and toluenes with two reagents—both involving electron transfer. The first uses Birch chemistry4 and the second uses low‐valent titanium reagents.5 Accordingly, we prepared a range of N‐benzylindole substrates (15–23; Scheme 2), to test for cleavage with silane and tert‐butoxide, and the outcomes are shown in Table 1. In each case, reactions afforded the debenzylated products, while blank reactions (no silane) led to excellent recovery of starting materials. The examples 15–22 also afforded volatile products from the benzyl unit. To counteract this, the naphthylmethyl substrate 23 was subjected to the reaction and afforded 1‐methylnaphthalene (30), in addition to 3‐methylindole (26), and recovered 23 (entry 18).
Table 1.
Cleavage of benzyl groups from indole derivatives.
| Entry | Substrate | Silane (3 or 0 equiv) | Base (3 equiv) | Yield [%] | |
|---|---|---|---|---|---|
| Product | Recovered Substrate | ||||
| 1 | 15 | Et3SiH | KOtBu | 24 (29) | – |
| 2 | 15 | ‐(blank)‐ | KOtBu | – | (85) |
| 3 | 16 | Et3SiH | KOtBu | 25 (49) + 29 (15) | – |
| 4 | 16 | ‐(blank)‐ | KOtBu | – | (99) |
| 5 | 17 | Et3SiH | KOtBu | 26 (73) | – |
| 6 | 17 | Et3SiH | NaOtBu[a] | – | (98) |
| 7 | 17 | ‐(blank)‐ | KOtBu | – | (88) |
| 8 | 18 | Et3SiH | KOtBu | 26 (76) | – |
| 9 | 18 | ‐(blank)‐ | KOtBu | – | (98) |
| 10 | 19 | Et3SiH | KOtBu | 26 (63) | Trace |
| 11 | 19 | ‐(blank)‐ | KOtBu | – | (86) |
| 12 | 20 | Et3SiH | KOtBu | 26 (47) | trace |
| 13 | 20 | ‐(blank)‐ | KOtBu | – | (93) |
| 14 | 21 | Et3SiH | KOtBu | 27 (80) | – |
| 15 | 21 | ‐(blank)‐ | KOtBu | – | (100) |
| 16 | 22 | Et3SiH | KOtBu | 28 (57) | (26) |
| 17 | 22 | ‐(blank)‐ | KOtBu | – | (99) |
| 18 | 23 | Et3SiH | KOtBu | 26 (55) + 30 (23) | (23) |
| 19 | 23 | ‐(blank)‐ | KOtBu | – | (88) |
Yields of products and recovered substrates are those for the isolated compounds. [a] As in Ref. 1, NaOtBu is not a successful substitute for KOtBu.
To understand the site of electron transfer in these reactions, we modelled the formation and reaction of two radical anions—those arising by electron transfer to the indole 17 and carbazole 22. In both cases (Figure 1), the SOMO showed spin density on the heterocycle, rather than on the benzyl group. These data is consistent with the greater delocalization available in either the bicyclic or tricyclic heterocycle for the transferred electron.
Figure 1.
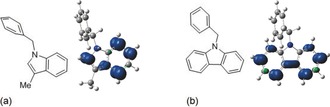
Representations of the spin density of the SOMO of the radical anion of N‐benzyl‐3‐methylindole 17 (a) and N‐benzylcarbazole 22 (b). Geometry optimizations and frequency calculations were carried out in Gaussian13 at M062X/6‐31++G(d,p) level of theory,14, 15 with solvation modelled implicitly using the C‐PCM model16 (For full computational details, see the Supporting Information).
We now use computational methods to compare the cleavage of the N‐benzyl group of 15 by an SET mechanism (Table 2) with the three potential electron donors 12 a–14 a. Here it is seen that electron transfer from 12 a to 15 is almost barrierless and is exergonic (entry 1; the scheme also shows facile fragmentation of the radical anion 31), while the electron‐transfer reactions from 13 a and 14 a (entries 2 and 3) show prohibitive energy profiles.
Table 2.
Energy profiles for candidate electron transfers to 15.
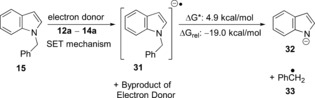
| Entry | Electron donor | Energy profile [kcal mol−1] | Byproduct of electron donor | Byproduct of electron donor | |
|---|---|---|---|---|---|
| 1 |

|
12 a | ΔG*=0.3 ΔG rel=−8.1 |
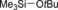
|
34 |
| 2 |
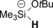
|
13 a | ΔG*=53.6 ΔG rel=49.4 |
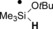
|
35 |
| 3 |

|
14 a | ΔG*=44.8 ΔG rel=38.7 |
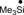
|
36 |
We also tested energy profiles for the debenzylation reaction with two possible competing pathways (Table 2; lower panels). The first of these recognizes that 13 a could be a very powerful hydride‐transfer agent and might facilitate an SN2 reaction, although an unusual one, at the benzylic carbon center. However, transfer of hydride from 13 a to 15 shows a barrier of 36.9 kcal mol−1 for the benzyl cleavage, and so this type of reaction will not occur under our reaction conditions in the laboratory. The second competing reaction type would involve an SH2 reaction by a R3Si radical at the benzylic carbon center. This path would also be an unexpected reaction, as radical displacements at tetrahedral carbon centers are almost unknown, and indeed the kinetic barrier (44.3 kcal mol−1) is again insurmountable. From these results, SET from 12 a is overwhelmingly the most likely of the computed candidate mechanisms for benzyl group cleavage. In effect, cleavage occurred to afford N‐methylaniline, 41, which was converted into the more easily isolated 42 following acetylation (56 % over 2 steps; Scheme 3). When the reaction was repeated, but in the absence of Et3SiH, no cleavage was observed, with the starting material 40 recovered (97 %). We next varied the protecting group on our indole substrates from benzyl to allyl. Given that the computational results showed electron transfer to the indole group in the substrates 17 and 22, rather than to the benzyl group, then the reagent should also to be able to cleave N‐allylindoles by an SET mechanism, because of the stabilization of the allyl radical leaving group.6 Accordingly, the substrates 43 and 45 were prepared. The indole products 26 and 46 were indeed formed from these substrates (35 % and 33 % respectively). The low yields may indicate the wealth of alternative reactions open to this reagent system. Indeed, a second product was isolated from the reaction of 43, namely o‐isopropylaniline (44; 18 %), although we have not explored the mechanism of its formation as yet. It was clear that the KOtBu/Et3SiH system is a more than competent electron‐donating system.
Scheme 3.
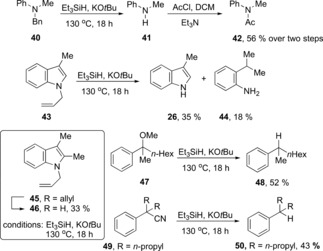
Reductive cleavage induced by the Et3SiH/KOtBu system.
In a more challenging probe for electron‐transfer potency, we subjected the benzyl methyl ether 47 to reduction by this system (Scheme 3). A close analogue of this substrate had proven a very tough substrate in previous studies.3h It did not undergo fragmentation until two electrons had been transferred. In this case, the reduced product 48 was produced in 52 % yield [a blank reaction afforded recovered starting material exclusively (62 %)]. Additionally, subjecting the nitrile 49 7 to the reaction afforded the hydrocarbon 50 as the sole product, consistent with electron transfer followed by loss of cyanide anion.
We calculated the oxidation potential of 12 a 8 to be E=−3.74 V vs. SCE (MeCN). This potential makes it much more powerful than alkali metals. Such a powerful electron donor should provide a good probe for the Marcus inverted region of SET reactions with substrates that show low reorganization energies, (e.g. polycyclic arenes).9 Stoltz, Grubbs et al. reported1d small amounts of partially reduced arenes from reduction of naphthalenes. In our hands, and in the presence of excess of KOtBu/Et3SiH, anthracene, phenanthrene, and naphthalene all afforded significant amounts of their dihydro counterparts (Scheme 4). These compounds would be expected products from Birch‐type electron‐transfer processes, but to probe the mechanism we undertook computational studies of electron transfer from 12 a to the hydrocarbons 51–53 to yield the corresponding radical anions 60–62. (Table 3) Here, the expected normal order of reactivity is 51>52>53.10 This order is also reflected in the ΔG rel values shown in Table 3. However, the reverse pattern is seen for the ΔG* values. SET to 51 from the radical anion 12 a shows an extraordinary barrier of 90 kcal mol−1,11 while reduction of 52 and 53 show progressively lower barriers; if this can be verified by detailed experimental studies, it will be a very rare intermolecular ground‐state illustration of the Marcus inverted region, (stronger driving force leads to retarded electron transfer).
Scheme 4.
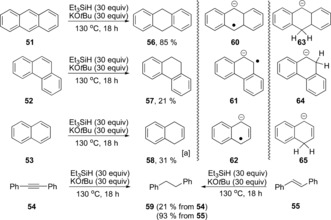
Reductions of polycylic arenes by KOtBu/Et3SiH. [a] Yield determined by NMR spectroscopy.
Table 3.
Energy profiles: SET from 12 a.
| Substrate | Energy profile [kcal mol−1] | Radical anion product |
|---|---|---|
| 51 | ΔG*: 90.0; ΔG rel: −37.8 | 60 |
| 52 | ΔG*: 28.3; ΔG rel: −25.0 | 61 |
| 53 | ΔG*: 25.7; ΔG rel: −22.3 | 62 |
In comparison, hydride transfer from 13 a to afford the corresponding anions 63–65 featured low barriers and favorable thermodynamics (Table 4). At least for the reduction of anthracene, hydride transfer from 13 a is indeed likely to occur. With the other substrates, hydride‐transfer reactions again show lower barriers than electron transfer from 12 a and this will of course be modulated by the concentration of the reducing species present. Finally, the alkyne 54 and stilbene 55 were reacted and gave (PhCH2)2 59 as the sole product (21 and 93 % respectively; Scheme 4).12
Table 4.
Energy profiles: Hydride transfer from 13 a.
| Substrate | Energy profile [kcal mol−1] | Anionic product |
|---|---|---|
| 51 | ΔG*: 16.7; ΔG rel: −29.4 | 63 |
| 52 | ΔG*: 20.0; ΔG rel: −14.8 | 64 |
| 53 | ΔG*: 21.7; ΔG rel: −13.2 | 65 |
In summary, the KOtBu/Et3SiH system provides access to a broad range of mechanisms for reductive chemistry, now including electron transfer and hydride delivery to arenes. The electron‐donor 12 b is identified as a uniquely powerful agent.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the University of Strathclyde, GSK, and EPSRC for funding, and SFC and WestCHEM for PECRE bursary funding, as well as EPSRC National Mass Spectrometry Service, Swansea for HRMS. Results were obtained using ARCHIE‐WeSt High Performance Computer (www.archie‐west.ac.uk). EPSRC grant nos. EP/K000586/1 and EP/N509371/1′.
A. J. Smith, A. Young, S. Rohrbach, E. F. O'Connor, M. Allison, H.-S. Wang, D. L. Poole, T. Tuttle, J. A. Murphy, Angew. Chem. Int. Ed. 2017, 56, 13747.
Contributor Information
Dr. Tell Tuttle, Email: tell.tuttle@strath.ac.uk
Prof. Dr. John A. Murphy, Email: john.murphy@strath.ac.uk.
References
- 1.
- 1a. Fedorov A., Toutov A. A., Swisher N. A., Grubbs R. H., Chem. Sci. 2013, 4, 1640–1645; [Google Scholar]
- 1b. Toutov A. A., Liu W.-B., Betz K. N., Fedorov A., Stoltz B. M., Grubbs R. H., Nature 2015, 518, 80–84; [DOI] [PubMed] [Google Scholar]
- 1c. Toutov A. A., Liu W.-B., Betz K. N., Stoltz B. M., Grubbs R. H., Nat. Protoc. 2016, 10, 1897–1903; [DOI] [PubMed] [Google Scholar]
- 1d.A. A. Toutov, M. Salata, A. Fedorov, Y.-F. Yang, Y. Liang, R. Cariou, K. N. Betz, E. P. A. Couzijn, J. W. Shabaker, K. N. Houk, R. H. Grubbs, Nat. Energy 2017, 2, issue DOI: https://doi.org/10.1038/nenergy.2017.8,
- 1e. Liu W.-B., Schuman D. P., Yang Y.-F., Toutov A. A., Liang Y., Klare H. F. T., Nesnas N., Oestreich M., Blackmond D. G., Virgil S. C., Banerjee S., Zare R. N., Grubbs R. H., Houk K. N., Stoltz B. M., J. Am. Chem. Soc. 2017, 139, 6867–6879; [DOI] [PubMed] [Google Scholar]
- 1f. Banerjee S., Yang Y.-F., Jenkins I. D., Liang Y., Toutov A. A., Liu W.-B., Schuman D. P., Grubbs R. H., Stoltz B. M., Krenske E. H., Houk K. N., Zare R. N., J. Am. Chem. Soc. 2017, 139, 6880–6887. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Barham J. P., Coulthard G., Kane R. G., Delgado N., John M. P., Murphy J. A., Angew. Chem. Int. Ed. 2016, 55, 4492–4496; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 4568–4572; [Google Scholar]
- 2b. Barham J. P., Coulthard G., Emery K., Doni E., Cumine F., Nocera G., John M. P., Berlouis L. E. A., McGuire T., Tuttle T., Murphy J. A., J. Am. Chem. Soc. 2016, 138, 7402–7410; [DOI] [PubMed] [Google Scholar]
- 2c. Zhou S., Anderson G. M., Mondal B., Doni E., Ironmonger V., Kranz M., Tuttle T., Murphy J. A., Chem. Sci. 2014, 5, 476–482; [Google Scholar]
- 2d. Zhou S., Doni E., Anderson G. M., Kane R. G., MacDougall S. W., Ironmonger V. M., Tuttle T., Murphy J. A., J. Am. Chem. Soc. 2014, 136, 17818–17826. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Hanson S. S., Doni E., Traboulsee K. T., Coulthard G., Murphy J. A., Dyker C. A., Angew. Chem. Int. Ed. 2015, 54, 11236–11239; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11388–11391; [Google Scholar]
- 3b. Doni E., Mondal B., O'Sullivan S., Tuttle T., Murphy J. A., J. Am. Chem. Soc. 2013, 135, 10934–10937; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Cahard E., Schoenebeck F., Garnier J., Cutulic S. P. Y., Zhou S., Murphy J. A., Angew. Chem. Int. Ed. 2012, 51, 3673–3676; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3733–3736; [Google Scholar]
- 3d. Murphy J. A., Schoenebeck F., Zhou S.-Z., Uenoyama Y., Miclo Y., Tuttle T., J. Am. Chem. Soc. 2007, 129, 13368–13369; [DOI] [PubMed] [Google Scholar]
- 3e. Murphy J. A., Zhou S.-Z., Thomson D. W., Schoenebeck F., Mohan M., Park S. R., Tuttle T., Berlouis L. E. A., Angew. Chem. Int. Ed. 2007, 46, 5178–5183; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 5270–5275; [Google Scholar]
- 3f. Murphy J. A., Khan T. A., Zhou S.-Z., Thomson D. W., Mahesh M., Angew. Chem. Int. Ed. 2005, 44, 1356–1360; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 1380–1384; [Google Scholar]
- 3g. O'Sullivan S., Doni E., Tuttle T., Murphy J. A., Angew. Chem. Int. Ed. 2014, 53, 474–478; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 484–488; [Google Scholar]
- 3h. Doni E., O‘Sullivan S., Murphy J. A., Angew. Chem. Int. Ed. 2013, 52, 2239–2242; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2295–2298. [Google Scholar]
- 4. Fujiwara J., Fukutani Y., Sano H., Maruoka K., Yamamoto H., J. Am. Chem. Soc. 1983, 105, 7177–7179. [Google Scholar]
- 5. Talukdar S., Nayak S. K., Benerji A., J. Org. Chem. 1998, 63, 4925–4929. [Google Scholar]
- 6.See Figure 3.10 in “Beyond Orbital Overlap”. Alabugin I. V., Stereoelectronic Effects: A Bridge Between Structure and Reactivity; Wiley: Chichester, 2016, pp 47. [Google Scholar]
- 7.
- 7a. Mattalia J. M., Marchi-Delapierre C., Hazimeh H., Chanon M., Arkivoc 2006, iv, 90–118; [Google Scholar]
- 7b. Fleming F. F., Zhang Z., Tetrahedron 2005, 61, 747–789; [Google Scholar]
- 7c. Sinz C. J., Rychnovsky S. D., Top. Curr. Chem. 2001, 216, 51–92; [Google Scholar]
- 7d. Beilstein J. M. J. Org. Chem. 2017, 13, 267–284; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7e. Kawamoto T., Geib S. J., Curran D. P., J. Am. Chem. Soc. 2015, 137, 8617–8622; [DOI] [PubMed] [Google Scholar]
- 7f. Doni E., Murphy J. A., Org. Chem. Front. 2014, 1, 1072–1076. [Google Scholar]
- 8. Roth H. G., Romero N. A., Nicewicz D. A., Synlett 2016, 27, 714–723. [Google Scholar]
- 9. Marcus R. A., Angew. Chem. Int. Ed. Engl. 1993, 32, 1111–1121; [Google Scholar]; Angew. Chem. 1993, 105, 1161–1172. [Google Scholar]
- 10.
- 10a. Tschurl M., Boesl U., Gilb S., J. Chem. Phys. 2006, 125, 194310; [DOI] [PubMed] [Google Scholar]
- 10b. Heinis T., Chowdhury S., Kebarle P., Org. Mass Spect. 1993, 28, 358–365. [Google Scholar]
- 11.Using Marcus theory, this is the inevitable result of extremely strong driving force ΔG rel and low reorganization energy, λ. See the Supporting Information.
- 12. Szostak M., Spain M., Procter D. J., J. Org. Chem. 2014, 79, 2522–2537. [DOI] [PubMed] [Google Scholar]
- 13.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox Gaussian 09, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- 14. Zhao Y., Truhlar D. G., Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- 15. Rassolov V. A., Ratner M. A., Pople J. A., Redfern P. C., Curtiss L. A., J. Comput. Chem. 2001, 22, 976–984. [Google Scholar]
- 16. Cossi M., Rega N., Scalmani G., Barone V., J. Comput. Chem. 2003, 24, 669–681. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary