

Abstract
We herein showcase the ability of NHC‐coordinated dinuclear NiI–NiI complexes to override fundamental reactivity limits of mononuclear (NHC)Ni0 catalysts in cross‐couplings. This is demonstrated with the development of a chemoselective trifluoromethylselenolation of aryl iodides catalyzed by a NiI dimer. A novel SeCF3‐bridged NiI dimer was isolated and shown to selectively react with Ar−I bonds. Our computational and experimental reactivity data suggest dinuclear NiI catalysis to be operative. The corresponding Ni0 species, on the other hand, suffers from preferred reaction with the product, ArSeCF3, over productive cross‐coupling and is hence inactive.
Keywords: catalysis, chemoselectivity, density functional calculations, fluorine, nickel
Despite the widespread existence of multinuclear metal sites in naturally occurring catalysts (enzymes), man‐made homogeneous catalysis is dominated by mononuclear metal cores.1 This might be due to our still limited understanding of the underlying synergism and reactivity of multimetallic assemblies. A prominent example is nickel, which is of significant current synthetic interest and predominantly investigated as a monomer in synthesis,2, 3 although it is featured in higher‐order clusters in several enzymes.4 Whereas the greater sustainability of nickel is advantageous, its high reactivity and mechanistic diversity can make it difficult to tame this metal in a synthetic context, impacting in particular chemoselectivity—a key requirement for applications in synthesis. The relative instabilities of NiII intermediates and their comparably low propensities towards transmetalation as required in traditional Ni0/NiII catalysis have been identified as an origin of this reactivity behavior, leading to side reactions, undesired side products, multiple potentially reactive species, as well as catalyst deactivation.2a,2b, 5
We hypothesized that dinuclear Ni catalysis could be particularly advantageous in this context as the elementary steps, that is, oxidative addition and transmetalation, would be formally reversed, circumventing the intermediacy of poorly reactive NiII species that are prone to side reactions (Figure 1). Our group recently showed this concept to be viable for palladium.6 However, whereas NiI complexes have been successfully synthesized,7 detected in catalytic transformations employing typical Ni0 catalysts,8 used as precatalysts,9 or implicated as mechanistic intermediates,10 unambiguous mechanistic support and a rationale for the direct catalytic involvement of NiI dimers in cross‐couplings have not been reported.
Figure 1.
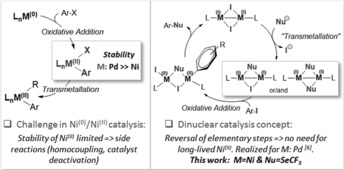
Catalysis with mononuclear M0/MII versus dinuclear MI–MI complexes.
Building on our research in the area of PdI dimer catalysis,6 which led to the development of a catalytic trifluoromethylselenolation of aryl iodides,6c we herein describe our efforts in exploring whether such a dinuclear catalysis concept is feasible also with the less precious and more sustainable element nickel.
The SeCF3 group features several agrochemically and pharmaceutically important properties in terms of the resulting membrane permeability and bioavailability.11 Consequently, there have been numerous activities in devising synthetic methods to access this compound class.12 The direct catalytic incorporation of the SeCF3 moiety is of particular interest as it may be used for late‐stage manipulations of molecules. The latter concept has, however, rarely been realized;13 it has been accomplished for aryl diazonium salts under Cu catalysis,14 and a coupling of aryl iodides with (Me4N)SeCF3 catalyzed by a dinuclear PdI complex has been developed by our group.6c
We started our investigations with assessing as to whether Ni0(cod)2 in combination with the NHC ligand 1,3‐bis(2,6‐diisopropylphenyl)imidazolin‐2‐ylidene (SIPr) could trigger the trifluoromethylselenolation of aryl halides. In the presence of this catalyst and ligand, 4‐iodoanisole (1) was converted into the corresponding ArSeCF3 product in 59 % yield (Figure 2). The analogous aryl bromide and chloride did not give the ArSeCF3 product. In all cases, the formation of the corresponding biaryls and dehalogenation were observed. The remainder was unreacted starting material. The latter observation may appear surprising at first given that Ni0 complexes are typically highly reactive, catalyzing even cross‐couplings of unactivated aryl ethers or aryl fluorides.2, 5 However, the formation of biaryl species hints toward a possible explanation. Such biaryl products can be an indication of a change in catalyst, arising from a ligand exchange between [LnNiII(Ar)(X)] intermediates to form [LnNiII(Ar)2] and [LnNiII(X)2]. Ultimately, biaryls are obtained upon reductive elimination. The resulting [LnNi0] species may then undergo comproportionation with [NiII(X)2] to [NiI]n.5a We speculated that the lack of significant conversion with ArBr and ArCl may be associated with the intermediacy of [NiI]n, rather than [Ni0].
Figure 2.
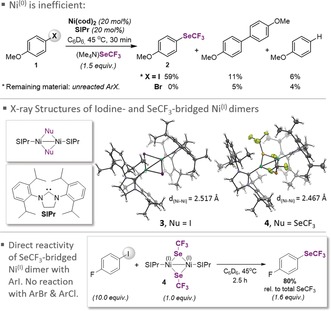
Direct reactivity of the NiI–NiI complex with ArI. X‐ray structures of iodine‐ and SeCF3‐bridged NiI dimers and comparison with Ni0.
Sigman and co‐workers have synthesized a Cl‐bridged SIPr‐derived NiI dimer and its unsaturated counterpart (i.e., with the IPr ligand).7b Matsubara and co‐workers very recently showed that the latter complex triggers Kumada cross‐couplings, and it was suggested that a NiI dimer or monomer may be involved in the process.15 Smaller NHC NiI monomers have also been shown to trigger C−C and C−N couplings.9 A pronounced ligand effect was observed by Louie et al. for the formation of NiI or NiII in reactions with (NHC)Ni0.16 While the origins are not understood, the data suggest that the NiI monomer or dimer species might function as precatalysts under these conditions. In this context, no rationale has been presented to date as to why a NiI may potentially be preferred over a Ni0 pathway. For phosphine‐based NiI complexes, our group and others have recently demonstrated that NiI is catalytically inactive or serves as a precatalyst.17 To obtain conclusive insight, we set out to prepare NiI complexes with iodine and SeCF3 ligands. We succeeded in the synthesis of an iodine‐ (3) and a SeCF3‐bridged (4) dinuclear NiI complex. The X‐ray structures are shown in Figure 2.
Given that we saw significantly more conversion with ArI but mainly biaryl formation with ArBr (Figure 2), we surmised that NiI species are formed in these cases and take over as the active catalyst for ArI, but may not be reactive enough for ArBr. To test this, we subjected our newly synthesized SeCF3‐bridged NiI dimer 4 (1 equiv) to ArI and ArBr (10 equiv). Whereas trifluoromethylselenolation was indeed observed for ArI (and 80 % of the total available SeCF3 was incorporated into ArI), there was no conversion for ArBr (Figure 2). Importantly, no biaryl species were detected in these reactions, suggesting that the NiI complex does not simply serve as a precursor to Ni0. Moreover, our kinetic studies under the same conditions gave first order in NiI dimer 4, in agreement with a direct reaction between the dimer and the aryl iodide. These data strongly suggest that NiI is a competent trifluoromethylselenolation species, and hence also likely a competent species in catalysis when generated from the iodine‐bridged NiI dimer 3 in the presence of (Me4N)SeCF3. Separate studies showed that a facile displacement of the iodine bridges in 3 with (Me4N)SeCF3 takes place to give 4, in analogy to our previously developed PdI–PdI chemistry.6c
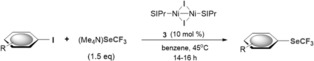
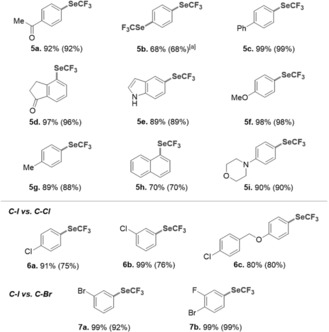
Pleasingly, with 10 mol % of [(SIPr)NiI(I)]2 (3) and (Me4N)SeCF3 (1.5 equiv) in benzene at 45 °C, a range of aryl iodides were successfully transformed into the corresponding ArSeCF3 products (Table 1). A number of electron‐rich and electron‐poor aryl iodides were functionalized in good to excellent yields. The method proved to be compatible with various functional groups, such as ketone (5 a and 5 d), methoxy (5 f), and amine (5 i) moieties, as well as the pharmaceutically interesting unprotected indole motif (5 e).
Table 1.
Scope of the trifluoromethylselenolation catalyzed by NiI dimer 3.
|
Reaction conditions: 3 (0.01 mmol, 10.9 mg), ArI (0.1 mmol), (Me4N)SeCF3 (0.15 mmol, 33 mg), benzene (1.0 mL). Yields determined by 19F NMR analysis with PhCF3 as an internal standard or upon isolation (in parentheses). [a] (Me4N)SeCF3 (3 equiv).
We hypothesized that the formally less electron‐rich NiI dimer might offer a platform for selective functionalizations and tested its potential to also trigger chemoselective catalytic C−SeCF3 bond formations. Pleasingly, we observed exclusive functionalization of C−I bonds in the presence of C−Br and C−Cl bonds (Table 1, bottom).
These data showcase the superiority of an isolated dinuclear NiI complex as a catalyst in the chemoselective SeCF3 functionalization of aryl iodides without the formation of side products. In this context, it was unclear why a NiI dimer would be the preferred reactive species in C−SeCF3 couplings over Ni0. To address this, we turned to computational studies.18
We initially assessed the feasibility of a [Ni0] catalyst to oxidatively add to PhI, PhBr, and PhCl with DFT methods.20 The activation barriers for oxidative addition follow the expected trend, that is, ΔG ≠ follows ArI<ArBr<ArCl. We previously demonstrated that another important factor for the efficiency and scope of Ni‐catalyzed functionalizations is the likelihood of the catalyst reacting with the desired product.19 In our case, an activated C−SeCF3 moiety was installed, which could also be prone to oxidative addition to [Ni]. Interestingly, we observed that the oxidative addition of PhSeCF3 to [Ni0] is characterized by a substantially lower activation barrier than addition of PhI, PhBr, and PhCl (Figure 3 and Table S1 in the Supporting Information). Depending on the level of theory (we considered M06L, M06, and PBE0‐D3),20 addition of PhSeCF3 is favored by ΔΔG ≠=4.0 to 6.6 kcal mol−1 over addition of PhI. These data suggest that [Ni0] should preferentially react with the product PhSeCF3 as soon as it is formed, rather than with the aryl halide substrate. The thereby generated [(SIPr)NiII(SeCF3)(Ar)] could then undergo side reactions, for example, the commonly occurring ligand exchange between two such NiII species to ultimately generate biaryl. In line with this, our experimental studies indeed gave the corresponding biaryls (7 %) when we subjected 20 mol % of Ni0(cod)2/SIPr to ArSeCF3 2 in benzene at 45 °C for 30 min. Thus the reason for the ineffectiveness of the Ni0(cod)2/SIPr process is the higher reactivity of the product, ArSeCF3, towards oxidative addition to Ni0 compared to the reactivity of the corresponding starting material.
Figure 3.
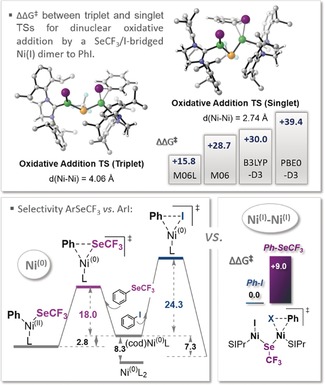
Top: Relative energy difference between triplet (favored) and singlet (disfavored) TSs for oxidative addition of ArI to a mixed NiI dimer (bearing a SCF3 and an I bridge). Bottom: Selectivity divergence for Ni0 versus NiI–NiI.20
With the origin of the ineffectiveness of Ni0 having been determined, we subsequently set out to assess the reactivity of the NiI dimer. We succeeded in the location of transition states for the direct oxidative addition of the NiI dimer to PhI (Figure 3).20 We optimized the dinuclear transition state as both closed‐shell singlet and open‐shell triplet states. While the singlet‐state TSs display a high degree of Ni−Ni bonding, the triplet‐state TSs show a larger distance between the two Ni centers (Figure 3) along with pronounced spin densities at both Ni centers, indicating open‐shell biradical character (see the Supporting Information). Our energy evaluation of the singlet versus triplet oxidative addition at various levels of theory (M06L, M06, and PBE0‐D3) suggested the triplet state to be consistently favored.20 As such, the computational data suggest that there will be a spin change from singlet (in the ground‐state dimer) to triplet (in the transition state). Following endergonic oxidative addition, a NiII−NiII intermediate may form and subsequently eliminate PhSeCF3 under formation of the mixed NiI dimer 8 bearing an iodine and a SeCF3 bridge. The latter species (8) is predicted to be more reactive than the doubly SeCF3‐bridged NiI dimer 4 and also favorably adds via the triplet transition state, leading to the conversion of another equivalent of ArI into ArSeCF3 (see the Supporting Information for the full path). The overall transformation was calculated to be exergonic (by ΔG rxn=−10.1 kcal mol−1 at M06L), and as such, to be thermodynamically driven.
As a mechanistic alternative, a NiI monomer pathway might be followed. If open‐shell NiI monomers were to be involved, we would expect EPR activity. However, our EPR investigations of the reaction mixture of the catalytic SeCF3 coupling of aryl iodides with 3, the substoichiometric reaction of NiI dimer 4 with ArI (as shown in Figure 2), as well as the NiI dimer itself in solution showed no EPR signals.
Lastly, we set out to investigate why the NiI dimer allows for productive catalysis, while Ni0 does not. To address this, we computationally studied the relative preference for oxidative addition to the product ArSeCF3 relative to ArI, ArBr, and ArCl once again. We employed a range of DFT methods (see the Supporting Information for details), and all consistently predicted the same reactivity trend. Interestingly, whereas Ni0 clearly preferred addition to the product ArSeCF3 by ΔΔG ≠=6.3 kcal mol−1 (with M06L), for NiI–NiI, a different reactivity pattern is seen, substantially favoring addition to the aryl halide over the product (by ΔΔG ≠=9.0 kcal mol−1 at M06L; see Figure 3). As such, remarkably, nickel in oxidation state I follows a different selectivity pattern than nickel in oxidation state 0.20
In conclusion, we have reported compelling data in support of NHC‐derived dinuclear Ni(I) catalysis in cross‐couplings with aryl iodides. The first iodine‐ (3) and SeCF3‐bridged (4) NiI dimers were synthesized, fully characterized, and complex 4 was shown to react directly with aryl iodides. Using the NiI dimer as the catalyst avoids the formation of undesired biaryl side products through the suppression of alternative pathways and circumvents mononuclear NiII intermediates, which are prone to side reactions. Selective functionalization of C−I bonds over C−Br, C−Cl, and alternative functional groups was possible. The corresponding Ni0 species was found to be inferior and inactive owing to its propensity to preferentially react with the product, ArSeCF3. Our computational and experimental data suggest fundamentally different reactivity trends, that is, for Ni0: ArSeCF3>ArI>ArBr≈ArCl and for NiI–NiI: ArI>ArBr>ArCl>ArSeCF3. These data provide an example of the superior reactivity of dinuclear NiI over mononuclear Ni0 catalysis and showcase the potential and importance of precisely controlling and harnessing the distinct metal oxidation states in catalysis.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the RWTH Aachen, the MIWF NRW, and the European Research Council (ERC‐637993) for funding. Calculations were performed with computing resources granted by JARA‐HPC from RWTH Aachen University under project “jara0091”. We are grateful to Kristina Deckers for technical assistance.
A. B. Dürr, H. C. Fisher, I. Kalvet, K.-N. Truong, F. Schoenebeck, Angew. Chem. Int. Ed. 2017, 56, 13431.
Contributor Information
Alexander B. Dürr, http://www.schoenebeck.oc.rwth‐aachen.de/
Prof. Dr. Franziska Schoenebeck, Email: franziska.schoenebeck@rwth-aachen.de.
References
- 1.
- 1a. Colacot T. J., New Trends in Cross-Coupling: Theory and Applications, RSC Catalysis Series, Cambridge, 2015; [Google Scholar]
- 1b. Johansson Seechurn C. C. C., Kitching M. O., Colacot T. J., Snieckus V., Angew. Chem. Int. Ed. 2012, 51, 5062; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 5150. [Google Scholar]
- 2.
- 2a.“Organonickel Chemistry”: Montgomery J., Organometallics in Synthesis, Wiley, Hoboken, 2013, p. 319; [Google Scholar]
- 2b. Tasker S. Z., Standley E. A., Jamison T. F., Nature 2014, 509, 299; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Jana R., Pathak T. P., Sigman M. S., Chem. Rev. 2011, 111, 1417; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2d. Rosen B. M., Quasdorf K. W., Wilson D. A., Zhang N., Resmerita A.-M., Garg N. K., Percec V., Chem. Rev. 2011, 111, 1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For reviews on the application of catalysts with metal–metal bonds, see:
- 3a. Powers I. G., Uyeda C., ACS Catal. 2017, 7, 936; [Google Scholar]
- 3b. Pye D. R., Mankad N. P., Chem. Sci. 2017, 8, 1705; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c.for a recent application of a Ni dimer in hydrosilylation, see: Steiman T. J., Uyeda C., J. Am. Chem. Soc. 2015, 137, 6104. [DOI] [PubMed] [Google Scholar]
- 4. J. R. Lancaster, Jr. , The Bioinorganic Chemistry of Nickel, VCH, New York, 1988, p. 337. [Google Scholar]
- 5.
- 5a. Kalvet I., Guo Q., Tizzard G. J., Schoenebeck F., ACS Catal. 2017, 7, 2126; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Dubinina G. G., Brennessel W. W., Miller J. L., Vicic D. A., Organometallics 2008, 27, 3933. [Google Scholar]
- 6.
- 6a. Kalvet I., Sperger T., Scattolin T., Magnin G., Schoenebeck F., Angew. Chem. Int. Ed. 2017, 56, 7078; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7184; [Google Scholar]
- 6b. Kalvet I., Magnin G., Schoenebeck F., Angew. Chem. Int. Ed. 2017, 56, 1581; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1603; [Google Scholar]
- 6c. Aufiero M., Sperger T., Tsang A. S.-K., Schoenebeck F., Angew. Chem. Int. Ed. 2015, 54, 10322; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 10462; [Google Scholar]
- 6d. Yin G., Kalvet I., Schoenebeck F., Angew. Chem. Int. Ed. 2015, 54, 6809; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6913; [Google Scholar]
- 6e. Bonney K. J., Proutiere F., Schoenebeck F., Chem. Sci. 2013, 4, 4434. [Google Scholar]
- 7.For examples with NHC ligands, see:
- 7a. Laskowski C. A., Bungum D. J., Baldwin S. M., Del Ciello S. A., Iluc V. M., Hillhouse G. L., J. Am. Chem. Soc. 2013, 135, 18272; [DOI] [PubMed] [Google Scholar]
- 7b. Dible B. R., Sigman M. S., Arif A. M., Inorg. Chem. 2005, 44, 3774. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Guard L. M., Mohadjer Beromi M., Brudvig G. W., Hazari N., Vinyard D. J., Angew. Chem. Int. Ed. 2015, 54, 13352; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13550; [Google Scholar]
- 8b. Cornella J., Gómez-Bengoa E., Martin R., J. Am. Chem. Soc. 2013, 135, 1997. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Nagao S., Matsumoto T., Koga Y., Matsubara K., Chem. Lett. 2011, 40, 1036; [Google Scholar]
- 9b. Miyazaki S., Koga Y., Matsumoto T., Matsubara K., Chem. Commun. 2010, 46, 1932; [DOI] [PubMed] [Google Scholar]
- 9c. Davies C. J. E., Page M. J., Ellul C. E., Mahon M. F., Whittlesey M. K., Chem. Commun. 2010, 46, 5151. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Gutierrez O., Tellis J. C., Primer D. N., Molander G. A., Kozlowski M., J. Am. Chem. Soc. 2015, 137, 4896; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10b. Zuo Z., Ahneman D. T., Chu L., Terrett J. A., Doyle A. G., MacMillan D. W. C., Science 2014, 345, 437; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10c. Li Z., Jiang Y.-Y., Fu Y., Chem. Eur. J. 2012, 18, 4345; [DOI] [PubMed] [Google Scholar]
- 10d. Phapale V. B., Buñuel E., García-Iglesias M., Cárdenas D. J., Angew. Chem. Int. Ed. 2007, 46, 8790; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 8946; [Google Scholar]
- 10e. Jones G. D., Martin J. L., McFarland C., Allen O. R., Hall R. E., Haley A. D., Brandon R. J., Konovalova T., Desrochers P. J., Pulay P., Vicic D. A., J. Am. Chem. Soc. 2006, 128, 13175. [DOI] [PubMed] [Google Scholar]
- 11.See Ref. [6c] and Glenadel Q., Ismalaj E., Billard T., Eur. J. Org. Chem. 2017, 530. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Ghiazza C., Tlili A., Billard T., Molecules 2017, 22, 833; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Potash S., Rozen S., J. Org. Chem. 2014, 79, 11205; [DOI] [PubMed] [Google Scholar]
- 12c. Chen C., Ouyang L., Lin Q., Liu Y., Hou C., Yuan Y., Weng Z., Chem. Eur. J. 2014, 20, 657; [DOI] [PubMed] [Google Scholar]
- 12d. Pooput C., W. R. Dolbier, Jr. , Medebielle M., J. Org. Chem. 2006, 71, 3564. [DOI] [PubMed] [Google Scholar]
- 13.While this manuscript was under review, a nickel-catalyzed reaction was reported; see: Han J.-B., Dong T., Vicic D. A., Zhang C.-P., Org. Lett. 2017, 19, 3919. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Matheis C., Wagner V., Goossen L. J., Chem. Eur. J. 2016, 22, 79; [DOI] [PubMed] [Google Scholar]
- 14b. Nikolaienko P., Rueping M., Chem. Eur. J. 2016, 22, 2620. [DOI] [PubMed] [Google Scholar]
- 15. Matsubara K., Yamamoto H., Miyazaki S., Inatomi T., Nonaka K., Koga Y., Yamada Y., Veiros L. F., Kirchner K., Organometallics 2017, 36, 255. [Google Scholar]
- 16. Zhang K., Conda-Sheridan M., Cooke S. R., Louie J., Organometallics 2011, 30, 2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.
- 17a. Mohadjer Beromi M., Nova A., Balcells D., Brasacchio A. M., Brudvig G. W., Guard L. M., Hazari N., Vinyard D. J., J. Am. Chem. Soc. 2017, 139, 922; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Yin G., Kalvet I., Englert U., Schoenebeck F., J. Am. Chem. Soc. 2015, 137, 4164; [DOI] [PubMed] [Google Scholar]
- 17c. Ge S., Green R. A., Hartwig J. F., J. Am. Chem. Soc. 2014, 136, 1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a.Gaussian 09, Revision D.01, M. J. Frisch et al.;
- 18b.for the appropriateness of the chosen computational method, see: Sperger T., Sanhueza I. A., Kalvet I., Schoenebeck F., Chem. Rev. 2015, 115, 9532. [DOI] [PubMed] [Google Scholar]
- 19. Dürr A. B., Yin G., Kalvet I., Napoly F., Schoenebeck F., Chem. Sci. 2016, 7, 1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geometries were optimized at the B3LYP or wB97XD level of theory with 6-31G(d) and SDD (for Ni, I). The TSs in Figure 3 result from ωB97XD optimization. Energies were calculated at CPCM (benzene) DFT/def2-TZVP. The reported energies are based on B3LYP geometries.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary