

Abstract
The conversion of CO2 with CH4 into liquid fuels and chemicals in a single‐step catalytic process that bypasses the production of syngas remains a challenge. In this study, liquid fuels and chemicals (e.g., acetic acid, methanol, ethanol, and formaldehyde) were synthesized in a one‐step process from CO2 and CH4 at room temperature (30 °C) and atmospheric pressure for the first time by using a novel plasma reactor with a water electrode. The total selectivity to oxygenates was approximately 50–60 %, with acetic acid being the major component at 40.2 % selectivity, the highest value reported for acetic acid thus far. Interestingly, the direct plasma synthesis of acetic acid from CH4 and CO2 is an ideal reaction with 100 % atom economy, but it is almost impossible by thermal catalysis owing to the significant thermodynamic barrier. The combination of plasma and catalyst in this process shows great potential for manipulating the distribution of liquid chemical products in a given process.
Keywords: carbon dioxide, heterogeneous catalysis, methane, plasma chemistry, reforming
Chemical transformations of CO2 into value‐added chemicals and fuels have been regarded as a key element for creating a sustainable low‐carbon economy in the chemical and energy industry. A particularly significant route that is currently being developed for CO2 utilization is catalytic CO2 hydrogenation. This process can produce a range of fuels and chemicals, including CO, formic acid, methanol, hydrocarbons, and alcohols; however, high H2 consumptions (CO2+3 H2→CH3OH+H2O) and high operating pressures (ca. 30–300 bar) are major challenges associated with this process.
Instead of using H2, the direct conversion of CO2 with CH4 (dry reforming of methane, DRM) into liquid fuels and chemicals (e.g., acetic acid) represents another promising route for both CO2 valorization and CH4 activation. CH4 is an ideal H supplier to replace H2 in CO2 hydrogenation as CH4 has a high H density and is available from a range of sources (e.g., natural gas, shale gas, biogas, and flared gas). Moreover, it is an inexpensive carbon source that can increase the atom utilization of CO2 hydrogenation owing to the stoichiometric ratio of C and O atoms, as well as reduce the formation of water.
Recently, Ge and co‐workers investigated the direct C−C coupling of CO2 and CH4 to form acetic acid on a Zn‐doped ceria catalyst by density functional theory (DFT) modeling;1 this is an attractive route as the direct conversion of CO2 and CH4 into acetic acid is a reaction with 100 % atom economy [Equation (1)]. However, this reaction is thermodynamically unfavorable under practical conditions. The conventional indirect catalytic process often proceeds through two steps (Scheme 1): 1) DRM to produce syngas (CO and H2) at high temperatures (>700 °C), and 2) conversion of syngas into liquid fuels and chemicals at high pressures. This indirect route for CO2 valorization and CH4 activation is inefficient as the DRM process for syngas production is highly endothermic and requires high temperatures and energy input [Equation (2)]. Catalyst deactivation due to carbon deposition is another challenge impacting the use of this reaction on a commercial scale. It is almost impossible to directly convert two stable and inert molecules (CO2 and CH4) into liquid fuels or chemicals in a one‐step catalytic process bypassing the production of syngas. A stepwise method was proposed to convert CO2 and CH4 into acetic acid over Cu/Co‐based catalysts,2 Pd/C, Pt/Al2O3,3 Pd/SiO2, and Rh/SiO2 4 by heterogeneous catalysis. The catalyst was first exposed to CH4, forming CHx species on the catalyst surface. Subsequently, the feed gas was changed from CH4 to CO2, and acetic acid was formed through the reaction of CO2 with CHx over the catalyst. This indirect process is complicated by the periodic change of reactants and the product collection.5
| (1) |
| (2) |
Scheme 1.
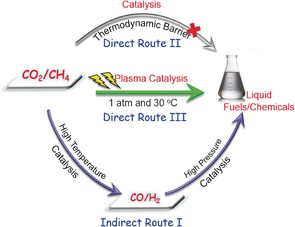
Direct and indirect processes for the conversion of CO2 and CH4 into liquid fuels and chemicals.
Non‐thermal plasmas (NTPs) offer a unique way to enable thermodynamically unfavorable chemical reactions at low temperatures owing to the non‐equilibrium character. The overall gas temperature in an NTP remains low while the generated electrons are highly energetic with a typical electron temperature of 1–10 eV, which is sufficient to activate inert molecules (e.g., CO2 and CH4) into reactive species, including radicals, excited atoms, molecules, and ions. These energetic species are capable of initiating a variety of chemical reactions. Although much effort has been devoted to the use of NTPs for the degradation of gas pollutants, far less has been done with regard to their use in the synthesis of fuels and chemicals.6 Previous work on DRM with NTPs mainly focused on syngas production,7 while very limited efforts have been devoted to the challenging one‐step conversion of CH4 and CO2 into liquid fuels and chemicals.8, 9 A few groups have reported on the formation of trace oxygenates (e.g., alcohols and acids) as side products in plasma DRM for syngas production.10 Thus far, the use of NTPs for the direct conversion of CO2 and CH4 into oxygenates has resulted in poor selectivities and yields.
Herein, we describe the development of a novel dielectric barrier discharge (DBD) reactor with a ground water electrode (see the Supporting Information, Schemes S1 and S2) for the one‐step conversion of CO2 and CH4 into oxygenates at room temperature (30 °C) and atmospheric pressure. This setup is unique and has not been reported previously. Figure 1 shows that no reaction occurred in the “catalyst only” mode at 30 °C without plasma. However, the use of an NTP enabled this thermodynamically unfavorable reaction to occur at room temperature and resulted in the production of liquid chemicals, including acetic acid, methanol, ethanol, and acetone, with acetic acid being the major product. Trace amounts of formic acid, propanol, and butanol were also detected in the condensed liquid. In the plasma process without a catalyst (“plasma only”), a total liquid selectivity of 59.1 % was achieved with selectivities of 33.7 %, 11.9 %, 11.9 %, and 1.6 % for acetic acid, ethanol, methanol, and acetone, respectively (Figure 1 a). The CO selectivity was only about 20.0 % (Figure 1 b), and the CH4 and CO2 conversions amounted to approximately 18.3 % and 15.4 %, respectively (Figure 1 c).
Figure 1.
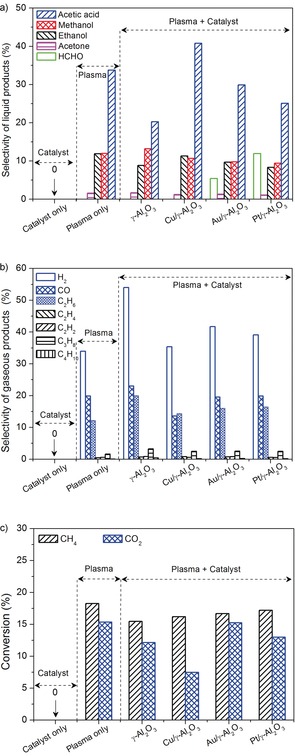
Effect of operating modes and catalysts on the reaction: a) Selectivities for oxygenates, b) selectivities for gaseous products, c) conversion of CH4 and CO2 (total flow rate 40 mL min−1, discharge power 10 W, catalyst ca. 2 g).
Combining the plasma process with a catalyst shows great potential for manipulating the production of different oxygenates under ambient conditions. Clearly, packing the Cu/γ‐Al2O3 catalyst in the DBD enhanced the selectivity for acetic acid to 40.2 %, compared to the plasma‐only mode and the plasma reaction using γ‐Al2O3 only (20.2 %). Acetic acid was the major product regardless of the catalyst used, followed by methanol and ethanol (Figure 1 a). HCHO was formed only when the supported noble metal catalysts were used in the plasma reaction, and the Pt/γ‐Al2O3 catalyst showed the highest selectivity to HCHO. Compared to the plasma‐only mode, placing the catalysts in the DBD gave similar gaseous product distributions, with H2, CO, and C2H6 being the major gaseous products (Figure 1 b). However, combining the NTP with the catalysts enhanced the H2 selectivity by 10–20 % (except for Cu/γ‐Al2O3), and slightly increased C2H6 production, but had a weak effect on the selectivity for CO (except for Cu/γ‐Al2O3, which decreased CO selectivity to 13.5 %) and other CxHy (i.e., C2H2, C2H4, C2H6, C3H8, and n‐C4H10). In addition, compared to the plasma‐only mode, the conversion of CO2 and CH4 slightly decreased with packing catalysts. This phenomenon can be attributed to the change in discharge behavior induced by the catalyst, which had a negative effect on the reaction (Figure S1). Interestingly, C6H12O4 (CAS No. 49653‐17‐0) was found on the inner reactor wall in the plasma‐catalyst mode (Figure S2). These results demonstrate the feasibility of using NTPs for the direct conversion of CH4 and CO2 into higher‐value liquid fuels and chemicals in a single‐step process under ambient conditions, bypassing the formation of syngas.
To understand the formation of the liquid chemicals, optical emission spectroscopy (OES) was used to investigate the species produced in the CH4/CO2 DBD (Figure 2). Hα and O atomic lines and CH, C2, CO2 +, CO2, and CO bands were identified in the emission spectra of the DBD, with CO, CH, and H being the major ones (Table S2).
Figure 2.
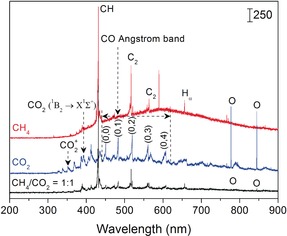
Optical emission spectra of CH4, CO2, and CH4/CO2 plasmas (total flow rate 40 mL min−1, CH4/CO2 ratio 1:1, discharge power 10 W, exposure time 2 s).
CO is mainly derived from reactions S1–S3 (Table S3) in the DBD. Our simulation showed that electron‐impact CO2 reactions produced about 95 % vibrationally excited CO2 (CO2(v)) compared to electronically excited CO2 as shown in Figure S3 and Table S4. O radicals generated from CO2 dissociation can attack CO2(v) molecules to produce CO (S1 and S2).11 Different from CH, CH3 derived from CH4 dissociation cannot be detected by OES, but recent simulations revealed that electron‐impact dissociation of CH4 leads to 79 % CH3 formation and only 15 % and 5 % CH2 and CH, respectively.12 Therefore, CH3 is the dominant species in the CH4/CO2 DBD. In addition to electrons (S4 in Table S3), reactive species such as OH, O, and H can also react with CH4 to produce CH3 radicals (S5–S7) in the CH4/CO2 DBD. Additionally, OH is an important species, especially for alcohol formation. In the CH4/CO2 DBD, OH could be produced indirectly by reactions S8–S13, with S8 and S9 as the major channels based on the reaction rate coefficients and E a.13 Special attention was given to S10, although a very low reaction rate coefficient of 1.4×10−29 cm3 molecule−1 s−1 and a high E a value of 111 kJ mol−1 were determined for ground‐state CO2 reacting with an H radical to produce an OH radical; this reaction (S10) can be accelerated by using CO2(v) instead of ground‐state CO2,14 and the use of vibrationally excited reagents is most effective in overcoming the activation barrier of the endothermic reaction.14, 15 Thus the reaction CO2(v)+H→CO+OH could be one of the major routes for OH formation under these conditions as CO2 is mainly present in vibrationally excited states (Figure S3).
Based on the analysis of the gaseous and condensed liquid products and the OES results, CO, CH3, and OH radicals are the key species in the CH4/CO2 plasma reaction. Therefore, possible reaction pathways for the formation of acetic acid, methanol, and ethanol under these conditions are proposed in Scheme 2.
Scheme 2.
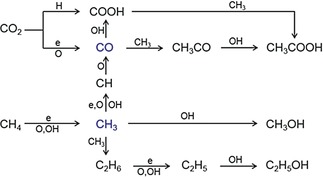
Possible reaction pathways for the formation of CH3COOH, CH3OH, and C2H5OH in the direct reforming of CH4 and CO2 with DBD.
Two possible reaction pathways could contribute to the formation of acetic acid. CO can react with a CH3 radical to form an acetyl radical (CH3CO) by reaction S14 in Table S3 with a low energy barrier of 28.77 kJ mol−1,16 followed by recombination with OH to produce acetic acid in reaction S15 with no energy barrier10g (see also Figures 3 and S4). Clearly, the selectivity to acetic acid increases initially and then decreases with the CH4/CO2 ratio, with optimal acetic acid formation at a CH4/CO2 ratio of 1:1. Correspondingly, the relative intensities of the CO band head and the O atomic line increased with a decrease in the CH4/CO2 ratio from 3:1 to 1:2 while that of the CH band head increased (Figure S4). This suggests that decreasing the CH4/CO2 molar ratio decreases the generation of CH3 radicals, but increases OH formation. A similar mechanism of acetic acid formation has been proposed on the basis of DFT modeling10g and by Eliasson and co‐workers.10i In addition, direct coupling of CH3 and carboxyl radicals (COOH) could also form acetic acid by reaction S16, while COOH radicals may be formed from reactions S17 and S18 in Table S3.10g
Figure 3.
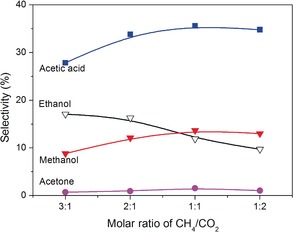
Effect of the CH4/CO2 molar ratio on the selectivity for oxygenates without a catalyst (total flow rate 40 mL min−1, discharge power 10 W).
Decreasing the CH4/CO2 molar ratio decreased the generation of CH3 radicals, but increased OH formation (Figure S4). Simultaneously, the formation of CH3OH increased initially with a decrease in the CH4/CO2 molar ratio and reached a peak at a CH4/CO2 molar ratio of 1:1. By contrast, the formation of C2H5OH decreased continuously as the CH4/CO2 molar ratio was decreased (Figure 3). These findings suggest that the production of CH3OH mainly depends on the generation of both CH3 and OH radicals while the formation of C2H5OH is more sensitive to the presence of CH3 radicals in the plasma reaction as C2H5OH formation requires twice the amount of CH3 radicals in comparison to the formation of CH3OH. As shown in Scheme 2, CH3OH can be directly formed from the coupling of CH3 and OH radicals with a high rate coefficient (S19 in Table S3),17 while C2H5OH formation requires several elementary reactions (S20–S24). The recombination of a CH3 radical with itself forms C2H6 (S20),18 which is followed by dehydrogenation to form a C2H5 radical by reactions S21–S23, with S21 as the primary reaction according to the reaction rates.13d, 19 The C2H5 radical finally recombines with OH to form C2H5OH with a high rate coefficient of 9.34×10−11 cm3 molecule−1 s−1 (S24).20
Clearly, adding catalysts to the plasma reaction influences the distribution of the formed oxygenates, especially for the formation of HCHO after addition of the Pt and Au catalysts, revealing the occurrence of surface reactions in addition to plasma gas phase reactions.21 In traditional catalysis, CO hydrogenation, CH3OH oxidation, and methylene (CH2) oxidation can lead to the generation of HCHO over noble‐metal catalysts.22 In this plasma process, adding noble‐metal catalysts in the plasma had almost no influence on the CO selectivity, but decreased the selectivity for CH3OH, C2H5OH, and CH3COOH and increased the selectivity for HCHO and C2H6 (Figure 1 a). Considering the major species that are present in the CH4/CO2 DBD, CHx (x=4, 3, and 2) could be the primary source for HCHO formation by oxidation reactions. Namely, CHx in the gas phase could be adsorbed onto the surface of the catalyst to form HCHO by the oxidation of CH2, ad (CHx,ad+O, H, OH→CH2,ad) and to produce C2H6 by self‐recombination of CH3 radicals instead of converting CH3 into CH3OH, C2H5OH, and CH3COOH. This could explain why the presence of the Au and Pt catalysts in the plasma decreased the formation of CH3OH, C2H5OH, and CH3COOH, but enhanced the production of C2H6 and HCHO (Figures 1 a and b). Possible pathways for the formation of the major oxygenates on the catalyst surface are proposed in Scheme S3. In addition, catalyst characterization (Figures S5–S8) suggested that the metal particle size and interactions between metal and support are not determining factors for the reaction performance (Figure 1), whereas the strength of the bonding of adsorbed intermediates to the catalyst surface, that is, the oxygen adsorption energy (ΔE O), could be a good activity descriptor towards the formation of different products in DRM.23
In conclusion, the one‐step room‐temperature synthesis of liquid fuels and chemicals from the direct reforming of CO2 with CH4 has been achieved by using a novel atmospheric‐pressure DBD reactor. The total selectivity for liquid chemicals was approximately 50–60 %, with acetic acid as the major product. The CH4/CO2 molar ratio and the type of catalyst can be used to manipulate the production of different oxygenates. These results clearly show that non‐thermal plasmas can be used to overcome the thermodynamic barrier for the direct transformation of CH4 and CO2 into a range of strategically important platform chemicals, especially for the production of acetic acid with 100 % atom economy. Additionally, combining the DBD with noble‐metal catalysts produced formaldehyde, which cannot be generated in the same plasma reaction without a catalyst. This finding suggests that new research should be directed at designing a catalyst with high selectivity towards a desirable product.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Support of this work by the EPSRC SUPERGEN Hydrogen & Fuel Cell (H2FC) Programme (EP/J016454/1) ECR Project (Ref. EACPR PS5768) is gratefully acknowledged.
L. Wang, Y. Yi, C. Wu, H. Guo, X. Tu, Angew. Chem. Int. Ed. 2017, 56, 13679.
References
- 1. Zhao Y., Cui C., Han J., Wang H., Zhu X., Ge Q., J. Am. Chem. Soc. 2016, 138, 10191–10198. [DOI] [PubMed] [Google Scholar]
- 2. Huang W., Xie K. C., Wang J. P., Gao Z. H., Yin L. H., Zhu Q. M., J. Catal. 2001, 201, 100–104. [Google Scholar]
- 3. Wilcox E. M., Roberts G. W., Spivey J. J., Catal. Today 2003, 88, 83–90. [Google Scholar]
- 4. Ding Y.-H., Huang W., Wang Y.-G., Fuel Process. Technol. 2007, 88, 319–324. [Google Scholar]
- 5.
- 5a. Olajire A. A., J. CO2 Util. 2013, 3–4, 74–92; [Google Scholar]
- 5b. Otto A., Grube T., Schiebahn S., Stolten D., Energy Environ. Sci. 2015, 8, 3283–3297; [Google Scholar]
- 5c. Dimitriou I., García-Gutiérrez P., Elder R. H., Cuéllar-Franca R. M., Azapagic A., Allen R. W., Energy Environ. Sci. 2015, 8, 1775–1789; [Google Scholar]
- 5d. Fan M.-S., Abdullah A. Z., Bhatia S., ChemCatChem 2009, 1, 192–208; [Google Scholar]
- 5e. Pakhare D., Spivey J., Chem. Soc. Rev. 2014, 43, 7813–7837; [DOI] [PubMed] [Google Scholar]
- 5f. Havran V., Dudukovic M. P., Lo C. S., Ind. Eng. Chem. Res. 2011, 50, 7089–7100. [Google Scholar]
- 6. Stere C. E., Anderson J. A., Chansai S., Delgado J. J., Goguet A., Graham W. G., Hardacre C., Taylor S., Tu X., Wang Z., Angew. Chem. Int. Ed. 2017, 56, 5579–5583; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 5671–5675. [Google Scholar]
- 7.
- 7a. Chung W.-C., Chang M.-B., Renewable Sustainable Energy Rev. 2016, 62, 13–31; [Google Scholar]
- 7b. Lebouvier A., Iwarere S. A., d'Argenlieu P., Ramjugernath D., Fulcheri L., Energy Fuels 2013, 27, 2712–2722; [Google Scholar]
- 7c. Tao X., Bai M., Li X., Long H., Shang S., Yin Y., Dai X., Prog. Energy Combust. Sci. 2011, 37, 113–124; [Google Scholar]
- 7d. Tu X., Whitehead J. C., Int. J. Hydrogen Energy 2014, 39, 9658–9669. [Google Scholar]
- 8. Zou J.-J., Zhang Y.-p., Liu C.-J., Li Y., Eliasson B., Plasma Chem. Plasma Process. 2003, 23, 69–82. [Google Scholar]
- 9. Scapinello M., Martini L. M., Tosi P., Plasma Processes Polym. 2014, 11, 624–628. [Google Scholar]
- 10.
- 10a. Kozlov K., Michel P., Wagner H.-E., Plasmas Polym. 2000, 5, 129–150; [Google Scholar]
- 10b. Dey G. R., Das T. N., Plasma Chem. Plasma Process. 2006, 26, 495–505; [Google Scholar]
- 10c. Gómez-Ramírez A., Rico V. J., Cotrino J., González-Elipe A. R., Lambert R. M., ACS Catal. 2014, 4, 402–408; [Google Scholar]
- 10d. Goujard V., Tatibouët J.-M., Batiot-Dupeyrat C., Appl. Catal. A 2009, 353, 228–235; [Google Scholar]
- 10e. Sentek J., Krawczyk K., Młotek M., Kalczewska M., Kroker T., Kolb T., Schenk A., Gericke K.-H., Schmidt-Szałowski K., Appl. Catal. B 2010, 94, 19–26; [Google Scholar]
- 10f. Krawczyk K., Młotek M., Ulejczyk B., Schmidt-Szałowski K., Fuel 2014, 117, 608–617; [Google Scholar]
- 10g. Martini L. M., Dilecce G., Guella G., Maranzana A., Tonachini G., Tosi P., Chem. Phys. Lett. 2014, 593, 55–60; [Google Scholar]
- 10h. Liu C., Wang J., Wang Y., Eliasson B., Fuel Chem. Div. Prepr. 2003, 48, 268; [Google Scholar]
- 10i. Wang J. G., Liu C. J., Eliassion B., Energy Fuels 2004, 18, 148–153. [Google Scholar]
- 11. Fridman A., Plasma Chemistry, Cambridge University Press, 2008. [Google Scholar]
- 12. De Bie C., Verheyde B., Martens T., van Dijk J., Paulussen S., Bogaerts A., Plasma Processes Polym. 2011, 8, 1033–1058. [Google Scholar]
- 13.
- 13a. Murrell J., Rodriguez J., J. Mol. Struct. THEOCHEM 1986, 139, 267–276; [Google Scholar]
- 13b. Robie D. C., Arepalli S., Presser N., Kitsopoulos T., Gordon R. J., J. Chem. Phys. 1990, 92, 7382–7393; [Google Scholar]
- 13c. Tsang W., Hampson R., J. Phys. Chem. Ref. Data 1986, 15, 1087–1279; [Google Scholar]
- 13d. Baulch D., Cobos C., Cox R., Esser C., Frank P., Just T., Kerr J., Pilling M., Troe J., Walker R., J. Phys. Chem. Ref. Data 1992, 21, 411–734; [Google Scholar]
- 13e. Karkach S. P., Osherov V. I., J. Chem. Phys. 1999, 110, 11918–11927. [Google Scholar]
- 14. Rusanov V., Fridman A., Sholin G., Phys. Usp. 1981, 24, 447–474. [Google Scholar]
- 15. Polanyi J. C., Science 1987, 236, 680–690. [DOI] [PubMed] [Google Scholar]
- 16. Baulch D., Cobos C., Cox R., Frank P., Hayman G., Just T., Kerr J., Murrells T., Pilling M., Troe J., J. Phys. Chem. Ref. Data 1994, 23, 847–848. [Google Scholar]
- 17. Jasper A. W., Klippenstein S. J., Harding L. B., Ruscic B., J. Phys. Chem. A 2007, 111, 3932–3950. [DOI] [PubMed] [Google Scholar]
- 18. Gomer R., Kistiakowsky G., J. Chem. Phys. 1951, 19, 85–91. [Google Scholar]
- 19. Atkinson R., Baulch D., Cox R., R. Hampson, Jr. , Kerr J., Rossi M., Troe J., J. Phys. Chem. Ref. Data 1997, 26, 521–1011. [Google Scholar]
- 20. Sivaramakrishnan R., Su M.-C., Michael J., Klippenstein S., Harding L., Ruscic B., J. Phys. Chem. A 2010, 114, 9425–9439. [DOI] [PubMed] [Google Scholar]
- 21. Wang L., Zhao Y., Liu C., Gong W., Guo H., Chem. Commun. 2013, 49, 3787–3789. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Lin Z.-Z., Chen X., Mater. Des. 2016, 107, 82–89; [Google Scholar]
- 22b. Yin S., Wang Z., Bernstein E. R., Phys. Chem. Chem. Phys. 2013, 15, 4699–4706; [DOI] [PubMed] [Google Scholar]
- 22c. Sobczak I., Kozlowska M., Ziolek M., J. Mol. Catal. A 2014, 390, 114–124; [Google Scholar]
- 22d. Czelej K., Cwieka K., Colmenares J. C., Kurzydlowski K. J., Langmuir 2016, 32, 7493–7502; [DOI] [PubMed] [Google Scholar]
- 22e. Niu J., Ran J., Wang R., Du X., Comput. Theor. Chem. 2015, 1067, 40–47. [Google Scholar]
- 23.
- 23a. Kattel S., Yan B., Yang Y., Chen J. G., Liu P., J. Am. Chem. Soc. 2016, 138, 12440–12450; [DOI] [PubMed] [Google Scholar]
- 23b. Behrens M., Studt F., Kasatkin I., Kühl S., Hävecker M., Abild-Pedersen F., Zander S., Girgsdies F., Kurr P., Kniep B.-L., Science 2012, 336, 893–897; [DOI] [PubMed] [Google Scholar]
- 23c. Studt F., Sharafutdinov I., Abild-Pedersen F., Elkjær C. F., Hummelshøj J. S., Dahl S., Chorkendorff I., Nørskov J. K., Nat. Chem. 2014, 6, 320–324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary