

Abstract
Tertiary silane 1H, 2‐[(diphenylsilyl)methyl]‐6‐methylpyridine, reacts with tris(pentafluorophenyl)borane (BCF) to form the intramolecular pyridine‐stabilized silylium 1+‐HBCF. The corresponding 2‐[(diphenylsilyl)methyl]pyridine, lacking the methyl‐group on the pyridine ring, forms classic N(py)→B adduct 2H‐BCF featuring an intact silane Si−H fragment. Complex 1+‐HBCF promotes cleavage of the C≡O triple bond in carbon monoxide with double C−Csp2 bond formation, leading to complex 3 featuring a B‐(diarylmethyl)‐B‐aryl‐boryloxysilane fragment. Reaction with pinacol generates bis(pentafluorophenyl)methane 4 as isolable product, proving the transition‐metal‐free deoxygenation of carbon monoxide by this main‐group system. Experimental data and DFT calculations support the existence of an equilibrium between the silylium–hydroborate ion pair and the silane–borane mixture that is responsible for the observed reactivity.
Keywords: boron, C−C bond formation, CO, silicon, small molecule activation
The coordination chemistry and subsequent reactivity of carbon monoxide with transition‐metal complexes is well‐developed and forms the platform for current‐day applications of CO as C 1 building block, including large‐scale industrial processes (e.g., acetic acid production, hydroformylation).1 However, the C−O linkage is typically preserved in these applications. This is in stark contrast to heterogeneous Fischer–Tropsch catalysis, where CO (with H2) is utilized as a true C 1 building block to make new C−C bonds concomitant with deoxygenation of CO (i.e., C≡O cleavage).2 In this context, also the valorization of carbon dioxide is currently attracting much attention.3 Partial deoxygenation strategies to convert carbon dioxide with main‐group elements (e.g., hydrosilylation, hydroboration) are investigated.4 Since the pioneering work of Brown on the carbonylation of alkylboranes,5 the transition‐metal‐free chemistry of carbon monoxide with main‐group compounds is also rapidly developing. To generate stable or observable adducts of CO, mainly boron‐based compounds6 and diaminocarbenes are utilized,7 but only a handful of compounds have proven capable of activating CO to the extent that new C−X bonds (X=O, N, C, H) can be formed. Insertion of CO in the B−B single bond of azaboriridines,8a,8b or the M−C bond of alanes8c,8d and gallanes8e is known, and reductive coupling of CO has recently been achieved.9
Well‐defined homogeneous transition‐metal complexes that are capable of CO triple‐bond scission normally require highly reactive co‐reagents and/or strongly reducing conditions in order to enforce (stepwise) C−O cleavage.10 Recently, a mononuclear Mo platform was shown to generate C2O1 species by stoichiometric CO triple‐bond scission combined with C−C bond formation, aided by the use of silyl chlorides as co‐reagents, which inadvertently generates siloxanes as by‐product.11 In contrast, however, selective transition‐metal‐free deoxygenation of CO is virtually unknown.
Combinations of an electrophilic borane and a phosphine‐based donor, (referred to as FLPs (Frustrated Lewis Pairs),12 have been used to activate CO. The group of Erker described the trapping of the CO adduct of Piers borane13 (OC→B(H)(C6F5)2) with intramolecular phosphine–borane FLP systems, generating a new C−H bond (Scheme 1, top).14 Stephan and co‐workers reported that a 2:1 mixture of tris(pentafluorophenyl)borane (B(C6F5)3=BCF) and P(tBu)3 leads to the complete splitting of CO (from syngas), via a boron formyl intermediate, to afford A featuring one new C−Csp2 bond, together with cyclic product B (Scheme 1, middle).15a The group of Scheschkewitz reported on the reaction between a lithium disilenide and CO (1:2 ratio) to generate a highly nonsymmetric species in which three out of four CO molecules have undergone C≡O cleavage.15b These reports represent the only previous examples of main‐group‐element promoted C≡O cleavage and concurrent C−Csp2 bond formation, to the best of our knowledge. No mechanistic information is available to shed light on the formation to A. Also, more extensive CO functionalization (i.e., additional C−C bond formation) would be very interesting.
Scheme 1.
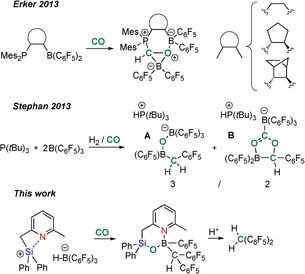
Top: Rare example of CO activation using main‐group (FLP) systems, including C−H and single C−Csp2 bond formation, respectively. Bottom: CO triple‐bond activation of intramolecularly stabilized silylium hydroborate system with double C−Csp2 bond formation.
Inspired by these recent advances and realizing (i) the ability of triphenylboron hydrides to activate CO2 and carbonyl groups owing to the reactive B−H bond,16 and (ii) the well‐known ability of mixtures of silanes and B(C6F5)3 to promote hydrosilylation reactions of unsaturated substrates via formation of silyl cations,17, 18 we sought to develop a reactive Si−B system that would be capable of CO activation (Scheme 1, bottom). Although the chemistry and catalysis with electrophilic silylium species is rapidly gaining attention,19 examples of frustrated Lewis pair‐type chemistry involving silicon are relatively rare.20
To address both challenges, we herein report a strategy to stabilize a silylium species with a weak donor such as pyridine.21 whilst retaining the inherent reactivity of the Si center. This strategy has enabled transition‐metal‐free rupture of the triple bond in carbon monoxide and subsequent double C−Csp2 bond formation by two C6F5 transfers using a simple and novel pyridine‐stabilized silylium hydroborate system. Through a combination of experimental data and supported by DFT calculations, the role of each component in this hitherto unknown reaction sequence is detailed.
Synthesis of 2‐[(diphenylsilyl)methyl]‐6‐methylpyridine 1H from lutidine is straightforward (Scheme 2). The Si−H hydrogen resonates at δ 5.01 ppm in the 1H NMR spectrum (1 J SiH=199.9 Hz). The hydrogen atoms of the methylene bridgehead (‐CH2‐) group appear as a doublet at δ 2.91 ppm through coupling with the Si−H hydrogen (3 J HH=3.7 Hz). The corresponding 29Si{1H} NMR signal appears as a singlet at δ −14.6 ppm. The analogous species 2H (2‐[(diphenylsilyl)methyl]pyridine) was also prepared in this way.
Scheme 2.
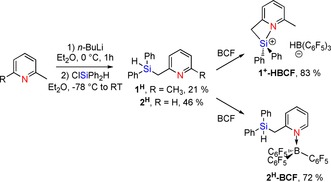
Synthesis of 1H and 2H and reaction with B(C6F5)3 (BCF) to pyridine‐stabilized silylium 1+‐HBCF and pyridine‐borane adduct 2H‐BCF, respectively.
Upon addition of an equimolar amount of tris(pentafluorophenyl)borane (BCF) to a solution of 2H, selective formation of species 2H‐BCF, featuring a direct N→B adduct, is observed with an intact hydrosilane unit, according to 1H and 29Si NMR spectroscopy (Scheme 2). The 19F NMR spectrum shows 15 different resonance signals due to hindered rotation around the N−B and B−C linkages, resulting in low overall symmetry. Single‐crystal X‐ray diffraction analysis confirms the molecular structure of species 2 (Figure 1). The analogous reaction between BCF and 1H in CD2Cl2 at room temperature immediately generates a new singlet at δ 23.2 ppm in the HSQC 29Si‐1H NMR spectrum, correlating with a broad singlet at δ −25.5 ppm in the 11B NMR spectrum. In the corresponding 1H NMR spectrum, the ‐CH2‐ hydrogens appear as a singlet at δ 3.43 ppm, indicating Si−H cleavage. In the 1H{11B} NMR spectrum, the formation of the hydroborate H−B(C6F5)3 − is evidenced by a broad singlet at δ 3.57 ppm that nearly disappears in the baseline in the 11B‐coupled spectrum. Furthermore, the para‐pyridine H‐atom is strongly shifted downfield upon hydride abstraction, which suggests an intramolecular N→Si donor–acceptor interaction in the product.22
Figure 1.
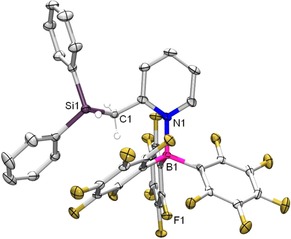
Displacement ellipsoid plot (50 % probability level) of 2H‐BCF. Hydrogen atoms are omitted for clarity, except those on C1 and Si1. Selected bond lengths (Å): N1−B1 1.6547(35); C1−Si1 1.8983(28); Si1−H1 1.366(26).
Abstraction of the hydride to generate cationic pyridine‐stabilized silylium fragment 1+‐HBCF (Scheme 2) was further ascertained by high resolution mass spectrometry (HR‐MS; see the Supporting Information). To the best of our knowledge, only one other example of a pyridine‐stabilized silylium derivative has been described to date (featuring a five‐membered ring), whereas 1+‐HBCF features an N→Si stabilization within a four‐membered ring.23 Apparently, introduction of a methyl group ortho to the pyridine nitrogen fully suppresses the py→B(C6F5)3 adduct in favor of formation of the [N→Si]+ hydroborate Lewis pair. This species can be considered isoelectronic with previously described pyridine‐stabilized organoboranes.24 Silylium salt 1+‐HBCF is isolated as an oil, preventing structure elucidation by single‐crystal X‐ray crystallography.
Upon pressurizing a solution of 1+‐HBCF in CD2Cl2 with 5 bar of CO for two hours at 50 °C, a single new resonance at δ −8.6 ppm is observed in the 29Si DEPT NMR spectrum, revealing a reaction to a new species 3 featuring a saturated tetracoordinated silicon (Scheme 3). HR‐MS reveals the incorporation of one molecule of CO or 13CO, respectively. A new signal is observed at δ 2.3 ppm in the 11B NMR spectrum, while a new singlet integrating for one hydrogen atom is observed at δ 4.75 ppm in the 1H NMR spectrum. This signal becomes a doublet (1 J CH=116.8 Hz) when the same reaction is performed with 13C‐labeled CO, supporting the formation of a direct C−H bond, with the hydrogen atom most likely stemming from the silane. In agreement with this observation, the proton‐coupled 13C NMR spectrum of the labeled compound displays a doublet (1 J CH=116.8 Hz) at δ 25.9 ppm. In the 13C{1H} NMR spectrum, this signal is broad (full‐width at half‐maximum=20.3 Hz) and nearly disappears in the baseline in the non‐labeled product, which suggests a direct scalar coupling with the 11B nucleus. The 19F NMR spectrum displays two sets of signals with an integration ratio of 2:1, suggestive of (double) C6F5 transfer from boron to the boron‐bound carbon atom.
Scheme 3.
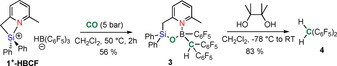
Reaction of 1+‐HBCF with CO to generate 3 and follow‐up hydrolysis with pinacol to release bis(pentafluorophenyl)methane.
Based on multinuclear NMR spectroscopy and literature data on related organoboron compounds,25 we conclude that 3 contains a (C6F5)B−C(H)(C6F5)2 fragment as well as a siloxyborate linkage. DFT calculated and benchmarked NMR chemical shifts support the presence of a four‐coordinated B‐nucleus with an additional pyridine→boron interaction, as in 3 (Scheme 3 and the Supporting Information). It can be noted that the hydrogens of the methylene bridge in 3 do not display a diastereotopic effect (low temperature 1H NMR shows only broadening of the CH2 resonance at −70 °C). This suggests that 3 is probably in equilibrium with its open form 3′ featuring a three‐coordinated B‐nucleus, as supported by DFT calculations (see the Supporting Information for details).26
As a result, the C≡O triple bond in carbon monoxide is completely cleaved, with the formation of one new C−H and two new C−C bonds. Although complex 3 proved highly air‐sensitive and the oily nature of 3 prohibited direct elucidation of the structure by single‐crystal X‐ray crystallography, a crystallization attempt at −20 °C furnished crystals of moderate quality of the oxidized derivative 3O, which confirms the proposed connectivity with a six‐membered ∼C‐Si‐O‐B‐N‐C∼ linkage (see the Supporting Information).27
Liberation of the organic C(H)(C6F5)2 fragment from 3 was achieved through alcoholysis with an equimolar amount of pinacol in dichloromethane (Scheme 3). Gratifyingly, this results in clean formation and isolation of bis(pentafluorophenyl)methane 4 (as well as the 13C‐labeled derivative, available from 13CO) in 83 % yield, confirming the generation of two new C−Csp2 bonds from CO in 3. Compound 4 is fully characterized by multinuclear NMR spectroscopy, high resolution mass spectrometry, micro‐analysis, and single‐crystal X‐ray diffraction (see the Supporting Information for details). The skeletal composition, the protocol for its formation and the subsequent reactivity of 3 are unique, to the best of our knowledge, as it represents the first example of double C−Csp2 bond formation from C−O cleavage by CO activation promoted by a main‐group derivative.
To get additional insights in the reaction mechanism and the requirement for silylium species 1+‐HBCF as reagent, several control experiments were carried out. Firstly, 1+‐BArF 4, the tetrakis(pentafluorophenyl)borate analogue of 1+‐HBCF, lacking a hydridic B−H fragment, was prepared by the reaction of 1H and (CPh3)B(C6F5)4 in 76 % yield and characterized by multinuclear NMR spectroscopy and HR‐MS analysis (see the Supporting Information for details). This compound is completely unreactive toward CO, which suggests that a borohydride moiety is necessary for carbon monoxide activation to occur (Scheme 4 a). Furthermore, the lithium hydroborate salt LiBH(C6F5)3 also gives no reaction under the same reaction conditions, thus showing that the pyridine–silylium fragment is also crucial for the reaction to proceed.
Scheme 4.
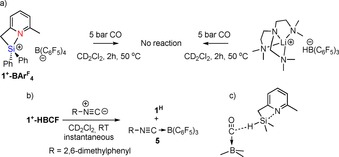
a) Control experiments to highlight the need for silicon, pyridine, and hydroborate in Lewis pair 1+‐HBCF to cleave CO. b) Reaction with isocyanide to give 5, demonstrating equilibrium between 1+‐HBCF and 1H+BCF. c) Proposed hydride transfer from 1H to OC→B(C6F5)3.
Next, we attempted to translate the observed reactivity with CO to the electronically related isocyanide moiety. Surprisingly, reaction of 1+‐HBCF with tert‐butylisocyanide at RT led to complete regeneration of the silane Si−H bond, as supported by 1H NMR spectroscopy, concomitant with formation of the tBuNC→B(C6F5)3 adduct 5 (Scheme 4 b). This observation suggests that 1+‐HBCF is also in equilibrium with the silane and the carbonyl adduct OC→B(C6F5)3 28 under CO pressure and that intact 1H rather than the silylium derivative may be responsible for the observed reactivity. At this stage, we hypothesized that the reaction could be initiated by a hydride transfer from the silane to the activated CO carbon of the boron‐adduct (Scheme 4 c). The hydridicity of the silane would be enhanced through intramolecular stabilization of the corresponding silylium fragment by the nearby pyridine group. In agreement with this proposed equilibrium between 1+‐HBCF and 1H, a mixture of methyldiphenylsilane (HSiMePh2), lacking the internal stabilizing pyridine ring, and B(C6F5)3 do not react with CO.
To provide a qualitative picture of the plausible mechanism of CO triple‐bond scission and formation of 3, featuring the highly unusual six‐membered B‐(diarylmethyl)‐B‐aryl‐boryloxysilane system, we performed DFT calculations (BP86, def2‐TZVP, disp3, m4 grid). The complete pathway from 1H to 3 is displayed in Figure 2. The separated Lewis acid–base pair (1H+BCF; set at 0 kcal mol−1 with free CO; starting point) and 1+‐HBCF (+2.6 kcal mol−1) are likely in equilibrium under the experimental reaction conditions. Activation of CO to give A most likely proceeds via the separated Lewis acid–base pair. The entropy penalty associated with formation of a van der Waals adduct of OC−B(C6F5)3 and 1H is largely compensated by relatively strong π–π stacking interactions. This pre‐organization facilitates hydride transfer, via a low TS1 barrier of +4.5 kcal mol−1 relative to 1H+BCF, to form zwitterionic silylium‐formylborate species B (the ion‐separated conformer B′ is 2.4 kcal mol−1 higher in energy than B). Intermediate C (−11.8 kcal mol−1), is formed upon interaction of the silyl cation with the oxygen atom of the formyl group. Subsequent facile transfer of a C6F5‐group from boron to carbon via transition state TS2 (+5.6 kcal mol−1 relative to 1H+BCF) generates the open form of the neutral boranylmethoxysilane derivative D (−19.6 kcal mol−1).29 The bora‐epoxide conformer D′ (+0.8 kcal mol−1 relative to D) undergoes a 1,2‐oxo‐shift via TS3 (+11.9 kcal mol−1 compared to D′) to form the open zwitterionic λ4‐boranyloxysilane E with a formal cationic carbon fragment (−9.6 kcal mol−1). Interestingly, bora‐epoxides have been proposed as intermediates in alkyl transfer from boron to carbon.30 This reactive species E can undergo direct transfer of a second C6F5‐group from boron to form G over transition state TS4 (+1.9 kcal mol−1 relative to E). Alternatively, formation of the closed zwitterionic pyridinium intermediate F (−26.3 kcal mol−1 relative to E) followed by transfer of a second C6F5‐group from boron would form the open form 3′ over transition state TS5. Given the strong driving force for formation of intermediate F, this path is deemed most probable, with the barrier to TS5 (+20.8 kcal mol−1) representing the rate‐limiting step of the reaction. The closed form 3, featuring an additional NPy→B interaction, is only 5.4 kcal mol−1 uphill relative to 3′.
Figure 2.
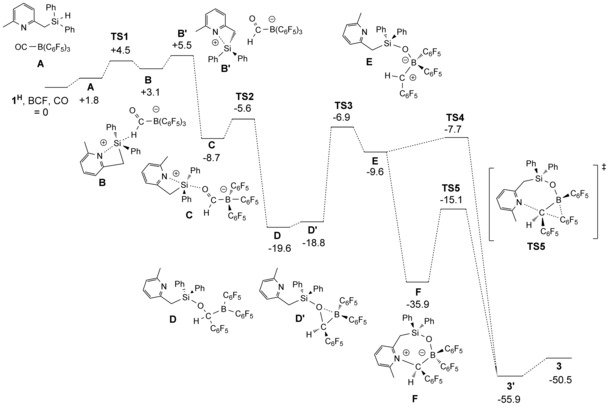
DFT‐calculated (BP86, def2‐TZVP, disp3, m4 grid) pathway for the conversion of 1H+BCF to 3. For details, see also the Supporting Information.
In summary, we have prepared silylium species 1+‐HBCF using the pyridine‐appended tertiary silane 1H and Lewis acid tris(pentafluorophenyl)borane. This FLP‐like compound is able to induce full cleavage of the C≡O triple bond to generate 3. This sequence also results in the formation of one new C−H and two new C−Csp2 bonds, as supported by multinuclear NMR spectroscopy and solid‐state characterization of the oxidized derivative. Alcoholysis of complex 3 with pinacol provides bis(pentafluorophenyl)methane as the organic C 1 product from transition‐metal‐free deoxygenation of CO. This study shows that pyridine‐appended silanes in combination with B‐based Lewis acids may provide suitable platforms to deoxygenate CO and to form new C−C bonds. Follow‐up research to explore the scope of Lewis acids available for this TM‐free transformation is ongoing.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This research is funded by the European Research Council through ERC Starting Grant EuReCat 279097 to JIvdV. We thank Ed Zuidinga for MS analysis and Dr. Andreas Ehlers and Jan‐Meine Ernsting for assistance with NMR spectroscopy.
M. Devillard, B. de Bruin, M. A. Siegler, J. I. van der Vlugt, Chem. Eur. J. 2017, 23, 13628.
References
- 1.
- 1a. Beller M., Wu X.-F., Transition Metal Catalyzed Carbonylation Processes, Springer, 2013; [Google Scholar]
- 1b. Gadge S. T., Bhanage B. M., RSC Adv. 2014, 4, 10367–10389. [Google Scholar]
- 2.
- 2a. Maitlis P. M., Zanotti V., Chem. Commun. 2009, 1619–1634; [DOI] [PubMed] [Google Scholar]
- 2b. Rofer-DePoorter C. K., Chem. Rev. 1981, 81, 447–474. [Google Scholar]
- 3.
- 3a. Klankermayer J., Wesselbaum S., Beydoun K., Leitner W., Angew. Chem. Int. Ed. 2016, 55, 7296–7343; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7416–7467; [Google Scholar]
- 3b. Aresta M., Dibenedetto A., Angelini A., Chem. Rev. 2014, 114, 1709–1749; [DOI] [PubMed] [Google Scholar]
- 3c. Appel A. M., Bercaw J. E., Bocarsly A. B., Dobbek H., DuBois D. L., Dupuis M., Ferry J. G., Fujita E., Hille R., Kenis P. J. A., Kerfeld C. A., Morris R. H., Peden C. H. F., Portis A. R., Ragsdale S. W., Rauchfuss T. B., Reek J. N. H., Seefeldt L. C., Thauer R. K., Waldrop G. L., Chem. Rev. 2013, 113, 6621–6658; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Wang W., Wang S., Ma X., Gong J., Chem. Soc. Rev. 2011, 40, 3703–3727. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Chauvier C., Cantat T., ACS Catal. 2017, 7, 2107–2115; [Google Scholar]
- 4b. Bontemps S., Coord. Chem. Rev. 2016, 308, 117–130; [Google Scholar]
- 4c. Maity A., Teets T. S., Chem. Rev. 2016, 116, 8873–8911; [DOI] [PubMed] [Google Scholar]
- 4d. Aresta M., CO2 as Chemical Feedstock, Wiley, Weinheim, 2010. [Google Scholar]
- 5. Brown H. C., Acc. Chem. Res. 1969, 2, 65–72. [Google Scholar]
- 6.
- 6a. Finze M., Bernhardt E., Terheiden A., Berkei M., Willner H., Christen D., Oberhammer H., Aubke F., J. Am. Chem. Soc. 2002, 124, 15385–15398; [DOI] [PubMed] [Google Scholar]
- 6b. Fukazawa A., Dutton J. L., Fan C., Mercier L. G., Houghton A. Y., Wu Q., Piers W. E., Parvez M., Chem. Sci. 2012, 3, 1814–1818; [Google Scholar]
- 6c. Braunschweig H., Dewhurst R. D., Hupp F., Nutz M., Radacki K., Tate C. W., Vargas A., Ye Q., Nature 2015, 522, 327–330. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Martin D., Moore C. E., Rheingold A. L., Bertrand G., Angew. Chem. Int. Ed. 2013, 52, 7014–7017; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7152–7155; [Google Scholar]
- 7b. Siemeling U., Färber C., Bruhn C., Leibold M., Selent D., Baumann W., von Hopffgarten M., Goedecke C., Frenking G., Chem. Sci. 2010, 1, 697–704. [Google Scholar]
- 8.
- 8a. Paetzold P., Redenz-Stormanns B., Boese R., Angew. Chem. Int. Ed. Engl. 1990, 29, 900–902; [Google Scholar]; Angew. Chem. 1990, 102, 910–911; [Google Scholar]
- 8b. Teichmann J., Stock H., Pritzkow H., Siebert W., Eur. J. Inorg. Chem. 1998, 459–453; [Google Scholar]
- 8c. Mason M. R., Song B., Kirschbaum K., J. Am. Chem. Soc. 2004, 126, 11812–11813; [DOI] [PubMed] [Google Scholar]
- 8d. Li X., Ni C., Song H., Cui C., Chem. Commun. 2006, 1763–1765; [DOI] [PubMed] [Google Scholar]
- 8e. Mason M. R., Song B., Han Y., Hu X., Inorg. Chim. Acta 2008, 361, 3332–3337. [Google Scholar]
- 9.
- 9a. Braunschweig H., Dellermann T., Dewhurst R. D., Ewing W. C., Hammond K., Jimenez-Halla J. O. C., Kramer T., Krummenacher I., Mies J., Phukan A. K., Vargas A., Nat. Chem. 2013, 5, 1025–1028; [DOI] [PubMed] [Google Scholar]
- 9b. Wang X., Zhu Z., Peng Y., Lei H., Fettinger J. C., Power P. P., J. Am. Chem. Soc. 2009, 131, 6912–6913; [DOI] [PubMed] [Google Scholar]
- 9c. Brown Z. D., Power P. P., Inorg. Chem. 2013, 52, 6248–6259. [DOI] [PubMed] [Google Scholar]
- 10. West N. M., Miller A. J. M., Labinger J. A., Bercaw J. E., Coord. Chem. Rev. 2011, 255, 881–898. [Google Scholar]
- 11.
- 11a. Buss J. A., Agapie T., Nature 2016, 529, 72–75; [DOI] [PubMed] [Google Scholar]
- 11b. Buss J. A., Agapie T., J. Am. Chem. Soc. 2016, 138, 16466–16477. [DOI] [PubMed] [Google Scholar]
- 12. Stephan D. W., J. Am. Chem. Soc. 2015, 137, 10018–10032. [DOI] [PubMed] [Google Scholar]
- 13. Parks D. J., Piers W. E., Yap G. P. A., Organometallics 1998, 17, 5492–5503. [Google Scholar]
- 14. Sajid M., Elmer L.-M., Rosorius C., Daniliuc C. G., Grimme S., Kehr G., Erker G., Angew. Chem. Int. Ed. 2013, 52, 2243–2246; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 2299–2302. [Google Scholar]
- 15.
- 15a. Dobrovetsky R., Stephan D. W., J. Am. Chem. Soc. 2013, 135, 4974–4977; [DOI] [PubMed] [Google Scholar]
- 15b. Majumdar M., Omlor I., Yildiz C. B., Azizoglu A., Huch V., Scheschkewitz D., Angew. Chem. Int. Ed. 2015, 54, 8746–8750; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 8870–8874. [Google Scholar]
- 16. Mukherjee D., Osseili H., Spaniol T. P., Okuda J., J. Am. Chem. Soc. 2016, 138, 10790–10793. [DOI] [PubMed] [Google Scholar]
- 17.Recent review: Oestreich M., Hermeke J., Mohr J., Chem. Soc. Rev. 2015, 44, 2202–2220. [DOI] [PubMed] [Google Scholar]
- 18. Houghton A. Y., Hurmalainen J., Mansikkamäki A., Piers W. E., Tuononen H. M., Nat. Chem. 2014, 6, 983–988. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Lipke M. C., Liberman-Martin A. L., Tilley T. D., Angew. Chem. Int. Ed. 2017, 56, 2260–2294; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 2298–2335; [Google Scholar]
- 19b. Klare H. F. T., Oestreich M., Dalton Trans. 2010, 39, 9176–9184; see also: [DOI] [PubMed] [Google Scholar]
- 19c. Khandelwal M., Wehmschulte R. J., Angew. Chem. Int. Ed. 2012, 51, 7323–7326; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7435–7439. [Google Scholar]
- 20.
- 20a. Schäfer A., Reissmann M., Schäfer A., Saak W., Haase D., Müller T., Angew. Chem. Int. Ed. 2011, 50, 12636–12638; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12845–12848; [Google Scholar]
- 20b. Waerder B., Pieper M., Körte L. A., Kinder T. A., Mix A., Neumann B., Stammler H.-G., Mitzel N. W., Angew. Chem. Int. Ed. 2015, 54, 13416–13419; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 13614–13617; [Google Scholar]
- 20c. Mo Z., Szilvási T., Zhou Y.-P., Yao S., Driess M., Angew. Chem. Int. Ed. 2017, 56, 3699–3702; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3753–3756; [Google Scholar]
- 20d. Nie W., Klare H. F. T., Oestreich M., Fröhlich R., Kehr G., Erker G., Z. Naturforsch. B 2012, 67, 987–994. [Google Scholar]
- 21.We prefer the R2Si+-Py notation over R2Si-Py+ for reasons of relative electronegativity of N vs. Si. DFT-calculated natural bond order (NBO) values support this assignment: Si=+1.77; N=−0.54.
- 22.
- 22a. Vergnaud J., Ayed T., Hussein K., Vendier L., Grellier M., Bouhadir G., Barthelat J.-C., Sabo-Etienne S., Bourissou D., Dalton Trans. 2007, 2370–2372; [DOI] [PubMed] [Google Scholar]
- 22b. Clark E. R., Del Grosso A., Ingleson M. J., Chem. Eur. J. 2013, 19, 2462–2466; [DOI] [PubMed] [Google Scholar]
- 22c. Körte L. A., Warner R., Vishnevskiy V. Y., Neumann B., Stammler H.-G., Mitzel N. W., Dalton Trans. 2015, 44, 9992–10002; [DOI] [PubMed] [Google Scholar]
- 22d. Zheng J., Lin Y.-J., Wang H., Dalton Trans. 2016, 45, 6088–6093; [DOI] [PubMed] [Google Scholar]
- 22e. Devillard M., Alvarez Lamsfus C., Vreeken V., Maron L., van der Vlugt J. I., Dalton Trans. 2016, 45, 10989–10998. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Nokami T., Soma R., Yamamoto Y., Kamei T., Itami K., Yoshida J.-I., Beilstein J. Org. Chem. 2007, 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b.For stabilization of hypervalent SiIV in a 4-membered ring, see: van den Ancker T. R., Raston C. L., Skelton B. W., White A. H., Organometallics 2000, 19, 4437–4444. [Google Scholar]
- 24.See ref. [18e] and: Gellrich U., Diskin-Posner Y., Shimon L. J. W., Milstein D., J. Am. Chem. Soc. 2016, 138, 13307–13313. [DOI] [PubMed] [Google Scholar]
- 25. Neu R. C., Jiang C., Stephan D. W., Dalton Trans. 2013, 42, 726–736. [DOI] [PubMed] [Google Scholar]
- 26.For lutidine-boron N→B equilibrated adducts and 11B NMR data, see:
- 26a. Geier S. J., Stephan D. W., J. Am. Chem. Soc. 2009, 131, 3476–3477; [DOI] [PubMed] [Google Scholar]
- 26b. Ferguson G., Murphy J. F. G. D., Sheehan J. P., Spalding T. R., Polyhedron 1993, 12, 859–864. [Google Scholar]
- 26c.See also: Nuclear Magnetic Resonance Spectroscopy of Boron Compounds (Eds.: H. Niith, B. Wrackmeyer), Springer, Berlin, 1978. [Google Scholar]
- 27.Despite multiple attempts to selectively oxidize 3 to generate 3O in a targeted manner, this compound was not obtained in pure form again.
- 28.This adduct decomposes above −120 °C, see ref. [5 a]. For DFT calculations, see:
- 28a. Jacobsen H., Berke H., Döring S., Kehr G., Erker G., Fröhlich R., Meyer O., Organometallics 1999, 18, 1724–1735; [Google Scholar]
- 28b.for a structurally related adduct OC→B(H)(C6F5)2, see: Sajid M., Kehr G., Daniliuc C. G., Kehr G., Angew. Chem. Int. Ed. 2014, 53, 1118–1121; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1136–1139. [Google Scholar]
- 29.Akin to 1,1-carboboration reactions, see:
- 29a. Wrackmeyer B., Khan E., Eur. J. Inorg. Chem. 2016, 300–312; [Google Scholar]
- 29b. Kehr G., Erker G., Chem. Commun. 2012, 48, 1839–1850. [DOI] [PubMed] [Google Scholar]
- 30.See ref. [4] and: Berkefeld A., Piers W. E., Parvez M., Castro L., Maron L., Eisenstein O., J. Am. Chem. Soc. 2012, 134, 10843–10851. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary