

Abstract
7‐Deazapurine (pyrrolo[2,3‐d]pyrimidine) nucleosides are important analogues of biogenic purine nucleosides with diverse biological activities. Replacement of the N7 atom with a carbon atom makes the five‐membered ring more electron rich and brings a possibility of attaching additional substituents at the C7 position. This often leads to derivatives with increased base‐pairing in DNA or RNA or better binding to enzymes. Several types of 7‐deazapurine nucleosides with potent cytostatic or cytotoxic effects have been identified. The most promising are 7‐hetaryl‐7‐deazaadenosines, which are activated in cancer cells by phosphorylation and get incorporated both to RNA (causing inhibition of proteosynthesis) and to DNA (causing DNA damage). Mechanism of action of other types of cytostatic nucleosides, 6‐hetaryl‐7‐deazapurine and thieno‐fused deazapurine ribonucleosides, is not yet known. Many 7‐deazaadenosine derivatives are potent inhibitors of adenosine kinases. Many types of sugar‐modified derivatives of 7‐deazapurine nucleosides are also strong antivirals. Most important are 2′‐C‐methylribo‐ or 2′‐C‐methyl‐2′‐fluororibonucleosides with anti‐HCV activities (several compounds underwent clinical trials). Some underexplored areas of potential interest are also outlined.
Keywords: antivirals, cytostatics, deazapurines, nucleosides, nucleotides
1. INTRODUCTION
Pyrrolo[2,3‐d]pyrimidine (7‐deazapurine) nucleosides is a class of compounds closely related to purine nucleosides. The shape of pyrrolo[2,3‐d]pyrimidine resembles purine and therefore 7‐deazapurine nucleosides can substitute purine nucleosides in DNA and RNA [for the reference to natural purines, we will use the (deaza)purine nomenclature and numbering instead of the IUPAC preferred pyrrolo[2,3‐d]pyrimidine nomenclature].
Some 7‐deazapurine nucleosides even occur in nature—both as nucleosides and as components of nucleic acids. Queuosine (1a)1, 2, 3 and archaeosine (2)4 are examples of 7‐deazapurine nucleosides isolated from tRNA of prokaryotic and eukaryotic organisms and archaea, respectively. Recently, the presence of 2′‐deoxyqueuosine (1b) was also discovered in bacterial DNA5 (Fig. 1). Several nucleoside antibiotics contain pyrrolo[2,3‐d]pyrimidine nucleobase and their biological activities will be mentioned later in the text. The biosynthetic pathway to 7‐deazapurines uses GTP as a precursor. More details about 7‐deazapurine biosynthesis can be found in a detailed review.6
Figure 1.
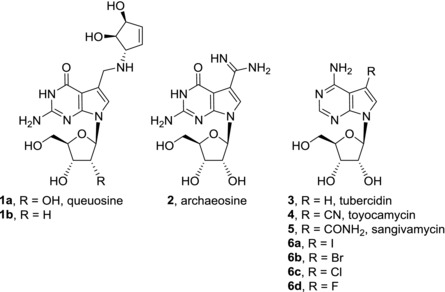
Naturally occurring deazapurine nucleosides (1–5) and related compounds
Replacement of the N7 nitrogen by carbon changes the electronic properties of the five‐membered ring, making it more electron‐rich and thus (at least in theory) more prone to cation–π or π–π interactions. Also, the presence of this additional C7 carbon gives a suitable position for attachment of substituents which (in DNA or RNA) point out to the major groove. It has been repeatedly shown that 7‐substituted 7‐deazapurine nucleoside triphosphates are good substrates for DNA or RNA polymerases and DNA containing 7‐substituted 7‐deazapurines forms stable duplexes (some 7‐alkynyl‐ or 7‐aryl‐7‐deazapurines nucleotides are even better substrates for polymerases than natural dATP or dGTP7, 8 and in DNA they stabilize duplexes9). For applications in medicinal chemistry, the position C7 offers a possibility for functionalization and many 7‐substituted 7‐deazapurine nucleosides display important biological activities which are summarized and discussed in this review. It should be noted that a general review on syntheses and biological activities of pyrrolopyrimidines was published recently,10 but it did not specifically cover nucleosides and completely missed many important classes of relevant compounds. The synthetic approaches to 7‐deazapurine nucleosides are covered in the above‐mentioned review10 or in a recent book.11 This review focuses on recent (2000–2016) advances in medicinal chemistry of 7‐deazapurine nucleosides and is based on SciFinder, Reaxys, and Web of Knowledge searches.
2. CYTOTOXIC NUCLEOSIDES
2.1. Natural compounds
Cytotoxic activities of naturally occurring 7‐deazapurine nucleosides were discovered already in 1960s. Tubercidin (3), toyocamycin (4), and sangivamycin (5) were isolated from Streptomyces cultures. All of them show potent cytotoxicity against cancer cell lines12, 13, 14, 15 but despite their structural similarities, their modes of action are different (IC50 values for compounds mentioned in this section are listed in Table 1). Tubercidin (3), toyocamycin (4), and sangivamycin (5) are phosphorylated by cellular kinases to their mono‐, di‐, and triphosphate forms and the resulting nucleotides are then incorporated into RNA and DNA which causes damage to nucleic acid functions.16, 17, 18, 19, 20 Furthermore, tubercidin (3) impairs numerous cellular processes such as pre‐mRNA processing and nuclear speckle formation,21 mitochondrial respiration, purine synthesis, rRNA processing, and methylation of tRNA.22 Tubercidin (3) is also a potent inhibitor of S‐adenosylhomocysteine hydrolase.23 Toyocamycin (4) showed inhibition of phosphatidylinositol kinase24, inhibitory effect on rRNA synthesis, and maturation.25, 26 In a more recent study, toyocamycin (4) was identified as a potent inhibitor of induced XBP1 (X‐box binding protein 1) mRNA cleavage. IRE1α‐XBP1 pathway is a key component of endoplasmic reticulum stress response. IRE1α‐XBP1 inhibition by toyocamycin (4) leads to apoptosis in endoplasmic reticulum‐stressed tumors and multiple myeloma.27 On the other hand, cytotoxic effect of sangivamycin (5) is mainly caused through potent and selective inhibition of protein kinase C.28 Recently, the mechanism of action of sangivamycin (5) in primary effusion lymphoma cells was studied showing that sangivamycin (5) acts through inhibition of Erk and Akt signaling in these cells.15 Sangivamycin (5) is also able to bind heat‐shock protein 70 (HSP70) (KD = 3.3 μM) which is a molecular chaperone with a proposed importance in oncology.29 Despite the attention paid to the naturally occurring 7‐deazapurine nucleosides, none of the compounds proceeded to clinical use.
Table 1.
Cytotoxic activities of 7‐deazapurine nucleosides
| Compound | IC50 [μM] (cell line) | Ref. |
|---|---|---|
| Natural compounds | ||
| Tubercidin (3) | 0.001 (A549) | 12 |
| Toyocamycin (4) | 0.012 (HTB‐81) | 13 |
| Sangivamycin (5) | 0.006 (A549) | 14 |
| 7‐Substituted 7‐deazapurine ribonucleosides | ||
| 7‐Iodotubercidin (6a) | 2.6a (HeLa) | 35 |
| 7‐Bromotubercidin (6b) | 2.9a (HeLa) | 35 |
| 7‐Chlorotubercidin (6c) | 13.3a (HeLa) | 35 |
| 7‐Fluorotubercidin (6d) | 1 (L‐1210) | 36 |
| AB61 (7a) | 0.01 (A549); 0.00036 (CCRF‐CEM) | 42 |
| 7b | 0.35 (A549); 0.10 (CCRF‐CEM); 0.003 (HCT15) | 12 |
| 7c | >20 (A549); > 10 (CCRF‐CEM) | 12 |
| 7d | 0.701 (A549); >10 (CCRF‐CEM); 0.152 (Du145) | 12 |
| 8 | 43.76 (A549); 64.44 (CCRF‐CEM) | 41 |
| 9a | 0.03 (A549); 0.02 (CCRF‐CEM); | 44 |
| 9b | 0.05 (A549); 0.05 (CCRF‐CEM); 0.01 (HCT116p53‐/‐) | 44 |
| 9c | 2.57 (A549); 0.18 (CCRF‐CEM); | 44 |
| 9d | 4.60 (A549); 2.91 (CCRF‐CEM); 0.15 (K562) | 44 |
| 9e | 0.11 (A549); 0.04 (CCRF‐CEM) | 44 |
| 10a‐c | >20 (A549); >20 (CCRF‐CEM) | 44 |
| 11a | 0.01 (CCRF‐CEM); 0.001 (Hs578) | 12 |
| 11b | 0.14 (A549); >20 (CCRF‐CEM); 0.03 (HCT116) | 44 |
| 11c | 0.39 (A549); 0.09 (CCRF‐CEM) | 44 |
| 11d | 19.2 (A549); 5.60 (CCRF‐CEM); 0.11 (K562‐TAX) | 44 |
| 12a | 0.4 (HeLa) | 45 |
| 12b | 0.2 (HeLa) | 45 |
| Sugar‐modified 7‐substituted 7‐deazapurine nucleosides | ||
| 13a | 4.25 (CCRF‐CEM) | 46 |
| 13b | NAb (CCRF‐CEM) | 46 |
| 13c | NA (CCRF‐CEM) | 46 |
| 13d | 7.50 (CCRF‐CEM) | 46 |
| 13e | >10 (CCRF‐CEM) | 47 |
| 13f | 3.44 (CCRF‐CEM) | 47 |
| 14a | 6.64 (CCRF‐CEM) | 46 |
| 14b | 6.08 (NCI‐H23) | 46 |
| 14c | NA (CCRF‐CEM) | 46 |
| 14d | 8.49 (Hs578) | 46 |
| 14e | >10 (CCRF‐CEM) | 47 |
| 14f | 6.63 (CCRF‐CEM) | 47 |
| 15 | 0.18 (CCRF‐CEM); 0.002 (Du145) | 46 |
| 16a | 0.64 (CCRF‐CEM); 0.15 (HepG2) | 47 |
| 16b | 0.42 (CCRF‐CEM); 0.35 (HepG2) | 47 |
| 17 | 3.5 (CCRF‐CEM) | 48 |
| 18 | 7.13 (A549); 16.63 (CCRF‐CEM) | 49 |
| 8‐Substituted deazapurine nucleosides | ||
| ARC (21a) | 0.97 (SW620); 0.49 (DM366); 0.20 (SK‐N‐AS) | 54, 55, 56 |
| Xylocydine (22a) | >50 (A549) | 64 |
| JRS‐15 (23b) | 12.42 (HepG2) | 64 |
| 24 | 2.6 (PA‐1) | 70 |
| 6‐Substituted 7‐deazapurine ribonucleosides and pronucleotides | ||
| 25a | 0.088 (A549); 0.31 (CCRF‐CEM); 0.007 (Du145) | 71 |
| 25b | 0.045 (A549); 0.29 (CCRF‐CEM); 0.009 (Du145) | 71 |
| 25c | >10 (A549); 4.0 (CCRF‐CEM); 1.1 (Du145) | 71 |
| 26a | 0.11 (A549); 1.4 (CCRF‐CEM); 0.005 (Du145) | 71 |
| 26b | 0.061 (A549); 0.27 (CCRF‐CEM); 0.009 (Du145) | 71 |
| 27a | >10 (A549); 3.8 (CCRF‐CEM) | 71 |
| 27b | 0.38 (A549); 2.7 (CCRF‐CEM); 0.013 (Du145) | 71 |
| 28a | 2.00 (CCRF‐CEM); 0.017 (HCT116) | 72 |
| 28b | 18.60 (CCRF‐CEM); 0.011 (Du145) | 72 |
| 28c | >10 (CCRF‐CEM) | 72 |
| 29a | 17.67 (CCRF‐CEM); 0.007 (Du145) | 72 |
| 29b | >10 (CCRF‐CEM); 0.022 (Du145) | 72 |
| 30a | 3.44 (CCRF‐CEM); 1.03 (HCT116) | 73 |
| 30b | 3.34 (CCRF‐CEM); 1.02 (HeLa S3) | 73 |
| 30c | 1.13 (CCRF‐CEM); 0.58 (HeLa S3) | 73 |
| 31a | 8.47 (CCRF‐CEM); 1.41 (HepG2) | 73 |
| 31b | >10 (CCRF‐CEM); 1.53 (HepG2) | 73 |
| 31c | 2.09 (CCRF‐CEM); 0.68 (HCT116) | 73 |
| 32a | 29.4 (A549); 2.22 (CCRF‐CEM) | 74 |
| 32b–d | >150 (A549); > 150 (CCRF‐CEM) | 74 |
| Sugar‐modified 6‐substituted 7‐deazapurine nucleosides | ||
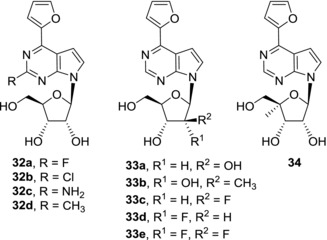
| 33a‐e | NA (CCRF‐CEM) | 75, 76 |
| 34 | NA (A549); NA (CCRF‐CEM) | 49 |
| Other 7‐deazapurine nucleosides | ||
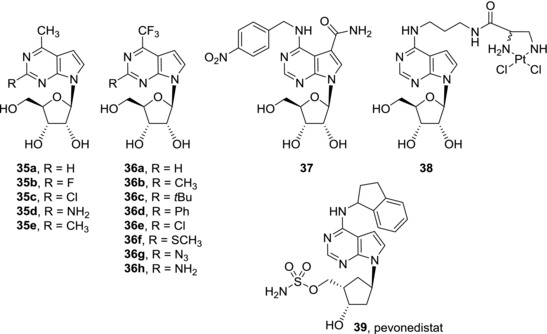
| 35a | 0.03 (A549); 0.01 (CCRF‐CEM) | 44 |
| 35b–e | >150 (A549); >150 (CCRF‐CEM) | 74 |
| 36a | 0.028 (HepG2); 0.090 (HeLa S3) | 77 |
| 36b–h | >50 (HepG2); >50 (HeLa S3) | 77 |
| 37 | 115 (CCRF‐CEM); 1.8 (HeLa) | 78 |
| 38 | 36.20 (A549) | 79 |
| Fused 7‐deazapurine nucleosides | ||
| 41a | 7.9 (CCRF‐CEM) | 91 |
| 41b | 20.2 (CCRF‐CEM); 2.0 (L‐1210) | 91, 92 |
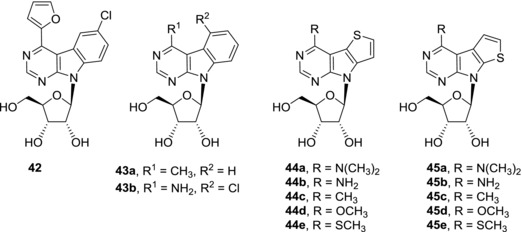
| 42 | 0.175 (HepG2) | 93 |
| 43a | 18 (CCRF‐CEM); 11 (HeLa S3) | 94 |
| 43b | 12 (CCRF‐CEM) | 94 |
| 44a | >50 (A549); 47.62 (CCRF‐CEM) | 95 |
| 44b | 11.9 (A549); 6.22 (CCRF‐CEM); 2.5 (U2OS) | 95 |
| 44c | 0.21 (A549); 0.02 (CCRF‐CEM) | 95 |
| 44d | 0.22 (A549); 0.13 (CCRF‐CEM); 0.11 (HL60) | 95 |
| 44e | 0.66 (A549); 0.027 (CCRF‐CEM) | 95 |
| 45a | >50 (A549); >50 (CCRF‐CEM) | 95 |
| 45b | 30.11 (A549); 0.3 (CCRF‐CEM) | 95 |
| 45c | 0.72 (A549); 0.03 (CCRF‐CEM) | 95 |
| 45d | 1.23 (A549); 0.22 (CCRF‐CEM) | 95 |
| 45e | 0.80 (A549); 0.20 (CCRF‐CEM) | 95 |
minimal cytotoxic concentration; bNA = inactive.
Cell lines mentioned in the table: A549, human lung carcinoma; CCRF‐CEM, human lymphoblastic leukemia; DM366, human melanoma; Du145, human prostate carcinoma; HCT116, human colorectal carcinoma; HCT116p53−/−, human colorectal carcinoma, p53 mutated; HCT15, human colorectal adenocarcinoma; HeLa, human cervical adenocarcinoma; HeLa S3, human cervical adenocarcinoma, HepG2, human hepatocellular carcinoma; HL60, human acute promyelocytic leukemia; Hs578, human breast carcinoma; HTB‐81, human prostate carcinoma; K562, human chronic myelogenous leukemia; K562‐TAX, human colorectal carcinoma, taxol‐resistant; L‐1210, mouse lymphocytic leukemia; NCI‐H23, human lung adenocarcinoma; PA‐1, human teratocarcinoma; SK‐N‐AS, human neuroblastoma; SW620, human colon adenocarcinoma; U2OS, human osteosarcoma.
2.2. Synthetic nucleosides with C7 and C8 substituents
First structure‐activity studies among 7‐deazapurine nucleosides focused on modifications of the sugar moiety of the nucleosides. Even though many sugar‐modified analogs of naturally occurring 7‐deazapurine nucleosides (e.g. deoxynucleosides, ara‐, and xylo‐diastereomers) were synthesized, modification of the sugar moiety typically led to a decrease in cytotoxic activity.13, 14, 30, 31, 32, 33 Therefore, nucleobase‐modified derivatives and compounds combining nucleobase and sugar modifications attracted more attention. 7‐Halogenated tubercidins (6a–d)34 differ in their cytotoxic activity depending on the nature of the halogen (Fig. 2). 7‐Iodo‐, bromo‐, and chlorotubercidin (6a–c) are less cytotoxic compared with tubercidin (3).35 On the other hand, 7‐fluorotubercidin (6d) showed superior cytotoxicity over tubercidin (3) and improved selectivity towards leukemic cells36 but its mechanism of action has not been studied. 7‐Iodotubercidin (6a) acts mainly as a potent inhibitor of adenosine kinase.37 For example, it showed promising submicromolar cytotoxicity in canine osteosarcoma cell lines which show dysregulation of kinase activity.38 7‐Iodotubercidin (6a) is also a p53 activator, and generates DNA damage and G2 cell cycle arrest through its incorporation into DNA.39 On the contrary, the cytotoxic effect of 7‐bromotubercidin (6b) is caused by incorporation of its triphosphate form into cellular RNA and inhibition of RNA synthesis.22
Figure 2.
Structure‐activity relationship among cytotoxic 7‐deazapurine ribonucleosides
In our laboratory, we developed a new group of potent nucleoside cytostatics by the attachment of heterocycles to C‐7 position of 7‐deazaadenosine12 (Fig. 3). It was shown that derivatives substituted with five‐membered heterocycles, i.e. thiophene or furan (7a‐b) exerted nanomolar in vitro cytotoxicity to broad panel of cancer and leukemia cell lines [similar to tubercidin (3)], while phenyl derivative 7c is apparently too bulky and therefore inactive. Similar results were reported by Schram and Townsend in their pioneering study on synthesis of 7‐hetaryl‐7‐deazapurine ribonucleosides.40 Derivatives with five‐membered rings showed mild antileukemic activity and six‐membered ring derivatives were inactive, however IC50 values are not given in this study. Electronic properties of the substituents are also important for the activity. The presence of an electron‐donating p‐methoxy group on a phenyl substituent made compound 7d active in submicromolar concentrations. Direct connection of thiophene and 7‐deazapurine moieties in 7a (known as AB61) is also crucial for the cytotoxic activity, as its analogue 8 with the thienyl group attached via a sulfur atom showed a substantial drop of activity (IC50 > 20 μM).41 The mechanism of action of AB61 (7a) was studied in detail42 (Fig. 3). AB61 (7a) is efficiently phosphorylated in cancer cell lines but not in fibroblasts which is the reason it shows selectivity toward cancer cells. The resulting ribonucleoside triphosphate (NTP) is incorporated both into cellular RNA and DNA where it causes block of translation and DNA damage, respectively. Interestingly, although tubercidin triphosphate was incorporated into RNA under the same experimental conditions, subsequent RNA translation proceeded smoothly. AB61 trisphosphate is a substrate for mitochondrial DNA polymerase γ and therefore AB61 (7a) might interfere with mtDNA replication and mitochondrial functions similarly as described for other nucleoside analogs.43 The in vivo studies of xenograft models confirmed the promising properties of AB61 (7a) for its further development as an anticancer agent.
Figure 3.
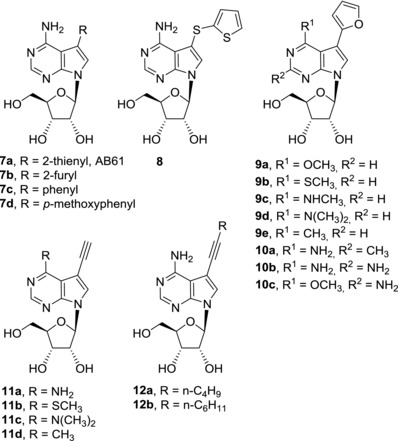
Cytotoxic C7‐substituted deazapurine ribonucleosides
Figure 4.
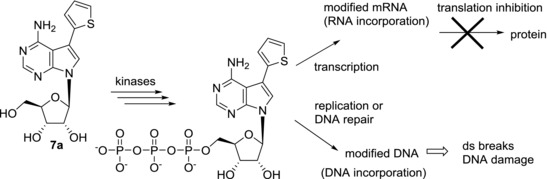
Scheme of the mechanism of action of AB61 (7a)
In order to further expand the series of 7‐hetaryl 7‐deazapurine ribonucleosides, a series of their C6 and C2 modified analogues was prepared44 (Fig. 3). Within the series, C6‐substituted 7‐(2‐furyl) derivatives (9a–e) showed the strongest cytotoxic effect (submicromolar), especially the derivatives with 6‐methoxy (9a), 6‐methylsulfanyl (9b), and 6‐methyl (9e) substituent. On the other hand, introduction of methyl (10a) or amino group (10b‐c) into position 2 resulted in complete loss of activity. Also, 7‐hetaryl‐7‐deazainosines and 7‐deazaguanosines were inactive.44 7‐Ethynyl‐7‐deazaadenosine (11a) can be considered as a structural analogue of toyocamycin (4). Its cytotoxic effect against cancer cell lines (nanomolar) is even more powerful than that of clinically used cytostatics doxorubicin and clofarabine or tubercidin (3).12 Substitution of 6‐amino group in 7‐ethynyl‐7‐deazaadenosine (11a) generally reduced its cytotoxic activity but 6‐methylsulfanyl (11b), 6‐dimethylamino (11c), and 6‐methyl (11d) derivatives still kept submicromolar cytotoxicity toward some cancer cell lines.44 Interestingly, 7‐ethynyl‐6‐methylsulfanyl‐7‐deazapurine ribonucleoside (11b) showed only mild cytotoxicity against normal fibroblasts while 6‐dimethylamino (11c) and 6‐methyl (11d) derivatives were poorly selective. 7‐Alkynyl‐7‐deazaadenosines (12a‐b) were similarly potent in vitro cytostatics as tubercidin (3) but their mechanisms of action are probably different. While inhibition of protein kinase A is considered as one of the cell growth inhibition mechanisms, e.g. of toyocamycin (4),32 derivative 12a is a protein kinase A activator.45
Introduction of sugar modifications in 2′‐position of the ribose moiety in 7‐hetaryl‐7‐deazaadenosines (13a‐f, 14a‐f) led to a decrease in cytotoxic activities46, 47 (Fig. 5). Still some of the arabinonucleosides (13a and 14a), 2′‐C‐methylribonucleosides (13d),46 2′‐deoxy‐2′‐fluoroarabinonucleosides (13d), and 2′‐deoxy‐2′,2′‐difluororibonucleosides (13f and 14f)47 possessed micromolar cytotoxicity. Again it seems that the mechanisms of action between parent ribonucleosides such as AB61 (7a) and their sugar‐modified analogues could be different. For instance, the triphosphate derived from 2′‐deoxy‐2′,2′‐difluororibonucleoside 13f was shown to be an inhibitor of human DNA polymerase α while AB61‐triphosphate was not an inhibitor of the enzyme.47 In contrary to 7‐hetaryl‐7‐deazaadenosines, a less profound decrease of activity was observed when cytotoxic activities of 7‐ethynyl‐7‐deazaadenosines (11a) and its 2′‐deoxy‐2′‐fluoro‐arabino‐analogue 15 were compared. Derivative 15 possessed submicromolar to nanomolar activities against cancer cell lines in vitro, however lower than those of the parent ribonucleoside 11a.46 Submicromolar cytotoxicities were observed among 2′‐fluorinated analogues of 5‐iodotubercidin 16a and 16b 47 and 7‐vinyl‐7‐deazaadenine 2′‐fluoronucleoside 17 also showed moderate (micromolar) cytotoxic activity.48 Introduction of 4′‐C‐methyl group into 7‐hetaryl‐7‐deazaadenosines led to complete loss of cytotoxic effect with the only exception of 2‐benzofuryl derivative 18 with modest (micromolar) activity.49 In conclusion, likewise among naturally occurring cytotoxic 7‐deazapurine nucleosides, modifications of the ribose moiety in 7‐substituted 7‐deazapurine nucleosides mostly result in significant decrease of cytotoxic activities.
Figure 5.
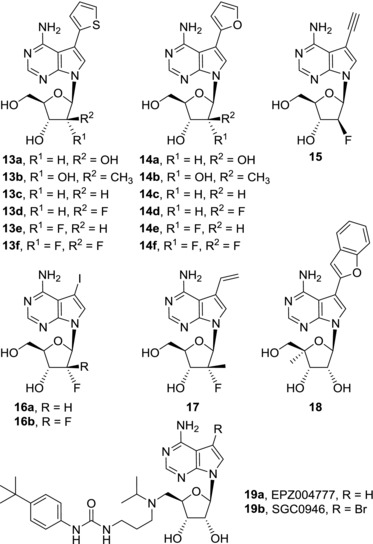
Sugar‐modified derivatives of 7‐substituted 7‐deazaadenosines
Structure‐based drug design was used in development of inhibitors of DOT1L methyltransferase. This enzyme is a protein methyltransferase that methylates histone H3 on lysine 79 (H3K79). Aberrant methylation of H3K79 by DOT1L is essential for development of MLL‐rearranged mixed lineage leukemia. The structure of EPZ004777 (19a), a selective subnanomolar inhibitor of DOT1L (Ki = 0.3 nM), was derived from structure of S‐adenosylhomocysteine (SAH), a cofactor by‐product of methylation catalyzed by DOT1L. Binding of EPZ004777 (19a) into SAH‐binding pocket of DOT1L was intensely studied. The residence time of EPZ004777 (19a) is much longer (τ ≈ 1 h) compared with SAH (τ = 10 s).50 EPZ004777 (19a) is capable of selective inhibition of proliferation of MLL‐rearranged cell lines and showed efficacy in vivo in a mouse xenograft model.51 Introduction of bromine atom into position 7 of 7‐deazapurine moiety led to compound SGC0946 (19b) which showed improved DOT1L inhibitory and MLL‐rearrangement‐related cytotoxic effect as compared with EPZ004777 (19a).52
In so‐called sangivamycin‐like molecules (SLMs), the carbamoyl group of sangivamycin (5) is replaced by other carboxylic‐group‐related substituents and/or additional substituents are attached to position 8 of the 7‐deazapurine moiety (Fig. 6). SLMs, including SLM6 (20) and ARC (21a), showed potent in vitro cytotoxicity against multiple myeloma cell lines.53 The efficacy of SLM6 (20) was also confirmed in vivo. Its cytotoxic effect is caused by inhibition of cyclin dependent kinase 9 (CDK9). ARC (21a), 8‐hydrazinosangivamycin, attracted attention for its promising cytotoxic activities against colorectal cancer,54 melanoma,55 and neuroblastoma cells.56 Synergistic effect was observed in treatment of cancer cell lines with ARC (21a) and a pan‐Bcl‐2 inhibitor ABT‐737 (for structure, see 57).58 ARC (21a) is a general transcriptional inhibitor acting through inhibition of positive transcription elongation factor b (P‐TEFb), a complex of CDK9/cyclin T1.59 ARC (21a) induces p53‐independent apoptosis in malignant cells but not in normal cells. It inhibits Akt signaling pathway56 and decreases expression of antiapoptotic proteins such as Mcl‐1.54, 55 Nevertheless, ARC (21a) was found inactive in xenograft models in vivo, presumably due to rapid serum clearance.60 It was shown that the activity and mechanistic aspects of ARC (21a) are identical to those of sangivamycin (5) so that the lack of efficacy of ARC (21a) is in accord with previous failures of sangivamycin (5) in clinical trials.60 Compound 21b, an 8‐substituted toyocamycin analogue, was found to bind HSP70 more efficiently than toyocamycin (4) and sangivamycin (5) (KD = 2.8 μM). As inhibition of HSP70 in cancer cell lines induces apoptosis,61 derivative 21b has a potential to possess cytotoxic activity but the data are not yet reported. Xylocydine (22a), also called BMK‐Y101, a sugar‐modified analogue of 8‐bromosangivamycin, is a selective inhibitor of cyclin dependent kinases (CDK1, CDK2, CDK7, and CDK9)62, 63 which demonstrated its antitumor potential in hepatocellular carcinoma both in vitro and in vivo, while being inactive against cervical, prostate, and hepatic carcinoma or lung adenocarcinoma.64 Further studies with leukemic HL‐60 cell line showed that xylocydine (22a) induces apoptosis in these cells by CDK1 and CDK4 inhibition and upregulation of protein p16INK4a, which is a CDK inhibitor.65 Isobutyryl ester prodrug of xylocydine, ibulocydine (22b), also causes inhibition of CDK7 and CDK9. Subsequent inhibition of RNA polymerase II phosphorylation leads to rapid down‐regulation anti‐apoptotic of proteins Mcl‐1, survivin, and XIAP, thus inducing apoptosis in hepatocellular carcinoma cell lines.66 Hepatocellular carcinoma cell lines can be sensitized to TRAIL (tumor necrosis factor‐related apoptosis‐induced ligand)‐induced apoptosis by treatment with ibulocydine (22b)67 that is also capable of inhibiting growth of hepatocellular carcinoma in a mouse xenograft model.66 Combination of radiotherapy and ibulocydine (22b) treatment showed promising results both in vitro (apoptotic cell death accompanied with activation of caspases, decrease in Bcl‐2/Bax expression, loss of mitochondrial membrane potential, and release of cytochrome c into cytosol) and in vivo (reduced tumor volume in lung cancer xenografts in mice).68 Replacement of an 8‐bromine atom of xylocydine (22a) by p‐tolyl, m‐ or p‐methoxyphenyl, and m‐ or p‐bromophenyl groups in compounds 23a led to loss of CDK1 and CDK2 inhibitory activity69 and also JRS‐15 (23b) with 8‐biphenylyl group is devoid of any CDK inhibiton.64 However, JRS‐15 (23b) showed cytotoxicity in broader panel of cancer cell lines compared to xylocydine (22a), even though these are only moderate (micromolar).64 Carbocyclic derivative 24 that shares 7‐deazapurine substitution pattern of xylocydine (22a) possesses mild cytotoxic activity against ovarian cancer cell line PA‐1.70 It is also important to mention that 8‐substituted 7‐deazapurine are likely to adopt syn‐conformation69 and binding modes to their molecular target may substantially differ from those of 8‐unsubstituted 7‐deazapurine nucleosides where anti‐conformation is preferred. In conclusion, nucleobase modifications of 7‐deazapurine moiety in positions 7 and 8 cannot only dramatically change the cytotoxic activities but it also affects mechanisms of action of particular nucleosides. Mechanisms of action of cytotoxic 7‐deazapurine nucleosides are typically quite complex and often include activation in cells to obtain corresponding nucleotides that can interfere e.g. with DNA and RNA synthesis. It is common that one compound targets more pathways in parallel, which makes it difficult to decide which of them is the most significant. Some 7‐deazapurine nucleosides target specific enzymes such as adenosine kinase, CDKs or protein kinase A and C. It is difficult to predict the mechanism of action of novel analogues of 7‐ and 8‐substituted 7‐deazapurine nucleosides because many compounds from this group significantly differ in their modes of action although their structures are very similar. Even though target‐based design was successfully applied in the development of DOT1L inhibitors, most of discoveries in development of cytotoxic 7‐deazapurine nucleosides are based on serendipity. More focused studies of binding of cytotoxic nucleotides to cellular DNA and RNA polymerases are required in the future in order to move structure‐activity studies into more target‐related research.
Figure 6.
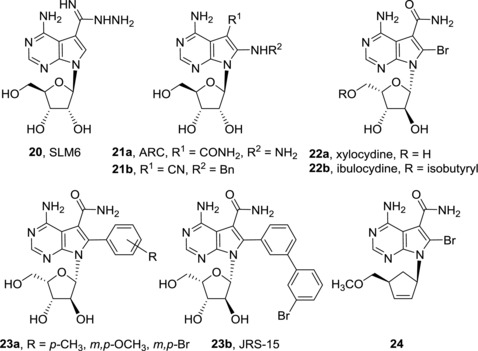
Structures of sangivamycin‐like molecules
2.3. Synthetic nucleosides with C6 substituents
Substitution of 6‐amino group of 7‐deazapurine nucleosides by carbon substituents brings further possibilities for structure‐activity relationship studies among 7‐deazapurine nucleosides. As the substitution at position 6 of 7‐deazapurine nucleosides affects, or in some cases even prevents, formation of stable Watson‐Crick base pairs, their mechanisms of action may differ from those of 7‐deazaadenine nucleosides. In our lab, we prepared a large series of 6‐(het)aryl‐7‐deazapurine ribonucleosides 25 and found that they show different cytotoxic activities based on the nature of the substituent (Fig. 7). Derivatives substituted with a small five‐membered heterocycles, e.g. furan (25a) or thiophene (25b), are nanomolar cytostatics against a broad range of cancer cell lines, while phenyl derivative 25c is mostly inactive.71 In this case, substitution of the phenyl ring did not bring substantial improvement of activity. Further substitution by fluorine in position 7 in derivatives 26a‐b did not affect the efficacy of the parent compounds, on the other hand 7‐chloroderivatives 27a‐b were significantly less active or inactive. The decrease of cytotoxic potency in 7‐chloroderivatives is in agreement with the fact that intracellular phosphorylation of 27a is much less efficient than that of unsubstituted derivative 25a, indicating intracellular phosphorylation may be limiting the activity. The mechanism of action of 6‐hetaryl‐7‐deazapurine ribonucleosides has not been fully examined yet, however, rapid and powerful cellular RNA synthesis inhibition was observed after treatment with 25a‐b and 26a.
Figure 7.
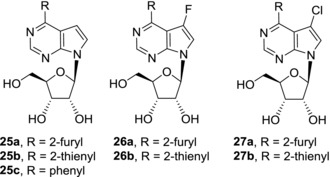
Structures of cytotoxic 6‐(het)aryl‐7‐deazapurine ribonucleosides
Since the efficient phosphorylation seemed to be crucial for cytotoxic activities of 6‐hetaryl‐7‐deazapurine ribonucleosides 25 and 26, phosphate prodrugs of these compounds were synthesized in efforts to further improve their cytotoxicity (Fig. 8). CycloSal pronucleotides of 6‐hetaryl‐7‐deazapurine (28) and 6‐hetaryl‐7‐fluoro‐7‐deazapurine (29) nucleosides however showed similar or slightly lower cytotoxic activities compared to parent nucleosides in most cancer cell lines.72 Again, derivatives with small furyl and thienyl substituents 28a‐b and 29a‐b were more potent than derivatives with bulky substituents such as benzofuryl 28c which were devoid of any cytotoxic effect. ProTide approach was also applied and phosphoramidate prodrugs based on 6‐hetaryl‐7‐deazapurine nucleosides were prepared.73 Nevertheless, nanomolar cytotoxic activities of the parent nucleosides dropped to micromolar in their ProTides, no matter if methyl (30a, 31a), ethyl (30b, 31b), or benzyl (30c, 31c) alanine esters were used. The decrease of activities was presumably caused by efflux of ProTides out of the cells.
Figure 8.
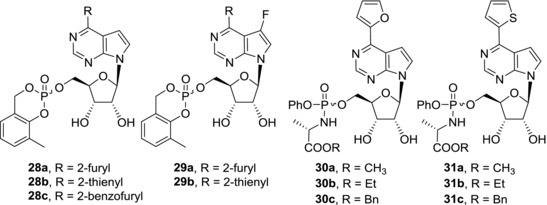
Phosphate‐prodrugs of 6‐(het)aryl‐7‐deazapurine ribonucleosides
Further nucleobase modifications of 6‐hetaryl‐7‐deazapurine nucleosides comprised of introduction of substituents into position 274 (Fig. 9). However, cytotoxicity screening showed significant drop of activities among 2‐substitued derivatives compared to their parent nucleosides. Only some modest (micromolar) activity of 2‐fluoro derivative 32a was observed, more bulky 2‐chloro‐ (32b), 2‐amino‐ (32c), and 2‐methyl (32d) derivatives were inactive. Also, the ribonucleoside moiety was shown to be crucial for keeping the potency of 6‐hetaryl‐7‐deazapurine nucleosides as all of the sugar‐modified derivatives, such as 33a‐33e and 34, were devoid of any cytotoxic activity.49, 75, 76 (Fig. 9). Structure‐activity studies of 6‐hetaryl‐7‐deazapurine nucleosides mentioned in this section and those of 7‐hetaryl‐7‐deazapurine nucleosides (Section 2.2) therefore are in agreement with the hypothesis that the modes of action of these classes of compounds are different.
Figure 9.
2‐Modified and sugar‐modified derivatives of 6‐hetaryl‐7‐deazapurine nucleosides
Replacement of the amino group of tubercidin (3) by a methyl group led to 6‐methyl‐7‐deazapurine ribonucleoside (35a) (Fig. 10). Compound 35a is a potent cytotoxic agent with low submicromolar activities against a broad range of cancer cell lines.44 Unfortunately, 6‐methyl‐7‐deazapurine ribonucleoside (35a) is poorly selective and showed similar cytotoxic effect also against normal fibroblasts. Substitution of compound 35a in position 2 is not tolerated as the resulting 2‐fluoro (35b), 2‐chloro‐ (35c), 2‐amino (35d), and 2‐methyl (35e) derivatives are completely inactive.74 6‐Trifluoromethyl‐7‐deazapurine ribonucleoside (36a) possesses similar cytotoxic activities against cancer cell lines as the 6‐methyl analogue 35a.77 Also in this case no 2‐modified derivatives 36b‐h showed any cytotoxicity.77
Figure 10.
Other examples of cytotoxic 7‐deazapurine ribonucleosides
Another option for modification in position 6 is N6‐alkylation of 7‐deazaadenine (Fig. 10). N6‐benzyl and N6‐nitrobenzyl derivatives of tubercidin (3), toyocamycin (4), and sangivamycin (5) were studied as inhibitors of nucleoside transporter 1 (hENT1) which is a protein that plays key role in nucleoside drug uptake. Among the compounds synthesized in the study, N6‐nitrobenzyl derivative of sangivamycin 37 showed both efficient (nanomolar) inhibition of the hENT1 transporter and mild (micromolar) cytotoxicity against several cancer cell lines.78 Conjugation of tubercidin (3) and cisplatin, a clinically used cytostatic, through N6‐nitrogen of tubercidin furnished derivative 38. Although compound 38 was able to react with purine residues in a similar way as cisplatin, its cytotoxic activity against cancer cell lines was only weak (micromolar) and dropped by one order of magnitude compared to cisplatin.79 Based on these two examples, it could seem that attachment of larger substituents to the N6‐amino group is not suitable for improvement of cytotoxic effect of 7‐deazapurine nucleosides but such conclusion would be too simplistic. Pevonedistat (39, MLN4924) is N6‐substituted carbocyclic 7‐deazapurine nucleoside analogue which shows mode of action distinct from those of other 7‐deazapurine nucleosides. Pevonedistat (39) is a potent and selective inhibitor of NEDD8‐activating enzyme (NAE) (IC50 = 4 nM) that regulates proteasome‐mediated protein degradation.80 Accumulation of specific proteins caused by inhibition of NAE leads to deregulation of S‐phase DNA synthesis and subsequently to apoptosis. Despite modest clinical activity of pevonedistat (39) in phase I study of patients with acute myeloid leukemia and myelodysplastic syndromes,81 it showed significant therapeutic effect in phase I trials against refractory multiple myeloma,82 metastatic melanoma,83 and advanced solid tumors.84 In conclusion, some of 6‐modified‐7‐deazapurine nucleosides show potent cytotoxic effect against cancer cell lines. With the exception of pevonedistat (39), their mode of action is not well understood yet and most of the reports focused only on structure‐activity studies. Nevertheless, it seems that the mechanisms of action of 6‐substituted‐7‐deazapurine nucleosides vary within the group and, at the same time, they are significantly different from those of 7‐deazaadenine nucleosides.
2.4. Fused nucleosides
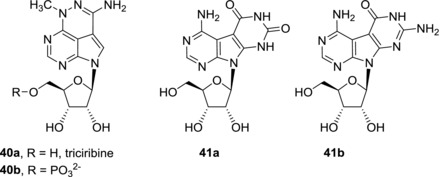
7‐Deazapurine nucleobase can be further extended by annulation with other aromatic rings leading to fused (tricyclic) nucleosides (Fig. 11). Triciribine (40a), also known as TCN or API‐2, was synthesized already in 1971.85 Triciribine (40a) suffers from poor solubility and therefore its soluble 5′‐monophosphate prodrug 40b (TCN‐P) is often used. Antineoplastic activities of triciribine (40a) and TCN‐P (40b) both in vitro and in vivo were studied in 1980s and early 1990s and were reviewed previously.86 Clinical trials were halted due to toxicity in high dosing of TCN‐P (40b) and mild efficacy presumably due to its low bioavailability. However, recently it was shown that the bioavailability can be significantly improved by application of phosphoramidate prodrug approach.87 Triciribine (40a) acts through selective inhibition of Akt kinase88 and therefore is effective in Akt‐overexpressing tumors such as pancreatic and ovarian. Recently, synergistic effects were observed in combinations of TCN‐P (40b) with other antineoplastic compounds such as gemcitabine89 and tipifarnib.90 Clinical studies of other combination therapies with TCN‐P (40b) are currently under way.
Figure 11.
Structures of triciribine and Janus‐type tricyclic nucleosides
Pyrimido‐fused nucleosides are also called dual bases or Janus‐type tricyclic nucleosides because they can present Watson‐Crick donor/acceptor array of one nucleobase on one face and different donor/acceptor array on the other face and therefore can form stable base pairs with two different complementary nucleobases. The Janus‐type nucleobases such as 41a‐b showed only moderate (micromolar) cytotoxic activities and later on they became conceived more as antivirals.91, 92
Most of the benzo‐fused 7‐deazapurine nucleosides prepared so far lack any cytotoxic activity against cancer cell lines (Fig. 12). Derivatives 42 and 43a‐b are the only examples of weakly active (micromolar) compounds.93, 94 On the other hand, thieno‐fused nucleosides, which are isosteric to above‐mentioned benzo‐fused nucleosides, showed interesting cytotoxic effects95 (Fig. 12). Substituents in position 6 of 7‐deazapurine part of the nucleobase play an important role in structure‐activity studies. While dimethylamino derivatives (44a, 45a) were inactive and amino derivatives (44b, 45b) possess only moderate (micromolar) cytotoxic activities, methyl (44c, 45c), methoxy (44d, 45d), and methylsulfanyl (44e, 45e) derivatives are potent cytotoxic compounds with submicromolar activities against cancer cell lines of different origin. The position of sulfur heteroatom has an impact on selectivity of the compounds. Series of compounds with thieno[2′,3′:4,5]pyrrolo[4,5‐d]pyrimidine nucleobase 44 was similarly toxic towards both cancer cells and normal fibroblasts but series of compounds with thieno[3′,2′:4,5]pyrrolo[4,5‐d]pyrimidine nucleobase 45 showed selectivity towards cancer cells. Initial studies of mode of action of compounds 44c‐e and 45c‐e revealed RNA synthesis inhibition and decrease in mitotic cell fraction in cell cycle analysis. Both compounds 44e and 45e are efficiently phosphorylated both in normal and cancer cells indicating that cell‐type specific phosphorylation is not a reason for the different selectivities of series 44 and 45.95 In conclusion, even though the family of fused nucleosides is relatively small, it has already brought interesting compound hits and deserves further development as a source of cytotoxic nucleosides. Detailed study of mechanism of action of these compounds is particularly needed and is under way.
Figure 12.
Structures of cytotoxic benzo‐ and thieno‐fused 7‐deazapurine nucleosides
3. ADENOSINE KINASE INHIBITORS
3.1. Inhibitors of mammalian adenosine kinases
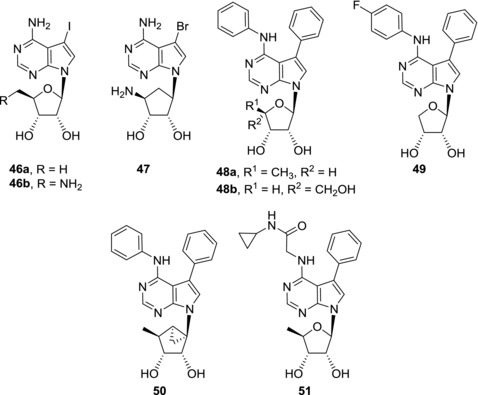
Adenosine kinase (ADK) is an enzyme that catalyzes the conversion of adenosine to adenosine‐5′‐O‐monophosphate (AMP). Extracellular adenosine is a ligand of adenosine receptors which regulates heart rate, neurotransmitter release in brain, and inflammatory response. Stimulation of adenosine receptors is connected with anticonvulsant, analgesic, and anti‐inflammatory activity. Inhibition of ADK leads to increased adenosine levels and so ADK inhibitors possess similar effect as adenosine receptor agonists. Moreover it was shown that ADK inhibitors show less side effects in anti‐seizure activity assays and therefore can be considered as a promising class of anticonvulsants.96 Data from Section 3 is summarized in Table 2. 5′‐Deoxy‐5‐iodotubercidin (46a) is a naturally occurring nucleoside isolated from a marine red alga (Fig. 13). 5′‐Deoxy‐5‐iodotubercidin (46a) is a potent inhibitor of mammalian adenosine kinases37, 97 (IC50 = 9 nM against human ADK) and does not bind to adenosine receptors.98 Also other 7‐halogenated 7‐deazaadenine nucleosides show ADK inhibitory activity. Although 5‐iodotubercidin (6a)37, 97 and 5‐bromotubercidin (6b)22, 97 are less potent ADK inhibitors than 5′‐deoxy‐5‐iodotubercidin (46a), they still possess submicromolar IC50 values. Both 5′‐deoxy‐5‐iodotubercidin (46a) and 5‐iodotubercidin (6a) showed anti‐seizure activities in vivo,96, 97 however, these compounds are not suitable for clinical use due to behavioral effects (decreased locomotor activity, hypothermia, and muscle flaccidity)98 and cytotoxicity,39 respectively. In order to obtain more suitable clinical candidates, a large structure‐activity study was performed.97 It was shown that the presence of a halogen atom in position 7 is crucial for the ADK inhibitory activity. The most potent activities were observed in derivatives with amino, chloro, or methylsulfanyl group in position 6 and 5′‐amino group. For instance, derivative 46b showed subnanomolar human ADK inhibition (IC50 = 0.6 nM). Compound 46b showed anti‐seizure activity in an in vivo model. However, it seems that anti‐seizure activities generally do not fully correlate with ADK inhibitory effects, presumably due to different pharmacological properties of the tested compouds.97
Table 2.
ADK inhibitory activity of 7‐deazapurine nucleosides
| Compound | IC50 [μM] (human ADK) | IC50 [μM] (Mtb‐ADK) | Ref. |
|---|---|---|---|
| 7‐Iodotubercidin (6a) | 0.026 | nda | 97 |
| 7‐Bromotubercidin (6b) | 0.12 | nd | 97 |
| 7c | >5 | >10 | 100 |
| 11a | 0.20 | 0.33 | 100 |
| 35a | >10 | 8.8 | 107 |
| 35b | >10 | >10 | 74 |
| 35c | >10 | >5 | 74 |
| 35d | >10 | >10 | 74 |
| 35e | >20 | >5 | 74 |
| 46a | 0.009 | nd | 97 |
| 46b | 0.0006 | nd | 97 |
| 47 | 0.00047 | nd | 101 |
| 48a | 0.0005 | nd | 101 |
| 48b | 0.00047 | nd | 102 |
| 49 | 0.006 | nd | 103 |
| 50 | 0.088 | nd | 103 |
| 51 | 0.003 | nd | 105 |
| 52 | >10 | 0.6 | 100 |
| 53a | >20 | 0.023 | 107 |
| 53b | >20 | 0.028 | 107 |
| 54a | 0.3 | 0.0075 | 107 |
| 54b | 2.1 | 0.0145 | 107 |
| 55 | >10 | 0.0012 | 74 |
nd, not determined
Figure 13.
Examples of 7‐deazapurine nucleosides as inhibitors of mammalian ADKs
ADK inhibitors are also promising analgetics. Carbocyclic 7‐bromo‐7‐deazapurine nucleoside 47 is a subnanomolar inhibitor of ADK (IC50 = 0.47 nM against human ADK) which showed antinociceptive activity in animal acute, inflammatory, and neuropathic pain models (Fig. 13).99
Replacement of iodine atom in 5‐iodotubercidin (6a) by ethynyl group led to drop of ADK inhibitory activity, however, resulting 7‐ethynyl‐7‐deazaadenosine (11a) still showed submicromolar ADK inhibition.100 On the other hand, tubercidin derivatives with larger hydrophobic groups in position 7, such as 7‐phenyl‐7‐deazaadenosine (7c), do not inhibit human ADK.100 Despite this fact, when 7‐phenyl group is combined with phenyl group attached to N6 nitrogen atom, the potent ADK inhibition in these so called diaryl derivatives is restored101 (Fig. 13). Further sugar modifications led to 5′‐deoxy derivative 48a 101 and α‐l‐lyxofuranosyl derivative 48b 102 that are subnanomolar human ADK inhibitors (IC50 = 0.5 and 0.47 nM, respectively). Compound 48a appeared to be a potent anticonvulsant in a rat seizure model,101 while compound 48b showed moderate anti‐inflammatory effect both in rat carrageenan paw edema model and rat skin lesion model.102 Also erythrofuranosyl diaryl‐derivative 49 is a nanomolar ADK inhibitor (IC50 = 6 nM).103 Derivative 49 showed analgesic activity in several animal pain models. The effect of compound 49 in rat formalin paw pain model was similar to that of standard opioid analgesic morphine.103 Recently, submicromolar ADK inhibition was also observed in carbocyclic diaryl nucleosides such as compound 50 (IC50 = 0.088 μM).104 However, in vivo activities of this class of compounds have not been studied yet. Despite potent activities of diaryl nucleosides, the compounds generally suffer from poor water solubility. Replacement of N6‐phenyl group by glycinamide substituent led to compound 51 which showed improved solubility and nanomolar ADK inhibitory effect at the same time (IC50 = 3 nM).105 In vivo animal pain models provided promising results as derivative 51 was shown to be more potent than morphine.105 Unfortunately, prolonged administration of compound 51 is connected with lethal toxicity and therefore further development of this compound was discontinued. In spite of this fact human ADK inhibitors still represent a very promising group of compounds and deserve attention for their antinociceptive, anti‐seizure and anti‐inflammatory activities.
3.2. Inhibitors of mycobacterial adenosine kinase
Many pathogens lack enzymes for de novo purine synthesis and use purine salvage pathway instead. In this pathway purine nucleotides are formed from nucleobases and phosphoribosyl pyrophosphate or purine nucleosides are phosphorylated by kinases, e.g. adenosine kinase. Targeting ADK of the pathogen can therefore lead to anti‐microbial compounds. Mycobacterium tuberculosis expresses enzymes from both salvage pathway and de novo purine synthesis pathway; however, the interdependence and regulation of these processes remain unclear.106 For instance, 7‐ethynyl‐7‐deazaadenosine (11a) is a submicromolar inhibitor of ADK of Mycobacterium tuberculosis (Mtb‐ADK) (IC50 = 0.33 μM) and showed antimycobacterial activities both against Mycobacterium bovis and Mycobacterium tuberculosis.100 Nevertheless, poor ADK inhibition selectivity and cytotoxicity of 11a are not desirable. On the other hand, 7‐hetaryl‐7‐deazaadenosines with bulky substituents have been already mentioned as compounds that are non‐cytotoxic and do not inhibit human ADK.12, 100 For instance, dibenzofuryl derivative 52 is a submicromolar inhibitor of Mtb‐ADK (IC50 = 0.6 μM) that is devoid of any cytotoxicity towards human fibroblasts and possesses micromolar antimicrobial activity against Mtb strains100 (Fig. 14). Also 6‐hetaryl‐7‐deazapurine ribonucleosides were shown to selectively inhibit with Mtb‐ADK in submicromolar concentrations.107 Derivatives with small hetaryl groups such as 53a and 53b (IC50 = 23 and 28 nM, respectively) are highly selective to Mtb‐ADK with selectivity index over 10,000. However, these compounds are cytotoxic and their antimycobacterial activities against Mycobacterium bovis are poor. On the other hand, derivatives with more bulky substituents such as benzothienyl (54a) and benzofuryl (54b) are very powerful Mtb‐ADK inhibitors (IC50 = 7.5 and 14.5 nM, respectively), they show good selectivity and are non‐cytotoxic.107 Nevertheless, these compounds lack antimycobacterial activity against Mycobacterium bovis. The reason for poor correlation between Mtb‐ADK inhibition and antimycobacterial activity could be poor penetration of the tested compounds through the mycobacterial cell wall or bypassing Mtb‐ADK inhibition through alternative purine synthesis pathways. The presence of a fluorine atom in 6‐hetaryl‐7‐deazapurine ribonucleosides can further improve both Mtb‐ADK inhibitory activity and selectivity. Benzofuryl derivative (55) is a nanomolar inhibitor of Mtb‐ADK (IC50 = 1.2 nM) but again derivative 55 showed only modest activity against Mtb.74 On the other hand, 6‐methyl‐7‐deazapurine ribonucleoside (35a) is a weak Mtb‐ADK inhibitor (IC50 = 8.8 μM) but showed strong submicromolar activity against Mycobacterium bovis.107 In this case, the mode of antimycobacterial activity is presumably independent of Mtb‐ADK and rather suggests a more general cytotoxic mechanism as compound 35a is highly cytotoxic also toward human cells. This is in agreement with the fact that its 2‐substituted analogues 35b–e showed neither antimycobacterial activity nor cytotoxicity.74 In conclusion, the most potent 7‐deazapurine nucleoside inhibitors of Mtb‐ADK are only weakly active in antimycobacterial assays. Poor cellular uptake could be one of the reasons for this fact, however, initial attempts to improve the cell penetration by lipophilic prodrugs failed.107 Therefore, showing that standard prodrug approaches for penetration for eukaryotic cells may not be effective in mycobacteria and requires further studies.
Figure 14.
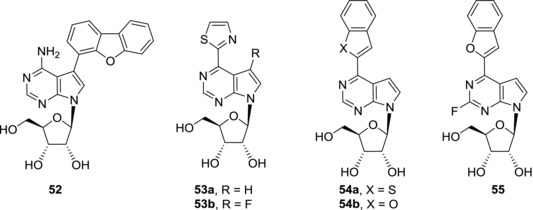
Examples of 7‐deazapurine nucleosides as inhibitors of Mtb‐ADK
4. NUCLEOSIDES WITH ANTIVIRAL ACTIVITIES
4.1. Nucleosides with anti‐HCV activities
4.1.1. Ribonucleosides
Hepatitis C virus (HCV) is an RNA virus of the family Flaviviridae. It uses RNA‐dependent RNA polymerase (NS5B) for replication of its genetic information. Because human cells do not express any RNA‐dependent RNA polymerase and HCV RNA polymerase (NS5B) is structurally different from human RNA and DNA polymerases, the viral enzyme became a target for new anti‐HCV compounds development. Thanks to their close resemblance to natural substrates of the viral RNA polymerases, many 7‐deazapurine nucleosides were studied as potential antiviral candidates not only for HCV but also other RNA viruses that will be mentioned later in the text. Structure‐activity studies of nucleobase‐modified analogues of naturally occurring 7‐deazapurine antibiotics such as tubercidin (3) and toyocamycin (4) brought several interesting structures with anti‐HCV activities (Fig. 15). Modifications of toyocamycin (4) in positions 6 and 7 led to nucleosides 56 and 57 that showed submicromolar activities in HCV replicon assays. EC50 and CC50 values for compounds mentioned in Section 4.1 are listed in Table 3. Compounds 56 and 57 are non‐cytotoxic and therefore they both represent good lead structures for anti‐HCV drug development.108 Also other 6‐ and 7‐modified‐7‐deazapurine ribonucleosides showed interesting in vitro anti‐HCV activities.44 7‐Furyl derivatives 9a and 9c showed submicromolar anti‐HCV activities in both HCV 2A and HCV 1B replicon assays. Both 9a and 9c were non‐cytotoxic to the HCV replicon cells but 9a showed submicromolar cytotoxicity towards normal fibroblasts and therefore it cannot be considered as suitable for further development as an anti‐HCV agent. Also 7‐ethynyl derivative 11c possessed submicromolar anti‐HCV activity and no cytotoxicity in HCV replicon assay but was cytotoxic to fibroblasts.44 On the other hand, 6‐N,N‐dimethylamino‐7‐deazapurine ribonucleoside (58) seems to be more suitable lead structure because it showed submicromolar activities and no cytotoxicity in HCV replicon assay and only weak cytotoxicity towards fibroblasts. Sangivamycin‐like molecule ARC (21a) that was developed as an anti‐cancer agent showed in vitro anti‐HCV activity while being non‐toxic to host cells.109 It was found out that anti‐HCV activity of ARC (21a) is not caused by transcription or translation inhibition but the molecular target of ARC (21a) remains unknown. Also another antineoplastic compound, triciribine (40a), showed moderate anti‐HCV activity (EC50 = 2 μM) and low cytotoxicity.110 Triciribine analogue 59 was more potent (EC50 = 1 μM) and remained non‐cytotoxic.110 Janus‐type nucleosides 41a‐b showed moderate anti‐HCV activities (EC50 = 5.7 and 3 μM, respectively) which was accompanied by cytotoxicity toward Vero, CEM, and PBM cell lines.91 Anti‐HCV screening of thieno‐fused 7‐deazapurine ribonucleosides showed that derivatives 44c,e and 45c‐e possess submicromolar activities and do not show any cytotoxic effect against the replicon cells.95 The anti‐HCV potency of these compounds is similar to that of anti‐HCV agent mericitabine. However, thieno‐fused 7‐deazapurine nucleosides are mostly cytotoxic to fibroblasts and only derivative 45c was devoid of this cytotoxic effect. In conclusion, despite interesting anti‐HCV activities of some 7‐deazapurine ribonucleosides, the anti‐HCV effect is often accompanied by cytotoxicity to normal cells and therefore further modifications of the lead structures are necessary in order to obtain suitable anti‐HCV drug candidates.
Figure 15.
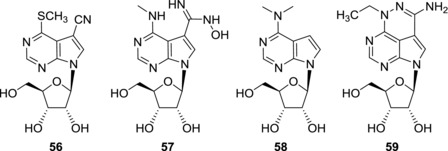
Examples of anti‐HCV 7‐deazapurine ribonucleosides
Table 3.
Anti‐HCV activities of 7‐deazapurine nucleosides
| Compound | EC50 [μM] (replicon)a | CC50 [μM] (replicon) | Ref. |
|---|---|---|---|
| 7‐Deazapurine ribonucleosides | |||
| 9a | 0.02 (1B); 0.03 (2A) | >44 (1B); > 44 (2A) | 44 |
| 9c | 0.06 (1B); 0.14 (2A) | >44 (1B); > 44 (2A) | 44 |
| 11c | 0.07 (1B); 0.10 (2A) | >44 (1B); > 44 (2A) | 44 |
| 40a | 2 | >300 | 110 |
| 41a | 5.7 | 5.7 | 91 |
| 41b | 3 | <10 | 91 |
| 44c | 0.11 (1B); 0.06 (2A) | >44 (1B); >44 (2A) | 95 |
| 44e | 0.47 (1B); 0.34 (2A) | >44 (1B); >44 (2A) | 95 |
| 45c | 0.06 (1B); 0.24 (2A) | >44 (1B); >44 (2A) | 95 |
| 45d | 0.23 (1B); 0.40 (2A) | >44 (1B); >44 (2A) | 95 |
| 45e | 0.13 (1B); 0.80 (2A) | >44 (1B); >44 (2A) | 95 |
| 56 | 0.4 | >250 | 108 |
| 57 | 0.6 | >200 | 108 |
| 58 | 0.27 (1B); 0.97 (2A) | >44 (1B); >44 (2A) | 44 |
| 59 | 1 | 150 | 110 |
| Sugar‐modified 7‐deazapurine nucleosides | |||
| 17 | 1.8 | 4.5 | 48 |
| 60a (MK‐0608) | 0.3 | >100 | 111 |
| 60b | 0.07 | >100 | 116 |
| 60c | 0.13 | ∼50 | 116 |
| 60d | 0.24 | 40 | 116 |
| 60e | 0.37 (1A); 0.38 (1B) | 19.75 (1A); 26.27 (1B) | 46 |
| 60f | 0.09 | nd | 117 |
| 61 | >50 | >100 | 111 |
| 62 | 3 | >100 | 118 |
| 63a | >50 | ndb | 119 |
| 63b | >50 | nd | 119 |
| 64 | 1.9 | nd | 120 |
| 65 | >100 | nd | 120 |
| 66 | 1.8 | nd | 120 |
| 67 | 23.8 | nd | 121 |
| 68 | 33.8 | nd | 121 |
| 69a | 18 | >300 | 110 |
| 69b | 15 | >200 | 110 |
| 70a | 2.66; >10 | 1165; >10 | 48, 124 |
| 70b | 1.1 | 10.4 | 48 |
| 70c | 24 | nd | 117 |
| 70d | 35 | nd | 120 |
| 71 | 0.9 | >50 | 125 |
| 72a | 0.11 | nd | 126 |
if replicon is not mentioned, the genotype is not specified in the original article;
nd, not determined
4.1.2. Sugar‐modified nucleosides
Decreased cytotoxic activities of sugar‐modified 7‐deazapurine nucleosides compared to corresponding ribonucleosides have already been mentioned. Because anti‐HCV activities of 7‐deazapurine ribonucleosides are often accompanied by cytotoxicity, sugar modifications seem to be suitable for restriction of the cytotoxic effect. Among the sugar‐modified nucleosides (such as arabinosides, 3′‐deoxyribonucleosides, and 2′‐O‐methylribonucleosides) 2′‐C‐methylribonucleosides (more precisely their NTPs) are usually the best HCV RNA polymerase inhibitors111 (Fig. 16). 7‐Deaza‐2′‐C‐methyladenosine (60a),112 also known as MK‐0608, was thoroughly studied as an anti‐HCV drug candidate.111 It showed submicromolar activity against HCV in a replicon assay and is non‐cytotoxic. MK‐0608 (60a) is efficiently phosphorylated in cells and the corresponding NTP is a potent inhibitor of HCV NS5B RNA polymerase. In fact, the NTP is incorporated into the growing RNA chain and acts as a chain terminator.111 A single mutation in the NS5B RNA polymerase, S282T, leads to a resistance to MK‐0608 (60a).111 The anti‐HCV activity of MK‐0608 (60a) was confirmed in HCV‐infected chimpanzees where significant reduction of viral load was observed but viral loads rebounded after dosing ended in all tested animals.113 Combinational therapy with MK‐0608 (60a) and an HCV NS3/4A protease inhibitor vaniprevir (MK‐7009), however, resulted in sustained virological response of viral negativity 6 months after treatment in chimpanzees.114 Combinational treatment with MK‐0608 (60a) was also successful in HCV‐infected human hepatocyte chimeric mice. The combination of MK‐0608 (60a) and another HCV NS3/4A protease inhibitor telaprevir led to sustained virological response but only in high dose regimen or in triple combination therapy with MK‐0608 (60a), telaprevir, and interferon.115 In spite of these promising results, clinical trials with MK‐0608 (60a) have been halted for undisclosed reasons.
Figure 16.
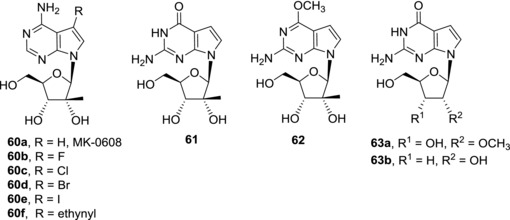
Examples of sugar‐modified anti‐HCV 7‐deazapurine nucleosides
Halogen atoms are tolerated in position 7 of 7‐deaza‐2′‐C‐methyladenosine. All 7‐halogenated deaza‐2′‐C‐methyladenosines 60b‐e showed submicromolar activities in HCV replicon assays.46, 116 The anti‐HCV activity of the derivatives decreases with the increasing size of the halogen, on the other hand cytotoxicity was found to increase. Therefore only fluoro‐derivative 60b shows desirable properties and fourfold higher potency compared to 7‐unsubtituted derivative MK‐0608 (60a).116 The increased potency is presumably caused by improved cellular uptake of 60b. Introduction of a 7‐ethynyl group in compound 60f brought significant anti‐HCV activity but unfortunately cytotoxicity of this derivative was not reported. The corresponding nucleoside triphosphate efficiently inhibited HCV RNA polymerase (IC50 = 0.75 μM).117
Effective cellular permeation and intracellular phosphorylation play a crucial role in development of anti‐HCV agents targeting HCV RNA polymerase. Although 7‐deazaguanine nucleosides (in their triphosphate forms) are potent inhibitors of NS5B RNA polymerase, often more powerful than corresponding 7‐adenine nucleosides,111 they perform poorly in the HCV replicon assays. For instance, 7‐deaza‐2′‐C‐methylguanosine 61 was shown to be completely inactive.111 Even its prodrug, 6‐O‐methyl derivative 62 that should be more lipophilic and therefore should enter the cells more easily, is devoid of any anti‐HCV activity.118 Also phosphate prodrug approach failed to bring promising anti‐HCV compounds as the prodrugs were poorly active and/or cytotoxic.118 Also other sugar‐modified 7‐deazaguanine nucleosides, such as 2′‐O‐methylribonucleoside 63a and 3′‐deoxyribonucleoside 63b were strong HCV RNA polymerase inhibitors as NTPs111, 119 but again their phosphate prodrugs were only weakly active or inactive.119
7‐Hetaryl‐7‐deaza‐2′‐C‐methyladenosines generally show modest (micromolar) anti‐HCV activities46, 120, 121 (Fig. 17). The strongest anti‐HCV effect (EC50 = 1.9 μM) was observed for oxadiazolyl derivative 64.120 The corresponding NTP is a submicromolar inhibitor of HCV RNA polymerase (IC50 = 0.5 μM). Derivative 64 is orally available in rats and high levels of NTP were detected in rat liver. In contrary to 7‐deazaguanine nucleosides, pyrazolyl derivative 65, which was inactive in HCV replicon assay, was turned to a low micromolar anti‐HCV compound 66 (EC50 = 1.8 μM) using ProTide prodrug approach.120 Unfortunately, cytotoxicity data were not reported for compounds 64–66.
Figure 17.
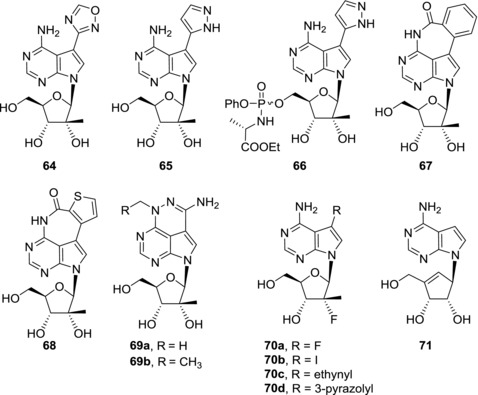
Examples of anti‐HCV derivatives of 7‐(hetaryl)‐7‐deaza‐2′‐C‐methyladenosines and 7‐deazaneplanocin A
Tetracyclic 2′‐C‐methylribonucleosides 67 and 68 are excellent inhibitors of NS5B RNA polymerase when converted to NTPs (IC50 = 0.10 and 0.12 μM, respectively), but their in vitro anti‐HCV activity is only moderate and again, the cytotoxicity data are missing.121 Also 2′‐C‐methylribonucleoside derivatives of triciribine 69a and 69b are only weakly active in HCV replicon assay and significant drop in activities compared to those of corresponding ribonucleosides 40a and 59 was observed.110
Nucleosides with fluorine atoms attached to sugar moiety are also common among compounds with anti‐HCV activity (Fig. 17). Currently, the only approved NS5B polymerase inhibitor, sofosbuvir, is a phosphate prodrug of a 2′‐deoxy‐2′‐fluoro‐2′‐C‐methylribonucleoside.122, 123 Therefore, 7‐Deazapurine 2′‐deoxy‐2′‐fluoro‐2′‐C‐methylribonucleosides were also prepared. 7‐Fluoro‐7‐deaza‐2′‐deoxy‐2′‐fluoro‐2′‐C‐methyladenosine (70a) showed promising activity in HCV replicon assay (EC50 = 2.7 μM) and low cytotoxicity124 but was found inactive in a different study.48 A structure‐activity relationship study of 7‐deazapurine 2′‐deoxy‐2′‐fluoro‐2′‐C‐methylribonucleosides identified only 7‐iodo derivative 70b and 7‐vinyl derivative 17 as compounds that possess moderate anti‐HCV activity, however, derivative 17 was also cytotoxic.48 NTPs derived from 7‐ethynyl derivative 70c 117 and 7‐(pyrazol‐3‐yl) derivative 70d 120 are both excellent inhibitors of HCV RNA polymerase (IC50 = 0.4 and 0.1 μM, respectively) but the nucleosides are inactive or poorly active in cell‐based assay. In contrary to sofosbuvir, in this case again the ProTide prodrug approach failed to improve the anti‐HCV activities of the parent nucleosides.
7‐Deazaneplanocin A (71), a carbocyclic nucleoside, showed micromolar activity in HCV replicon assay and was non‐cytotoxic in 100 μM concentration.125 The potency of 7‐deazaneplanocin A (71) is similar or even better than that of known anti‐HCV compounds 2′‐fluoro‐2′‐deoxy‐2′‐C‐methylcytosine and 2′‐C‐methylcytosine. 7‐Substituted analogues of 7‐deazaneplanocin A showed either weaker anti‐HCV activity or increased cytotoxicity so that unsubstituted derivative 71 seems to be the most promising lead compound in this series.
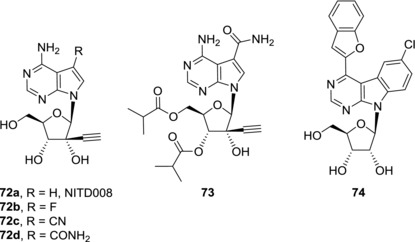
7‐Deaza‐2′‐ethynyl‐adenosine (72a), also known as NITD008 (Fig. 18), was developed as an anti‐dengue compound.126 Because HCV and dengue virus both belong to Flaviviridae, NITD008 (72a) also possesses anti‐HCV activity.126 Despite its anti‐HCV potency, further research related to NITD008 (72a) was focused on its anti‐dengue effect only.
Figure 18.
Examples of anti‐DENV 7‐deazapurine nucleosides
Despite the effort made in development of new HCV RNA polymerase inhibitors as anti‐HCV agents, none of the 7‐deazapurine nucleosides proceeded to clinical use. One of the reasons for this fact could be toxicities caused by off target effects as many of other nucleoside NS5B polymerase inhibitors showed to be substrates of human RNA polymerase II and human mitochondrial RNA polymerase.127 Finding proper balance between NS5B polymerase inhibition and selectivity, efficient cellular phosphorylation and cell penetration as well as cytotoxicity remains difficult. Still many of the HCV RNA polymerase inhibitors could be useful in development of compounds active against related RNA viruses from Flaviviridae family thanks to structural similarities among viral RNA polymerases. Efforts made in this field will be mentioned in the following sections.
4.2. Nucleosides with anti‐dengue activities
Dengue fever is a mosquito‐borne infectious disease caused by dengue virus (DENV). DENV is a membrane‐enveloped positive‐strand RNA virus that also belongs to Flaviviridae. Currently, there is no specific dengue treatment available. DENV RNA polymerase is one of suitable targets for development of anti‐dengue compounds. Incorporation of unnatural nucleotides into viral RNA could inhibit viral RNA replication and/or hamper function of the viral RNA. Data from Section 4.2 is summarized in Table 4. 6‐Methyl‐7‐deazapurine ribonucleoside (35a) showed potent anti‐dengue activity both in a replicon assay (EC50 = 0.877 μM) and in an infectivity assay.128 Unfortunately, cytotoxicities against host cells were not reported in this study, however, 6‐methyl‐7‐deazapurine ribonucleoside (35a) was shown to be highly cytotoxic towards human fibroblasts107 and therefore anti‐dengue activity caused by unspecific cytotoxicity of the compound 35a cannot be ruled out. On the other hand, 7‐deaza‐2′‐C‐methyladenosine (60a) was able to reduce viral loads in a mouse dengue fever viremia model but its mechanism of action against DENV has not been studied.129 Replacement of the 2′‐C‐methyl group by ethynyl group led to derivative NITD008 (72a). NITD008 (72a) showed submicromolar activity against DENV in vitro (EC50 = 0.64 μM) and was devoid of cytotoxicity against host cells (Fig. 18).126 Further studies showed that NITD008 (72a) is phosphorylated in vivo to its NTP130 which then acts as a chain inhibitor of RNA‐dependent RNA polymerase of DENV.126, 131 NITD008 (72a) is orally available, reduced viral loads in a mouse model and protected infected animals from death but showed toxicity when treatment was longer than one week.126 Compound 72b, a 7‐fluoro analogue of NITD008, showed slightly increased anti‐dengue activity in vitro compared to NITD008 (72a) (EC50 = 0.42 μM) but also increased cytotoxicity.130 On the other hand, compounds with CN (72c) and CONH2 (72d) substitutions in position 7 showed lower anti‐dengue activities (EC50 = 3.1 and 2.0 μM, respectively).130 Compound 72d suffers from poor oral bioavailability in mice and rats.132 In order to improve its gastrointestinal absorption and cell penetration, its prodrug, compound 73, was prepared. Derivative 73 showed both better anti‐dengue activity in vitro (EC50 = 0.54 ‐ 0.71 μM, depending on DENV strain) and improved bioavailability than the parent nucleoside 72d.132 Compound 73 efficiently inhibits viral RNA synthesis and was found to be more potent than NITD008 (72a) in an in vivo mouse model. Nevertheless, in vivo toxicity in higher doses precluded further development of compound 73 as an anti‐dengue agent. Some benzo‐fused 7‐deazapurine ribonucleosides also showed submicromolar activities against DENV but in case of compounds 42 94 and 43b 94 the anti‐dengue effect was presumably caused by unspecific cytotoxicity towards host cells. Only benzofuryl derivative 74 possesses submicromolar anti‐dengue activity in vitro (EC50 = 0.976 μM) and is non‐cytotoxic at the same time.93 Compound 74 therefore represents a good lead structure for further development of compounds with anti‐dengue effect.
Table 4.
Anti‐dengue activities of 7‐deazapurine nucleosides
| Compound | EC50 [μM] (assay) | CC50 [μM] | Ref. |
|---|---|---|---|
| 35a | 0.877 (replicon) | nda | 128 |
| 42 | 0.238 (replicon) | nd | 93 |
| 43b | 0.85 (Vero cells) | 1.14 | 95 |
| 72a | 0.64; 0.7 (CFI) | nd; >100 | 126, 130 |
| 72b | 0.42 (CFI) | 44 | 130 |
| 72c | 3.1 (CFI) | >100 | 130 |
| 72d | 2.0 (CFI) | 62 | 130 |
| 73 | 0.54‐0.71 (CFI) | nd | 132 |
| 74 | 0.976 (replicon) | nd | 93 |
nd, not determined
Even though none of the 7‐deazapurine nucleosides reached clinical trials, the preclinical studies showed that inhibitors of viral RNA synthesis are able to gain in vivo antiviral effect and further development of structurally related compounds could lead to clinical candidates.
4.3. Nucleosides with antiviral activities against other viruses
7‐Deazapurine nucleosides were subjected to antiviral activity screening against other viruses than HCV and DENV. The largest structure‐activity relationship studies against both DNA and RNA viruses were mostly performed in the 1980s.31, 35, 133, 134, 135, 136, 137 Nevertheless, most of the derivatives that showed antiviral effects were also cytotoxic. Therefore, only the compounds with sufficient selectivity between antiviral activity and cytotoxicity will be mentioned here. Xylotubercidin (75) possesses potent activity against herpes simplex virus (HSV‐1 and HSV‐2) (EC50 = 0.75 and 0.26 μM, respectively) and showed in vivo anti‐HSV effect in mice, however, the therapeutic window is rather narrow because at higher doses of xylotubercidin (75) its toxicity to treated animals became apparent138 (Fig. 19). Significant activity against hepatitis B virus (HBV) was observed for fluoroderivative 76 which is able to efficiently inhibit episomal viral replication (EC50 = 2.5 μM) and was well tolerated in mice.139, 140 On the other hand, compound 76 significantly affected metabolism and morphology of mitochondria and therefore it is likely to be hepatotoxic.141 Ethynyl derivative of 7‐deazaneplanocin A 77 showed submicromolar anti‐HBV activity (EC50 = 2.5 μM) and no cytotoxicity (CC50 > 300 μM).125 Although the mechanism of action of compound 77 has not been studied yet, this derivative represents an interesting lead structure as it was also active against lamivudine‐ and adefovir‐associated HBV mutants.
Figure 19.
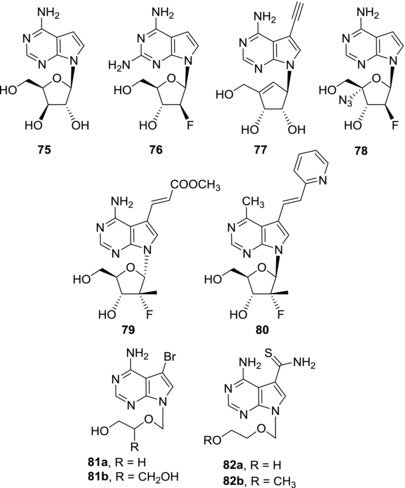
Examples of 7‐deazapurine nucleosides and acyclic nucleosides active against other viruses
Several fluorinated 7‐deazapurine nucleosides showed activity against human immunodeficiency virus (HIV). Submicromolar anti‐HIV activities and no cytotoxicity were observed for derivatives 78 143 (EC50 = 0.5 μM) and 79 49 (EC50 = 0.71 μM) indicating that this class of nucleosides might bring more interesting anti‐HIV candidates in the future. Compound 80 bearing a 7‐(pyridine‐2‐yl)vinyl group showed promising results against influenza A virus (EC50/H1N1 strain = 5.88 μM and EC50/H3N2 strain = 6.95 μM) and low toxicity (CC50 > 100 μM).143 This derivative could serve as a basis for further lead optimization studies with an aim of developing novel anti‐influenza A virus agents.
Antiviral activities of 7‐deaza‐2′‐C‐methyladenosine (MK‐0608, 60a) against HCV and DENV have been already mentioned. However, MK‐0608 (60a) was also found active against other RNA viruses such as human rhinovirus type C (HRV‐C)144, two viruses from Flaviviridae family: tick‐borne encephalitis virus (TBEV)145, 146 (EC50 = 1.1 μM) and recently also Zika virus (ZIKV). MK‐0608 (60a) showed promising anti‐ZIKV activity both in vitro147, 148 (EC50 = 8.92 μM) and in an in vivo mouse model.148 5′‐Triphosphate form of MK‐0608 (60a) was identified as an inhibitor of ZIKV‐RNA‐dependent RNA polymerase (IC50 = 7.9 μM).149 As well, NITD008 (72a) is not only a potent anti‐dengue compound but also was shown to inhibit proliferation of enterovirus 71 (EV71) (CPE50 = 0.625 μM), a causative agent of Hand‐Foot‐and‐Mouth Disease150, TBEV (CPE EC50 = 0.9 μM), and other three tick‐borne viruses from Flaviviridae family.151 Co‐treatment with NITD008 (72a) and an anti‐inflammatory drug vorinostat (SAHA), a histone deacetylase inhibitor, was successful in a mouse model of West Nile virus (WNV) infection.152 Structurally similar prodrug 73 was also found to be active against many members of the Flaviviridae family, such as yellow fever virus and WNV.132
Acyclic analogues of 7‐deazapurine nucleosides and nucleotides were also screened for antiviral activities but generally these compounds showed only poor activities, especially when compared with the corresponding purine analogues.153, 154, 155, 156 Nevertheless, some acyclic analogues of 7‐bromo‐7‐deazaadenosine (81a‐b)157, 158, 159 and thiosangivamycin (82a‐b)160, 161, 162, 163 possess selective activity against human cytomegalovirus that is comparable or better than that of ganciclovir.
In conclusion, 7‐deazapurine nucleosides have a potential to provide new structures for development of antivirals against both DNA and RNA viruses and representatives of this class of compounds should be covered in antiviral screenings of compound libraries. 7‐Deazapurine nucleosides seem in particular promising as compounds with antiviral activities against RNA viruses from Flaviviridae family.
5. CONCLUSIONS
Replacement of N7 atom by CH in purine nucleosides is a crucial structural change, which can lead to diverse biologically active nucleoside analogues. The combination of the electronic effect of the electron‐rich pyrrole ring with the possibility of attachment of an additional substituent at position 7 does not interfere with base pairing with complementary base or recognition of the adenosine moiety by enzymes and could even increase the binding due to more efficient π–π or cation–π stacking interactions (though the only proven examples of such interactions are from our recent works on DNA polymerase incorporations of 7‐aryl‐substituted 7‐deazapurine nucloetides which were found7, 8 to be better substrates for polymerases than natural dATP or dGTP due to increased cation–π stacking in the active site of the polymerase). Due to wide range of relevant biological activities, the 7‐deazapurine moiety can be regarded as a privileged scaffold in design of antitumor or antiviral nucleosides.
Several types of deazapurine nucleosides exert promising cytostatic activities and some examples are under preclinical developments. The 7‐hetaryl‐7‐deazaadenosines (AB61 and analogues) get specifically phosphorylated in the cancer cells and get incorporated both to RNA (where they cause inhibition of the proteosynthesis) and to DNA (where they cause DNA damage). Mechanisms of the other types of cytostatic hetaryl‐substituted or thieno‐fused deazapurine nucleosides are yet under study. Many 7‐deazaadenosine derivatives are inhibitors of either human or Mtb‐ADK but their antimycobacterial activities were rather moderate. Many sugar‐modified 7‐deazapurine nucleosides exert antiviral activities. In particular, 2′‐C‐methyl‐ribonucleosides and 2′‐C‐methyl‐2′‐fluororibonucleosides showed promising activities against HCV and several underwent clinical trials, although none of them made it to FDA approval so far.
There is still a lot of potential in this interesting class of compounds to investigate. Most of the derivatives were derivatives of 7‐deazaadenosine, which are easier to synthesize. Much less attention was paid to derivatives of 7‐deazaguanosine, 7‐deazainosine, or 7‐deazaisoguanine. Undoubtedly, there is also a lot of space in other types of tri‐ and perhaps even tetracyclic hetero‐fused deazapurine nucleosides and in other fused heterocycles (e.g. analogues of triciribine), but their synthesis is very challenging. Combination of a modified deazapurine base with modified sugars is also an underexplored area with some potential, especially for antiviral activities against new emerging viruses. Little is known about the inhibition of protein kinases by these compounds, as well as about their interactions with adenosine receptors. 7‐Deazapurine analogues of some nucleotide cofactors could be also of interest both as drug candidates and as tools in chemical biology. We hope that this review will ignite higher attention to this important class of molecules.
ACKNOWLEDGMENT
This work was supported by the Academy of Sciences of the Czech Republic (RVO 61388963 and the Praemium Academiae award to M. H.), by the Czech Science Foundation (16‐0011785 to M. H.) and by Gilead Sciences, Inc.
Biographies
Pavla Perlíková (née Spáčilová), PhD, received her PhD from the Charles University in Prague in 2012 and took her postdoctoral stay at the University of Southern Denmark. She is now a staff scientist at IOCB Prague. Her research interests include medicinal chemistry of modified nucleosides and nucleic acids. She (co‐)authored over 20 papers and two patents.
Michal Hocek, PhD, DSc. received his PhD from the Czech Academy of Sciences in 1996 and took his postdoctoral stay at the Université Catholique de Louvain, Belgium. He is now a senior group leader at IOCB Prague and a full professor at the Charles University in Prague. His research interests include bioorganic and medicinal chemistry of nucleosides, nucleotides and nucleic acids. He (co‐)authored over 205 papers and five patents.
Perlíková P, Hocek M. Pyrrolo[2,3‐d]pyrimidine (7‐deazapurine) as a privileged scaffold in design of antitumor and antiviral nucleosides. Med Res Rev. 2017;37:1429–1460. https://doi.org/10.1002/med.21465
REFERENCES
- 1. Morris RC, Elliott MS. Queuosine modification of tRNA: a case for convergent evolution. Mol Genet Metab. 2001;74(1‐2):147–159. [DOI] [PubMed] [Google Scholar]
- 2. Iwata‐Reuyl D. Biosynthesis of the 7‐deazaguanosine hypermodified nucleosides of transfer RNA. Bioorg Chem. 2003;31(1):24–43. [DOI] [PubMed] [Google Scholar]
- 3. Ohgi T, Kondo T, Goto T. Total Synthesis of Optically Pure Nucleoside Q. Determination of Absolute Configuration of Natural Nucleoside Q. J Am Chem Soc. 1979;101(13):3629–3633. [Google Scholar]
- 4. Gregson JM, Crain PF, Edmonds CG, et al. Structure of the Archaeal Transfer RNA Nucleoside G*‐15 (2‐Amino‐4,7‐dihydro‐4‐oxo‐7‐β‐d‐ribofuranosyl‐1H‐pyrrolo[2,3‐d]pyrimidine‐5‐carboximidamide (Archaeosine)). J Biol Chem. 1993;268(14):10076–10086. [PubMed] [Google Scholar]
- 5. Thiaville JJ, Kellner SM, Yuan Y, et al. Novel genomic island modifies DNA with 7‐deazaguanine derivatives. Proc Natl Acad Sci USA. 2016;113(11):E1452–E1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McCarty RM, Bandarian V. Biosynthesis of pyrrolopyrimidines. Bioorg Chem. 2012;43:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kielkowski P, Fanfrlík J, Hocek M. 7‐Aryl‐7‐deazaadenine 2'‐deoxyribonucleoside triphosphates (dNTPs): better substrates for DNA polymerases than dATP in competitive incorporations. Angew Chem Int Ed. 2014;53(29):7552–7555. [DOI] [PubMed] [Google Scholar]
- 8. Cahová H, Panattoni A, Kielkowski P, Fanfrlík J, Hocek M. 5‐Substituted Pyrimidine and 7‐Substituted 7‐Deazapurine dNTPs as Substrates for DNA Polymerases in Competitive Primer Extension in the Presence of Natural dNTPs. ACS Chem Biol. 2016;11(11):3165–3171. [DOI] [PubMed] [Google Scholar]
- 9. Seela F, Sirivolu VR. DNA containing side chains with terminal triple bonds: Base‐pair stability and functionalization of alkynylated pyrimidines and 7‐deazapurines. Chem Biodivers. 2006;3(5):509–514. [DOI] [PubMed] [Google Scholar]
- 10. De Coen LM HeugebaertTS, Garcia D, Stevens CV. Synthetic Entries to and Biological Activity of Pyrrolopyrimidines. Chem Rev. 2016;116(1):80–139. [DOI] [PubMed] [Google Scholar]
- 11. Merino P, ed. Chemical synthesis of nucleoside analogues. Hoboken: John Wiley & Sons; 2013. [Google Scholar]
- 12. Bourderioux A, Nauš P, Perlíková P, et al. Synthesis and significant cytostatic activity of 7‐hetaryl‐7‐deazaadenosines. J Med Chem. 2011;54(15):5498–5507. [DOI] [PubMed] [Google Scholar]
- 13. Gunic E, Girardet J‐L, Pietrzkowski Z, Esler C, Wang G. Synthesis and Cytotoxicity of 4’‐C‐ and 5’‐C‐Substituted Toyocamycins. Bioorg Med Chem. 2001;9(1):163–170. [DOI] [PubMed] [Google Scholar]
- 14. Huang B‐G, Bobek M. Synthesis and in vitro antitumor activity of some amino‐deoxy 7‐hexofuranosylpyrrolo[2,3‐d]pyrimidines. Carbohydr Res. 1998;308(3‐4):319–328. [DOI] [PubMed] [Google Scholar]
- 15. Wakao K, Watanabe T, Takadama T, et al. Sangivamycin induces apoptosis by suppressing Erk signaling in primary effusion lymphoma cells. Biochem Biophys Res Commun. 2014;444(2):135–140. [DOI] [PubMed] [Google Scholar]
- 16. Acs G, Reich E, Mori M. Biological and biochemical properties of the analogue antibiotic tubercidin. Proc Natl Acad Sci USA. 1964;52(2):493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suhadolnik RJ, Uematsu T, Uematsu H. Toyocamycin: phosphorylation and incorporation into RNA and DNA and biochemical properties of the triphosphate. Biochim Biophys Acta. 1967;146(1):41–49. [DOI] [PubMed] [Google Scholar]
- 18. Glazer RI, Hartman KD. Cytokinetic and biochemical effects of sangivamycin in human colon carcinoma cells in culture. Mol Pharmacol. 1981;20(3):657–661. [PubMed] [Google Scholar]
- 19. Glazer RI, Hartman KD. In vitro translational activity of messenger RNA following treatment of human colon carcinoma cells with sangivamycin. Mol Pharmacol. 1983;24(3):509–512. [PubMed] [Google Scholar]
- 20. Hardesty CT, Chaney NA, Waravdekar VS, Mead JAR. The disposition of the antitumor agent, sangivamycin, in mice. Cancer Res. 1974;34(5):1005–1009. [PubMed] [Google Scholar]
- 21. Kurogi Y, Matsuo Y, Mihara Y, et al. Identification of a chemical inhibitor for nuclear speckle formation: implications for the function of nuclear speckles in regulation of alternative pre‐mRNA splicing. Biochem Biophys Res Commun. 2014;446(1):119–124. [DOI] [PubMed] [Google Scholar]
- 22. Brdar B, Reich E. Biochemical and biological properties of 5‐bromotubercidin: differential effects on cellular DNA‐directed and viral RNA‐directed RNA synthesis. Bioorg Med Chem. 2008;16(3):1481–1492. [DOI] [PubMed] [Google Scholar]
- 23. Fabianovska‐Majewska K, Duley JA. Simmonds. Effects of novel anti‐viral adenosine analogues on the activity of S‐adenosylhomocysteine hydrolase from human liver. Biochem Pharmacol. 1994;48(5):897–903. [DOI] [PubMed] [Google Scholar]
- 24. Nishioka H, Sawa T, Hamada M, Shimura N, Imoto M, Umezawa K. Inhibition of phosphatidylinositol kinase by toyocamycin. J Antibiot. 1990;43(12):1586–1589. [DOI] [PubMed] [Google Scholar]
- 25. Tavitian A, Uretsky SC, Acs G. Selective inhibition of ribosomal RNA synthesis in mammalian cells. Biochim Biophys Acta. 1968;157(1):33–42. [DOI] [PubMed] [Google Scholar]
- 26. Iapalucci‐Espinoza S, Cereghini S, Franze‐Fernandez MT. Regulation of ribosomal RNA synthesis in mammalian cells: Effect of toyocamycin. Biochemistry. 1977;16(13):2885–2889. [DOI] [PubMed] [Google Scholar]
- 27. Ri M, Tashiro E, Oikawa D, et al. Identification of Toyocamycin, an agent cytotoxic for multiple myeloma cells, as a potent inhibitor of ER stress‐induced XBP1 mRNA splicing. Blood Cancer J. 2012;2(7):e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Loomis CR, Bell RM. Sangivamycin, a nucleoside analogue, is a potent inhibitor of protein kinase C. J Biol Chem. 1988;263(4):1682–1692. [PubMed] [Google Scholar]
- 29. Cheeseman MD, Westwood IM, Barbeau O, et al. Exploiting Protein Conformational Change to Optimize Adenosine‐Derived Inhibitors of HSP70. J Med Chem. 2016;59(10):4625–4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maruyama T, Wotring LL, Townsend LB. Pyrrolopyrimidine nucleosides. 18. Synthesis and chemotherapeutic activity of 4‐amino‐7‐(3‐deoxy‐β‐D‐ribofuranosyl)pyrrolo[2,3‐d]pyrimidine‐5‐carboxamide (3’‐deoxysangivamycin) and 4‐amino‐7‐(2‐deoxy‐β‐d‐ribofuranosyl)pyrrolo‐[2,3‐d]pyrimidine‐5‐carboxamide (2'‐deoxysangivamycin). J Med Chem. 1983;26(1):25–29. [DOI] [PubMed] [Google Scholar]
- 31. De Clercq E, Balzarini J, Madej D, Hansske F, Robins MJ. Nucleic acid related compounds. 51. Synthesis and biological properties of sugar‐modified analogues of the nucleoside antibiotics tubercidin, toyocamycin, sangivamycin, and formycin. J Med Chem. 1987;30(3):481–486. [DOI] [PubMed] [Google Scholar]
- 32. Bobek M, Bloch A. Relationship between the structure of sangivamycin‐derived nucleosides and their effect on leukemic cell growth and on protein kinase A and C activity. Nucleos Nucleot. 1994;13(1‐3):429–435. [Google Scholar]
- 33. Seela F, Zulauf M, Chen SF. Pyrrolo[2,3‐d]pyrimidine nucleosides: synthesis and antitumor activity of 7‐substituted 7‐deaza‐2'‐deoxyadenosines. Nucleos Nucleot Nucl. 2000;19(1‐2):237–251. [DOI] [PubMed] [Google Scholar]
- 34. Seela F, Ming X. 7‐Functionalized 7‐deazapurine β‐D and β‐L‐ribonucleosides related to tubercidin and 7‐deazainosine: glycosylation of pyrrolo[2,3‐d]pyrimidines with 1‐O‐acetyl‐2,3,5‐tri‐O‐benzoyl‐β‐D or β‐L‐ribofuranose. Tetrahedron. 2007;63(39):9850–9861. [Google Scholar]
- 35. Bergstrom DE, Brattesani AJ, Ogawa MK, et al. Antiviral Activity of C‐5 substituted tubercidin analogues. J Med Chem. 1984;27(3):285–292. [DOI] [PubMed] [Google Scholar]
- 36. Wang X, Seth PP, Ranken R, Swayze EE, Migawa MT. Synthesis and biological activity of 5‐fluorotubercidin. Nucleos Nucleot Nucl. 2004;23(1‐2):161–170. [DOI] [PubMed] [Google Scholar]
- 37. Davies LP, Jamieson DD, Baird‐Lambert JA, Kazlauskas R. Halogenated pyrrolopyrimidine analogues of adenosine from marine organisms: pharmacological activities and potent inhibition of adenosine kinase. Biochem Pharmacol. 1984;33(3):347–355. [DOI] [PubMed] [Google Scholar]
- 38. Mauchle U, Selvarajah GT, Mol JA, Kirpensteijn J, Verheije MH. Identification of anti‐proliferative kinase inhibitors as potential therapeutic agents to treat canine osteosarcoma. Vet J. 2015;205(2):281–287. [DOI] [PubMed] [Google Scholar]
- 39. Zhang X, Jia D, Liu H, et al. Identification of 5‐Iodotubercidin as a genotoxic drug with anti‐cancer potential. PLoS One. 2013;8(5):e62527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schram KH, Townsend LB. Pyrrolopyrimidine nucleosides VIII. Synthesis of sangivamycin derivatives possessing exocyclic heterocycles at C5. J Carb Nucleos Nucl. 1974;1(1):39–54. [Google Scholar]
- 41. Klečka M, Poštová Slavětínská L, Tloušťová E, Džubák P, Hajdúch M, Hocek M. Synthesis and cytostatic activity of 7‐arylsulfanyl‐7‐deazapurine bases and ribonucleosides. Med Chem Commun. 2015;6(4):576–580. [Google Scholar]
- 42. Perlíková P, Rylová G, Nauš P, et al. 7‐(2‐thienyl)‐7‐Deazaadenosine (AB61), a new potent nucleoside cytostatic with a complex mode of action. Mol Cancer Ther. 2016;15(5):922–937. [DOI] [PubMed] [Google Scholar]
- 43. Liyanage SU, Hurren R, Voisin V, et al. Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML. Blood. 2017;129(19):2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nauš P, Caletková O, Konečný P, et al. Synthesis, cytostatic, antimicrobial, and anti‐HCV activity of 6‐substituted 7‐(het)aryl‐7‐deazapurine ribonucleosides. J Med Chem. 2014;57(3):1097–1110. [DOI] [PubMed] [Google Scholar]
- 45. Zhang L, Zhang Y, Li X, Zhang L. Study on the synthesis and PKA‐I binding activities of 5‐alkynyl tubercidin analogues. Bioorg Med Chem. 2002;10(4):907–912. [DOI] [PubMed] [Google Scholar]
- 46. Nauš P, Perlíková P, Bourderioux A, et al. Sugar‐modified derivatives of cytostatic 7‐(het)aryl‐7‐deazaadenosines: 2'‐C‐methylribonucleosides, 2'‐deoxy‐2'‐fluoroarabinonucleosides, arabinonucleosides and 2'‐deoxyribonucleosides. Bioorg Med Chem. 2012;20(17):5202–5214. [DOI] [PubMed] [Google Scholar]
- 47. Perlíková P, Eberlin L, Ménova P, et al. Synthesis and cytostatic and antiviral activities of 2'‐deoxy‐2',2'‐difluororibo‐ and 2'‐deoxy‐2'‐fluororibonucleosides derived from 7‐(Het)aryl‐7‐deazaadenines. Chem Med Chem. 2013;8(5):832–846. [DOI] [PubMed] [Google Scholar]
- 48. Shi J, Zhou L, Zhang H, et al. Synthesis and antiviral activity of 2'‐deoxy‐2'‐fluoro‐2'‐C‐methyl‐7‐deazapurine nucleosides, their phosphoramidate prodrugs and 5'‐triphosphates. Bioorg Med Chem Lett. 2011;21(23):7094–7098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nauš P, Caletková O, Perlíková P, et al. Synthesis and biological profiling of 6‐ or 7‐(het)aryl‐7‐deazapurine 4'‐C‐methylribonucleosides. Bioorg Med Chem. 2015;23(23):7422–7438. [DOI] [PubMed] [Google Scholar]
- 50. Basavapathruni A, Jin L, Daigle SR, et al. Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem Biol Drug Des. 2012;80(6):971–980. [DOI] [PubMed] [Google Scholar]
- 51. Daigle SR, Olhava EJ, Therkelsen CA, et al. Selective killing of mixed lineage leukemia cells by a potent small‐molecule DOT1L inhibitor. Cancer Cell. 2011;20(1):53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yu W, Chory EJ, Wernimont AK, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3:1288. [DOI] [PubMed] [Google Scholar]
- 53. Dolloff NG, Allen JE, Dicker DT, et al. Sangivamycin‐like molecule 6 exhibits potent anti‐multiple myeloma activity through inhibition of cyclin‐dependent kinase‐9. Mol Cancer Ther. 2012;11(11):2321–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bhat UG, Gartel AL. Differential sensitivity of human colon cancer cell lines to the nucleoside analogs ARC and DRB. Int J Cancer. 2008;122(6):1426–1429. [DOI] [PubMed] [Google Scholar]
- 55. Bhat UG, Zipfel PA, Tyler DS, Gartel AL. Novel anticancer compounds induce apoptosis in melanoma cells. Cell Cycle. 2008;7(12):1851–1855. [DOI] [PubMed] [Google Scholar]
- 56. Radhakrishnan SK, Halasi M, Bhat UG, Kurmasheva RT, Houghton PJ, Gartel AL. Proapoptotic compound ARC targets Akt and N‐myc in neuroblastoma cells. Oncogene. 2008;27(5):694–699. [DOI] [PubMed] [Google Scholar]
- 57. Oltersdorf T, Elmore SW, Shoemaker AR, et al. An inhibitor of Bcl‐2 family proteins induces regression of solid tumors. Nature. 2005;435(7042):677–681. [DOI] [PubMed] [Google Scholar]
- 58. Bhat UG, Pandit B, Gartel AL. ARC synergizes with ABT‐737 to induce apoptosis in human cancer cells. Mol Cancer Ther. 2010;9(6):1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Radhakrishnan SK, Gartel AL. A novel transcriptional inhibitor induces apoptosis in tumor cells and exhibits antiangiogenic activity. Cancer Res. 2006;66(6):3264–3270. [DOI] [PubMed] [Google Scholar]
- 60. Stockwin LH, Yu SX, Stotler H, Hollingshead MG, Newton DL. ARC (NSC 188491) has identical activity to Sangivamycin (NSC 65346) including inhibition of both P‐TEFb and PKC. BMC Cancer. 2009;9:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Powers MV, Clarke PA, Workman P. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor‐specific apoptosis. Cancer Cell. 2008;14(3):250–262. [DOI] [PubMed] [Google Scholar]
- 62. Ham YM, Choi KJ, Song SY, Jin YH, Chun MW, Lee SK. Xylocydine, a novel inhibitor of cyclin‐dependent kinases, prevents the tumor necrosis factor‐related apoptosis‐inducing ligand‐induced apoptotic cell death of SK‐HEP‐1 cells. J Pharmacol Exp Ther. 2004;308(3):814–819. [DOI] [PubMed] [Google Scholar]
- 63. Cho SJ, Lee SS, Kim YJ, et al. Xylocydine, a novel Cdk inhibitor, is an effective inducer of apoptosis in hepatocellular carcinoma cells in vitro and in vivo. Cancer Lett. 2010;287(2):196–206. [DOI] [PubMed] [Google Scholar]
- 64. Sun C, Guo XX, Zhu D, et al. Apoptosis is induced in cancer cells via the mitochondrial pathway by the novel xylocydine‐derived compound JRS‐15. Int J Mol Sci. 2013;14(1):850–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Choi BY, Lee CH. Cell cycle arrest and cytochrome c‐mediated apoptotic induction by MCS‐5A is associated with up‐regulation of p16(INK4a) in HL‐60 cells. Bioorg Med Chem Lett. 2010;20(13):3880–3884. [DOI] [PubMed] [Google Scholar]
- 66. Cho SJ, Kim YJ, Surh YJ, Kim BM, Lee SK. Ibulocydine is a novel prodrug Cdk inhibitor that effectively induces apoptosis in hepatocellular carcinoma cells. J Biol Chem. 2011;286(22):19662–19671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Park SS, Jwa E, Shin SH, et al. Ibulocydine sensitizes human hepatocellular carcinoma cells to TRAIL‐induced apoptosis via calpain‐mediated Bax cleavage. Int J Biochem Cell Biol. 2017;83:47–55. [DOI] [PubMed] [Google Scholar]
- 68. Park SS, Kim YJ, Ju EJ, et al. Ibulocydine sensitizes human cancers to radiotherapy by induction of mitochondria‐mediated apoptosis. Radiother Oncol. 2014;112(2):295–301. [DOI] [PubMed] [Google Scholar]
- 69. Xiao C, Sun C, Han W, et al. Synthesis of 6‐(het) ary Xylocydine analogues and evaluating their inhibitory activities of CDK1 and CDK2 in vitro. Bioorg Med Chem. 2011;19(23):7100–7110. [DOI] [PubMed] [Google Scholar]
- 70. Lee J, Seo H, Yoon S, Choi K, Lee C‐H, Rhee H. The synthesis and evaluation of new carbocyclic pyrrolo[2,3‐d]pyrimidine nucleoside analogs. Heterocycles. 2014;89(7):1606–1619. [Google Scholar]
- 71. Nauš P, Pohl R, Votruba I, et al. 6‐(Het)aryl‐7‐deazapurine ribonucleosides as novel potent cytostatic agents. J Med Chem. 2010;53(1):460–470. [DOI] [PubMed] [Google Scholar]
- 72. Spáčilová P, Nauš P, Pohl R, et al. CycloSal‐phosphate pronucleotides of cytostatic 6‐(Het)aryl‐7‐deazapurine ribonucleosides: Synthesis, cytostatic activity, and inhibition of adenosine kinases. Chem Med Chem. 2010;5(8):1386–1396. [DOI] [PubMed] [Google Scholar]
- 73. Perlíková P, Pohl R, Votruba I, et al. Phosphoramidate pronucleotides of cytostatic 6‐aryl‐7‐deazapurine ribonucleosides. Bioorg Med Chem. 2011;19(1):229–242. [DOI] [PubMed] [Google Scholar]
- 74. Malnuit V, Poštová Slavětínská L, Nauš P, et al. 2‐Substituted 6‐(Het)aryl‐7‐deazapurine ribonucleosides: synthesis, inhibition of adenosine kinases, and antimycobacterial activity. Chem Med Chem. 2015;10(6):1079–1093. [DOI] [PubMed] [Google Scholar]
- 75. Nauš P, Perlíková P, Pohl R, Hocek M. Sugar‐modified derivatives of cytostatic 6‐(het)aryl‐7‐deazapurine nucleosides: 2'‐C‐methylribonucleosides, arabinonucleosides, and 2'‐deoxy‐2'‐fluoroarabinonucleosides. Collect Czech Chem Commun. 2011;76(8):957–988. [Google Scholar]
- 76. Perlíková P, Jornet Martínez N, Slavětínská L, Hocek M. Synthesis of 2'‐deoxy‐2'‐fluororibo‐ and 2'‐deoxy‐2',2'‐difluororibonucleosides derived from 6‐(het)aryl‐7‐deazapurines. Tetrahedron. 2012;68(39):8300–8310. [Google Scholar]
- 77. Drexler J, Groth U. Trifluoromethylated nucleosides: A building block approach to cytotoxic adenosine analogues. Eur J Org Chem. 2014;2014(28):6314–6320. [Google Scholar]
- 78. Rayala R, Theard P, Ortiz H, et al. Synthesis of purine and 7‐deazapurine nucleoside analogues of 6‐N‐(4‐Nitrobenzyl)adenosine; inhibition of nucleoside transport and proliferation of cancer cells. Chem Med Chem. 2014;9(9):2186–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. D'Errico S, Oliviero G, Borbone N, et al. Synthesis and evaluation of the antiproliferative properties of a tethered tubercidin‐platinum(II) complex. Eur J Org Chem. 2015;2015(34):7550–7556. [Google Scholar]
- 80. Soucy TA, Smith PG, Milhollen MA, et al. An inhibitor of NEDD8‐activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732–736. [DOI] [PubMed] [Google Scholar]
- 81. Swords RT, Erba HP, DeAngelo DJ, et al. Pevonedistat (MLN4924), a first‐in‐class NEDD8‐activating enzyme inhibitor, in patients with acute myeloid leukaemia and myelodysplastic syndromes: a phase 1 study. Br J Haematol. 2015;169(4):534–543. [DOI] [PubMed] [Google Scholar]
- 82. Shah JJ, Jakubowiak AJ, O'Connor OA, et al. Phase I study of the novel investigational NEDD8‐activating enzyme inhibitor pevonedistat (MLN4924) in patients with relapsed/refractory multiple myeloma or lymphoma. Clin Cancer Res. 2016;22(1):34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bhatia S, Pavlick AC, Boasberg P, et al. A phase I study of the investigational NEDD8‐activating enzyme inhibitor pevonedistat (TAK‐924/MLN4924) in patients with metastatic melanoma. Invest New Drugs. 2016;34(4):439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sarantopoulos J, Shapiro GI, Cohen RB, et al. Phase I study of the investigational NEDD8‐activating enzyme inhibitor pevonedistat (TAK‐924/MLN4924) in patients with advanced solid tumors. Clin Cancer Res. 2016;22(4):847–857. [DOI] [PubMed] [Google Scholar]
- 85. Schram KH, Townsend LB. The synthesis of 6‐amino‐4‐methyl‐8‐(β‐d‐ribofuranosyl)(4‐H,8‐H)pyrrolo[4,3,2‐d]pyrimido[4,5‐c]pyridazine, a new tricyclic nucleoside. Tetrahedron Lett. 1971;12(49):4757–4760. [Google Scholar]
- 86. Porcari AR, Townsend LB. An improved total synthesis of triciribine: a tricyclic nucleoside with antineoplastic and antiviral properties. Nucleos Nucleot Nucl. 2004;23(1‐2):31–39. [DOI] [PubMed] [Google Scholar]
- 87. Shen W, Kim J‐S, Hilfinger J. Expedient Total Synthesis of Triciribine and Its Prodrugs. Synthetic Commun. 2012;42(3):358–374. [Google Scholar]
- 88. Yang L, Dan HC, Sun M, et al. Akt/protein kinase B signaling inhibitor‐2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004;64(13):6394–6399. [DOI] [PubMed] [Google Scholar]
- 89. Kim R, Yamauchi T, Husain K, Sebti S, Malafa M. Triciribine phosphate monohydrate, an Akt inhibitor, enhances gemcitabine activity in pancreatic cancer cells. Anticancer Res. 2015;35(9):4599–4604. [PubMed] [Google Scholar]
- 90. Balasis ME, Forinash KD, Chen YA, et al. Combination of farnesyl transferase and Akt inhibitors is synergistic in breast cancer cells and causes significant breast tumor regression in ErbB2 transgenic mice. Clin Cancer Res. 2011;17(9):2852–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhou L, Amblard F, Zhang H, et al. Synthesis and evaluation of Janus type nucleosides as potential HCV NS5B polymerase inhibitors. Bioorg Med Chem Lett. 2013;23(11):3385–3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chung FL, Schram KH, Panzica RP, Earl RA, Wotring LL, Townsend LB. Synthesis of certain [6:5:6] linear tricyclic nucleosides as potential antitumor agents. J Med Chem. 1980;23(11):1158–1166. [DOI] [PubMed] [Google Scholar]
- 93. Tichý M, Pohl R, Xu HY, et al. Synthesis and antiviral activity of 4,6‐disubstituted pyrimido[4,5‐b]indole ribonucleosides. Bioorg Med Chem. 2012;20(20):6123–6133. [DOI] [PubMed] [Google Scholar]
- 94. Tichý M, Pohl R, Tloušťová E, et al. Synthesis and biological activity of benzo‐fused 7‐deazaadenosine analogues. 5‐ And 6‐substituted 4‐amino‐ or 4‐alkylpyrimido[4,5‐b]indole ribonucleosides. Bioorg Med Chem. 2013;21(17):5362–5372. [DOI] [PubMed] [Google Scholar]
- 95. Tichý M, Smoleń S, Tloušťová E, et al. Synthesis and cytostatic and antiviral profiling of thieno‐fused 7‐deazapurine ribonucleosides. J Med Chem. 2017;60(6):2411–2424. [DOI] [PubMed] [Google Scholar]
- 96. Wiesner JB, Ugarkar BG, Castellino AJ, et al. Adenosine kinase inhibitors as a novel approach to anticonvulsant therapy. J Pharmacol Exp Ther. 1999;289(3):1669–1677. [PubMed] [Google Scholar]
- 97. Ugarkar BG, DaRe JM, Kopcho JJ, et al. Adenosine kinase inhibitors. 1. Synthesis, enzyme inhibition, and antiseizure activity of 5‐iodotubercidin analogues. J Med Chem. 2000;43(15):2883–2893. [DOI] [PubMed] [Google Scholar]
- 98. Davies LP, Baird‐Lambert J, Marwood JF. Studies on several pyrrolo[2,3‐d]pyrimidine analogues of adenosine which lack significant agonist activity at A1 and A2 receptors but have potent pharmacological activity in vivo. Biochem Pharmacol. 1986;35(18):3021–3029. [DOI] [PubMed] [Google Scholar]
- 99. Jarvis MF, Yu H, McGaraughty S, et al. Analgesic and anti‐inflammatory effects of A‐286501, a novel orally active adenosine kinase inhibitor. Pain. 2002;96(1‐2):107–118. [DOI] [PubMed] [Google Scholar]
- 100. Snášel J, Nauš P, Dostál J, et al. Structural basis for inhibition of mycobacterial and human adenosine kinase by 7‐substituted 7‐(Het)aryl‐7‐deazaadenine ribonucleosides. J Med Chem. 2014;57(20):8268–8279. [DOI] [PubMed] [Google Scholar]
- 101. Ugarkar BG, Castellino AJ, DaRe JM, et al. Adenosine kinase inhibitors. 2. Synthesis, enzyme inhibition, and antiseizure activity of diaryltubercidin analogues. J Med Chem. 2000;43(15):2894–2905. [DOI] [PubMed] [Google Scholar]
- 102. Ugarkar BG, Castellino AJ, DaRe JS, et al. Adenosine kinase inhibitors. 3. Synthesis, SAR, and anti‐inflammatory activity of a series of l‐lyxofuranosyl nucleosides. J Med Chem. 2003;46(22):4750–4760. [DOI] [PubMed] [Google Scholar]
- 103. Boyer SH, Ugarkar BG, Solbach J, et al. Adenosine kinase inhibitors. 5. Synthesis, enzyme inhibition, and analgesic activity of diaryl‐erythro‐furanosyltubercidin analogues. J Med Chem. 2005;48(20):6430–6441. [DOI] [PubMed] [Google Scholar]
- 104. Toti KS, Osborne D, Ciancetta A, Boison D, Jacobson KA. South (S)‐ and north (N)‐methanocarba‐7‐deazaadenosine analogues as inhibitors of human adenosine kinase. J Med Chem. 2016;59(14):6860–6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Bookser BC, Ugarkar BG, Matelich MC, et al. Adenosine kinase inhibitors. 6. Synthesis, water solubility, and antinociceptive activity of 5‐phenyl‐7‐(5‐deoxy‐β‐D‐ribofuranosyl)pyrrolo[2,3‐d]pyrimidines substituted at C4 with glycinamides and related compounds. J Med Chem. 2005;48(24):7808–7820. [DOI] [PubMed] [Google Scholar]
- 106. Parker WB, Long MC. Purine metabolism in Mycobacterium tuberculosis as a target for drug development. Curr Pharm Des. 2007;13(6):599–608. [DOI] [PubMed] [Google Scholar]
- 107. Perlíková P, Konečný P, Nauš P, et al. 6‐Alkyl‐, 6‐aryl‐ or 6‐hetaryl‐7‐deazapurine ribonucleosides as inhibitors of human or MTB adenosine kinase and potential antimycobacterial agents. Med Chem Comm. 2013;4(11):1497–1500. [Google Scholar]
- 108. Varaprasad CV, Ramasamy KS, Girardet JL, et al. Synthesis of pyrrolo[2,3‐d]pyrimidine nucleoside derivatives as potential anti‐HCV agents. Bioorg Chem. 2007;35(1):25–34. [DOI] [PubMed] [Google Scholar]
- 109. Nekhai S, Bhat UG, Ammosova T, et al. A novel anticancer agent ARC antagonizes HIV‐1 and HCV. Oncogene. 2007;26(26):3899–3903. [DOI] [PubMed] [Google Scholar]
- 110. Smith KL, Lai VC, Prigaro BJ, et al. Synthesis of new 2'‐beta‐C‐methyl related triciribine analogues as anti‐HCV agents. Bioorg Med Chem Lett. 2004;14(13):3517–3520. [DOI] [PubMed] [Google Scholar]
- 111. Olsen DB, Eldrup AB, Bartholomew L, et al. A 7‐deaza‐adenosine analog is a potent and selective inhibitor of hepatitis C virus replication with excellent pharmacokinetic properties. Antimicrob Agents Chemother. 2004;48(10):3944–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Seela F, Budow S, Xu K, Peng X, Eickmeier H. 7‐Halogenated 7‐deazapurine 2′‐C‐methylribonucleosides. Collect Czech Chem Commun. 2011;76(12):1413–1431. [Google Scholar]
- 113. Carroll SS, Ludmerer S, Handt L, et al. Robust antiviral efficacy upon administration of a nucleoside analog to hepatitis C virus‐infected chimpanzees. Antimicrob Agents Chemother. 2009;53(3):926–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Olsen DB, Davies ME, Handt L, et al. Sustained viral response in a hepatitis C virus‐infected chimpanzee via a combination of direct‐acting antiviral agents. Antimicrob Agents Chemother. 2011;55(2):937–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ohara E, Hiraga N, Imamura M, et al. Elimination of hepatitis C virus by short term NS3‐4A and NS5B inhibitor combination therapy in human hepatocyte chimeric mice. J Hepatol. 2011;54(5):872–878. [DOI] [PubMed] [Google Scholar]
- 116. Eldrup AB, Prhavc M, Brooks J, et al. Structure activity relationship of heterobase‐modified 2'‐C‐methyl ribonucleosides as inhibitors of hepatitis C virus RNA replication. J Med Chem. 2004;47(21):5284–5297. [DOI] [PubMed] [Google Scholar]
- 117. Prhavc M, Dyatkina N, Keicher J, et al. Synthesis and biological activity of 7‐deaza‐7‐ethynyl‐2'‐deoxy‐2'‐fluoro‐2'‐C‐methyladenosine and its 2'‐C‐methyl‐ribo analogue. Nucleic Acids Symp Ser (Oxf). 2008(52):643‐644. [DOI] [PubMed] [Google Scholar]
- 118. Bourdin C, McGuigan C, Brancale A, et al. Synthesis and evaluation against hepatitis C virus of 7‐deaza analogues of 2'‐C‐methyl‐6‐O‐methyl guanosine nucleoside and l‐Alanine ester phosphoramidates. Bioorg Med Chem Lett. 2013;23(7):2260–2264. [DOI] [PubMed] [Google Scholar]
- 119. Prakash TP, Prhavc M, Eldrup AB, et al. Synthesis and evaluation of S‐acyl‐2‐thioethyl esters of modified nucleoside 5'‐monophosphates as inhibitors of hepatitis C virus RNA replication. J Med Chem. 2005;48(4):1199–1210. [DOI] [PubMed] [Google Scholar]
- 120. Di Francesco ME, Avolio S, Pompei M, et al. Synthesis and antiviral properties of novel 7‐heterocyclic substituted 7‐deaza‐adenine nucleoside inhibitors of Hepatitis C NS5B polymerase. Bioorg Med Chem. 2012;20(15):4801–4811. [DOI] [PubMed] [Google Scholar]
- 121. Di Francesco ME, Avolio S, Dessole G, et al. Synthesis and antiviral properties of novel tetracyclic nucleoside inhibitors of hepatitis C NS5B polymerase. Nucleos Nucleot Nucl. 2012;31(8):592–607. [DOI] [PubMed] [Google Scholar]
- 122. Sofia MJ, Bao D, Chang W, et al. Discovery of a β‐d‐2'‐deoxy‐2'‐α‐fluoro‐2'‐β‐C‐methyluridine nucleotide prodrug (PSI‐7977) for the treatment of hepatitis C virus. J Med Chem. 2010;53(19):7202–7218. [DOI] [PubMed] [Google Scholar]
- 123. Appleby TC, Perry JK, Murakami E, et al. Viral replication. Structural basis for RNA replication by the hepatitis C virus polymerase. Science. 2015;347(6223):771–775. [DOI] [PubMed] [Google Scholar]
- 124. Hu W, Wang P, Song C, et al. Synthesis and anti‐HCV activity of a new 2'‐deoxy‐2'‐fluoro‐2'‐C‐methyl nucleoside analogue. Bioorg Med Chem Lett. 2010;20(24):7297–7298. [DOI] [PubMed] [Google Scholar]
- 125. Kim H‐J, Sharon A, Bal C, et al. Synthesis, anti‐hepatitis B virus and anti‐hepatitis C virus activities of 7‐deazaneplanocin A analogues in vitro. J Med Chem. 2009;52(1):206–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Yin Z, Chen YL, Schul W, et al. An adenosine nucleoside inhibitor of dengue virus. Proc Natl Acad Sci USA. 2009;106(48):20435–20439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Arnold JJ, Sharma SD, Feng JY, et al. Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleosides. PLoS Pathog. 2012;8(11):e1003030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Wu R, Smidansky ED, Oh HS, et al. Synthesis of a 6‐methyl‐7‐deaza analogue of adenosine that potently inhibits replication of polio and dengue viruses. J Med Chem. 2010;53(22):7958–7966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Schul W, Liu W, Xu HY, Flamand M, Vasudevan SG. A dengue fever viremia model in mice shows reduction in viral replication and suppression of the inflammatory response after treatment with antiviral drugs. J Infect Dis. 2007;195(5):665–674. [DOI] [PubMed] [Google Scholar]
- 130. Chen YL, Yin Z, Duraiswamy J, et al. Inhibition of dengue virus RNA synthesis by an adenosine nucleoside. Antimicrob Agents Chemother. 2010;54(7):2932–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Latour DR, Jekle A, Javanbakht H, et al. Biochemical characterization of the inhibition of the dengue virus RNA polymerase by β‐D‐2'‐ethynyl‐7‐deaza‐adenosine triphosphate. Antiviral Res. 2010;87(2):213–222. [DOI] [PubMed] [Google Scholar]
- 132. Chen YL, Yin Z, Lakshminarayana SB, et al. Inhibition of dengue virus by an ester prodrug of an adenosine analog. Antimicrob Agents Chemother. 2010;54(8):3255–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Cottam HB, Kazimierczuk Z, Geary S, McKernan PA, Revankar GR, Robins RK. Synthesis and biological activity of certain 6‐substituted and 2,6‐disubstituted 2'‐deoxytubercidins prepared via the stereospecific sodium salt glycosylation procedure. J Med Chem. 1985;28(10):1461–1467. [DOI] [PubMed] [Google Scholar]
- 134. De Clercq E, Bernaerts R, Bergstrom DE, Robins MJ, Montgomery JA, Holy A. Anti‐rhino virus activity of purine nucleoside analogs. Antimicrob Agents Chemother. 1986;29(3):482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Smee DF, McKernan PA, Alaghamandan HA, et al. Antiviral activities of 2'‐deoxyribofuranosyl and arabinofuranosyl analogs of sangivamycin against retro‐ and DNA viruses. Antiviral Res. 1988;10(6):263–277. [DOI] [PubMed] [Google Scholar]
- 136. Krawczyk SH, Renau TE, Nassiri MR, et al. Synthesis and evaluation of certain thiosangivamycin analogs as potential inhibitors of cell proliferation and human cytomegalovirus. J Med Chem. 1995;38(20):4115–4119. [DOI] [PubMed] [Google Scholar]
- 137. Seela F, Peng X. Pyrrolo[2,3‐d]pyrimidine β‐L‐nucleosides containing 7‐deazaadenine, 2‐amino‐7‐deazaadenine, 7‐deazaguanine, 7‐deazaisoguanine, and 7‐deazaxanthine. Collect Czech Chem Commun. 2006;71(7):956–977. [Google Scholar]
- 138. De Clercq E, Robins MJ. Xylotubercidin against herpes simplex virus type 2 in mice. Antimicrob Agents Chemother. 1986;30(5):719–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Bhattacharya BK, Ojwang JO, Rando RF, Huffman JH, Revankar GR. Synthesis and anti‐DNA viral activities in vitro of certain 2,4‐disubstituted‐7‐(2‐deoxy‐2‐fluoro‐β‐d‐arabinofuranosyl)pyrrolo[2,3‐d]pyrimidine nucleosides. J Med Chem. 1995;38(20):3957–3966. [DOI] [PubMed] [Google Scholar]
- 140. Ojwang JO, Bhattacharya BK, Marshall HB, Korba BE, Revankar GR, Rando RF. Inhibition of episomal hepatitis B virus DNA in vitro by 2,4‐diamino‐7‐(2‐deoxy‐2‐fluoro‐β‐D‐arabinofuranosyl)‐pyrrolo[2,3‐d]pyrimidine. Antimicrob Agents Chemother. 1995;39(11):2570–2573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Cui L, Schinazi RF, Gosselin G, et al. Effect of β‐enantiomeric and racemic nucleoside analogues on mitochondrial functions in HepG2 cells. Implications for predicting drug hepatotoxicity. Biochem Pharmacol. 1996;52(10):1577–1584. [DOI] [PubMed] [Google Scholar]
- 142. Guo X, Li Y, Tao L, et al. Synthesis and anti‐HIV‐1 activity of 4‐substituted‐7‐(2'‐deoxy‐2'‐fluoro‐4'‐azido‐β‐d‐ribofuranosyl)pyrrolo[2,3‐d]pyrimidine analogues. Bioorg Med Chem Lett. 2011;21(22):6770–6772. [DOI] [PubMed] [Google Scholar]
- 143. Lin C, Sun C, Liu X, et al. Design, synthesis, and in vitro biological evaluation of novel 6‐methyl‐7‐substituted‐7‐deaza purine nucleoside analogs as anti‐influenza A agents. Antiviral Res. 2016;129:13–20. [DOI] [PubMed] [Google Scholar]
- 144. Mello C, Aguayo E, Rodriguez M, et al. Multiple classes of antiviral agents exhibit in vitro activity against human rhinovirus type C. Antimicrob Agents Chemother. 2014;58(3):1546–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Eyer L, Valdés JJ, Gil VA, et al. Nucleoside inhibitors of tick‐borne encephalitis virus. Antimicrob Agents Chemother. 2015;59(9):5483–5493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Eyer L, Šmídková M, Nencka R, et al. Structure‐activity relationships of nucleoside analogues for inhibition of tick‐borne encephalitis virus. Antiviral Res. 2016;133:119–129. [DOI] [PubMed] [Google Scholar]
- 147. Eyer L, Nencka R, Huvarova I, et al. Nucleoside inhibitors of Zika virus. J Infect Dis. 2016;214(5):707–711. [DOI] [PubMed] [Google Scholar]
- 148. Zmurko J, Marques RE, Schols D, Verbeken E, Kaptein SJ, Neyts J. The viral polymerase inhibitor 7‐deaza‐2'‐C‐methyladenosine is a potent inhibitor of in vitro Zika virus replication and delays disease progression in a robust mouse infection model. PLoS Negl Trop Dis. 2016;10(5):e0004695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Hercík K, Kozak J, Šála M, et al. Adenosine triphosphate analogs can efficiently inhibit the Zika virus RNA‐dependent RNA polymerase. Antiviral Res. 2017;137:131–133. [DOI] [PubMed] [Google Scholar]
- 150. Shang L, Wang Y, Qing J, et al. An adenosine nucleoside analogue NITD008 inhibits EV71 proliferation. Antiviral Res. 2014;112:47–58. [DOI] [PubMed] [Google Scholar]
- 151. Lo MK, Shi PY, Chen YL, Flint M, Spiropoulou CF. In vitro antiviral activity of adenosine analog NITD008 against tick‐borne flaviviruses. Antiviral Res. 2016;130:46–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Nelson J, Roe K, Orillo B, Shi PY, Verma S. Combined treatment of adenosine nucleoside inhibitor NITD008 and histone deacetylase inhibitor vorinostat represents an immunotherapy strategy to ameliorate West Nile virus infection. Antiviral Res. 2015;122:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Reymen D, Naesens L, Balzarini J, Holý A, Dvořáková H, De Clercq E. Antiviral activity of selected acyclic nucleoside analogues against human Herpes virus 6. Antiviral Res. 1995;28(4):343–357. [DOI] [PubMed] [Google Scholar]
- 154. Coen N, Duraffour S, Naesens L, et al. Evaluation of novel acyclic nucleoside phosphonates against human and animal gamma Herpes viruses revealed an altered metabolism of cyclic prodrugs upon Epstein‐Barr virus reactivation in P3HR‐1 cells. J Virol. 2013;87(22):12422–12432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Holý A, Günter J, Dvořáková H, et al. Structure‐antiviral activity relationship in the series of pyrimidine and purine N‐[2‐(2‐phosphonomethoxy)ethyl] nucleotide analogues. 1. Derivatives substituted at the carbon atoms of the base. J Med Chem. 1999;42(12):2064–2086. [DOI] [PubMed] [Google Scholar]
- 156. Saxena NK, Hagenow BM, Genzlinger G, Turk SR, Drach JC, Townsend LB. Synthesis and antiviral activity of certain 4‐substituted and 2,4‐disubstituted 7‐[(2‐hydroxyethoxy)methyl]pyrrolo[2,3‐d]pyrimidines. J Med Chem. 1988;31(8):1501–1506. [DOI] [PubMed] [Google Scholar]
- 157. Pudlo JS, Saxena NK, Nassiri MR, Turk SR, Drach JC, Townsend LB. Synthesis and antiviral activity of certain 4‐ and 4,5‐disubstituted 7‐[(2‐hydroxyethoxy)methyl]pyrrolo[2,3‐d]pyrimidines. J Med Chem. 1988;31(11):2086–2092. [DOI] [PubMed] [Google Scholar]
- 158. Pudlo JS, Nassiri MR, Kern ER, Wotring LL, Drach JC, Townsend LB. Synthesis, antiproliferative, and antiviral activity of certain 4‐substituted and 4,5‐disubstituted 7‐[(1,3‐dihydroxy‐2‐propoxy)methyl]pyrrolo[2,3‐d]pyrimidines. J Med Chem. 1990;33(7):1984–1992. [DOI] [PubMed] [Google Scholar]
- 159. Nassiri MR, Turk SR, Birch GM, et al. Activity of acyclic halogenated tubercidin analogs against human cytomegalovirus and in uninfected cells. Antiviral Res. 1991;16(2):135–150. [DOI] [PubMed] [Google Scholar]
- 160. Gupta PK, Nassiri MR, Coleman LA, Wotring LL, Drach JC, Townsend LB. Synthesis, cytotoxicity, and antiviral activity of certain 7‐[(2‐hydroxyethoxy)methyl]pyrrolo[2,3‐d]pyrimidine nucleosides related to toyocamycin and sangivamycin. J Med Chem. 1989;32(7):1420–1425. [DOI] [PubMed] [Google Scholar]
- 161. Renau TE, Wotring LL, Drach JC, Townsend LB. Synthesis of non‐nucleoside analogs of toyocamycin, sangivamycin, and thiosangivamycin: influence of various 7‐substituents on antiviral activity. J Med Chem. 1996;39(4):873–880. [DOI] [PubMed] [Google Scholar]
- 162. Renau TE, Nassiri MR, Swayze EE, Kern ER, Townsend LB, Drach JC. Improved synthesis and biological evaluation of an acyclic thiosangivamycin active against human cytomegalovirus. Antiviral Res. 1992;19(1):15–28. [DOI] [PubMed] [Google Scholar]
- 163. Jacobson JG, Renau TE, Nassiri MR, et al. Non‐nucleoside pyrrolopyrimidines with a unique mechanism of action against human cytomegalovirus. Antimicrob Agents Chemother. 1999;43(8):1888–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]