

Abstract
Transcription factor 4 (TCF4 also known as ITF2 or E2-2) is a basic helix-loop-helix (bHLH) protein associated with Pitt–Hopkins syndrome, intellectual disability, and schizophrenia (SCZ). Here, we show that TCF4-dependent transcription in cortical neurons cultured from embryonic rats of both sexes is induced by neuronal activity via soluble adenylyl cyclase and protein kinase A (PKA) signaling. PKA phosphorylates TCF4 directly and a PKA phosphorylation site in TCF4 is necessary for its transcriptional activity in cultured neurons and in the developing brain in vivo. We also demonstrate that Gadd45g (growth arrest and DNA damage inducible gamma) is a direct target of neuronal-activity-induced, TCF4-dependent transcriptional regulation and that TCF4 missense variations identified in SCZ patients alter the transcriptional activity of TCF4 in neurons. This study identifies a new role for TCF4 as a neuronal-activity-regulated transcription factor, offering a novel perspective on the association of TCF4 with cognitive disorders.
SIGNIFICANCE STATEMENT The importance of the basic helix-loop-helix transcription factor transcription factor 4 (TCF4) in the nervous system is underlined by its association with common and rare cognitive disorders. In the current study, we show that TCF4-controlled transcription in primary cortical neurons is induced by neuronal activity and protein kinase A. Our results support the hypotheses that dysregulation of neuronal-activity-dependent signaling plays a significant part in the etiology of neuropsychiatric and neurodevelopmental disorders.
Keywords: bHLH, E2-2, ITF2, neuronal activity, Pitt–Hopkins syndrome, schizophrenia
Introduction
Transcription factor 4 (TCF4, also called ITF2, SEF2, and E2-2) is a dosage-sensitive gene with emerging functions in the nervous system (Quednow et al., 2012; Sweatt, 2013; Forrest et al., 2014). It is one of the three mammalian genes coding for ubiquitous class A basic helix-loop-helix (bHLH) factors, also referred to as E-proteins, that regulate transcription via binding Ephrussi box (E-box) DNA elements (CANNTG) as homodimers or as heterodimers with tissue-specific bHLH factors (Massari and Murre, 2000). TCF4 is transcribed from multiple promoters, resulting in a repertoire of functionally different protein isoforms with distinct N termini (Sepp et al., 2011).
TCF4 knock-out mice die at birth and reduction of tcf4 expression in zebrafish leads to a general delay in embryogenesis, indicating its importance in development (Zhuang et al., 1996; Bergqvist et al., 2000; Brockschmidt et al., 2011). The correct E-protein dose is crucial for survival even after development has been completed, as demonstrated in fruit flies (Tamberg et al., 2015). The wide spatiotemporal expression pattern of TCF4 in the mammalian nervous system suggests its involvement in all stages of brain development, including proliferation, differentiation, migration, and synaptogenesis, as well as in adult brain plasticity (Quednow et al., 2014). Consistently, in vitro studies with neural cells have provided support for its role in proliferation (Chen et al., 2014; Hill et al., 2017), cell cycle arrest (Schmidt-Edelkraut et al., 2014), survival, and epithelial–mesenchymal transition and ribosome biogenesis (Forrest et al., 2013; Slomnicki et al., 2016). In vivo evidence exists for the involvement of TCF4 in cell cycle exit during postnatal neurogenesis in forebrain (Fischer et al., 2014), migration of pontine nucleus and cortical neurons (Flora et al., 2007; Chen et al., 2016), formation of prefrontal cortical minicolumns (Page et al., 2017), and neurite branching restriction in olfactory neurons (D'Rozario et al., 2016). In addition, enhanced spontaneous activity of prefrontal neurons has been demonstrated recently in rats with in utero TCF4 gain-of-function (Page et al., 2017), whereas decreased excitability of prefrontal neurons and enhanced long-term potentiation in the CA1 area of the hippocampus have been reported in rats with in utero suppression of Tcf4 and/or Tcf4 heterozygous knock-out mice, respectively (Kennedy et al., 2016; Rannals et al., 2016). Behavioral analyses of Tcf4 heterozygous knock-out mice and transgenic mice with mild Tcf4 overexpression in forebrain have revealed impaired sensorimotor gating and compromised learning and memory (Brzózka et al., 2010; Kennedy et al., 2016). Moreover, gating and cognitive functions are influenced by common TCF4 variants in humans (Quednow et al., 2014).
Mutations in one of the TCF4 alleles cause Pitt–Hopkins syndrome (PTHS), a rare congenital disorder characterized by severe mental and motor deficits, gastrointestinal problems, and autistic-like behavior (Sweatt, 2013). PTHS-associated TCF4 alleles vary from hypomorphic to dominant-negative (DN) and missense mutations are congregated in the C-terminal bHLH coding exon (Forrest et al., 2012; Sepp et al., 2012; Tamberg et al., 2015). Upstream TCF4 mutations that do not affect all alternative isoforms have been linked to mild to moderate nonsyndromic intellectual disability (Kharbanda et al., 2016; Maduro et al., 2016). Single nucleotide polymorphisms in TCF4 are among the genome-wide significant markers associated with schizophrenia (SCZ) (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014) and rare TCF4 coding variants outside of the bHLH exon have been identified in sporadic SCZ cases (Hu et al., 2014; Basmanav et al., 2015). Therefore, TCF4 is a pleiotropic gene that links common and rare cognitive disorders. One of the shared causes for different cognitive disorders, including intellectual disability, autism spectrum disorder, bipolar disorder, and SCZ, may be disruption of synaptic activity-induced signaling and transcription (West and Greenberg, 2011; Ebert and Greenberg, 2013; Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). This pathway is critical for normal brain development, its responses to external stimuli, as well as for learning and memory. Here, we show that TCF4-controlled transcription in neurons is induced by neuronal activity via the cAMP pathway and is altered by TCF4 missense variations identified in SCZ patients.
Materials and Methods
Constructs and siRNAs.
The pcDNA3.1 constructs encoding TCF4 isoforms or ASCL1, pCAG-TCF4-A−, pCAG-GFP, reporter vectors pGL4.29[luc2P/12μE5/Hygro], pGL4[luc2P/12μE5/TK/Hygro], pGL4[hRlucP/min/Hygro], and pGL4.83[hRlucP/EF1α/Puro] have been described previously (Sepp et al., 2011; Sepp et al., 2012; Page et al., 2017). For pcDNA-EF1α-TCF4-B−, pcDNA-EF1α-TCF4-A−, pcDNA-EF1α-TCF4-A−-V5/His, pcDNA-EF1α, pcDNA-SRα-ASCL1, and pcDNA-SRα the cytomegalovirus (CMV) promoter in the respective pcDNA3.1 constructs was replaced with elongation factor 1 α (EF1α) promoter from pGL4.83[hRlucP/EF1α/Puro] or synthetic SRα promoter (AY613994: 3403–4025). pACT (Promega) and pCMV-Tag2 (Agilent Technologies) vectors were used for the generation of constructs encoding for VP16- or Flag-fused N-terminal TCF4 deletants TCF4-M217(I−), TCF4-G316(NaeI), TCF4-S363(XbaI), TCF4-M430(NcoI), and TCF4-P498(PaeI), named according to the first included amino acid of TCF4-B−. C-terminally HA-tagged constitutively active and dominant negative forms of PKCα coding constructs pHACE-PKCαCAT (Addgene plasmid #21234) and pHACE-PKCαDN (Addgene plasmid #21235) have been described previously (Soh and Weinstein, 2003). Coding sequences of constitutively active S218D, S222D mutant of MEK (MAPK/ERK kinase) obtained from pBabe-Puro-MEK-DD (Addgene plasmid #15268) (Boehm et al., 2007), PKA catalytic subunit Cα obtained from pCαEV (Addgene plasmid #15310) (Uhler and McKnight, 1987) and a DN mutant of the PKA RI subunit (Clegg et al., 1987) were inserted into pQM-CMV-E2-N vector (Icosagen) behind the E2-tag. Sequences encoding constitutively active forms of CAMK2B (M1-C290) and CAMK4 (M1-K316) were PCR amplified from pWZL-Neu-Myr-Flag-CAMK2B (Addgene plasmid #20439) and pWZL-Neu-Myr-Flag-CAMK4 (Addgene plasmid #20441), respectively (Boehm et al., 2007), and inserted into pCMV-Tag2 (Stratagene) behind the Flag-tag. For pGL4.83[hRlucP/PGK1/Puro] mouse 3-phosphoglycerate kinase 1 (PGK1) promoter sequence (ChrX:103382066-103382573 according to NCBI37) was inserted into pGL4.83[hRlucP/Puro] (Promega). For pGL4.15[luc2P/Gadd45g/Hygro] human growth arrest, a DNA-damage-inducible protein 45 gamma (GADD45G) promoter sequence (chr9:92219712-92219956 according to hg19) was inserted into pGL4.15[luc2P/Hygro] (Promega). Mutagenesis of the GADD45G promoter and TCF4 was performed using complementary primers containing the respective mutation and Phusion High-Fidelity DNA polymerase (Finnzymes). pG5luc vector containing GAL4 binding sites is from Promega and pEGFP-C1 from Clontech. The shRNAs-encoding AAV cis-constructs were prepared by inserting annealed oligos into rAAV-U6-shRNA-mCherry (Lau and Bading, 2009). The shRNAs targeted the following sequences: rat TCF4 exon 12 5′-CCGGAACAGACAGTATAATG-3′, rat TCF4 exon 20 5′-CGTGCGAGAAAGGAACCTGA-3′, and scrambled 5′-CACTACCGTTGTTATAGGTG-3′. The scrambled siRNA Silencer Negative control #1 and TCF4-specific siRNAs 12 I, 12 II, 20 II (Sepp et al., 2011) and 20 I (forward: 5′-GUGCGAGAAAGGAACCUGATT-3′, reverse: 5′-UCAGGUUCCUUUCUCGCACTT-3′) were from Ambion.
Cell culture and transfections.
Human embryonic kidney HEK-293 (RRID:CVCL_0045) and HEK-293FT (RRID:CVCL_6911) cells were grown in MEM (PAA Laboratories) or DMEM (PAN-Biotech), respectively, supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 0.1 mg/ml streptomycin (all from PAA Laboratories). For transfection of HEK-293 cells, 0.375 μg of DNA and 0.75 μl of LipoD293 reagent (SignaGen) were used per well of a 48-well plate or scaled up accordingly. For cotransfections, equal amounts of pGL4.29[luc2P/12μE5/Hygro], pGL4[hRlucP/min/Hygro], and effector constructs were used. For transfection of HEK-293FT cells, 10 μg of pFlag-TCF4 or empty vector, 10 μg of pQM-CMV-E2-PKA-Cα, and 40 μg of PEI (Sigma-Aldrich) were used per 10 cm plate.
Rat hippocampal and cortical mixed neuronal cultures from Sprague Dawley rat embryonic day 22.5 (E22.5) embryos were obtained and maintained as described previously (Pruunsild et al., 2011). Animal procedures were approved by the local ethics committee. For most experiments, neuronal cultures were transfected at 6–7 d in vitro (DIV) in Neurobasal A medium (Invitrogen); for experiments with bicuculline and 4-aminopyridine (4-AP), the tranfections were performed at 10 DIV in transfection medium (Bading et al., 1993) consisting 9:1 of salt-glucose-glycine solution containing the following (in mm): 10 HEPES, pH 7.4, 114 NaCl, 26.1 NaHCO3, 5.3 KCl, 1 MgCl2, 2 CaCl2, 1 glycine, 30 glucose, and 0.5 sodium pyruvate, along with minimum Eagle's medium (with Earle's salt, without l-glutamine; Capricorn Scientific) supplemented with insulin (6.3 μg/ml), transferrin (5.7 μg/ml), and sodium selenite (7.5 ng/ml) (ITS; Sigma-Aldrich). 0.25 μg of effector protein(s) coding construct(s), 0.25 μg of firefly luciferase construct pGL4.29[luc2P/12μE5/TK/Hygro], pG5luc or pGL4.15[luc2P/GADD45G/Hygro], 10 or 20 ng of Renilla luciferase construct pGL4.83[hRlucP/EF1α/Puro] or pGL4.83[hRlucP/PGK1/Puro], and 1 μl of Lipofectamine 2000 reagent (Invitrogen) were used per well of a 48-well plate. For cotransfection of effectors 0.125 μg of pcDNA-EF1α-TCF4-A− or pcDNA-EF1α and 0.125 μg of pcDNA-SRα-ASCL1 or pcDNA-SRα were used. In siRNA cotransfection experiments, 4.8 pmol of siRNA, 0.5 μg of DNA and 1 μl of RNAiMAX reagent (Invitrogen) were used per well of a 48-well plate. Forty hours after transfection, 25 mm KCl, 1 mm dbcAMP (dibutyryl cAMP; Serva), or 1 μm phorbol 12,13-dibutyrate (PDBu; Sigma-Aldrich) was added to the culture medium for 8 h. Treatments with 50 μm bicuculline (Sigma-Aldrich) and 0.5 mm 4-AP (Tocris Bioscience) were performed 3 d after transfection for 8 h. When indicated, 5 μm nifedipine, 1 μm tetrodotoxin, 10 μm MK801, 10 μm KN-62, 10 μm H89, 10 μm Go 6983, 2 μm Go 6976, 10 μm UO126, and 10 μm TBB (all from Tocris Bioscience), 5 μm STO-609 (Sigma-Aldrich), 50 μm ddAdo (Santa Cruz Biotechnology), 50 μm APV or 30 μm KH7 (both from Cayman Chemicals) were added to the culture medium 15 min before addition of KCl or bicuculline and 4-AP.
Nucleofection and transduction of neurons.
Amaxa nucleofection of neurons was performed as described previously (Pruunsild et al., 2011) using the Amaxa Rat Neuron Nucleofector Kit and O-003 program of the Nucleofector II (Lonza). The chimeric particles of adeno-associated viruses (AAV) 1 and 2 were produced as described previously (Koppel et al., 2015) using AAV1 helper pRV1, AAV2 helper pH21, adenovirus helper pFdelta6, and Tcf4 specific or scrambled shRNAs encoding rAAV-U6-shRNA-mCherry cis-constructs. Primary neurons were transduced at 2 DIV. Nucleofected or AAV-transduced neurons were treated with 25 mm KCl for 6 h and harvested at 5 or 8 DIV, respectively.
In utero electroporations and histochemistry.
In utero electroporation of pregnant Wistar rats and histochemistry were performed as described previously (Page et al., 2017). Briefly, pCAG-GFP DNA, alone or with either pCAG-TCF4-A− or pCAG-TCF4-A−-S448A, was injected into single ventricle of an E16 embryo and targeted to medial prefrontal cortex by three electrode pulses (65 V, 100 ms). P21 male and female animals were transcardially perfused with 4% paraformaldehyde in PBS and brains were dissected and postfixed for 24 h. Coronal vibratome sections were prepared and analyzed by confocal microscopy (LSM 700; Zeiss). Images were analyzed in ImageJ (RRID:SCR_003070). The distribution of cells was quantified by binning pixel intensities perpendicular to the medial surface and the coefficient of variance (CV) of these values was used as a measure of cellular distribution, with low CV indicating a uniform distribution and high CV indicating clustering of cells.
Immunocytochemistry.
Immunocytochemistry was performed as described previously (Sepp et al., 2011) using the following antibodies: rabbit polyclonal anti-TCF4 (CeMines, 1:200), rabbit polyclonal anti-V5 (Sigma-Aldrich catalog #V8137; RRID:AB_261889, 1:2000), mouse monoclonal anti-tubulin III (Merck-Millipore catalog #MAB1637, RRID:AB_2210524, 1:2000), and goat anti mouse/rabbit IgG conjugated with Alexa Fluor 405, 488, 546, or 568 (Invitrogen, 1:2000). DNA was stained with DRAQ5 (1:5000; Biostatus Limited) or DAPI (Invitrogen). Samples were mounted in ProLong Gold reagent (Invitrogen) and analyzed by confocal microscopy (LSM Duo; Zeiss).
Protein electrophoresis and Western blotting.
Preparation of whole-cell lysates in RIPA buffer (50 mm Tris HCl pH 8.0, 150 mm NaCl, 1% NP-40, 0.5% Na-DOC, 0.1% SDS, 1 mm DTT, 1 mm PMSF, Protease Inhibitors Cocktail Complete; Roche) and Western blotting have been described previously (Sepp et al., 2012). The following antibodies were used: rabbit polyclonal anti-TCF4 (CeMines, 1:1000), rabbit polyclonal anti-Flag (Sigma-Aldrich catalog #F7425, RRID:AB_439687, 1 μg/ml), rabbit polyclonal anti-HA (Sigma-Aldrich catalog #H6908, RRID:AB_260070, 1:1000), monoclonal anti-V5 (Thermo Fisher Scientific catalog #R960-25, RRID:AB_2556564, 1:5000), mouse monoclonal anti-E2 (5E11; Icosagen catalog #A-100-100, RRID:AB_11133493, 1:5000), mouse monoclonal anti-VP16 (2GV-4; Eurogentec, 1:5000), and HRP-conjugated goat anti mouse/rabbit IgG (Pierce, 1:5000).
Luciferase and electrophoretic mobility shift assays.
Luciferase assays were performed as described previously (Sepp et al., 2011) using Passive Lysis Buffer (Promega) and the Dual-Glo Luciferase Assay (Promega). HEK-293 cell were lysed at 24 h and neurons at 48 h after transfection. Electrophoretic mobility shift assay was performed as described previously (Sepp et al., 2012) using in vitro translated proteins produced with TnT Quick Coupled Transcription/Translation System (Promega) and 32P-labeled μE5 E-box oligos.
Immunoprecipitations and in vitro kinase assay.
Coimmunoprecipitation was performed as described previously (Kazantseva et al., 2009). HEK-293FT cells overexpressing Flag-TCF4 and/or E2-PKA-Cα proteins were lysed 2 d after transfection and 1 mg of protein lysate was subjected to immunoprecipitation with 20 μl of anti-Flag M2 Affinity Gel (Sigma-Aldrich catalog #A2220, RRID:AB_10063035). ChIPs were performed as described previously (Kannike et al., 2014) using DIV 7 cultures of rat primary neurons, TCF4 (Cemines), or acetyl-histone H4 antibodies (Millipore catalog #06-866, RRID:AB_310270), and protein A-Sepharose slurry (GE Healthcare). PKA kinase assay was performed as described previously (Cox et al., 2000) with modifications. Briefly, HEK-293 transfected with pFlag-TCF4 constructs were lysed in RIPA buffer, diluted 1:9 in IP buffer (50 mm Tris-HCl pH 7.4, 150 mm NaCl, 1 mm EDTA, 1% Triton X-100, 1 mm DTT, Protease Inhibitor Cocktail Complete; Roche), and Flag-TCF4 proteins were immunoprecipitated using 12 μl of anti-Flag M2 Affinity Gel per 1 mg of protein lysate. HEK-293 transfected with E2-tagged PKA-Cα encoding construct were lysed in HO buffer [50 mm HEPES pH 7.5, 100 mm NaCl, 1% NP-40, 2 mm EDTA, 1 mm DTT, Protease Inhibitor Cocktail Complete (Roche), and phosphatase inhibitor cocktail PhosStop (Roche)] and E2-PKA-Cα was immunoprecipitated using 1 μg of mouse monoclonal anti-E2 (5E11; Icosagen catalog #A-100-100, RRID:AB_11133493) and 12 μl of Protein A Sepharose CL-4B (GE Healthcare) per 1 mg of protein lysate. Immune complexes were washed 4 times with IP or HO buffer, respectively, once with kinase buffer (50 mm HEPES, pH 7.5, 10 mm MgCl2, 1 mm EGTA, 0.014% Tween 20), and resuspended in kinase buffer. Kinase reactions were performed by mixing Flag-TCF4 and E2-PKA-Cα immune complexes with 5 μm ATP and 1.67 μm [γ-32P]ATP (6000 Ci/mmol) and incubated at 30°C for 30 min. The samples were resolved in 10% SDS-PAGE and phosphorylated proteins were visualized by autoradiography.
Reverse transcription and quantitative PCR.
RNA extraction, reverse transcription, and quantitative PCR using cDNA or ChIP gDNA as template were performed as described previously (Sepp et al., 2012). The following primers were used: Gadd45g forward: 5′-TGTCTGACCGCTGGCGTCTA-3′, reverse: 5′-AAGGTCACATTGTCAGGGTCCACA-3′; Tcf4 exons 10–11 (Sepp et al., 2011), Hprt1 and an unrelated rat chromosomal region (Kannike et al., 2014), and Gadd45g promoter F: 5′-GCTCCAGTCTGCCTGGTAACA-3′, R: 5′-GCCTCAAATCTGCCGCTTTTGT-3′.
Experimental design and statistical analyses.
Based on our previously published data from similar experiments and standards in the field, the sample sizes were selected as follows: three to five biological replicates for reporter assays and mRNA levels quantifications and seven biological replicates for in utero transfections. Biological replicates represent independent animals, primary cultures from different litters or cultures of cell lines at separate times. For each independent experiment, luciferase assays and qPCRs were performed in technical duplicates or triplicates, respectively. For most experiments, balanced design was used; however, for the analyses of TCF4 N-terminal deletion mutants and pharmacological modulators of signaling pathways, the experiments were performed in subseries that resulted in larger sample sizes for conditions used as controls. In Figure 3d, n = 7 for vector and VP16-TCF4-M217 (I−), n = 4 for VP16-TCF4-G316 and VP16-TCF4-S363, and n = 3 for VP16-TCF4-M430 and VP16-TCF4-P498. In Figure 3e, n = 8 for vector, n = 4 for VP16-TCF4-S363 and VP16-TCF4-M430, and n = 3 for VP16-TCF4-G316 and VP16-TCF4-P498. In Figure 4, a and c, in case of CMV-TCF4 n = 12 for CNTR and KCl only; n = 7 for vehicle + KCl; n = 4 for STO-609, Go 6976, ddAdo, KH7, and vehicle only; and n = 3 for KN-62, H89, Go 6983, U0126, TBB, dbcAMP, and PDBu; in case of EF1α-TCF4 n = 11 for CNTR and KCl only, n = 8 for dbcAMP; n = 7 for vehicle + KCl; n = 5 for Go 6976; n = 4 for STO-609, Go 6983, and vehicle only; and n = 3 for KN-62, H89, U0126, TBB, ddAdo, KH7, and PDBu. The normalized data from luciferase assays and qRT-PCR analyses were subjected to log transformation, mean centering, and unit variance scaling to obtain normal and comparable distributions. ChIP-qPCR data were log-transformed; the CV values of cell distribution were not transformed. Statistical significance was tested using Prism 6 software (GraphPad, RRID:SCR_002798) by unpaired two-sided t tests or by one-way or two-way between-subjects ANOVA followed by Bonferroni's or Dunnett's multiple-comparisons tests as specified in the Results section and figure legends. The degrees of freedom, p-values and effect sizes (ηp2 or r2 values for ANOVA or t tests, respectively) are reported in the Results section for each experiment. The post hoc tests and the comparisons made are specified in the figure legends. If needed, the calculated means were back transformed for graphical presentation. Error bars in the figures indicate SEM.
Figure 3.

Mapping of the TCF4 regions required for its regulation by neuronal activity. a, b, d, e, Reporter assays with primary neurons (a, b, e) or HEK293 cells (d) transfected with CMV promoter containing constructs encoding TCF4 isoforms (a), E2-tagged GAL4 fusion proteins (b), or VP16-TCF4-I− deletants (d, e) as indicated. Firefly luciferase constructs carrying μE5 E-boxes (a, d, e) or GAL4-binding sites (b) and Renilla luciferase construct with EF1α promoter were cotransfected. Neurons were left untreated or treated with KCl for 8 h. Data are presented as fold induced levels above the signals obtained from empty vector transfected untreated cells (a, b, d) or as fold induction of luciferase activity in KCl-treated neurons compared with untreated neurons (e). Shown are the means ± SEM from at least three independent experiments (see Materials and Methods for details). Statistical significance shown with asterisks is relative to the reporter levels measured from untreated or KCl-treated cells of the first data point in each chart or between the bars connected with lines. *p < 0.05; **p < 0.01; ***p < 0.001; two-way ANOVA followed by Bonferroni post hoc tests for comparisons across treatment and transfections (a, b) or one-way ANOVA followed by Dunnett's post hoc tests for comparisons with vector control (d, e). c, Western blot analysis of VP16-TCF4-I− deletants expressed in HEK-293 cell and electrophoretic mobility shift assay (EMSA) for binding of μE5 E-box by in vitro translated VP16-TCF4-I− deletant proteins. AD, Activation domain; AV MLP, adenovirus major late promoter; GBS, GAL4 binding site; min, minimal promoter; RLU, relative luciferase units; TK, thymidine kinase promoter.
Figure 4.

Identification of signaling pathways involved in the induction of TCF4-dependent transcription by neuronal activity. Shown is the effect of treatment with pharmacological inhibitors (a), coexpression of constitutively active kinases (b), treatment with kinase activators (c), and coexpression of DN kinase subunits (d) on TCF4-A−-mediated transcription. Neurons were transfected with firefly luciferase construct carrying 12 μE5 E-boxes in front of TK promoter, Renilla luciferase construct with EF1α or PGK1 promoter, and TCF4-A− vector with CMV or EF1α promoter, respectively. Constructs coding for various constitutively active and DN protein kinase subunits were cotransfected in b and d, respectively. The transfected neurons were left untreated or treated with the indicated compounds for 8 h. Data are presented as fold induced levels above the signals obtained from empty vector transfected and untreated cells in a–c and as fold induction of luciferase activity in KCl-treated neurons compared with untreated neurons in d. Shown are the mean results from at least three independent experiments (see Materials and Methods for details) performed in duplicates. Error bars indicate SEM. Figures a and c are to scale and dashed lines indicate reporter levels in nontreated and KCl-treated TCF4-A−-expressing cells. Statistical significance shown with asterisks is relative to the first data point in each chart. *p < 0.05; **p < 0.01; ***p < 0.001; one-way ANOVA followed by Dunnett's post hoc tests. RLU, Relative luciferase units. E, Western blot analysis of constitutively active and dominant negative protein kinase subunits overexpressed in HEK-293 cells.
Results
TCF4-controlled transcription is activity regulated in neurons
Previously, we have described numerous human TCF4 isoforms that differ in their N termini and internal sequences due to alternative 5′ exon usage and splicing (Sepp et al., 2011). TCF4 isoforms are briefly characterized by the following: the DNA-binding bHLH domain and the transactivation domain AD2 are present in all isoforms; the transactivation domain AD1 and the nuclear localization signal (NLS) are present only in isoforms with longer N termini; the NLS is absent from TCF4 Δ isoforms; and 4 extra amino acids upsteam of the bHLH domain differentiate “+” isoforms from the “−” isoforms (Fig. 1a). To study the functioning of TCF4 isoforms in neurons we performed μE5 (CACCTG) E-box-dependent reporter assays with selected isoforms (TCF4-A to TCF4-I) in DIV 8–9 rat primary neurons left untreated or treated with 25 mm KCl for 8 h to model neuronal activity. As shown in Figure 1a, the activation of E-box-controlled transcription by all studied TCF4 isoforms in neurons was induced by KCl-mediated depolarization of neuronal membranes (two-way ANOVA, treatment × transfection interaction F(13,84) = 8.083, p < 0.0001, ηp(interaction, treatment, transfection)2 = 0.556, 0.917, 0.744; n = 4). In depolarized neurons strong (>20-fold), transactivation was achieved with isoforms TCF4-B+, TCF4-B−, TCF4-BΔ+, and TCF4-A+, medium (10- to 20-fold) with isoforms TCF4-BΔ−, TCF4-C−, TCF4-CΔ−, TCF4-D−, TCF4-A−, and TCF4-F−, and weak (<10-fold) with isoforms TCF4-H−, TCF4-I−, and TCF4-E− (Fig. 1a).
Figure 1.

Identification of TCF4 as a neuronal activity-regulated transcription factor. The effects of membrane depolarization (a, b) and increased synaptic activity (d) on the induction of TCF4-controlled transcription in primary neurons are shown. Rat primary neurons were transfected with TCF4 isoforms encoding vectors, firefly luciferase construct carrying 12 μE5 E-boxes in front of TK promoter, and Renilla luciferase construct with EF1α promoter (a) or PGK1 promoter (b, d). For overexpression of TCF4 isoforms, vectors containing CMV (a, b, d) or EF1α (b) promoter were used. Transfected neurons were left untreated or treated with KCl (a, b) or biculline/4-AP (d) for 8 h. In b and d, tetrodotoxin, nifedipine, or APV/MK801 was used to block action potentials, VGCCs, or NMDARs, respectively. Luciferase activities were measured and data are presented as fold induced levels above the signals obtained from empty vector transfected untreated cells (a, b) or TCF4-A−-expressing untreated cells (d). Shown are the mean results from four independent experiments performed in duplicates. Error bars indicate SEM. *p < 0.05; **p < 0.01; ***p < 0.001; two-way ANOVA followed by Bonferroni post hoc tests for comparisons across treatment (a) or by Dunnett's post hoc tests for comparisons with cells treated only with KCl (b) or bicuculline/4-AP (d). AD, Activation domain; RLU, relative luciferase units. c, Western blot analysis of C-terminally V5/His-tagged TCF4-A− protein overexpressed from CMV or EF1α promoter in primary neurons left untreated or treated with KCl for 8 h.
Next, we aimed to verify that the depolarization-induced rise in TCF4-mediated transcription requires Ca2+ influx to neurons and is not caused by neuronal-activity-dependent upregulation of CMV promoter (Wheeler and Cooper, 2001), which was used above for overexpression of TCF4 isoforms. In these experiments, we used TCF4-A−, one of the dominant isoforms expressed in the nervous system (Sepp et al., 2011). We overexpressed TCF4-A− from CMV promoter or from the neuronal-activity-nonresponsive EF1α promoter and included a control condition in which depolarization was performed in the presence of nifedipine, a blocker of L-type voltage-gated Ca2+ channels (VGCCs). As shown in Figure 1, b and c, EF1α -TCF4-mediated transcription in neurons was increased by membrane depolarization similarly to CMV-TCF4-mediated transcription, although, in contrast to CMV-TCF4-A−, the levels of EF1α promoter-controlled TCF4-A− protein remained stable after KCl treatment. The induction of TCF4-controlled transcription was effectively inhibited by nifedipine in both experimental setups (one-way ANOVA, CMV transfection F(2,6) = 168.8, p < 0.0001, ηp2 = 0.983, n = 3; EF1α transfection F(2,9) = 83.98, p < 0.0001, ηp2 = 0.949, n = 4). Collectively, these data show that activation of E-box-controlled transcription by TCF4 in primary neurons is upregulated by membrane depolarization independent of isoformal specificity or expression levels of TCF4.
To demonstrate that not only depolarization of neurons but also excitatory synaptic activity results in upregulation of TCF4-controlled transcription, we stimulated action potential firing in DIV 13 primary neuron cultures by application of the GABA type A receptor antagonist bicuculline together with the K+ channel blocker 4-AP (Hardingham et al., 2001). TCF4-A−-mediated transcription was increased in response to bicuculline/4-AP treatment and this induction was inhibited in the presence of the Na+ channel blocker tetrodotoxin (which prevents action potential generation), the L-type VGCC blocker nifedipine, and/or antagonists of N-methyl-d-aspartate subtype of glutamate receptors (NMDARs) 2-amino-5-phosphonovalerate (APV) and MK801 (Fig. 1d, one-way ANOVA, F(5,18) = 24.18, p < 0.0001, ηp2 = 0.870, n = 4). This confirms that TCF4 is activity regulated in neurons and indicates that Ca2+ influx through L-type VGCCs and NMDARs is involved in triggering TCF4-controlled transcription in response to synaptic activity.
Endogenous and overexpressed TCF4 proteins are present in the nuclei of primary neurons
Next, we aimed to gain insights into the mechanisms that underlie neuronal-activity-dependent regulation of TCF4. We have shown previously that TCF4 is present in the nuclei of neurons in adult human hippocampal and cerebellar tissue sections (Sepp et al., 2011). However, in a recent study, the majority of TCF4 immunoreactivity was found in the neuronal cytoplasmic soma and dendrites of adult mouse brain (D'Rozario et al., 2016). To investigate whether the weak transactivation ability of TCF4 in primary neurons in basal conditions could be explained by the absence of TCF4 from the neuronal nuclei, we studied the distribution of TCF4 in primary neurons transfected with Tcf4-specific or scrambled siRNAs. In mock and scrambled siRNA-transfected neurons, we detected most of the TCF4-like signal in the nuclei, whereas in neurons transfected with three different Tcf4-specific siRNAs, the nuclear staining was severely reduced (Fig. 2a). Upon overexpression, the NLS-containing TCF4 isoforms localized to the nuclei, whereas NLS-lacking isoforms distributed to the nuclei and cytoplasm (Fig. 2b). These data demonstrate that, in primary neurons endogenous as well as overexpressed TCF4, proteins are present in the nuclei, corroborating our previous findings in adult human brain tissue and HEK-293 cells (Sepp et al., 2011). The differences between our results and findings by D'Rozario et al. (2016) could arise from the usage of different TCF4 antibodies. Here, we have confirmed the specificity of the endogenous nuclear signal in primary neurons, but the specificity of the endogenous cytoplasmic TCF4-like signal requires further validation, for example, by using TCF4 knock-out mice. We conclude that the regulation of TCF4 by neuronal activity cannot be attributed to its signal-dependent nuclear import.
Figure 2.

Intracellular distribution of endogenous and overexpressed V5-tagged TCF4 proteins in rat primary neurons. a, Neurons were cotransfected with Tcf4 exon 12 or 20 specific siRNAs and an EGFP construct for identification of transfected cells (marked with asterisks); mock and scrambled siRNA transfections were performed in control. Arrows indicate nonspecific staining of glial processes with TCF4 antibodies. b, Neurons were transfected with TCF4–V5 constructs. Localization pattern is indicated at the right. n, Nuclear; n + c, nuclear and cytoplasmic. In a and b, tubulin III and DNA were stained to label neurons and nuclei (encircled in a), respectively.
Region between AD2 and bHLH is critical for neuronal-activity-dependent regulation of TCF4 functions
Because all assessed TCF4 isoforms were responsive to neuronal activity, we reasoned that the effect could be dictated by the common domains AD2 and/or bHLH. To elucidate the role of AD2, we compared the activities of a deletion mutant of TCF4-B− that lacks AD2 but retains AD1 and the AD2-containing isoform TCF4-A−. In contrast to TCF4-A−, TCF4-B−ΔAD2 did not increase E-box-dependent reporter activity significantly above the vector control levels in depolarized neurons (Fig. 3a; two-way ANOVA, treatment × transfection interaction F(2,12) = 8.055, p = 0.0061, ηp(interaction, treatment, transfection)2 = 0.573, 0.811, 0.827; n = 3). To study the activation domains independently of the bHLH domain, we used heterologic constructs in which AD1 or AD2 is fused with the GAL4 DNA-binding domain and the E2-tag. GAL4-AD2-E2, but not GAL4-AD1-E2, raised reporter transcription from GAL4 binding sites carrying promoter above the control levels. However, this upregulation was not activity dependent because an ∼3.5-fold increase in reporter levels was recorded in GAL4-AD2-E2-expressing neurons in both basal and depolarized conditions (Fig. 3b; two-way ANOVA, treatment × transfection interaction F(2,18) = 3.762, p = 0.0431, ηp(interaction, treatment, transfection)2 = 0.295, 0.580, 0.980; n = 3). These results indicate that AD2 is required for transactivation in neurons, but is not sufficient for neuronal-activity-dependent regulation of TCF4 functions.
Next, we made serial deletions of TCF4 N-terminal sequences starting with the shortest isoform TCF4-I− and added SV40 LargeT NLS and the constitutively active VP16 transactivation domain to the N terminus of the deletants. VP16-TCF4-M217(I−), VP16-TCF4-G316, VP16-TCF4-S363, VP16-TCF4-M430, and VP16-TCF4-P498 (named according to the first included TCF4 aa using the TCF4-B sequence as a reference) were expressed at comparable levels in HEK-293 cells and bound μE5 E-box in vitro (Fig. 3c). In HEK-293 cells, all VP16-TCF4 deletant proteins upregulated E-box-controlled transcription, even though the transactivation decreased concomitant with increased deletions in TCF4 (Fig. 3d; one-way ANOVA, transfection F(5,22) = 1264, p < 0.0001, ηp2 = 0.997, n ≥ 3). Conversely, in rat primary neurons, overexpression of the shortest TCF4 deletant protein VP16-TCF4-P498 did not elicit induction of reporter activity in KCl-treated cells compared with untreated cells, whereas all other deletant proteins, including VP16-TCF4-M430, were able to mediate membrane depolarization-mediated induction of reporter activity (Fig. 3e; one-way ANOVA, transfection F(4,17) = 41.38, p < 0.0001, ηp2 = 0.907, n ≥ 3). These results indicate that the TCF4 protein region between aa M430 and P498, in conjunction with the bHLH domain, is critical for activity-dependent regulation of TCF4.
PKA participates in the regulation of TCF4 functions by neuronal activity
In neurons, a membrane-depolarization-induced rise in intracellular Ca2+ levels activates many protein kinases, including Ca2+-calmodulin kinases (CAMK), protein kinase A (PKA), protein kinase C (PKC), and mitogen-activated protein kinases (MAPKs) (Kotaleski and Blackwell, 2010). To elucidate which signaling pathways play a role in KCl-induced capacity of TCF4 to activate transcription in neurons, we tested whether inhibition of any of these kinases interferes with the induction. To exclude promoter-driven effects of the compounds on TCF4 levels, we performed parallel reporter experiments with TCF4-A− constructs carrying the CMV or EF1α promoter. Independent of the promoter used for TCF4-A− overexpression, KCl-induced reporter levels were reduced significantly by the CAMK2 inhibitor KN-62 and the PKA inhibitor H89, but not by the CAMKK inhibitor STO-609, the Ca2+-dependent PKC isoform inhibitor Go 6976, or an unrelated casein kinase inhibitor TBB (Fig. 4a; one-way ANOVA, CMV transfection F(10,39) = 54.48, p < 0.0001, ηp2 = 0.933, n ≥ 3; EF1α transfection F(10,39) = 10.44, p < 0.0001, ηp2 = 0.728, n ≥ 3). Inconsistent results were obtained with the pan-PKC inhibitor Go 6983 and the MEK inhibitor U0126 in the two experimental settings, possibly pointing to nonspecific effects. Next, we tested whether coexpression of constitutively active (CAT) forms of CAMK2B, CAMK4 (Sun et al., 1994), PKA (Uhler and McKnight, 1987), PKC (Soh and Weinstein, 2003), or MEK (Huang and Erikson, 1994) is able to trigger TCF4-dependent transcription in neurons grown in basal conditions. The results of this assay showed that constitutively active PKA catalytic subunit α and, to lesser extent, also PKCα CAT, was able to induce the reporter activity (Fig. 4b; one-way ANOVA, transfection F(5,12) = 75.66, p < 0.0001, ηp2 = 0.969, n = 3). We then focused on PKA and PKC and studied the effects of chemical activators and DN forms of these kinases on TCF4-dependent transcription in neurons. As demonstrated in Figure 4c, the reporter activity was upregulated by treatment with PKA activator dibutyryl cAMP (dbcAMP) and not with PKC activator PDBu independent of the promoter used for TCF4-A− overexpression (one-way ANOVA, CMV transfection F(3,18) = 33.69, p < 0.0001, ηp2 = 0.849, n ≥ 3; EF1α transfection F(3,22) = 10.45, p = 0.0002, ηp2 = 0.588, n ≥ 3). Consistently, induction of TCF4-dependent transcription by KCl-treatment was reduced by coexpression of the DN PKA-regulative subunit R1 (Clegg et al., 1987) and not by coexpression of DN PKCα (Soh and Weinstein, 2003) in neurons (Fig. 4d; one-way ANOVA, transfection F(2,6) = 14.40, p = 0.0051, ηp2 = 0.828, n = 3). We validated all CAT and DN forms of kinases by Western blot analyses performed with the appropriate tag-specific antibodies on proteins overexpressed in HEK-293 cells (Fig. 4e).
cAMP is generated by nine transmembrane adenylyl cyclases (tmACs) and one soluble AC (sAC) in mammals. To discern between the contributions of different ACs to regulation of TCF4 by membrane depolarization in neurons, we selectively inhibited tmACs and sAC with ddAdo (2′,3′-dideoxyadenosine) and KH7, respectively. Inhibition of sAC and not tmACs interfered with the induction of TCF4-dependent transcription in depolarized neurons independent of the promoter used for TCF4-A− overexpression (Fig. 4a). From the above results, we concluded that signaling via sAC and PKA is needed for membrane depolarization induced TCF4-dependent transcription in primary neurons. Nevertheless, other kinases, including CAMK2 and PKC, could also play a role.
TCF4 S448 is phosphorylated by PKA and contributes to the regulation of TCF4 by neuronal activity
We next investigated whether TCF4 is a substrate for PKA. To evaluate this, we performed in vitro immune complex kinase assays using immunoprecipitated E2-PKA-Cα from transfected HEK-293 cells as the enzyme and immunoprecipitated Flag-TCF4-M217(I−) or its deletion mutants Flag-TCF4-G316, Flag-TCF4-S363, Flag-TCF4-M430, or Flag-TCF4-P498 from transfected HEK-293 cells as the substrates in the presence of [γ-32P]ATP. All Flag-TCF4 deletant proteins were immunoprecipitated at comparable levels and all except Flag-TCF4-P498 were phosphorylated in the presence of E2-PKA-Cα (Fig. 5a). These results suggest that PKA phosphorylates TCF4 in the region between aa M430 and P498. We subsequently used NetPhos 2.0 (Blom et al., 2004) to predict phosphorylation sites in this region and identified S448 and S464 as potential PKA targets. Both of these serines are conserved in mammals, reptiles, and amphibians and S448 also in teleost fishes (Fig. 5b). To determine whether S448 and/or S464 are phosphorylated by PKA in vitro, we substituted one or both of the serines with alanines in the context of Flag-TCF4-M217(I−) and performed immune complex kinase assays as described above. Compared with the WT protein, phosphorylation by PKA was severely reduced when S448 was mutated, slightly reduced when S464 was mutated, and abolished in case of the double mutant (Fig. 5c). This indicates that, in TCF4, S448 is the major and S464 a minor site for phosphorylation by PKA. In addition, E2-PKA-Cα overexpressed in HEK-293FT cells was coimmunoprecipitated with Flag-TCF4-M217(I−) and to lesser extent also with Flag-TCF4-P498 (Fig. 5d), demonstrating that TCF4 interacts with PKA, but the region containing the phosphorylation sites is not absolutely required for the association.
Figure 5.
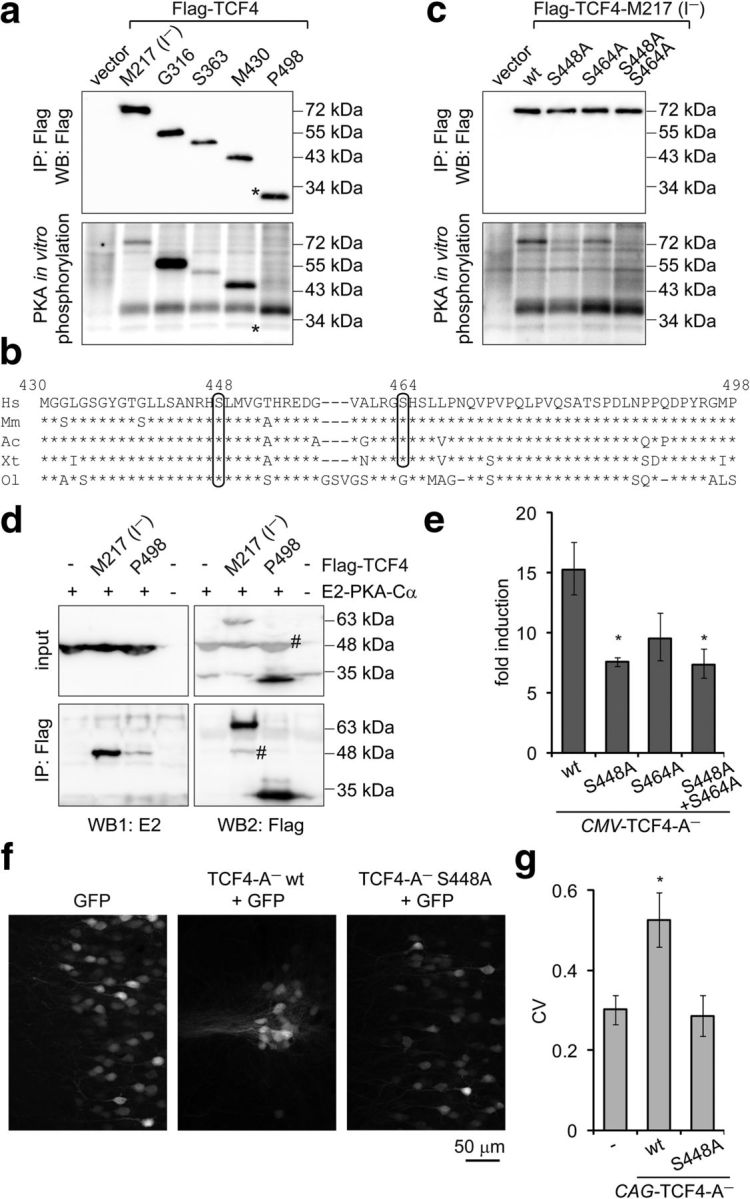
Identification and functional analysis of PKA phosphorylation sites in TCF4 in vitro and in vivo. a, c, Western blot analysis and in vitro phosphorylation assay with Flag-TCF4-I− deletant (a) and point mutant (c) proteins overexpressed in HEK-293 cells and immunoprecipitated with Flag antibodies. Kinase E2-PKA-Cα was overexpressed in HEK-293 cells and immunoprecipitated with E2 antibodies. The position of Flag-TCF4–P498 is indicated with an asterisk (*). b, Alignment of Homo sapiens (Hs) TCF4 protein region M430–P498 with the respective regions in TCF4 of Mus musculus (Mm), Arolis carolinensis (Ac), Xenopus tropicalis (Xt), and Oryzias latipes (Ol). The predicted PKA phosphorylation sites S448 and S464 are circled. d, Interactions of E2-PKA-Cα with Flag-TCF4. The proteins were overexpressed in HEK-293FT cells as indicated and coimmunoprecipitated with Flag antibodies. Western blots with E2 and Flag antibodies were performed sequentially, the number sign (#) marks bands from previous layer. In a, c, and d, a representative of at least two independent experiments with similar results is shown. e, Reporter assay with primary neurons transfected with firefly luciferase construct carrying 12 μE5 E-boxes in front of TK promoter, Renilla luciferase construct with EF1α promoter, and WT or mutant TCF4-A− encoding constructs. Data are presented as fold induction of luciferase activity in 25 mm KCl-treated neurons compared with untreated neurons. f, Distribution of layer 2/3 pyramidal neurons expressing TCF4 and/or GFP protein in the mPFC. Rat embryos were electroporated in utero at E16 and coronal sections from P21 animals were subjected to confocal microscopy. g, Quantification of the data in f. Pixel intensities perpendicular to the medial surface were binned and the CV across all bins was calculated. The higher the CV value, the more clustered is the distribution of cells. Results from four (e) or seven (f, g; except for GFP, which is n = 4) independent experiments are shown. Error bars indicate SEM. *p < 0.05; one-way ANOVA followed by Dunnett's post hoc tests for comparisons with WT (e) or control GFP (g).
To elucidate whether the identified PKA phosphorylation sites in TCF4 are involved in membrane-depolarization-mediated induction of TCF4-dependent transcription in neurons, we performed reporter assays using neurons transfected with constructs encoding WT, S448A, S464A, or S448A and S464A double mutant TCF4-A−. Compared with WT TCF4-A− overexpression, the induction of E-box-controlled luciferase activity in response to KCl treatment was reduced significantly when S448A or the double mutant TCF4-A− was expressed in neurons (Fig. 5e; one-way ANOVA, transfection F(3,12) = 4.836, p = 0.0197, ηp2 = 0.547, n = 4). This indicates that S448 is required for full induction of TCF4-dependent transcription in depolarized neurons.
In utero electroporation of TCF4 has recently been demonstrated to severely disrupt the columnar organization of pyramidal cells in layer 2/3 of the rat medial prefrontal cortex (mPFC) by inducing clustering of transfected cells in a neuronal-activity-dependent manner (Page et al., 2017). To test whether S448 is required for altering the distribution of pyramidal cells by TCF4 in vivo, we overexpressed WT or S448A TCF4-A− in mPFC of rat embryos by in utero electroporation. Compared with overexpression of GFP alone, coexpression of WT TCF4-A− resulted in the formation of cellular aggregates in layer 2/3 mPFC, an effect that was abolished by mutating S448 (Fig. 5f,g; one-way ANOVA, transfection F(2,15) = 5.567, p = 0.0155, ηp2 = 0.426, n = 7 except for GFP, which was n = 4). Altogether, the above data identify TCF4 S448 as a PKA substrate and provide evidence for the importance of S448 in the neuronal-activity-dependent regulation of TCF4 in neurons in culture as well as in vivo.
TCF4 participates in the neuronal-activity-induced transcription of Gadd45g
We next aimed to extend our studies that established TCF4 as a Ca2+-responsive transcription factor to a natural endogenous target gene. For this, we tested whether TCF4 participates in the regulation of Gadd45g, which is known to be upregulated by neuronal activity (Zhang et al., 2009). Gadd45g is a direct target of ASCL1 (Huang et al., 2010; Castro et al., 2011), a class II bHLH factor that requires an E-protein dimerization partner such as TCF4 for efficient DNA binding. We cloned the human GADD45G promoter region from −215 to +30 bp from the putative transcription start site containing two conserved ASCL1-responsive E-boxes (Huang et al., 2010), into a reporter vector (Fig. 6a) and performed luciferase assays in primary neurons. Overexpression of ASCL1 alone, but not TCF4 alone, upregulated transcription from the GADD45G promoter in neurons left untreated or treated with KCl (Fig. 6b; two-way ANOVA, treatment F(1,16) = 182.6, p < 0.0001; transfection F(3,16) = 36.79, p < 0.0001, ηp(interaction, treatment, transfection)2 = 0.303, 0.919, 0.873; n = 4). Importantly, the effect of ASCL1 on GADD45G promoter activity in depolarized neurons was augmented by TCF4 overexpression (Fig. 6b) and diminished by cotransfection of four different TCF4-specific siRNAs (Fig. 6c; two-way ANOVA, treatment × transfection interaction F(4,40) = 8.151, p < 0.0001, ηp(interaction, treatment, transfection)2 = 0.449, 0.877, 0.824; n = 5). This demonstrates that TCF4 is involved in neuronal-activity-dependent upregulation of ASCL1-mediated transcription from the GADD45G promoter in neurons.
Figure 6.

Regulation of GADD45G transcription by TCF4 in neurons. a, Alignment of the cloned GADD45G promoter region in Homo sapiens (Hs) with the respective region in Rattus norvegicus (Rn). Arrows indicate the primers used in ChIP analysis and the putative transcription start site (TSS). The two conserved E-boxes 1 and 2 are shown in boxes with the introduced mutations indicated below. b–d, Reporter assays with cultured primary neurons transfected with WT (b–d) or mutant (d) GADD45G promoter firefly luciferase constructs and Renilla luciferase construct with PGK promoter for normalization. Empty vector, EF1α-TCF4-A− and/or SRα-ASCL1 vector (b, d) and SRα-ASCL1 vector together with TCF4-specific or scrambled siRNAs (c) were cotransfected as indicated. Transfected neurons were left untreated or treated with 25 mm KCl for 8 h. e, ChIP analysis of TCF4 binding to Gadd45g promoter in neurons. Chromatin was coimmunoprecipitated with TCF4 antibodies or with beads alone. The levels of bound Gadd45g promoter or an unrelated region (URR) on chromosome 1 were quantified by qPCR. The means of three independent experiments are presented as percentages of input DNA. f–h, Quantitative RT-PCR analysis of Gadd45g (f, h) and total Tcf4 (g) mRNA expression in primary neurons nucleofected with EGFP or VP16-TCF4-I− constructs (f) or transduced with scrambled or TCF4-specific shRNAs encoding AAV vectors (g, h). Neurons were left untreated or treated with 25 mm KCl for 6 h. b–d, f–h, Data are presented relative to the normalized reporter or mRNA levels measured in untreated control cells in each experiment. The mean results from two (f), three (b, d), four (g, h), or five (c) independent experiments are shown. Error bars indicate SEM. Statistical significance shown with asterisks is relative to the reporter or mRNA levels measured from untreated or KCl-treated neurons of the first data point of each chart or between the bars connected with lines. *p < 0.05; **p < 0.01; ***p < 0.001; two-way ANOVA followed by Bonferroni post hoc tests for comparisons across TCF4 and ASCL1 transfections (b, d) or antibody use and genomic regions (e) or Dunnett's post hoc tests for comparisons with untreated or KCl-treated scrambled si/shRNA samples (c, g, h). RLU, Relative luciferase units.
To determine whether TCF4 binds and regulates the GADD45G promoter via the conserved E-boxes in neurons, we performed reporter assays with promoter constructs containing mutations in one or both of the E1 and E2 E-boxes (Fig. 6a) and ChIP experiments using TCF4 antibodies. In depolarized neurons, TCF4-A− and ASCL1 coexpression increased transcription from WT and E1 mutant promoters compared with empty vector control. In contrast, the induction was insignificant in case of E2 mutant and lost in case of double mutants (Fig. 6d; two-way ANOVA, transfection × treatment interaction F(3,16) = 6.667, p = 0.0040, ηp(interaction, transfection, treatment)2 = 0.555, 0.729, 0.780; n = 4). In ChIP experiments, we detected enrichment of TCF4 on Gadd45g promoter compared with an unrelated region on Chr1 that served as an indicator of unspecific background (Fig. 6e; two-way ANOVA; antibody × genomic region interaction F(1,8) = 9.376, p = 0.0155, ηp(interaction, antibody, genomic region)2 = 0.540, 0.447, 0.344; n = 3). The above data demonstrate that TCF4 binds directly to the Gadd45g promoter in neurons, most probably by interacting with the proximal E2 E-box.
Finally, to find out whether TCF4 regulates the endogenous Gadd45g gene, we overexpressed VP16-fused TCF4-I− or Tcf4-specific shRNAs in primary neurons using nucleofection or transduction of AAV vectors, respectively. Compared with EGFP control, VP16-TCF4-I− increased Gadd45g levels both before and after 6 h depolarization (Fig. 6f; n = 2). Silencing of Tcf4 to ∼40% or ∼10% of control levels using two different shRNAs (Fig. 6g; two-way ANOVA, transfection F(2,24) = 776.0, p < 0.0001, ηp(interaction, treatment, transfection)2 = 0.066, 0.060, 0.985; n = 3) decreased Gadd45g transcription in depolarized but not in resting neurons (Fig. 6h; two-way ANOVA, treatment × transfection interaction F(2,24) = 10.84, p = 0.0004, ηp(interaction, treatment, transfection)2 = 0.475, 0.994, 0.428; n = 3). Altogether, these results demonstrate that Gadd45g is an endogenous target of TCF4 in neurons.
TCF4 missense variations identified in SCZ patients alter the transcriptional activity of TCF4 in neurons
The rare coding variants of TCF4 detected in sporadic SCZ cases include S102C, P156T, F211L, P299S, and G428V identified in single SCZ patients and A315V identified in 14 cases and 10 controls (Hu et al., 2014; Basmanav et al., 2015). The variations are located outside of the functional domains of TCF4, suggesting that their impact on TCF4 function might be modest and dependent on cellular context. To evaluate the potential pathogenicity of these variations in the light of the novel role of TCF4 as an activity-regulated transcription factor, we studied the impact of the substitutions on TCF4-dependent reporter transcription in primary neurons. This experiment was performed with the long TCF4 isoform B− because it comprises all six sites of substitution (Fig. 7). Compared with WT TCF4-B−, the P299S variant increased E-box-dependent reporter levels in neurons in basal condition 1.3-fold and the G428V variant raised reporter levels 1.4- or 1.5-fold in untreated or KCl-treated neurons, respectively (Fig. 7; two-way ANOVA, treatment F(1,55) = 1339, p < 0.0001, transfection F(6,44) = 9.920, p < 0.0001, ηp(interaction, treatment, transfection)2 = 0.107, 0.961, 0.520; n = 5 except for P156T+KCl, n = 4). In addition, we observed a trend toward a reduction in the fold induction of E-box-dependent reporter activity in KCl-treated neurons compared with untreated neurons for four TCF4 variants: P299S (84% of WT fold induction, unpaired two-sided t test, t(8) = 3.081, p = 0.0151, r2 = 0.543, n = 5), P156T (85%, t(7) = 2.272, p = 0.0573, r2 = 0.425, n = 4), S102C (77%, t(8) = 2.036, p = 0.0762, r2 = 0.341, n = 5), and F211V (84%, t(8) = 1.998, p = 0.0808, r2 = 0.333, n = 5). These results raise the possibility that aberrant regulation of TCF4 transcriptional activity in neurons could play a role in SCZ.
Figure 7.

The effect of SCZ-linked TCF4 missense variations on the transcriptional activity of TCF4 in neurons. Reporter assay with primary neurons transfected with WT or mutant EF1α-TCF4-B− vectors, firefly luciferase construct carrying 12 μE5 E-boxes in front of TK promoter, and Renilla luciferase construct with PGK1 promoter. Transfected neurons were left untreated or treated with 25 mm KCl for 8 h. Data are presented as fold induced levels above the signals obtained from untreated cells overexpressing WT TCF4-B−. Shown are the mean results from 5 independent experiments (except for P156T+KCl, in which n = 4). Error bars indicate SEM. *p < 0.05; **p < 0.01; two-way ANOVA followed by Dunnett's post hoc tests for comparisons with untreated or KCl-treated WT. RLU, Relative luciferase units.
Discussion
Here, we report that the PTHS- and SCZ-associated bHLH transcription factor TCF4 is regulated by neuronal activity. We provide evidence that this regulation requires sAC and PKA activities. Moreover, we demonstrate that TCF4 function in neurons is modified by missense variations implicated in SCZ. Our data suggest that the role of TCF4 in the etiology of SCZ and PTHS converges with the roles of many other psychiatric and neurodevelopmental disorder-associated genes on a common pathway; that is, neuronal-activity-dependent signaling (West and Greenberg, 2011; Ebert and Greenberg, 2013; Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014).
Synaptic-activity-induced signaling modulates gene transcription by altering the function, localization, or expression of transcription factors (Lyons and West, 2011). In the current study, we showed that the activity-dependent induction of E-box-controlled transcription by TCF4 in cultured cortical and hippocampal neurons is not mediated by signal-regulated nuclear import of TCF4 and is independent of its isoformal specificity. We found that only transactivation domain AD2, and not AD1 separately, functions in activating transcription in primary neurons. This is in accordance with the requirement of AD2 for TCF4 activity in the developing prefrontal cortex in vivo (Page et al., 2017), but different from non-neural cells, in which both AD1 and AD2 are able to mediate transcriptional activation and function synergistically (Sepp et al., 2011). Although AD2 was essential for TCF4-mediated transactivation in neurons, it was not involved in neuronal-activity-dependent regulation of TCF4. Instead, we found that, for activity responsiveness, the C-terminal ∼240 aa of TCF4 are sufficient. This region contains the bHLH domain and a conserved upstream serine (S448) that was required for full depolarization-mediated TCF4 activation in primary neurons and for the ability of TCF4 to induce prefrontal layer 2/3 neuron clustering in vivo. Therefore, it appears that S448 phosphorylation is a central mechanism by which stimulus-dependent regulation of TCF4 is achieved.
Activity-dependent transcription in neurons is driven by several signaling pathways initiated predominantly by Ca2+ influx through VGCCs and/or NMDARs (Hagenston and Bading, 2011). Here, we found that Ca2+ signaling triggered by both of these Ca2+ entry routes is conveyed to TCF4. By combining pharmacological and genetic approaches, we demonstrated that the induction of TCF4-controlled transcription by membrane depolarization depends on the cAMP–PKA pathway. This result was corroborated by interaction of TCF4 with the catalytic subunit of PKA and direct phosphorylation of TCF4 at the critical S448 and S464 by PKA in vitro. Although the cAMP–PKA pathway has long been known to be involved in Ca2+-regulated synaptic function, plasticity, and the late phase of long-term potentiation, it has generally been considered to play a modulatory role in activity-dependent gene regulation in neurons (Poser and Storm, 2001). For instance, the prototypical neuronal activity-responsive transcriptional regulator CREB (cAMP response element binding protein) is primarily stimulated by the CAMK and MAPK pathways in neurons (Hagenston and Bading, 2011), whereas PKA influences it indirectly, for example, by supporting MAPK nuclear translocation and preserving nuclear localization of CREB-regulated transcriptional coactivator 1 (Poser and Storm, 2001; Ch'ng et al., 2012). In contrast, we found here that TCF4 activity in neurons depends predominantly on the cAMP-PKA pathway, although roles for other kinases, especially for CAMK2 and PKC, cannot be ruled out. The significance of cAMP-PKA signaling in TCF4 regulation was further substantiated by our finding that of the two sources of cellular cAMP, sAC activity, but not tmAC activity, was required for depolarization-induced TCF4-dependent transcription in neurons. sAC is a free Ca2+ and bicarbonate-responsive enzyme that localizes to different cellular compartments, including the nucleus, and has been shown to act as a sensor of neural activity in retinal ganglion cells (Stiles et al., 2014). Together, our results reveal a novel signaling axis in cortical neurons that links Ca2+ influx into neurons with activation of sAC, PKA and TCF4-dependent transcription.
The mechanisms by which phosphorylation of TCF4 by PKA regulates its transactivational capacity remain undefined. Previous studies have shown that phosphorylation can affect several properties of bHLH proteins, including their stability (Hong et al., 2011; Jo et al., 2011; Lin and Lee, 2012) and dimerization specificity (Sloan et al., 1996; Lluís et al., 2005). These regulatory mechanisms and differential modulation of DNA binding of E-protein homodimers and heterodimers by Ca2+-calmodulin (Hauser et al., 2008) provide the means for signal-responsive regulation of target genes by specific bHLH dimers. Here, we demonstrate that, in primary neurons, depolarization activates TCF4:ASCL1 heterodimers, which participate in the induction Gadd45g expression. Gadd45g codes for a member of growth arrest and DNA damage-inducible 45 protein family that are implicated in active DNA demethylation, adult cognitive function, and neuropsychiatric dysfunction (Sultan and Sweatt, 2013). Identification of a functional E-box and binding of TCF4 to its endogenous promoter establish Gadd45g as a direct target of TCF4. These results complement previous studies that have assigned serum response factor (SRF) and CREB as regulators of activity-dependent Gadd45g transcription (Zhang et al., 2009; Tan et al., 2012; Kuzniewska et al., 2016) and validate TCF4 as an activity-responsive transcription factor in the context of a natural target gene. Our findings are substantiated by recent studies that have implicated TCF4 in the regulation of synaptic stimuli-induced processes (Kennedy et al., 2016; Page et al., 2017). Kennedy et al. (2016) studied gene expression in the hippocampi of heterozygous TCF4 knock-out mice and found broad dysregulation of memory-related genes as well as differences in the induction of immediate early genes in response to experiential learning. These changes were accompanied by deficits in synaptic plasticity, learning, and memory. Page et al. (2017) demonstrated that TCF4-dependent transcription is involved in the activity-dependent formation of cortical minicolumns and in the regulation of early neuronal activity. Considering the latter data together with the results of the current study, we propose that TCF4 is involved in a positive feedback loop where it acts as sensor of neuronal activity that enhances spontaneous activity by regulating transcription.
We have shown previously that most of the PTHS-associated missense mutations impede the activation of E-box-controlled transcription by TCF4 in depolarized neurons (Sepp et al., 2012). Here, we found that two (G428V and P299S) of the six rare TCF4 missense variations identified in SCZ patients (Hu et al., 2014; Basmanav et al., 2015) increase TCF4 transcriptional activity in depolarized and/or resting neurons. In addition, several variants (P299S, P156T, S102C, and F211V) displayed a trend toward reduced signal-dependent induction of TCF4 activity. Given the polygenic nature of SCZ etiology, detecting variation-elicited functional changes is not expected to be fully correlated with a SCZ case diagnosis (Kim et al., 2017). Our results indicate that at least a subset of SCZ-linked TCF4 variants have functional consequences. The mechanisms underlying these effects remain uncharacterized, but might involve changes in TCF4 transactivation ability and/or response to neuronal activity. In contrast, most PTHS-associated missense mutations studied so far damage the ability of TCF4 to form stable dimers or bind DNA (Forrest et al., 2012; Sepp et al., 2012). Our finding that PTHS- and SCZ-linked missense substitutions have divergent effects on TCF4 functions is consistent with the accumulating evidence that both loss and gain of TCF4 expression or activity are related to cognitive diseases and even partial TCF4 deletions and duplications may result in disturbances (Sepp et al., 2012; Talkowski et al., 2012; Ye et al., 2012; Kharbanda et al., 2016). Even so, several aspects about the role of TCF4 in SCZ remain uncertain; for example, the causality of the rare coding variants of TCF4 in SCZ has not been established and the functional implications of the SCZ-associated common SNPs in TCF4 are unknown. It has been suggested that SCZ might be linked to increased TCF4 expression in the nervous system (Brzózka et al., 2010; Brennand et al., 2011; Guella et al., 2013; Quednow et al., 2014), although this is not supported by all studies (Umeda-Yano et al., 2014). We propose that the impaired cognitive functions seen in PTHS and SCZ patients could be caused in part by aberrant TCF4-dependent transcriptional response to neuronal activity that in turn could lead to incorrect neuronal connectivity and/or excitatory-inhibitory balance.
In summary, we have identified TCF4 as an activity-dependent transcriptional activator in neurons, providing a mechanistic rationale for the association of TCF4 with disorders affecting cognitive function and development.
Footnotes
This work was supported by Estonian Research Council (institutional research funding IUT19-18 Grants 7257 and 8844), the National R&D program “Health” (Grant AR12098), the European Union through the European Regional Development Fund (Project No. 2014-2020.4.01.15-0012) and H2020-MSCA-RISE-2016 (EU734791), the Estonian Academy of Sciences, and the Pitt Hopkins Research Foundation and Million Dollar Bike Ride Pilot Grant Program for Rare Disease Research at UPenn Orphan Disease Center (Grants MDBR-16-122-PHP and MDBR-17-127-Pitt Hopkins). B.J.M. was supported by the Lieber Institute of the National Institutes of Health (Grant R56MH104593), the National Institutes of Health (Grant R01MH110487), a NARSAD Young Investigator Award, and a Pitt Hopkins Research Foundation Award. We thank Indrek Koppel for constructive discussions and critical reading of the manuscript; Epp Väli and Maila Rähn for technical assistance; Buket Basmanav for sharing data about TCF4 mutations in szhizophrenia patients; and Hilmar Bading for pRV1, pH21, pFdelta6, and rAAV-U6-shRNA-mCherry constructs.
The authors declare no competing financial interests.
References
- Bading H, Ginty DD, Greenberg ME (1993) Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260:181–186. 10.1126/science.8097060 [DOI] [PubMed] [Google Scholar]
- Basmanav FB, et al. (2015) Investigation of the role of TCF4 rare sequence variants in schizophrenia. Am J Med Genet B Neuropsychiatr Genet 168B:354–362. 10.1002/ajmg.b.32318 [DOI] [PubMed] [Google Scholar]
- Bergqvist I, Eriksson M, Saarikettu J, Eriksson B, Corneliussen B, Grundström T, Holmberg D (2000) The basic helix-loop-helix transcription factor E2-2 is involved in T lymphocyte development. Eur J Immunol 30:2857–2863. 10.1002/1521-4141(200010)30:10%3C2857::AID-IMMU2857%3E3.0.CO%3B2-G [DOI] [PubMed] [Google Scholar]
- Blom N, Sicheritz-Pontén T, Gupta R, Gammeltoft S, Brunak S (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4:1633–1649. 10.1002/pmic.200300771 [DOI] [PubMed] [Google Scholar]
- Boehm JS, et al. (2007) Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell 129:1065–1079. 10.1016/j.cell.2007.03.052 [DOI] [PubMed] [Google Scholar]
- Brennand KJ, Simone A, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, Li Y, Mu Y, Chen G, Yu D, McCarthy S, Sebat J, Gage FH (2011) Modelling schizophrenia using human induced pluripotent stem cells. Nature 473:221–225. 10.1038/nature09915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockschmidt A, Filippi A, Charbel Issa P, Nelles M, Urbach H, Eter N, Driever W, Weber RG (2011) Neurologic and ocular phenotype in Pitt–Hopkins syndrome and a zebrafish model. Hum Genet 130:645–655. 10.1007/s00439-011-0999-4 [DOI] [PubMed] [Google Scholar]
- Brzózka MM, Radyushkin K, Wichert SP, Ehrenreich H, Rossner MJ (2010) Cognitive and sensorimotor gating impairments in transgenic mice overexpressing the schizophrenia susceptibility gene Tcf4 in the brain. Biol Psychiatry 68:33–40. 10.1016/j.biopsych.2010.03.015 [DOI] [PubMed] [Google Scholar]
- Castro DS, Martynoga B, Parras C, Ramesh V, Pacary E, Johnston C, Drechsel D, Lebel-Potter M, Garcia LG, Hunt C, Dolle D, Bithell A, Ettwiller L, Buckley N, Guillemot F (2011) A novel function of the proneural factor Ascl1 in progenitor proliferation identified by genome-wide characterization of its targets. Genes Dev 25:930–945. 10.1101/gad.627811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ES, Gigek CO, Rosenfeld JA, Diallo AB, Maussion G, Chen GG, Vaillancourt K, Lopez JP, Crapper L, Poujol R, Shaffer LG, Bourque G, Ernst C (2014) Molecular convergence of neurodevelopmental disorders. Am J Hum Genet 95:490–508. 10.1016/j.ajhg.2014.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Wu Q, Zhang Y, Lu T, Yue W, Zhang D (2016) Tcf4 controls neuronal migration of the cerebral cortex through regulation of Bmp7. Front Mol Neurosci 9:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ch'ng TH, Uzgil B, Lin P, Avliyakulov NK, O'Dell TJ, Martin KC (2012) Activity-dependent transport of the transcriptional coactivator CRTC1 from synapse to nucleus. Cell 150:207–221. 10.1016/j.cell.2012.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clegg CH, Correll LA, Cadd GG, McKnight GS (1987) Inhibition of intracellular cAMP-dependent protein kinase using mutant genes of the regulatory type I subunit. J Biol Chem 262:13111–13119. [PubMed] [Google Scholar]
- Cox ME, Deeble PD, Bissonette EA, Parsons SJ (2000) Activated 3′,5′-cyclic AMP-dependent protein kinase is sufficient to induce neuroendocrine-like differentiation of the LNCaP prostate tumor cell line. J Biol Chem 275:13812–13818. 10.1074/jbc.275.18.13812 [DOI] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics Consortium (2013) Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381:1371–1379. 10.1016/S0140-6736(12)62129-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Rozario M, Zhang T, Waddell EA, Zhang Y, Sahin C, Sharoni M, Hu T, Nayal M, Kutty K, Liebl F, Hu W, Marenda DR (2016) Type I bHLH proteins daughterless and Tcf4 restrict neurite branching and synapse formation by repressing neurexin in postmitotic neurons. Cell Rep 15:386–397. 10.1016/j.celrep.2016.03.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert DH, Greenberg ME (2013) Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493:327–337. 10.1038/nature11860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer B, Azim K, Hurtado-Chong A, Ramelli S, Fernández M, Raineteau O (2014) E-proteins orchestrate the progression of neural stem cell differentiation in the postnatal forebrain. Neural Dev 9:23. 10.1186/1749-8104-9-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flora A, Garcia JJ, Thaller C, Zoghbi HY (2007) The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proc Natl Acad Sci U S A 104:15382–15387. 10.1073/pnas.0707456104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest MP, Waite AJ, Martin-Rendon E, Blake DJ (2013) Knockdown of human TCF4 affects multiple signaling pathways involved in cell survival, epithelial to mesenchymal transition and neuronal differentiation. PLoS One 8:e73169. 10.1371/journal.pone.0073169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest MP, Hill MJ, Quantock AJ, Martin-Rendon E, Blake DJ (2014) The emerging roles of TCF4 in disease and development. Trends Mol Med 20:322–331. 10.1016/j.molmed.2014.01.010 [DOI] [PubMed] [Google Scholar]
- Forrest M, Chapman RM, Doyle AM, Tinsley CL, Waite A, Blake DJ (2012) Functional analysis of TCF4 missense mutations that cause Pitt–Hopkins syndrome. Hum Mutat 33:1676–1686. 10.1002/humu.22160 [DOI] [PubMed] [Google Scholar]
- Guella I, Sequeira A, Rollins B, Morgan L, Torri F, van Erp TG, Myers RM, Barchas JD, Schatzberg AF, Watson SJ, Akil H, Bunney WE, Potkin SG, Macciardi F, Vawter MP (2013) Analysis of miR-137 expression and rs1625579 in dorsolateral prefrontal cortex. J Psychiatr Res 47:1215–1221. 10.1016/j.jpsychires.2013.05.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenston AM, Bading H (2011) Calcium signaling in synapse-to-nucleus communication. Cold Spring Harb Perspect Biol 3:a004564. 10.1101/cshperspect.a004564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H (2001) Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci 4:261–267. 10.1038/85109 [DOI] [PubMed] [Google Scholar]
- Hauser J, Saarikettu J, Grundström T (2008) Calcium regulation of myogenesis by differential calmodulin inhibition of basic helix-loop-helix transcription factors. Mol Biol Cell 19:2509–2519. 10.1091/mbc.E07-09-0886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill MJ, Killick R, Navarrete K, Maruszak A, McLaughlin GM, Williams BP, Bray NJ (2017) Knockdown of the schizophrenia susceptibility gene TCF4 alters gene expression and proliferation of progenitor cells from the developing human neocortex. J Psychiatry Neurosci 42:181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J, Zhou J, Fu J, He T, Qin J, Wang L, Liao L, Xu J (2011) Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res 71:3980–3990. 10.1158/0008-5472.CAN-10-2914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HS, Kubish GM, Redmond TM, Turner DL, Thompson RC, Murphy GG, Uhler MD (2010) Direct transcriptional induction of Gadd45gamma by Ascl1 during neuronal differentiation. Mol Cell Neurosci 44:282–296. 10.1016/j.mcn.2010.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Erikson RL (1994) Constitutive activation of Mek1 by mutation of serine phosphorylation sites. Proc Natl Acad Sci U S A 91:8960–8963. 10.1073/pnas.91.19.8960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Zhang B, Liu W, Paciga S, He W, Lanz TA, Kleiman R, Dougherty B, Hall SK, McIntosh AM, Lawrie SM, Power A, John SL, Blackwood D, St Clair D, Brandon NJ (2014) A survey of rare coding variants in candidate genes in schizophrenia by deep sequencing. Mol Psychiatry 19:857–858. 10.1038/mp.2013.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo C, Cho SJ, Jo SA (2011) Mitogen-activated protein kinase kinase 1 (MEK1) stabilizes MyoD through direct phosphorylation at tyrosine 156 during myogenic differentiation. J Biol Chem 286:18903–18913. 10.1074/jbc.M111.225128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannike K, Sepp M, Zuccato C, Cattaneo E, Timmusk T (2014) Forkhead transcription factor FOXO3a levels are increased in Huntington disease because of overactivated positive autofeedback loop. J Biol Chem 289:32845–32857. 10.1074/jbc.M114.612424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantseva A, Sepp M, Kazantseva J, Sadam H, Pruunsild P, Timmusk T, Neuman T, Palm K (2009) N-terminally truncated BAF57 isoforms contribute to the diversity of SWI/SNF complexes in neurons. J Neurochem 109:807–818. 10.1111/j.1471-4159.2009.06005.x [DOI] [PubMed] [Google Scholar]
- Kennedy AJ, Rahn EJ, Paulukaitis BS, Savell KE, Kordasiewicz HB, Wang J, Lewis JW, Posey J, Strange SK, Guzman-Karlsson MC, Phillips SE, Decker K, Motley ST, Swayze EE, Ecker DJ, Michael TP, Day JJ, Sweatt JD (2016) Tcf4 regulates synaptic plasticity, DNA methylation, and memory function. Cell Rep 16:2666–2685. 10.1016/j.celrep.2016.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharbanda M, Kannike K, Lampe A, Berg J, Timmusk T, Sepp M (2016) Partial deletion of TCF4 in three generation family with non-syndromic intellectual disability, without features of Pitt–Hopkins syndrome. Eur J Med Genet 59:310–314. 10.1016/j.ejmg.2016.04.003 [DOI] [PubMed] [Google Scholar]
- Kim MJ, Biag J, Fass DM, Lewis MC, Zhang Q, Fleishman M, Gangwar SP, Machius M, Fromer M, Purcell SM, McCarroll SA, Rudenko G, Premont RT, Scolnick EM, Haggarty SJ (2016) Functional analysis of rare variants found in schizophrenia implicates a critical role for GIT1-PAK3 signaling in neuroplasticity. Mol Psychiatry 22:417–429. 10.1038/mp.2016.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel I, Tuvikene J, Lekk I, Timmusk T (2015) Efficient use of a translation start codon in BDNF exon I. J Neurochem 134:1015–1025. 10.1111/jnc.13124 [DOI] [PubMed] [Google Scholar]
- Kotaleski JH, Blackwell KT (2010) Modelling the molecular mechanisms of synaptic plasticity using systems biology approaches. Nat Rev Neurosci 11:239–251. 10.1038/nrn2807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzniewska B, Nader K, Dabrowski M, Kaczmarek L, Kalita K (2016) Adult deletion of SRF increases epileptogenesis and decreases activity-induced gene expression. Mol Neurobiol 53:1478–1493. 10.1007/s12035-014-9089-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau D, Bading H (2009) Synaptic activity-mediated suppression of p53 and induction of nuclear calcium-regulated neuroprotective genes promote survival through inhibition of mitochondrial permeability transition. J Neurosci 29:4420–4429. 10.1523/JNEUROSCI.0802-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Lee EH (2012) JNK1 inhibits GluR1 expression and GluR1-mediated calcium influx through phosphorylation and stabilization of Hes-1. J Neurosci 32:1826–1846. 10.1523/JNEUROSCI.3380-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lluís F, Ballestar E, Suelves M, Esteller M, Muñoz-Cánoves P (2005) E47 phosphorylation by p38 MAPK promotes MyoD/E47 association and muscle-specific gene transcription. EMBO J 24:974–984. 10.1038/sj.emboj.7600528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons MR, West AE (2011) Mechanisms of specificity in neuronal activity-regulated gene transcription. Prog Neurobiol 94:259–295. 10.1016/j.pneurobio.2011.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maduro V, Pusey BN, Cherukuri PF, Atkins P, du Souich C, Rupps R, Limbos M, Adams DR, Bhatt SS, Eydoux P, Links AE, Lehman A, Malicdan MC, Mason CE, Morimoto M, Mullikin JC, Sear A, Van Karnebeek C, Stankiewicz P, Gahl WA, Toro C, Boerkoel CF (2016) Complex translocation disrupting TCF4 and altering TCF4 isoform expression segregates as mild autosomal dominant intellectual disability. Orphanet J Rare Dis 11:62. 10.1186/s13023-016-0439-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massari ME, Murre C (2000) Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol 20:429–440. 10.1128/MCB.20.2.429-440.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page SC, Hamersky GR, Gallo RA, Rannals MD, Calcaterra NE, Campbell MN, Mayfield B, Briley A, Phan BN, Jaffe AE, Maher BJ (2017) The schizophrenia- and autism-associated gene, transcription factor 4 regulates the columnar distribution of layer 2/3 prefrontal pyramidal neurons in an activity-dependent manner. Mol Psychiatry. Advance online publication. Retrieved September 29, 2017 10.1038/mp.2017.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poser S, Storm DR (2001) Role of Ca2+-stimulated adenylyl cyclases in LTP and memory formation. Int J Dev Neurosci 19:387–394. 10.1016/S0736-5748(00)00094-0 [DOI] [PubMed] [Google Scholar]
- Pruunsild P, Sepp M, Orav E, Koppel I, Timmusk T (2011) Identification of cis-elements and transcription factors regulating neuronal-activity-dependent transcription of human BDNF gene. J Neurosci 31:3295–3308. 10.1523/JNEUROSCI.4540-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quednow BB, et al. (2012) Schizophrenia risk polymorphisms in the TCF4 gene interact with smoking in the modulation of auditory sensory gating. Proc Natl Acad Sci U S A 109:6271–6276. 10.1073/pnas.1118051109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quednow BB, Brzózka MM, Rossner MJ (2014) Transcription factor 4 (TCF4) and schizophrenia: integrating the animal and the human perspective. Cell Mol Life Sci 71:2815–2835. 10.1007/s00018-013-1553-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rannals MD, Hamersky GR, Page SC, Campbell MN, Briley A, Gallo RA, Phan BN, Hyde TM, Kleinman JE, Shin JH, Jaffe AE, Weinberger DR, Maher BJ (2016) Psychiatric risk gene transcription factor 4 regulates intrinsic excitability of prefrontal neurons via repression of SCN10a and KCNQ1. Neuron 90:43–55. 10.1016/j.neuron.2016.02.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014) Biological insights from 108 schizophrenia-associated genetic loci. Nature 511:421–427. 10.1038/nature13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Edelkraut U, Daniel G, Hoffmann A, Spengler D (2014) Zac1 regulates cell cycle arrest in neuronal progenitors via Tcf4. Mol Cell Biol 34:1020–1030. 10.1128/MCB.01195-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepp M, Kannike K, Eesmaa A, Urb M, Timmusk T (2011) Functional diversity of human basic helix-loop-helix transcription factor TCF4 isoforms generated by alternative 5′ exon usage and splicing. PLoS One 6:e22138. 10.1371/journal.pone.0022138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepp M, Pruunsild P, Timmusk T (2012) Pitt–Hopkins syndrome-associated mutations in TCF4 lead to variable impairment of the transcription factor function ranging from hypomorphic to dominant-negative effects. Hum Mol Genet 21:2873–2888. 10.1093/hmg/dds112 [DOI] [PubMed] [Google Scholar]
- Sloan SR, Shen CP, McCarrick-Walmsley R, Kadesch T (1996) Phosphorylation of E47 as a potential determinant of B-cell-specific activity. Mol Cell Biol 16:6900–6908. 10.1128/MCB.16.12.6900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomnicki LP, Malinowska A, Kistowski M, Palusinski A, Zheng JJ, Sepp M, Timmusk T, Dadlez M, Hetman M (2016) Nucleolar enrichment of brain proteins with critical roles in human neurodevelopment. Mol Cell Proteomics 15:2055–2075. 10.1074/mcp.M115.051920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh JW, Weinstein IB (2003) Roles of specific isoforms of protein kinase C in the transcriptional control of cyclin D1 and related genes. J Biol Chem 278:34709–34716. 10.1074/jbc.M302016200 [DOI] [PubMed] [Google Scholar]
- Stiles TL, Kapiloff MS, Goldberg JL (2014) The role of soluble adenylyl cyclase in neurite outgrowth. Biochim Biophys Acta 1842:2561–2568. 10.1016/j.bbadis.2014.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultan FA, Sweatt JD (2013) The role of the Gadd45 family in the nervous system: a focus on neurodevelopment, neuronal injury, and cognitive neuroepigenetics. Adv Exp Med Biol 793:81–119. 10.1007/978-1-4614-8289-5_6 [DOI] [PubMed] [Google Scholar]
- Sun P, Enslen H, Myung PS, Maurer RA (1994) Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev 8:2527–2539. 10.1101/gad.8.21.2527 [DOI] [PubMed] [Google Scholar]
- Sweatt JD. (2013) Pitt–Hopkins Syndrome: intellectual disability due to loss of TCF4-regulated gene transcription. Exp Mol Med 45:e21. 10.1038/emm.2013.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkowski ME, et al. (2012) Sequencing chromosomal abnormalities reveals neurodevelopmental loci that confer risk across diagnostic boundaries. Cell 149:525–537. 10.1016/j.cell.2012.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamberg L, Sepp M, Timmusk T, Palgi M (2015) Introducing Pitt–Hopkins syndrome-associated mutations of TCF4 to Drosophila daughterless. Biol Open 4:1762–1771. 10.1242/bio.014696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan YW, Zhang SJ, Hoffmann T, Bading H (2012) Increasing levels of wild-type CREB up-regulates several activity-regulated inhibitor of death (AID) genes and promotes neuronal survival. BMC Neurosci 13:48. 10.1186/1471-2202-13-48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhler MD, McKnight GS (1987) Expression of cDNAs for two isoforms of the catalytic subunit of cAMP-dependent protein kinase. J Biol Chem 262:15202–15207. [PubMed] [Google Scholar]
- Umeda-Yano S, Hashimoto R, Yamamori H, Weickert CS, Yasuda Y, Ohi K, Fujimoto M, Ito A, Takeda M (2014) Expression analysis of the genes identified in GWAS of the postmortem brain tissues from patients with schizophrenia. Neurosci Lett 568:12–16. 10.1016/j.neulet.2014.03.031 [DOI] [PubMed] [Google Scholar]
- West AE, Greenberg ME (2011) Neuronal activity-regulated gene transcription in synapse development and cognitive function. Cold Spring Harb Perspect Biol 3:pii:a005744. 10.1101/cshperspect.a005744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DG, Cooper E (2001) Depolarization strongly induces human cytomegalovirus major immediate-early promoter/enhancer activity in neurons. J Biol Chem 276:31978–31985. 10.1074/jbc.M103667200 [DOI] [PubMed] [Google Scholar]
- Ye T, Lipska BK, Tao R, Hyde TM, Wang L, Li C, Choi KH, Straub RE, Kleinman JE, Weinberger DR (2012) Analysis of copy number variations in brain DNA from patients with schizophrenia and other psychiatric disorders. Biol Psychiatry 72:651–654. 10.1016/j.biopsych.2012.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, Zou M, Lu L, Lau D, Ditzel DA, Delucinge-Vivier C, Aso Y, Descombes P, Bading H (2009) Nuclear calcium signaling controls expression of a large gene pool: identification of a gene program for acquired neuroprotection induced by synaptic activity. PLoS Genet 5:e1000604. 10.1371/journal.pgen.1000604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Cheng P, Weintraub H (1996) B-lymphocyte development is regulated by the combined dosage of three basic helix-loop-helix genes, E2A, E2-2, and HEB. Mol Cell Biol 16:2898–2905. 10.1128/MCB.16.6.2898 [DOI] [PMC free article] [PubMed] [Google Scholar]