

A 12-year-old Caucasian boy with X-linked chronic granulomatous disease (CGD) was evaluated for a matched unrelated hematopoietic progenitor cell transplantation, currently the standard definitive treatment of CGD. Large deletions on the X chromosome affect the CYBB/gp91phox gene, encoding 1 of the 4 major subunits of NADPH oxidase, and are a common cause of this form of CGD. Because of these defects, phagocytes cannot make hydrogen peroxide and other chemicals needed to kill certain bacteria and molds and the patients are prone to infections and the formation of inflammatory granuloma. The CYBB/gp91phox gene is located adjacent to another gene, XK, and the deletion of both of these contiguous genes is not uncommon.1 The XK gene encodes the Kx protein (Kx blood group system, ISBT number 19) required for proper expression of the Kell proteins (Kell blood group system, ISBT number 6).
In all peripheral blood films (Figure), a fraction of the red blood cells (RBC) exhibited membrane appendages that are characteristic for acanthocytes. These appendages, irregularly spaced over the surface, can number more than 10, look club-like with broad bases and blunted ends, rather than pointed tips, vaguely reminiscent of amoebic pseudopods. Typically, only 5% to 50% possess these appendages, representing older RBCs having experienced more membrane deforming shear forces and accumulated appendages.
Figure 1.
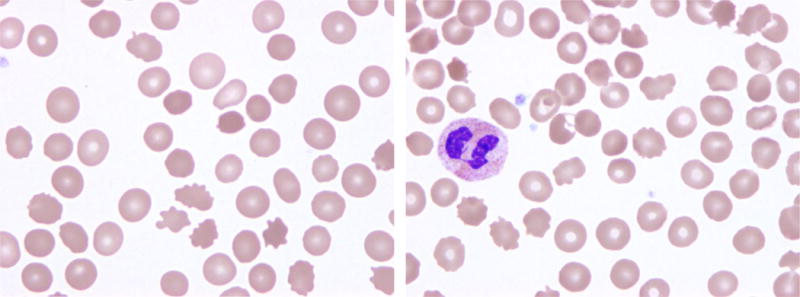
Acanthocytes (from the Greek word acantha, meaning ‘thorn’; “spur cells”) should be distinguised from echinocytes (Greek achinos, ‘sea urchin’; “burr cells”).2 The echinocytes have shorter, more numerous, and more evenly spaced appendages with sharper ends.3 Echinocytes are found in common clinical conditions, such as liver2 and kidney disease, and frequently as a technical artifact of sample storage if preparation of the blood smear is delayed.3 In contrast, typical acanthocytes are only found in the rare diseases CGD and McLeod syndrome, caused by the lack of a structural protein, or sometimes in apoliprotein B deficiency, caused by increased membrane cholesterol concentration.
In serology, K, Kpa, and Kpb, tested by macroscopic agglutination were negative, as were the Km antigen (a compound epitope formed by Kell and Kx protein interaction) and the Kx antigen. Microscopically the k antigen was observed, typical for low Kell antigen expression in the absence of the Kx protein, all diagnostic for the McLeod phenotype. A defective or deleted XK gene, aside from causing the McLeod phenotype, can cause clinical disease, termed McLeod syndrome, which patients begin to manifest in their 50s as peripheral neuropathy.4 Of note, the K0 phenotye (read “K-null”), distinct from the McLeod phenotype, is caused by a homozygous or compound heterozygous defect of the KEL gene resulting in the absence of all Kell protein. RBCs of the K0 phenotype carry the Kx antigen; are of normal morphology (acanthocytes are not expected to occur); and not suitable for transfusion to patients with McLeod phenotype, which is exceedingly rare in patients and even rarer in healthy blood donors. We collected 6 autologous red cell units over 1 year. Fortunately, the transplantation was largely uneventful; he never required red cell transfusion and is doing well during the 3 ¾ years’ follow up.
Acknowledgments
This work was supported by the Intramural Research Program (project ID Z99 CL999999) of the NIH Clinical Center.
Footnotes
Conflict of interest disclosure: The authors declared having no competing financial interest relevant to this article.
Statement of Disclaimer: The opinions expressed in this review are those of the authors and do not necessarily represent the views or policies of the National Institutes of Health, the Department of Health and Human Services, or the U.S. Federal Government.
References
- 1.Watkins CE, Litchfield J, Song E, Jaishankar GB, Misra N, Holla N, Duffourc M, Krishnaswamy G. Chronic granulomatous disease, the McLeod phenotype and the contiguous gene deletion syndrome-a review. Clin Mol Allergy. 2011;9:13. doi: 10.1186/1476-7961-9-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Martin M, Lesesve JF. Spur cell anemia associated with primary biliary cirrhosis. Transfusion. 2013;53:260. doi: 10.1111/j.1537-2995.2012.03733.x. [DOI] [PubMed] [Google Scholar]
- 3.Foglia A. The acanthocyte-echinocyte differential: the example of chorea-acanthocytosis. Swiss Med Wkly. 2010;140:w13039. doi: 10.4414/smw.2010.13039. [DOI] [PubMed] [Google Scholar]
- 4.Jung HH, Danek A, Walker RH. Neuroacanthocytosis syndromes. Orphanet J Rare Dis. 2011;6:68. doi: 10.1186/1750-1172-6-68. [DOI] [PMC free article] [PubMed] [Google Scholar]